

Abstract
Infection of primary cultures of human immature monocyte-derived dendritic cells (moDC) with the paramyxovirus Simian Virus 5 (SV5) results in extensive cytopathic effect (CPE) and induction of apoptosis, but DC maturation pathways are not activated. In this study, we investigated the relationship between SV5-induced apoptosis and the lack of DC maturation. Reducing CPE and apoptosis in SV5 infected immature DC by the addition of a pancaspase inhibitor resulted in only low level expression of maturation markers CD40, CD80 and CD86, suggesting that SV5 infection either actively blocked maturation pathways or failed to provide sufficient signals to activate maturation. To distinguish between these hypotheses, SV5-infected immature DC were challenged with agonists that stimulate toll-like receptors (TLRs). Treatment with the TLR-4 agonist LPS or TLR-6 agonist FSL1 enhanced cell surface expression of CD40, CD80 and CD86 on SV5 infected cells to levels approaching that of mock infected TLR-treated moDC, but treatment with agonists for TLR-2, -3, -5, or -8 had little effect. Addition of TLR-4 or -6 agonists to SV5-infected DC also dramatically reduced CPE and apoptosis, but the levels of viral protein and virus yield were not affected. Similarly, SV5 infected immature moDC were matured by treatment with IL-1beta, and these mature infected cells also showed reduced CPE and apoptosis. In the presence of NFkB inhibitors, TLR-4 and -6 agonists did not promote maturation or reduce apoptosis of SV5-infected DC, indicating that maturation and cell survival were both dependent on signaling through NFkB dependent pathways. Our results suggest a model whereby SV5 replication induces apoptosis in immature DC but fails to provide strong maturation signals, while activation of NFkB-dependent pathways by exogenous ligands can lead to moDC maturation and override SV5-induced cell death.
INTRODUCTION
Dendritic cells (DC) are pivotal professional antigen presenting cells that are capable of recognizing microbial products and promoting innate and adaptive immune responses (Banchereau et al., 2000; Reis e Sousa, 2001). Two major human DC populations have been described: CD11c− plasmacytoid (pDC) and CD11c+ conventional DC (cDC). pDC can respond to virus infection by producing type I interferon (IFN) which can limit virus growth (Hengel et al., 2005), but are thought to be relatively poor at antigen presentation (Barchet et al., 2005). By contrast, CD11c+ cDC are very efficient at processing and presenting antigen, and can serve as potent activators of other immune cells. Upon sensing viral components, immature DC are triggered to undergo a series of cell signaling events that result in conversion of an immature phenotype into a mature form that is capable of activating naïve T cell functions such as proliferation, cytokine secretion and cytolytic activity (Banchereau et al., 2000; Reis e Sousa, 2001). These DC maturation events include an increased cell surface expression of costimulatory molecules such as CD40, CD80, CD86, downregulation of antigen capture receptors and increased capacity to secrete immunomodulatory cytokines such as interleukin-12 (IL-12), IL-6, IL-8 and TNF-α (Banchereau et al. 2000; Reis e Sousa, 2001).
Virus-induced DC maturation can occur through recognition of components of the incoming virion particle or as newly synthesized viral components (Biron, 1999; Boehme and Compton, 2004; Klagge and Schneider-Schaulies, 1999). A major pathway for virus-induced DC maturation includes the Toll-like receptor (TLR) pathway (Iwasaki and Medzhitov, 2004; Pasare and Medzhitov, 2004; Takeda and Akira, 2004). Individual TLRs recognize a specific type of microbial component and initiate a cascade of intracellular signaling events that results in activation of the transcription factor NFkB and induction of host genes associated with antigen presentation and inflammatory responses. For recognition of RNA viruses, important TLRs include TLR-3, which recognizes dsRNA (Alexopoulou et al., 2001) and TLR-7 and TLR-8, which recognize ssRNA (Diebold et al., 2004; Heil et al., 2004; Lund et al., 2004). For some RNA viruses such as mouse mammary tumor virus, measles virus and respiratory syncytial virus (RSV), viral glycoproteins have been reported to activate TLR-2 or -4 (reviewed in Boehme and Compton, 2004). Immature DC can also be activated by alternative mechanisms that do not depend on TLR signaling (Lopez et al. 2006), but the pathways for TLR-independent recognition of virus infection are not completely understood.
