

Abstract
The adaptor appendage domains are believed to act as binding platforms for coated vesicle accessory proteins. Using glutathione S-transferase pulldowns from pig brain cytosol, we find three proteins that can bind to the appendage domains of both the AP-1 γ subunit and the GGAs: γ-synergin and two novel proteins, p56 and p200. p56 elicited better antibodies than p200 and was generally more tractable. Although p56 and γ-synergin bind to both GGA and γ appendages in vitro, immunofluorescence labeling of nocodazole-treated cells shows that p56 colocalizes with GGAs on TGN46-positive membranes, whereas γ-synergin colocalizes with AP-1 primarily on a different membrane compartment. Furthermore, in AP-1–deficient cells, p56 remains membrane-associated whereas γ-synergin becomes cytosolic. Thus, p56 and γ-synergin show very strong preferences for GGAs and AP-1, respectively, in vivo. However, the GGA and γ appendages share the same fold as determined by x-ray crystallography, and mutagenesis reveals that the same amino acids contribute to their binding sites. By overexpressing wild-type GGA and γ appendage domains in cells, we can drive p56 and γ-synergin, respectively, into the cytosol, suggesting a possible mechanism for selectively disrupting the two pathways.
INTRODUCTION
Adaptor proteins (APs) are vesicle coat components that were first identified as polypeptides enriched in purified clathrin-coated vesicles. Two types of adaptor complexes were originally described, AP-1 and AP-2, which localize to the trans-Golgi network (TGN) and/or endosomal compartments and to the plasma membrane, respectively. More recently two other types of adaptor complexes have been identified, AP-3 and AP-4. All four complexes are heterotetramers, consisting of two large subunits, γ/α/δ/ε and β1/β2/β3/β4, a medium-sized or μ subunit, and a small or σ subunit (Robinson and Bonifacino, 2001). The COOH-terminal domains of the two large subunits project from the complexes like appendages or ears and are thought to act as binding platforms for accessory proteins. These domains have been most extensively studied in the AP-2 complex, where a number of proteins have been shown to bind either directly or indirectly to the appendage domain of the α subunit. Several of these proteins in turn make connections with other molecules, including components of the actin cytoskeleton, lipids, cargo proteins, and proteins involved in signaling pathways (Slepnev and De Camilli, 2000). Some of the binding partners for the α appendage are also able to bind to the β2 appendage (Owen et al., 2000). The first binding partner to be identified for the appendage domain of the AP-1 γ subunit was the EH domain-containing protein γ-synergin (Page et al., 1999). Glutathione S-transferase (GST) pulldowns indicate that the γ appendage can bind to additional proteins in vitro, although it is less clear whether these proteins are associated with AP-1 in vivo (Hirst et al., 2000; Kent et al., 2002).
The structures of the α, β2, and γ appendages have all been solved by x-ray crystallography (Owen et al., 1999; Traub et al., 1999; Owen et al., 2000; Kent et al., 2002). Together with the recently determined structure of the AP-2 core (Collins et al., 2002), these structures provide a detailed model of how adaptor complexes are assembled at the molecular level. Despite little sequence homology, the α and β2 appendages share a similar overall structure consisting of two subdomains: an NH2-terminal β sandwich subdomain, which acts as a presentation scaffold for a COOH-terminal “platform” subdomain. The COOH-terminal platform contains a hydrophobic pocket that recognizes DΦF/W sequences in the various appendage domain binding partners. The γ appendage has virtually no sequence homology to either the α or β appendages, and it is approximately half their size. However, it has an almost identical fold to the NH2-terminal subdomain of both the α and β appendages. This is particularly surprising because functionally the γ appendage is more similar to the α and β COOH-terminal subdomain, which binds to the accessory proteins. Mutagenesis studies have revealed an entirely different mechanism for accessory protein binding by the γ appendage, involving residues situated at the junction between the two sheets of the β sandwich (Kent et al., 2002).
Recently, another family of adaptor proteins has been identified, the GGAs. There are three GGA genes in mammals, encoding proteins that colocalize with each other, and two GGA genes in yeast, encoding proteins that seem to be functionally interchangeable (Boman et al., 2000; Dell'Angelica et al., 2000; Hirst et al., 2000). Studies in both mammalian cells and yeast indicate that the GGAs facilitate the trafficking of cargo receptors, including (in mammalian cells) the mannose 6-phosphate receptors (MPRs) for lysosomal enzymes, from the TGN to an endosomal compartment (Robinson and Bonifacino, 2001). AP-1 has also been implicated in this pathway, raising the question of why the cell needs both types of adaptors. Several recent studies suggest that AP-1 may in fact be more important in retrieving proteins such as the MPRs from an endosomal compartment for recycling back to the TGN (Meyer et al., 2000, 2001; Valdivia et al., 2002).
Unlike the AP complexes, the GGAs seem to be monomeric. However, they are able to incorporate all of the functions of an adaptor complex into a single polypeptide, making use of distinct domains involved in cargo selection, membrane association, and clathrin binding (Puertollano et al., 2001a,b). At the COOH-terminal end is a domain related to the appendage domain of the γ subunit of AP-1. The homology between the GGA and γ appendage domains suggests that the two may recruit some of the same partners, and indeed GST pulldowns by using pig brain cytosol as a source of potential binding partners suggest that the GGA appendages bind to a subset of those proteins that bind to the γ appendage (Hirst et al., 2000). Western blotting indicates that one of these proteins is γ-synergin; however, the relevance of this interaction in vivo is still not clear (Hirst et al., 2000; Takatsu et al., 2000). In yeast, deleting the COOH-terminal appendage domain impairs GGA function, although such proteins are still partially functional (Hirst et al., 2001; Mullins and Bonifacino, 2001). Chimeric constructs, in which the GGA appendage has been replaced by the γ appendage, are fully functional, further supporting the view that the two domains may recruit some of the same proteins (Hirst et al., 2001).
In our previous study, we were able to identify only three of the bands that came down with the γ appendage and none that came down with the GGA appendages. We have now identified the remaining proteins. These include two different isoforms of γ-synergin, presumably related to each other by alternative splicing, and two novel proteins, p200 and p56. Here, we present an initial characterization of p56 and also address the question of how accurately the GST pulldowns reflect interactions that occur in vivo. In addition, we present the structure of the GGA appendage domain and use mutagenesis to identify amino acids that are essential in both the γ and GGA appendages for binding to accessory proteins.
MATERIALS AND METHODS
Constructs and Pulldowns
Most molecular biology techniques were carried out as described by Sambrook et al. (1989). Reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. All of the polymerase chain reaction (PCR) products made for this study were sequenced to ensure that no errors had been introduced. The GST-coupled γ appendage, GGA1 appendage, and GGA2 appendage constructs and their use in GST pulldowns have been described previously (Hirst et al., 2000). Gels of the pulldowns were stained with Coomassie Blue and the bands excised and analyzed using both matrix-assisted laser desorption ionization/time of flight (MALDI TOF) and nanoelectrospray mass spectrometry by MDS Protana (Odense, Denmark). p200 was able to be identified by MALDI TOF alone, whereas γ-synergin and p56 were only able to be identified by nanoelectrospray. Several point mutations (A563D, V564D, V570E, and L572E) were introduced into the GGA1 appendage by PCR, and these sequences were inserted into pGEX-4T for pulldowns as described above. Green fluorescent protein (GFP)-tagged appendage constructs were made using the vectors pEGFPC1 (for GGA1 and GGA2) and pEGFPC2 (for γ) (BD Biosciences Clontech, Palo Alto, CA).
p56 was originally identified as hypothetical protein FLJ11088. Nine extended sequence tags (ESTs) with sequence overlapping that of FLJ11088 were obtained from the IMAGE Consortium (MRC Geneservice Hinxton, United Kingdom) and sequenced to assemble a full-length contig: IMAGE clones 1307406, 1736582, 1866579, 2256993, 3062538, 3231483, 2604610, 1751619, and 743755. Because none of these clones had inserts encoding the full-length protein, for expression studies inserts from clones 1866579 and 1751619 were ligated together by using a naturally occurring BamHI site. The full-length sequence of p56 is available from GenBank/EMBL/DDBJ database under accession no. AY289196. For expression in mammalian cells, a myc tag was inserted into the NH2-terminal end by PCR and the construct was cloned into pcDNA3. For expression in Escherichia coli, a 6xHis tag was inserted, using either pQE30 (for the experiment shown in Figure 2d) or pTrcHisA (for raising antibodies against p56). For mapping the domains of p56, PCR was used to amplify the appropriate regions and the products were ligated into pTrcHisA. His-tagged constructs were purified using Ni-nitrilotriacetic acid agarose beads (QIAGEN, Valencia, CA). Cross-linking was performed by incubating 10 μg of fusion protein in 50 μl of phosphate-buffered saline containing 2 mM dithiobis(succinimidyl propionate) (DSP) for 1.5 h. The sample was then boiled in nonreducing sample buffer and subjected to SDS-PAGE followed by Western blotting.
Figure 2.

Biochemical characterization of p56. (a) Genomic structure of p56. The mRNA is alternatively spliced in at least four different ways. The most common isoform of p56, which was used for all of the expression studies in tissue culture cells, consists of exons 1, 3, 4, and 7–16 (dashed line, top). There is an inframe stop codon upstream from the initiator methionine in exon 3. A shorter isoform, which seems to be expressed mainly in testis (based on analysis of the EST database), consists of exons 5, 6, and 7–16 (gray line, top). In-frame stop codons are found in exon 5. There is also an entry in the database from ovary (BM545687) encoding a larger protein, which consists of exons 1, 2, 3, 4, 6, and 7 onwards (solid line, bottom). This protein has the same putative start as the common isoform, but includes an extra 5′UTR exon and an extra exon in the coding region. This exon (exon 6) includes the NH2-terminal sequence of the shorter isoform. In addition, there are entries in the database from both placenta (AL544393) and cervix (BI086262), consisting of exon 1 and exon 7 onwards (dotted line, bottom). The open reading frame is maintained in exon 1, with no start codon, suggesting that the initiator methionine may be in another exon further upstream. Introns range in size from 94bp to 67kb. (b) Amino acid sequence of the common isoform of human p56. While our manuscript was under review, full-length sequences of mouse p56 were deposited in the database under accession numbers BAB25348 (common isoform) and AAH28682 (testis isoform). (c) p56 is predicted to form coiled coils. The p56 sequence was analyzed using the COILS program. The x-axis shows the amino acid number; the y-axis shows the probability of coiled coil formation. The four domains used for the mapping experiment in e are indicated. (d) p56 binds directly to the γ and GGA appendages. Full-length p56 was expressed in E. coli as a His-tagged protein and blots were overlaid with either GST alone, GST-γ appendage, GST-GGA1 appendage, or GST-GGA2 appendage. The major band runs at the expected position, the lower band is presumably a breakdown product, and the higher band (asterisk) may be an SDS-resistant homodimer. (e) Mapping of the appendage binding domain. The four domains indicated in b were expressed as His/Xpress-tagged proteins, purified, and subjected to SDS-PAGE and Western blotting. Domain 1 contains amino acids 1–119, domain 2 contains amino acids 126–214, domain 3 contains amino acids 210–413, and domain 3 + 4 contains amino acids 210–441. Blots were probed either with anti-Xpress, to show the positions of the proteins, or with the GST-GGA1 appendage construct. The domain 3 + 4 construct was expressed at lower levels than the other constructs. Domain 1 binds to the GGA appendage. Note the higher molecular mass band in the lane containing domain 3 (asterisk), which may be an SDS-resistant homodimer. (f) Peptide competition for binding. A 15-residue peptide derived from the NH2-terminal domain of the common isoform of p56, DDDDFGGFEAAETFD, was added to the incubation mixtures. Top, a blot of domain 1 was probed with the GST-GGA1 appendage construct with increasing concentrations of peptide. Bottom, the GST-GGA1 appendage construct was used to pull down proteins from pig brain cytosol with increasing concentrations of peptide, and a Western blot of the pulldowns was probed with anti-p56. In both cases, competition is essentially complete at 0.5 mM peptide. (g) Cross-linking of domain 3. Domain 3 was run on a nonreducing gel, either with or without first treating with the cross-linker DSP, and the Western blot was probed with anti-Xpress. The anomalous mobility of the noncross-linked band is presumably due to the gel system. The band in the cross-linked lane is approximately twice the apparent molecular mass of the band in the noncross-linked lane, suggesting that it has been cross-linked into a homodimer. (h) Model of p56. The protein is predicted to form a coiled coil homodimer by using domains 2 and 3. The NH2-terminal domain (domain 1) interacts with the GGA appendage domains. The COOH-terminal domain (domain 4), which is the most conserved part of the protein, may interact with another binding partner(s).
Antibodies
Antibodies were raised in rabbits against the two novel proteins identified in this study, p200 and p56. For the p200 antibodies, clone KIAA1414 (generously provided by the Kazusa DNA Research Institute, Chiba, Japan) was used as a template to amplify the coding sequence for amino acids 946-1217 by PCR, and the product was ligated into pGEX4T-1 (Amersham Biosciences, Piscataway, NJ). The resulting GST fusion protein was soluble and was purified as specified by the manufacturer. Rabbits were immunized with the GST-p200 construct and with the His-p56 construct described above, and the antisera were affinity purified as described previously (Page et al., 1999).
Western blotting was performed using the above-mentioned antibodies, as well as antibodies against γ-synergin (Page et al., 1999) and the Xpress epitope (Invitrogen, Carlsbad, CA), followed by 125I-protein A. Ligand blotting was performed using either GST alone or one of the GST constructs, followed by anti-GST and 125I-protein A (Amersham Biosciences) (Page et al., 1999). For peptide competition experiments, a 15-residue peptide was synthesized by Sigma Genosys (The Woodlands, TX) and added to the incubation mixture in the appropriate buffer at the indicated concentrations. Immunofluorescence was performed on either methanol-acetone fixed cells or paraformaldehyde-fixed cells as described previously, by using anti-p56 (this study), anti-GGA1 (Hirst et al., 2000), anti-γ-synergin (Page et al., 1999), anti-FLAG (Sigma-Aldrich), anti-myc monoclonal antibody 9E10 (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-γ-adaptin (Seaman et al., 1996), and mouse monoclonal anti-γ-adaptin monoclonal antibody 100/3 (Sigma-Aldrich). Secondary antibodies were purchased from Molecular Probes (Eugene, OR). Cells were viewed using an Axiophot fluorescence microscope (Carl Zeiss, Jena, Germany) equipped with a charge-coupled device camera (Princeton Scientific Instruments, Monmouth Junction, NJ). Photographs were recorded using IP Labs software and then moved into Adobe Photoshop.
Cells
COS cells were used for most immunofluorescence experiments. For some experiments, they were transiently transfected with myc-tagged p56 by using FuGENE 6 (Roche Diagnostics, Mannheim, Germany) and fixed the following day. COS cells were also transiently transfected with the GFP-appendage constructs described above. For some experiments, normal rat kidney cells stably transfected with FLAG-tagged GGA2 were used (Hirst et al., 2000). To investigate the distribution of proteins in AP-1–deficient cells, fibroblasts from a μ1A knockout mouse were used, and the same cell line stably transfected with wild-type μ1A was used as a control (Meyer et al., 2000). To examine the distribution of various proteins in microtubule-disrupted cells, the cells were treated with 20 μg/ml nocodazole for 2 h at 37°C before fixation. To investigate the distribution of p56 after brefeldin A (BFA) treatment, cells were incubated with 5 μg/ml BFA for 2 min and then fixed immediately.