Clearance of a viral infection and the subsequent development of long term adaptive immunity depends on appropriate innate and adaptive responses at the time of infection (Biron, 1999), and DC play a pivotal role in linking these two arms of immunity (Banchereau et al, 2000; Reis e Sousa, 2001). As such, many viruses are poor inducers of maturation pathways in infected immature DC, resulting in limited production of cytokines and upregulation of costimulatory molecules (Klagge and Schneider-Schaulies, 1999). Examples of this include herpes simplex virus, cytomegalovirus (CMV), vaccinia virus, measles virus, parainfluenza virus type 3, Lassa fever virus, hantavirus, polio virus and Ebola virus (Baize et al, 2004; Bosio et al., 2003; Engelmayer et al., 1999; Grosjean et al, 1997; Moutaftsi et al, 2002; Plotnicky-Gilquin et al., 2001; Raftery et al, 2002; Schneider-Schaulies et al., 2003; Wahid et al, 2005). This lack of maturation following infection of immature DC could result from a virus-induced suppression of signaling pathways. For example, CMV infection of immature DC fails to induce maturation, but infected cells are also refractory to maturation induced by external stimuli (Moutaftsi et al, 2002). However, many viral infections of immature DC are highly cytopathic. Thus, in these cases, it is not clear if the lack of maturation following infection is due to active suppression of host cell signaling pathways or to a more general cytopathic effect (CPE) that indirectly limits maturation.
Our work and the work of others have shown that in human epithelial- and fibroblast-derived cells, the paramyxovirus SV5 is unusual among closely related viruses by being a poor inducer of host cell death pathways (Choppin, 1964; Lin et al; 2003; He et al., 2002; Wansley and Parks, 2002; Wansley et al., 2003). Similar results demonstrating a noncytopathic phenotype for WT SV5 were found with primary cultures of human epithelial cells (Wansley et al., 2005). Remarkably, the outcome of WT SV5 infection of primary cultures of human myeloid-derived immature DC (moDC) is very different, as we have recently shown that SV5 establishes a productive infection of immature moDC that is highly cytopathic with activation of apoptosis (Arimilli et al, 2006). Thus, host cell death pathways are differentially activated by WT SV5 infections of epithelial cells (limited CPE) versus immature moDC (high CPE).
In addition to being highly cytopathic in primary cultures of human immature DC, we have previously shown that WT SV5 is a poor inducer of CD40, CD80, and CD86 maturation markers and cytokine secretion (Arimilli et al, 2006). We hypothesed that the lack of maturation of SV5 infected immature moDC could result from two nonexlusive mechanisms: a cytopathic effect that indirectly precludes DC maturation or an active mechanism for suppression of maturation signaling pathways. Here we have examined the relationship between SV5 induced DC death pathways and the activation of DC maturation pathways. Our results suggest a model whereby the lack of maturation of SV5 infected DC is not due to apoptosis per se or to a global inhibition of maturation pathways, but rather a failure to provide sufficiently strong maturation signals. Activation of NFkB-dependent pathways by exogenous ligands can lead to maturation of infected cells and override SV5-induced cell death without inhibiting virus replication.