Crystallization of the GGA1 Appendage
A cDNA fragment encoding residues 494–639 of human GGA1 was cloned by standard PCR procedures and expressed in E. coli with an NH2-terminal 6xHis tag. The protein was purified by Ni-nitrilotriacetic acid affinity chromatography, followed by gel filtration in 5 mM HEPES (pH 7.5), 100 mM NaCl. The protein was concentrated to 37 mg/ml and crystallized by sitting drop vapor diffusion in 100 mM sodium citrate (pH 5.6), 1.7 M ammonium sulfate. Crystals grew to maximum dimensions of 1 × 0.8 × 0.25 mm within 24 h and belong to space group P3221 with unit cell dimensions a = 65.4 Å, b = 65.4 Å, c = 142.7 Å, α = β = 90°, and γ = 120°. In our crystals, we observe an extensive interface between two separate GGA1 proteins in the asymmetric unit; however, we do not see any evidence for dimerization in solution by using either gel filtration or dynamic light scattering, suggesting that this is not an in vivo natural dimeric interaction.
Data Collection and Structure Determination
Crystals were mounted in mother liquor containing 20% (vol/vol) glycerol, and data collected at 100 K by using a Rigaku rotating anode x-ray source fitted with a MAR345 image plate detector. A data set was collected to 2.3-Å resolution, integrated with MOSFLM (Leslie, 1992), and scaled using CCP4 programs (Dodson et al., 1997). The statistics are given in Table 1. Initial phases were determined by molecular replacement by using the γ ear as a trial model (PDB ID 1GYU; Kent et al., 2002). The position of the first molecule in the asymmetric unit was determined using Amore (r = 26.1; R-factor = 54.0). The position of the first molecule was then fixed and initial rotation function solutions used to search for the refined rotation and translation parameters of the second chain. The final solution, with two molecules in the asymmetric unit, had an r value of 38.3 and an R-factor of 49.4 and produced a readily interpretable electron density map after rigid body refinement in REFMAC5 (Murshudov et al., 1997), clearly showing regions where the map differed from the input model. The model was rebuilt in O (Jones et al., 1991) and refined using REFMAC5. Simulated annealing omit maps were routinely calculated in central nervous system (Brunger et al., 1998) to minimize the introduction of model bias. The model was refined to an Rcryst of 0.230 and Rfree of 0.305, after which translational liberational shear refinement was implemented in REFMAC5 (Winn et al., 2001), treating the two chains as separate rigid groups. This resulted in a reduction of Rcryst and Rfree to 0.219 and 0.279, respectively. The final model consists of two GGA1 appendage chains, A489-A639 (including five residues of the N-terminal 6xHis tag), B495-B639, and 175 water molecules. One residue (S499 of chain A) falls in the disallowed region of the Ramachandran plot. This residue has good electron density (average atomic B-factor = 17.3 Å2), and the distorted main-chain seems to be caused by a close crystal contact.
Table 1.
Statistics of data collection and structure refinementa
| Data collection | |
| Resolution (Å) | 57.0-2.3 (2.36-2.30) |
| Rmergeb | 0.053 (0.306) |
| Rmeasc | 0.058 (0.306) |
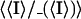
|
23.7 (5.9) |
| Completeness (%) | 99.0 (99.0) |
| Multiplicity | 5.6 (5.7) |
| Wilson plot B (Å2) | 45 |
| Refinement | |
| Resolution | 15-2.3 (2.36-2.30) |
| Rcryst (Rfree)d | 0.219 (0.279) |
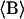 (Å2) (Å2)
|
23.1 |
| No. reflections (No. in Rfree) | 15310 (813) |
| No. atoms (No. water atoms) | 2499 (175) |
| Rmsd bond lengths (Å) | 0.019 |
| Rmsd bond angles (°) | 1.72 |
| Ramachandran plot (%)e | |
| Most favoured region | 89.0 |
| Additionally allowed | 10.6 |
| Disallowed (residue name) | 0.4 (Ser-A499) |
Unless otherwise stated, values in parentheses apply to the high-resolution shell
Rmerge = _i | Ih-Ihi | / _iIh where Ih is the mean intensity of reflection h
Rmeas = _ · (n/n-1)_i | Ih-Ihi | / _iIh, the multiplicity weighted Rmerge (Diederichs and Karplus, 1997)
R = _(Fp-Fcalc)/_Fp
From PROCHECK (Laskowski et al., 1993)
RESULTS
Identification of Novel Appendage Binding Partners
We have previously shown that constructs consisting of GST fused to the appendage domains of either γ or GGAs bring down a number of proteins from pig brain cytosol, three of which (bands 2, 3, and 7) seem to interact with both γ and GGA appendages (Hirst et al., 2000; Figure 1a). Using MALDI-TOF mass spectometry, we were able in our previous study to identify three of the bands in the pulldowns as MAP1A (band 1), rabaptin-5 (band 4), and Hsc70 (band 6). We have now analyzed the remaining bands by nanoelectrospray as well as MALDI-TOF mass spectometry. Band 2, or p200, was matched with KIAA1414, a protein of unknown function. Band 3 was found to be γ-synergin. Although γ-synergin sequences from both rat and human are available in the database, the pig protein is presumably too divergent to be identified by MALDI-TOF (the human and rat sequences are only ∼85% identical). Band 5, a doublet that is present only in the γ appendage pulldown, also matched γ-synergin. Band 7, or p56, matched another novel protein, FLJ11088.
Figure 1.

Identification of proteins that bind to the γ and GGA appendages in vitro. Pig brain cytosol was incubated with GST alone or with GST fusion proteins containing the appendage domains of γ-adaptin (GST-γ), GGA1 (GST-GGA1), or GGA2 (GST-GGA2). The Coomassie Blue-stained gel and the proteins identified by mass spectometry are shown in a. Western blots of the appropriate region of the gel labeled with the indicated antibodies are shown in b–d. p200 and p56 both come down with all three constructs, as does the larger isoform of γ-synergin. However, the smaller isoform of γ-synergin (which runs as a doublet) only comes down with the γ appendage construct.
To confirm these identities, antibodies were raised against recombinant p200 and p56 and used together with our previously characterized antibody against γ-synergin to probe Western blots of the pulldowns. Figure 1, b–d, shows that all three antibodies give the expected pattern. Anti-p200 and anti-p56 both label bands of the appropriate size in the γ and GGA pulldowns, but do not label bands in the pulldown with GST alone, or in pulldowns with α appendage (our unpublished data). The anti-γ-synergin antibody labels bands of ∼160 and ∼105 kDa in the γ pulldown lane, but only the higher molecular mass band in the GGA pulldown lanes. These two forms of γ-synergin are most likely alternatively spliced variants of the protein, because we have previously demonstrated that γ-synergin mRNA is alternatively spliced in several different places, particularly in brain (Page et al., 1999). Thus, we now have identities for all three of the major bands brought down by the GGA appendages.
p56 Domains
Of the two novel proteins identified in this study, p56 proved to be much more tractable than p200. The full-length sequence of p56 was determined by finding ESTs that overlap with FLJ11088 and with each other and sequencing several of the clones. ESTs encoding p56 can be found from a variety of tissues and organs, indicating that the gene is expressed ubiquitously. There are at least four alternatively spliced isoforms with different NH2 termini, as shown diagrammatically in Figure 2a. The isoform that occurs most frequently in the database, which we call the common isoform, consists of exons 1, 3, 4, and 7–16, and its sequence is shown in Figure 2b. Downstream from the alternatively spliced NH2-terminal domain, there is a long stretch of predicted coiled coil structure (Figure 2c).
To find out whether p56 binds directly to the appendage domains, we probed Western blots of bacterially expressed His-tagged p56 with GST coupled to γ, GGA1, or GGA2 appendage domains. Figure 2d shows that recombinant p56 is labeled with all three constructs but not with GST alone, confirming that the interaction is direct. To determine which part of p56 binds to the appendages, different regions of the common isoform were expressed in bacteria as His/Xpress-tagged constructs, indicated diagrammatically in Figure 2c. Figure 2e shows that domain 1 (amino acids 1–119), upstream from the coiled coil domains, can be labeled by the GGA1 appendage domain. None of the other domains were labeled, indicating that the NH2-terminal domain is both necessary and sufficient for this interaction.
To map the binding site more precisely, we first compared the common isoform with a second isoform, consisting of exons 5, 6, and 7–16, which seems to be expressed mainly in testis. Only the common isoform was found to bind to a GGA1 appendage domain construct (our unpublished data), indicating that amino acids 1–36, encoded by exons 3 and 4, contain the binding site. A comparison of the mouse and human sequences revealed that amino acids 1–17 are identical, whereas amino acids 18–36 are poorly conserved. Because it seemed likely that the binding site would be conserved, a 15-residue peptide, consisting of amino acids 2–16, was tested for its ability to compete for appendage domain binding to p56. Figure 2f shows that the peptide inhibits binding by 50% at concentrations of between 10 and 100 μM in both overlays and pulldown assays. The inhibition is somewhat more robust than that reported for DPF/W-containing peptides, which need to be added at concentrations of ∼500 μM to compete effectively for binding between the α appendage and its partners (Owen et al., 1999).
We also investigated the properties of the predicted coiled coil portion of the protein. Figure 2, d and e, shows that both the full-length His-tagged construct and domain 3 occur on blots as a major band of the expected size, a small amount of breakdown product, and a higher molecular mass band that is exactly twice the apparent molecular mass of the major band (see asterisks). Because coiled coil domains are often involved in dimerization, this suggests that the protein may be assembling into SDS-resistant homodimers. Further evidence suggesting that p56 can form homodimers is shown in Figure 2g. The His-tagged domain 3 construct was treated with the cross-linker DSP before running on a nonreducing gel, next to a lane containing noncross-linked domain 3. The band seen in the noncross-linked lane disappears in the cross-linked lane and is replaced by a more diffuse band with approximately twice the apparent molecular mass. These results suggest that the central portion of p56 may form a coiled coil homodimer, leaving the NH2-terminal domain free to interact with GGA and/or γ appendages, whereas the small COOH-terminal domain may interact with another binding partner. A schematic diagram of p56 is shown in Figure 2h.
Localization of p56
To determine whether p56 is associated with AP-1 and/or GGAs under physiological conditions, immunofluorescence double labeling was carried out. Figure 3a shows that the antibody we raised against p56 gives strong labeling of a band of the appropriate size when used to probe blots of total homogenates from pig brain and COS cells. When used for immunofluorescence on COS cells, this antibody produces a perinuclear pattern (Figure 3b). We also inserted an NH2-terminal myc tag into p56 and observed the same type of pattern in transiently expressing cells labeled with an anti-myc antibody (Figure 3c). Double labeling shows complete coincidence between tagged p56 (c) and tagged plus endogenous p56 (b) in the transfected cells. Treating cells with the drug BFA for 2 min caused p56 to redistribute into a grainy cytoplasmic pattern (Figure 3d). Thus, p56 behaves in a similar manner to both AP-1 and the GGAs in BFA-treated cells (Hirst et al., 2000).
Figure 3.

Localization of p56. (a) Western blot showing that the anti-p56 antibody specifically labels a band of the expected size in both pig brain and COS cell homogenates. The lower molecular mass band may be a degradation product or a splice variant. (b and c) Double labeling of COS cells for endogenous plus tagged (b) and myc-tagged (c) p56. The two labeling patterns show complete colocalization in the transfected cells. (d) p56 is sensitive to BFA. COS cells were treated with BFA for 2 min and then labeled with the antibody against the endogenous protein. (e and f) Tagged GGA2 (e) and endogenous p56 (f) show good colocalization, although there is some background with the tagged GGA2. (g and h) Endogenous GGA1 (g) and tagged p56 (h) show excellent colocalization. (i–l) γ-Adaptin (i) and endogenous p56 (j), or γ-adaptin (k) and tagged p56 (l) have similar labeling patterns but do not colocalize as well as GGAs and p56. Bar, 25 μm (b–d); 20 μm (e–l).
To double label cells for GGAs and p56, we had to transfect the cells with a tagged version of one of the two proteins, because only rabbit antibodies are available against either of them. Figure 3, e and f, shows double labeling for FLAG-tagged GGA2 (e) and endogenous p56 (f). The perinuclear patterns are very similar, although there is some background labeling seen with the anti-FLAG antibody. Myc-tagged p56 produced less background (Figure 3h), and again very similar patterns were seen compared with endogenous GGA1 (Figure 3g). Figure 3, i–l, shows cells double labeled for γ-adaptin (i and k) and either endogenous (j) or myc-tagged (l) p56. The patterns overlap, but they are less completely coincident than in cells double labeled for GGAs and p56, suggesting that p56 may preferentially interact with GGAs in vivo. However, it can be very difficult to resolve different patterns when comparing two proteins that both have a perinuclear distribution, because so many membranes are concentrated in this part of the cell. Thus, we needed to use other methods to compare the distributions of these proteins.
Preferential Binding of p56 and γ-Synergin to Different Partners
The first method that we used to compare the various labeling patterns was to treat the cells with the microtubule-disrupting drug nocodazole (Figure 4). This causes membranes whose distribution is dependent upon an intact network of microtubules to scatter throughout the cell. Under these conditions, AP-1 (a; red in c) and the GGAs (b; green in c) generally have nonoverlapping distributions, indicating that for the most part they are recruited onto distinct membrane compartments. To try to identify these compartments, we double labeled the cells with antibodies against TGN46 (e and h; green in f and i) and either γ-adaptin (d; red in f) or GGAs (g; red in i). The GGAs show much better colocalization with TGN46 than AP-1, consistent with the hypothesis that GGAs act at the TGN, whereas AP-1 may act mainly at a post-TGN compartment. Double labeling for γ-adaptin (j; red in l) and tagged p56 (k; green in l) showed little overlap between the two proteins. However, excellent overlap was seen between GGAs (m; red in o) and tagged p56 (n; green in o). Conversely, when we double labeled for γ-adaptin (p; red in r) and γ-synergin (q; green in r) in nocodazole-treated cells, we saw essentially complete colocalization.
Figure 4.

Localization of proteins in nocodazole-treated cells. COS cells were treated with 20 μg/ml nocodazole for 2 h to disperse membranes associated with the microtubule-organizing center. (a–c) Cells were double labeled with anti-γ-adaptin (a; red in c) and anti-GGA1 (b; green in c). The two adaptors have distinct punctate patterns, with only 28% of the spots in this cell colocalizing. (d–f) Cells were double labeled with anti-γ-adaptin (d; red in f) and anti-TGN46 (e; green in f). The two proteins have distinct patterns (18% colocalization), indicating that most of the AP-1 is not associated with TGN membranes as defined by the presence of TGN46. (g–i) Cells were double labeled with anti-GGA3 (g; red in i) and anti-TGN46 (h; green in i). The two proteins show good colocalization (71% of the spots are labeled with both antibodies), indicating that GGA3 is associated with TGN membranes. Similar results were seen with anti-GGA1 and with anti-p56 (our unpublished observations). (j–l) Transiently transfected cells expressing myc-tagged p56 were double labeled with anti-γ-adaptin (j; red in l) and anti-myc (k; green in l). The two patterns are distinct (12% colocalization). (m–o) Transiently transfected cells expressing myc-tagged p56 were double labeled with anti-GGA1 (m; red in o) and anti-myc (n; green in o). The two patterns show essentially complete colocalization on membranes (98%). (p–r) Cells were double labeled with anti-γ-adaptin (p; red in r) and anti-γ-synergin (q; green in r). The two patterns show excellent colocalization (99%). Bar, 20 μm.