RESULTS
A pan-caspase inhibitor reduces apoptosis in SV5 infected human moDC, but results in limited activation of maturation markers
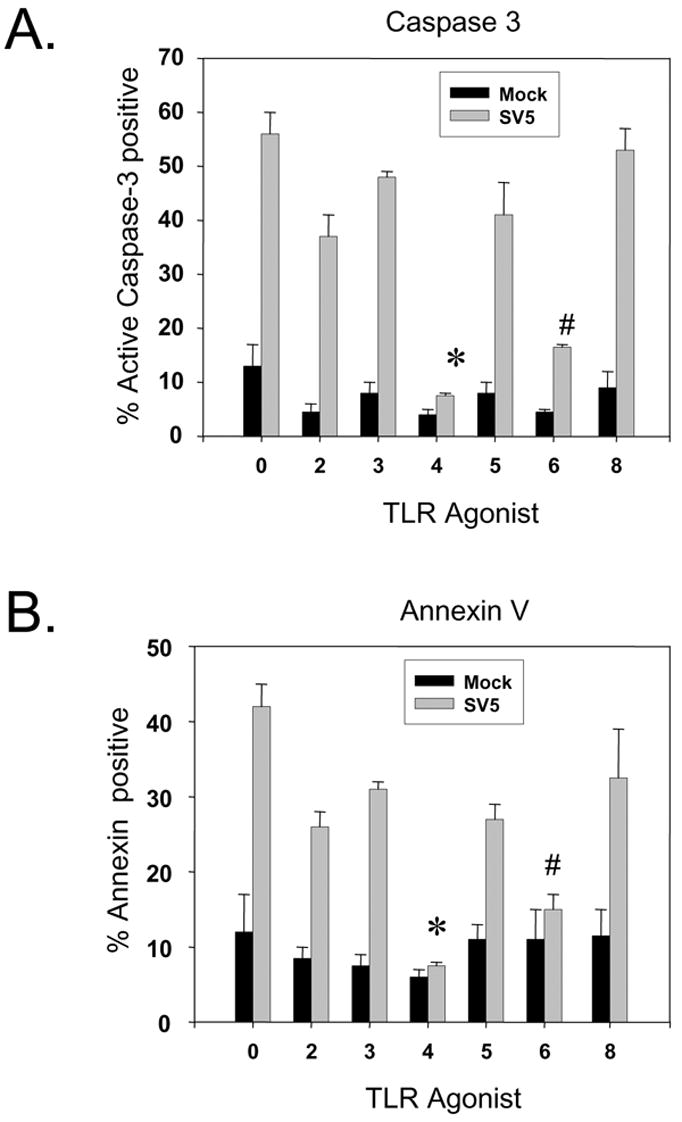
Primary cultures of immature monocyte-derived DC (moDC) were generated by selection of CD14-positive cells from PBMC followed by culturing for six days with recombinant GM-CSF and IL-4 as detailed previously (Arimilli et al., 2006). Greater than 95% of this immature DC population was CD11c+ (not shown). To analyze SV5-induced cytopathic effects (CPE) and apoptosis, immature moDC were infected with WT recombinant SV5 (rSV5) or rSV5-EGFP, a WT rSV5 engineered to encode EGFP as an additional gene between the viral HN and L genes as described previously (He et al., 1997). At 22 h pi, cells were examined by microscopy for CPE and EGFP expression. As shown in Fig. 1A, the majority of mock infected immature moDC were found as adherent healthy cells with some rounded cells that were still attached to the dish. By contrast, immature moDC infected with WT rSV5 showed increased levels of cellular debris, and many cells were detached from the dish and floating. Likewise, infections with rSV5-EGFP showed that nearly all cells were expressing EGFP (EGFP panel, Fig. 1A), and the CPE was indistinguishable from that seen with WT rSV5. When analyzed by flow cytometry, a low percentage of mock infected immature moDC showed staining with an antibody specific for cleaved active caspase-3 (Fig. 1B). By contrast, a much larger percentage of immature moDC infected with either WT rSV5 or rSV5-EGFP showed high levels of cleaved caspase-3. The percentage of cells with active caspase-3 staining was reduced when infected cells were cultured with the pancaspase inhibitor z-VAD-FMK. We have not detected differences in any properties of WT rSV5 and rSV5-EGFP in immature DC, including the kinetics of viral gene expression or appearance of apoptotic markers (see below).
Figure 1. WT SV5 and rSV5-EGFP are cytopathic in primary cultures of immature moDC.