Because it is formally possible that nocodazole may have indirect effects on protein binding, we also used a second approach to investigate the relative importance of AP-1 and GGAs in the localization of p56 and γ-synergin. This was to compare the distribution of all four proteins in cells with no membrane-associated AP-1 complexes. The cells were derived from a μ1A (ubiquitously expressed AP-1 medium chain) knockout mouse (Meyer et al., 2000). Although the knockout is embryonic lethal, a fibroblast line was generated from the embryos. The fibroblasts were shown to form partial AP-1 complexes containing γ-adaptin, but such complexes are unable to be recruited onto membranes (Figure 5a). Transfecting the cells with wild-type μ1A restores the ability of the cells to form functional, correctly localized AP-1 (Figure 5b).
Figure 5.

Localization of proteins in μ1A-deficient cells. Fibroblasts lacking μ1A (μ1A deficient) as well as the same cell line stably transfected with wild-type μ1A (μ1A rescued) were labeled with antibodies against either γ-adaptin (a and b), GGA1 (c and d), p56 (e and f), or γ-synergin (g and h). Both γ-adaptin and γ-synergin are cytosolic rather than membrane associated in the μ1A-deficient cells. However, the distribution of GGA1 and p56 is not affected, indicating that γ-synergin requires AP-1 to get onto the membrane, but GGA1 and p56 do not. Bar, 20 μm.
We looked first at the localization of the GGAs in the μ1A knockout and rescued cells. Figure 5, c and d, shows that GGAs have a similar distribution whether or not the cells are able to recruit AP-1 onto membranes. p56 also looks the same in the knockout and rescued cells (Figure 5, e and f). However, γ-synergin becomes completely cytosolic in the knockout cells (Figure 5g), although it has a normal distribution in the rescued cells (Figure 5h). Thus, membrane-associated GGAs are unable to recruit γ-synergin onto membranes in cells where AP-1 is cytosolic; however, the distribution of p56 is not affected in such cells.
Together, these studies indicate that although p56 and γ-synergin are able to interact with both GGA and γ appendages in vitro, under physiological conditions p56 shows a strong preference for the GGAs, whereas γ-synergin shows a strong preference for γ-adaptin.
Structure of the GGA Appendage Domain
Sequence alignments of the GGA and γ appendages show that the two domains are significantly homologous to each other (Hirst et al., 2000; Figure 6c). To compare the structures of the two appendage domains, we have expressed the GGA1 appendage (residues 494–639) in E. coli and determined its structure by x-ray crystallography. Crystals of the GGA1 appendage diffract to better than 2.3-Å resolution, and the structure was solved by molecular replacement using the γ appendage structure as a starting model (Kent et al., 2002; PDB ID 1GYU). Like the γ appendage, the GGA1 appendage is an eight-stranded β sandwich (Figure 6a). Overlaying the structures of the GGA1 appendage (green), the γ appendage (orange), and the α appendage (magenta) shows that the GGA1 appendage is structurally almost identical to the γ appendage, and also shows a high degree of similarity to the NH2-terminal subdomain of the α appendage (Figure 6b).
Figure 6.

Structure of the GGA1 appendage domain and comparison with the γ and α appendages. (a) Structure of the GGA1 appendage shown as a ribbon diagram. The residues that have been mutated in GGA1 for protein–protein interaction studies are indicated as ball and stick representations. (b) Cα overlay of the GGA1 appendage (green) with the γ appendage (orange) (Kent et al., 2002) and the α appendage (magenta) (Owen et al., 1999). The γ appendage overlays the GGA1 appendage with an rmsd of 1.2 Å > 113 Cα atoms. Figures were made with AESOP (Collins et al., 2002). (c) Structure-based sequence alignment of GGA1 and γ appendage domains. The secondary structure of the GGA1 appendage is shown as green arrows, and conserved residues are highlighted in gray. The region shown to bind p56 by mutagenesis is boxed in red.
The finding that the GGA and γ appendages can bind to a common subset of proteins suggests that the two domains use a similar mechanism to interact with accessory proteins. Structure-based mutagenesis of the γ appendage has allowed the identification of residues involved in the binding of γ-synergin and Eps15 (Kent et al., 2002). To test whether the GGA1 appendage can interact with its own ligand, p56, in the same way, we have constructed a set of mutants based on the γ-adaptin results. The residues that have been shown to mediate protein interactions by the γ appendage are situated within a hydrophobic cleft that lies at the interface between the two β sheets, with A753 and L762 the most important residues (Figure 6, a and c). We have mutated the corresponding residues of GGA1 to negatively charged amino acids (A563D and L572E) and have also mutated two of the neighboring hydrophobic residues (V564D and V570E). Circular dichroism analysis indicates that the mutants all have an identical fold to the native protein (our unpublished data). Figure 7a shows that all four of these mutations abolish binding of the GGA1 appendage to p56 in GST pulldowns, indicating that GGAs bind to protein ligands in an identical manner to the AP-1 γ subunit. The results also show that the interaction is governed in part by hydrophobic interactions, similar to the binding of the α and β appendage domains to short peptides with the sequence DΦF (Owen et al., 1999).
Figure 7.

Mutagenesis of the GGA1 appendage domain and expression of wild-type and mutant GFP constructs. (a) Wild-type and mutant GGA1 appendage domain constructs were fused to GST and tested for their ability to bind p56 from pig brain cytosol in pulldowns. Controls include α and γ appendages, as well as a mutant form of γ (A753D) that prevents binding to other accessory proteins (Owen et al., 1999; Kent et al., 2002). Mutations in hydrophobic residues positioned between β strands 4 and 5 abolish the interaction with p56. (b and c) COS cells were transiently transfected with GFP coupled to wild-type GGA1 appendage domain (b) and labeled with anti-p56 (c). Transfected cells are marked with asterisks. Even in low-expressing cells, the p56 has become essentially completely cytosolic. Similar results were obtained with a GFP-GGA2 appendage domain construct (our unpublished data). (d and e) COS cells were transiently transfected with GFP coupled to the A563D mutant GGA1 appendage domain (d) and labeled with anti-p56 (e). The mutant construct has little or no effect on the distribution of p56. (f and g) COS cells were transiently transfected with GFP coupled to the wild-type GGA1 appendage domain (f) and labeled with anti-γ-synergin (g). At moderate expression levels, the construct has little effect on the distribution of γ-synergin. h and i, COS cells were transiently transfected with GFP coupled to the wild-type γ appendage domain (h) and labeled with anti-γ-synergin (i). The γ appendage domain construct causes the γ-synergin to become cytosolic. (j and k) COS cells were transiently transfected with GFP coupled to the wild-type γ appendage domain (h) and labeled with anti-p56 (i). At moderate expression levels, the γ appendage domain construct has little effect on the distribution of p56. Bar, 20 μm.
Distribution of p56 and γ-Synergin in Cells Overexpressing Appendage Domains
It has been previously shown that overexpressing the appendage domain of the α subunit of the AP-2 complex has a dominant negative effect and blocks clathrin-mediated endocytosis, presumably by sequestering binding partners that are required for this event (Owen et al., 1999). To determine whether overexpressing the GGA or γ appendages might have a similar effect, we transfected cells with GFP coupled to either the wild-type GGA1 appendage domain, the A563D mutant GGA1 appendage domain, or the wild-type γ appendage domain. The cells were then double labeled for either p56 or γ-synergin.
Figure 7, b and c, shows that in cells expressing the GFP-GGA wild-type construct (b), p56 is predominantly cytosolic (c), presumably because the construct is expressed at higher levels than endogenous GGAs and sequesters most of the p56. In contrast, in cells expressing the A563D mutant GGA construct (d), p56 looks normal (e). Moderate expression of the wild-type GGA construct (f) has little effect on the distribution of γ-synergin (g). However, moderate expression of the γ appendage construct (h) causes γ-synergin (i) to redistribute to the cytosol. Conversely, moderate expression of the γ construct has little effect on p56 (j and k). Thus, these experiments provide further evidence that the distribution of p56 and γ-synergin is primarily determined by the distribution of the appendage domains of GGAs and AP-1, respectively. In addition, they suggest a possible means whereby one might be able to selectively block either the GGA pathway or the AP-1 pathway and then look at effects on the trafficking of other proteins.
DISCUSSION
The discovery of the GGAs was reported in 2000 by several groups who had independently used different approaches to find these proteins (Boman et al., 2000; Dell'Angelica et al., 2000; Hirst et al., 2000; Poussu et al., 2000; Takatsu et al., 2000). Our own approach had been to search databases for proteins with homology to known adaptor subunits (Hirst et al., 2000). We identified the GGAs as proteins with COOH-terminal domains that were related to the appendage domain of the AP-1 γ subunit. At the time, we proposed that the GGA and γ appendage domains might bind some of the same partners. The sequence homology also suggested that the two domains might share a similar structure. Here, we show that both hypotheses are correct. We have identified the three bands visible by Coomassie Blue staining that come down with both GGA and γ appendages in GST pulldowns and have shown that they are indeed the same proteins: p200, γ-synergin, and p56. We have also shown that both appendage domains have a β sandwich fold and that they interact with protein ligands in an identical manner. Binding by both GGA and γ appendages to accessory proteins is governed by conserved residues lying within a shallow cleft where the two β sheets come together. These interactions are probably mediated in part by a hydrophobic contact, because mutation of hydrophobic residues completely abolishes binding. Thus, this study clearly indicates why the two appendage domains are able to bind to an overlapping set of proteins.
Of the three proteins that bind to both GGA and γ appendages in the pulldowns from pig brain cytosol, the one we know least about is p200. It contains no obvious domains or motifs, and we were unable to raise antibodies against it that were specific enough for immunolocalization studies. Epitope tagging has also proved problematic because of the large size of the protein and our inability so far to clone the 5′ end. It is the most well conserved of the three proteins, with homologs in flies, worms, and yeast, suggesting that it may have a more fundamental role than either γ-synergin or p56. However, when we delete the p200 homolog in yeast, we see no apparent phenotype. The cells are completely viable, and the sorting and processing of carboxypeptidase Y and α-factor, both of which are aberrant in GGA-deficient cells (Hirst et al., 2000 and 2001), remain normal in p200-deficient cells (Hirst, unpublished observations). This suggests that p200 may be functionally redundant in yeast. Although Saccharomyces cerevisiae contains no other obvious homologues of p200, there may be another protein or proteins that can perform the same role.
γ-Synergin is the only one of the three shared binding partners that had already been identified and characterized. In our previous study, we showed by Western blotting that γ-synergin could be pulled down by both γ and GGA appendages (Hirst et al., 2000). We have now confirmed this result by mass spectrometry and have shown that γ-synergin corresponds to two of the Coomassie Blue-stained bands in the pulldowns. In our previous study, we also proposed that the interaction between γ-synergin and the GGA appendages might not take place in vivo. This was based on our finding that γ-synergin could be coimmunoprecipitated with γ-adaptin but not with GGA1 or GGA2 and that it completely colocalized with γ-adaptin but not with the GGAs by immunofluorescence. More recently, Nakayama and coworkers have shown that γ-synergin interacts with GGAs in the yeast two-hybrid system, and they also saw what seemed to be significant colocalization between tagged γ-synergin and GGAs by immunofluorescence, leading them to propose that the interaction was in fact physiologically relevant (Takatsu et al., 2000). In the present study, we have further addressed this question. We find essentially complete colocalization between γ-synergin and AP-1 in nocodazole-treated cells; however, there is little colocalization between AP-1 and the GGAs under these conditions. Furthermore, in μ1A-deficient cells, which have membrane-associated GGAs but no membrane-associated AP-1, γ-synergin is completely cytosolic. Thus, γ-synergin seems to show a strong preference for the γ appendage in vivo.
The opposite result was obtained for p56. This protein bound equally well to the GGA and γ appendages in both pulldowns and overlay assays. However, in nocodazole-treated cells it colocalized with the GGAs but not with AP-1. Even in transfected cells that were overexpressing p56, the excess protein remained in the cytosol and was not recruited onto AP-1–positive membranes. In addition, the distribution of p56 was not affected in μ1A-deficient cells.
Why do p56 and γ-synergin show such strong preferences for different appendage domains in vivo, when they can interact with both in vitro? One possibility is that conditions inside the cell, such as the presence of other proteins, may affect their binding. Alternatively, the large excess of appendage domain constructs in GST pulldowns and overlay assays may mask binding preferences that become apparent in the context of the whole cell, where both AP-1 and GGAs are expressed at relatively low levels. As a first step toward understanding the molecular basis for the binding preferences of the two appendages, we have narrowed down the appendage domain-binding site on p56 to a 15-residue peptide. Interestingly, this peptide contains a sequence, DDFGGF, which is related to the DDFXDF motif that has been proposed as a candidate sequence for binding to the γ appendage (Page et al., 1999; Nogi et al., 2002). Moreover, while our manuscript was under review, Payne and colleagues identified the sequence (D/E)2–3FXXΦ as a γ/GGA appendage binding motif in yeast (Duncan et al., 2003). The presence of hydrophobic and acidic residues in these sequences fits in well with mutagenesis studies on the two appendage domains, which show that both the hydrophobic cleft (Kent et al., 2002; present study) and conserved basic residues adjacent to the cleft (Nogi et al., 2002), contribute to binding.
But if the two appendages can bind to the same motif by using the same residues, how might specificity be determined? This may be the function of residues adjacent to the “strong” binding sites on the two appendages. An analogous situation is seen with the μ subunits of the four AP complexes. All of the μ subunits interact with xxYxxΦx motifs in the cytoplasmic tails of membrane proteins, but each subunit has a distinct set of preferences (Ohno et al., 1998; Owen and Evans, 1998; Owen et al., 2001). Structural studies on μ2 reveal that there is a strong interaction between the Y and Φ side chains and residues that are highly conserved among the different μ chains, whereas specificity is likely to arise from interactions between the “x” residues and nonconserved regions of the μ subunits outside the binding pocket. Similarly, although the consensus sequence for binding to GGA and γ appendages may be the same, interactions between amino acids outside the consensus sequence and nonconserved residues in the two appendage domains may determine binding preferences. The answers to these questions must await the structural characterization of appendage-peptide complexes. We are currently working on cocrystallizing the GGA appendage together with the 15-residue peptide.
Very recently, another protein has been reported that can interact with both γ and GGA appendages in vitro. We found this protein in γ appendage domain pulldowns from A431 cell cytosol and named it epsinR (Hirst et al., 2003). Three other laboratories have independently identified the same protein, which has also been called enthoprotin (Wasiak et al., 2002) and Clint (Kalthoff et al., 2002) as well as epsinR (Mills et al., 2003). Yeast homologs of epsinR, Ent3p and Ent5p, have also recently been identified in two-hybrid screens for γ and GGA appendage domain binding partners (Duncan et al., 2003). We find that epsinR behaves somewhat differently from either γ-synergin or p56, in that its membrane association is independent of both AP-1 and GGAs, depending instead upon an NH2-terminal ENTH domain (Hirst et al., 2003). In nocodazole-treated cells epsinR colocalizes to some extent with both AP-1 and GGAs (Hirst et al., 2003), indicating that it interacts with both appendage domains under physiological conditions. Indeed, the high degree of similarity between the GGA and γ appendages, and the observation that they are functionally interchangeable in yeast, suggests that they are likely to share at least some binding partners in vivo.