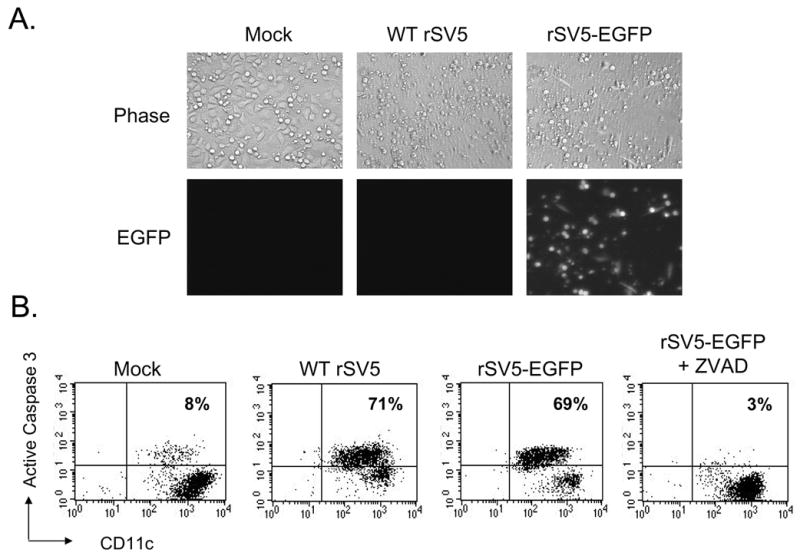
A) Immature moDC from a representative donor were mock infected or infected at an moi of 5 with WT rSV5 or rSV5-EGFP. Phase-contrast and EGFP expression were visualized at 22 h pi. B) Immature moDC infected with the indicated viruses were analyzed by flow cytometry at 22 h pi for intracellular levels of activated caspase-3 and cell surface CD11c. z-VAD-FMK was included in one culture of infected DC at 100 uM. Numbers in the upper quadrant indicate the percent of CD11c+ cells that stained positive for cleaved caspase-3. Representative of three experiments.
We have previously shown that at 24 h pi, SV5-infected human immature moDC do not show an increase in surface expression of maturation markers such as CD40, CD80 or CD86 (Arimilli et al, 2006). To determine if inhibiting SV5-induced apoptosis leads to increased surface maturation marker expression, mock infected or infected immature moDC were cultured in the presence of the pancaspase inhibitor z-VAD-FMK. As shown in Fig. 2, the pancaspase inhibitor reduced levels of cleaved caspase-3 in SV5-infected immature moDC. However, the levels of cell surface maturation markers CD40, CD80 and CD86 were not above that seen for inhibitor-treated mock infected cells and were much lower than that see with LPS-treated control cells (Figs. 2B–D). Uninfected control DC that were treated with a TLR-3 agonist gave CD40, and CD86 levels that were 3–4 fold over mock treated cells (see below). These data indicate that the lack of maturation of SV5 infected moDC cannot be solely accounted for by the induction of apoptosis.
Figure 2. Pan-caspase inhibitor decreases apoptosis in SV5 infected immature moDC, but results in only low levels of cell surface maturation markers.