What is the function of p56? In our model shown in Figure 2h, we propose that it may connect the GGAs to another molecule or molecules via the short COOH-terminal domain, which together with the NH2-terminal GGA-binding domain is the most highly conserved part of p56 when human and mouse sequences are compared. Interestingly, this domain shows some homology to a GRIP domain, a Golgi-targeting domain found in a number of coiled coil proteins (Munro and Nichols, 1999), although we have been unable to show any membrane localization when we express it on its own. According to our model, p56 would be similar to proteins such as myosin or p115 (Nakamura et al., 1997), by using its NH2 and COOH terminal ends to bring two proteins, or possibly a protein and a lipid, together, whereas the central predicted coiled coil domain would help to increase the efficiency of binding by forming a homodimer. p56 would also be similar in this respect to several of the other appendage binding partners. All of the proteins that have so far been shown to bind directly to the α appendage also bind at least one other protein or lipid, setting up a complex network of interactions at the site of endocytic coated vesicle formation (Slepnev and De Camilli, 2000). γ-Synergin is also believed to interact with additional proteins (e.g., SCAMP1), by using its Eps15 homology (DH) domain (Page et al., 1999; Fernandez-Chacon et al., 2000), and epsinR interacts in vitro with several phosphoinositides, including PtdIns(4)P (Hirst et al., 2003; Mills et al., 2003). Experiments are currently in progress to look for additional binding partners for p56, by using the short COOH-terminal domain as bait. Intriguingly, at least two of the alternatively spliced isoforms of p56 lack the GGA appendage binding domain, but still contain the coiled coil portion and the COOH-terminal domain. These proteins may play a regulatory role, competing with the more abundant, GGA-binding isoform for other molecules.
When we transfected cells with either GGA or γ appendage domains coupled to GFP, we were able to change the distribution of p56 and γ-synergin, respectively. Even relatively low expression levels had a strong effect, particularly in the case of the GGA appendage and p56. At very high expression levels, we started to see effects on the other partner (our unpublished data). Overexpressing the α appendage has been shown to inhibit clathrin-mediated endocytosis (Owen et al., 1999), so it is possible that the GGA and γ appendage domains, when expressed on their own at appropriate levels, may selectively disrupt GGA-mediated and AP-1–mediated pathways, respectively. There is currently some confusion over precisely what the GGAs and AP-1 are actually doing. Originally, AP-1 was assumed to facilitate the trafficking of proteins such as the MPRs from the TGN to an endosomal compartment. The discovery of the GGAs, and the compelling evidence for their role in TGN-to-endosome trafficking in both mammals and yeast, has caused the role of AP-1 to be reassessed. Gene knockout experiments in both mammals and yeast suggest that in fact AP-1 may primarily be involved in the retrograde trafficking of certain cargo proteins from an endosomal compartment back to the TGN (Meyer et al., 2000, 2001; Valdivia et al., 2002). Therefore, one current hypothesis is that GGAs and AP-1 facilitate traffic in opposite directions. Recently, an alternative hypothesis was proposed, suggesting that the function of the GGAs might be, at least in part, to “hand over” cargo to AP-1 (Doray et al., 2002). The double-labeling experiments on nocodazole-treated cells reported in this study are consistent with both hypotheses, because both hypotheses predict that GGAs should act before AP-1. We find that GGAs and p56 show good colocalization with the TGN marker TGN46, as well as with other proteins associated with the Golgi stack (Figure 4, g–i; our unpublished observations). In contrast, AP-1 shows little colocalization with Golgi markers (Figure 4, d–f), suggesting that it is primarily associated with a post-TGN compartment. So far, we have not seen convincing colocalization in nocodazole-treated cells between AP-1 and any of the marker proteins we have investigated, so the identity of this compartment is not yet known. However, the use of GGA and γ appendage domains as dominant negatives may help to establish the precise functions not only of accessory proteins such as p56 and γ-synergin but also of the GGAs and of AP-1.
With the exception of their appendage domains, the GGAs have always been assumed to be structurally distinct from the heterotetrameric adaptor complexes (Boehm and Bonifacino, 2001; Robinson and Bonifacino, 2001). However, this work, in combination with the recently solved structure of the AP-2 core complex (Collins et al., 2002), reveals an interesting relationship between the GGAs and the large subunits of adaptor complexes. The GGAs are composed of four domains: the VHS domain, the GAT domain, the hinge-like domain, and the appendage domain. The trunks of the adaptor large subunits are α-helical structures with a high degree of similarity to the VHS domain of the GGAs (Misra et al., 2002; Shiba et al., 2002). Secondary structure predictions of the GAT domain suggest that this region also has a predominantly α-helical secondary structure (our unpublished observations). In the present study, we have demonstrated conclusively that the appendage domain of the GGAs is structurally related to the appendages of the adaptor large subunits. When pooled, these data show that the GGAs and adaptor large subunits share a very similar structural organization despite a lack of sequence homology: an NH2-terminal α-helical structure connected by a flexible, clathrin-binding linker to a β sheet appendage domain. The GGAs may thus be similar, both structurally and functionally, to the ancestral large chain from which both adaptor and coatomer subunits are thought to have derived.
Acknowledgments
We thank the Kazusa Institute and the IMAGE Consortium for the p200 and p56 cDNAs. We are grateful to Juan Bonifacino for the anti-GGA3 antibody. We thank Nicola Berg for performing some of the immunofluorescence experiments. We also thank Paul Luzio, Matthew Seaman, Rainer Duden, John Kilmartin, Sean Munro, and members of the Robinson laboratory for reading the manuscript and for helpful discussions. This work was supported by grants from the Wellcome Trust and the Medical Research Council.
References
- Boehm, M., and Bonifacino, J.S. (2001). Adaptins: the final recount. Mol. Biol. Cell 12, 2907-2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boman, A.L., Zhang, C.J., Zhu, X., and Kahn, R.A. (2000). A family of ADP-ribosylation factor effectors that can alter membrane transport through the trans-Golgi. Mol. Biol. Cell 11, 1241-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger, A.T., et al. (1998). Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- Collins, B.M., McCoy, A.J., Kent, H.M., Evans, P.R., and Owen, D.J. (2002). Molecular architecture and functional model of the endocytic AP2 complex. Cell 109, 523-535. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica, E.C., Puertollano, R., Mullins, C., Aguilar, R.C., Vargas, J.D., Hartnell, L.M., and Bonifacino, J.S. (2000). GGAs: a family of ADP ribosylation factor-binding proteins related to adaptors and associated with the Golgi complex. J. Cell Biol. 149, 81-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs, K., and Karplus, P.A. (1997). Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4, 269-275. [DOI] [PubMed] [Google Scholar]
- Dodson, E.J., Winn, M., and Ralph, A. (1997). Collaborative computational project, number 4: providing programs for crystallography. Methods Enzymol. 227, 620-634. [DOI] [PubMed] [Google Scholar]
- Doray, B., Ghosh, P., Griffith, J., Geuze, H.J., and Kornfeld, S. (2002). Cooperation of GGAs and AP-1 in packaging MPRs at the trans-Golgi network. Science 297, 1700-1703. [DOI] [PubMed] [Google Scholar]
- Duncan, M.C., Costaguta, G., and Payne, G.S. (2003). Yeast epsin-related proteins required for Golgi-endosome traffic define a γ-adaptin ear-binding motif. Nat. Cell Biol. 5, 77-81. [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon, R., Achiriloaie, M., Janz, R., Albanesi, J.P., and Sudhof, T.C. (2000). SCAMP1 function in endocytosis. J. Biol. Chem. 275, 12752-12756. [DOI] [PubMed] [Google Scholar]
- Hirst, J., Lui, W.W.Y., Bright, N.A., Totty, N., Seaman, M.N.J., and Robinson, M.S. (2000). A family of proteins with γ-adaptin and VHS domains that facilitate trafficking between the TGN and the vacuole/lysosome. J. Cell Biol. 149, 67-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst, J., Lindsay, M., and Robinson, M.S. (2001). GGAs: roles of the different domains and comparison with AP-1 and clathrin. Mol. Biol. Cell 12, 3573-3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirst, J., Motley, A., Harasaki, K., Peak Chew, S.Y., and Robinson, M.S. (2003). EpsinR: an ENTH domain-containing protein that interacts with AP-1. Mol. Biol. Cell 14, 625-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowen, S.W., and Kjeldgaard, M. (1991). Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- Kalthoff, C., Groos, S., Mahrhold, S., and Ungewickell, E.J. (2002). Clint: a novel clathrin-binding ENTH-domain protein at the Golgi. Mol. Biol. Cell 13, 4060-4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, H.M., McMahon, H.T., Evans, P.R., Benmerah, A., and Owen, D.J. (2002). γ-Adaptin appendage domain: structure and binding site for Eps15 and γ-synergin. Structure 10, 1-20. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283-291. [Google Scholar]
- Leslie, A.G.W. (1992). Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP + ESF-EAMCB newsletter on protein crystallography. 26
- Meyer, C., Eskelinen, E.L., Guruprasad, M.R., von Figura, K., and Schu, P. (2001). μ1A deficiency induces a profound increase in MPR300/IGF-II receptor internalization rate. J. Cell Sci. 114, 4469-4476. [DOI] [PubMed] [Google Scholar]
- Meyer, C., Zizioli, D., Lausmann, S., Eskelinen, E.L., Hamann, J., Saftig, P., von Figura, K., and Schu, P. (2000). μ1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phsophate receptors. EMBO J. 19, 2193-2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills, I.G., Praefcke, G.J.K., Vallis, Y., Peter, B.J., Olesen, L.E., Gallop, J.L., Butler, P.J.G., Evans, P.R., and McMahon, H.T. (2003). EpsinR: an AP-1/clathrin interacting protein involved in vesicle trafficking. J. Cell Biol. 160, 213-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra, S., Puertollano, R., Kato, Y., Bonifacino, J.S., and Hurley, J.H. (2002). Structural basis for acidic-cluster-dileucine sorting-signal recognition by VHS domains. Nature 21, 933-937. [DOI] [PubMed] [Google Scholar]
- Mullins, C., and Bonifacino, J.S. (2001). Structural requirements for function of yeast GGAs in vacuolar protein sorting, α-factor maturation, and interactions with clathrin. Mol. Cell. Biol. 21, 7981-7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro, S., and Nichols, B.J. (1999). The GRIP domain –a novel Golgi targeting domain found in several coiled-coil proteins.Curr. Biol. 9, 377-380. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240-255. [DOI] [PubMed] [Google Scholar]
- Nakamura, N., Lowe, M., Levine, T.P., Rabouille, C., and Warren, G. (1997). The vesicle docking protein p115 binds GM130, a cis-Golgi matrix protein, in a mitotically regulated manner. Cell 89, 445-455. [DOI] [PubMed] [Google Scholar]
- Nogi, T., et al. (2002). Structural basis for the accessory protein recruitment by the γ-adaptin ear domain. Nat. Struct. Biol. 9, 527-531. [DOI] [PubMed] [Google Scholar]
- Ohno, H., Aguilar, R.C., Yeh, D., Taura, D., Saito, T., and Bonifacino, J.S. (1998). The medium subunits of adaptor complexes recognize distinct but overlapping sets of tyrosine-based sorting signals. J. Biol. Chem. 273, 25915-25921. [DOI] [PubMed] [Google Scholar]
- Owen, D.J., and Evans, P.R. (1998). A structural explanation for the recognition of tyrosine-based endocytic signals. Science 282, 1327-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen, D.J., Setiadi, H., Evans, P.R., McEver, R.P., and Green, S.A. (2001). A third specificity-determining site in μ2 adaptin for sequences upstream of Yxx phi sorting motifs. Traffic 2, 105-110. [DOI] [PubMed] [Google Scholar]
- Owen, D.J., Vallis, Y., Noble, M.E., Hunter, J.B., Dafforn, T.R., Evans, P.R., and McMahon, H.T. (1999). A structural explanation for the binding of multiple ligands by the α-adaptin appendage domain. Cell 97, 805-815. [DOI] [PubMed] [Google Scholar]
- Owen, D.J., Vallis, Y., Pearse, B.M.F., McMahon, H.T., and Evans, P.R. (2000). The structure and function of the β2-adaptin appendage domain. EMBO J. 19, 4216-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page, L.J., Sowerby, P.J., Lui, W.W.Y., and Robinson, M.S. (1999). γ-Synergin: an EH domain-containing protein that interacts with γ-adaptin. J. Cell Biol. 146, 993-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poussu, A., Lohi, O., and Lehto, V.P. (2000). Vear, a novel Golgi-associated protein with VHS and γ-adaptin “ear” domains. J. Biol. Chem. 275, 7176-7183. [DOI] [PubMed] [Google Scholar]
- Puertollano, R., Aguilar, R.C., Gorshkova, I., Crouch, R.J., and Bonifacino, J.S. (2001a). Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science 292, 1663-1665. [DOI] [PubMed] [Google Scholar]
- Puertollano, R., Randazzo, P.A., Presley, J.F., Hartnell, L.M., and Bonifacino, J.S. (2001b). The GGAs promote ARF-dependent recruitment of clathrin to the TGN. Cell 105, 93-102. [DOI] [PubMed] [Google Scholar]
- Robinson, M.S., and Bonifacino, J.S. (2001). Adaptor-related proteins. Curr. Opin. Cell Biol. 13, 444-453. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritsch, E.F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Seaman, M.N.J., Sowerby, P.J., and Robinson, M.S. (1996). Cytosolic and membrane-associated proteins involved in the recruitment of AP-1 adaptors onto the trans-Golgi network. J. Biol. Chem. 271, 25446-25451. [DOI] [PubMed] [Google Scholar]
- Shiba, T., et al. (2002). Structural basis for recognition of acidic-cluster dileucine sequence by GGA1. Nature 21, 937-941. [DOI] [PubMed] [Google Scholar]
- Slepnev, V.I., and De Camilli, P. (2000). Accessory factors in clathrin-dependent synaptic vesicle endocytosis. Nat. Rev. Neurosci. 1, 161-172. [DOI] [PubMed] [Google Scholar]
- Takatsu, H., Yoshino, K., and Nakayama, K. (2000). Adaptor γ ear homology domain conserved in γ-adaptin and GGA proteins that interact with γ-synergin. Biochem. Biophys. Res. Commun. 271, 719-725. [DOI] [PubMed] [Google Scholar]
- Traub, L.M., Downs, M.A., Westrich, J.L., and Fremont, D.H. (1999). Crystal structure of the α appendage of AP-2 reveals a recruitment platform for clathrin-coat assembly. Proc. Natl. Acad. Sci. USA 96, 8907-8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia, R.H., Baggott, D., Chuang, J.S., and Schekman, R.W. (2002). The yeast clathrin adaptor protein complex 1 is required for the efficient retention of a subset of late Golgi membrane proteins. Dev. Cell 2, 283-294. [DOI] [PubMed] [Google Scholar]
- Wasiak, S., Legendre-Guillemin, V., Puertollano, R., Blondeau, F., Girard, M., de Heuvel, E., Boismenu, D., Bell, A.W., Bonifacino, J.S., and McPherson, P.S. (2002). Enthoprotin: a novel clathrin-associated protein identified through subcellular proteomics. J. Cell Biol. 158, 855-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn, M.D., Isupov, M.N., and Murshudov, G.N. (2001). Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D 57, 122-133. [DOI] [PubMed] [Google Scholar]