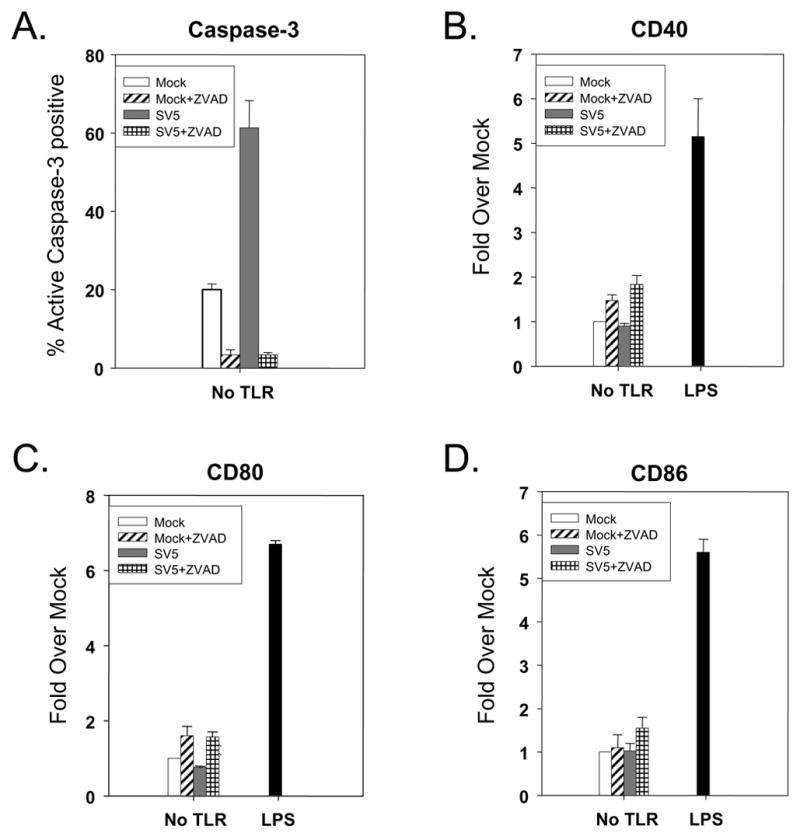
Immature moDC were mock infected or infected at an moi of 5 with WT rSV5 and then treated with or without 100 uM z-VAD-FMK. Levels of intracellular cleaved caspase-3 (panel A) and the indicated cell surface maturation markers (panels B–D) were assayed by flow cytometry at 24 h pi. Cells treated with 200 ng/ml of LPS were used as a positive control. Results are the average of three experiments from three individual donors (with standard error bars). Results in panels B–D are expressed as a fold change relative to mock infected, mock treated sample set at 1.
Maturation of SV5-infected moDC in response to treatment with TLR-4 and -6 agonists
Previous results with epithelial and fibroblast cell lines have shown that SV5 infection induces the loss of STAT1, resulting in a block in IFN signaling pathways (Didcock et al., 1999b). This raised the hypothesis that the lack of induction of maturation markers following WT rSV5 infection was due to virus-induced inhibition of maturation pathways. As a first step in testing this hypothesis, we determined the kinetics of viral protein expression, STAT1 degradation and appearance of apoptosis markers. Immature moDC were mock infected or infected at high moi with WT rSV5 and cell lysates were analyzed by western blotting at 0, 6, 12 and 22 h pi. As shown for a representative donor in Fig. 3A, SV5 P protein was detected by 6 h pi, and accumulated to higher levels by 12 and 22 h pi. Similarly, STAT1 levels were greatly reduced by 6 h pi and were at the limit of detection by 22 h pi.
Figure 3. Timecourse of STAT1 degradation, viral protein expression, apoptotic markers and maturation markers during SV5 infection of immature moDC.
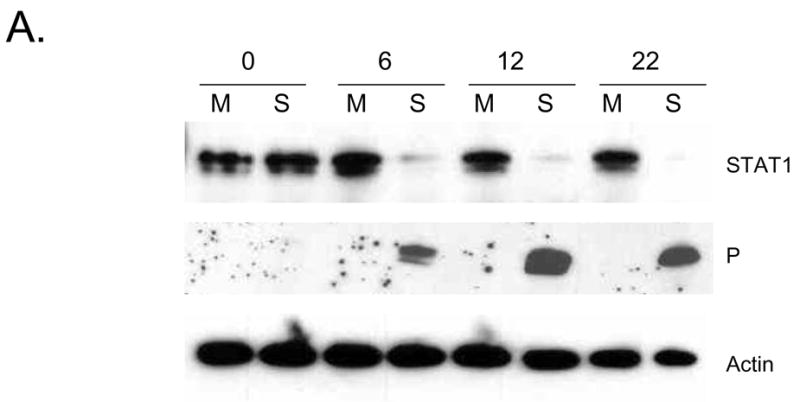
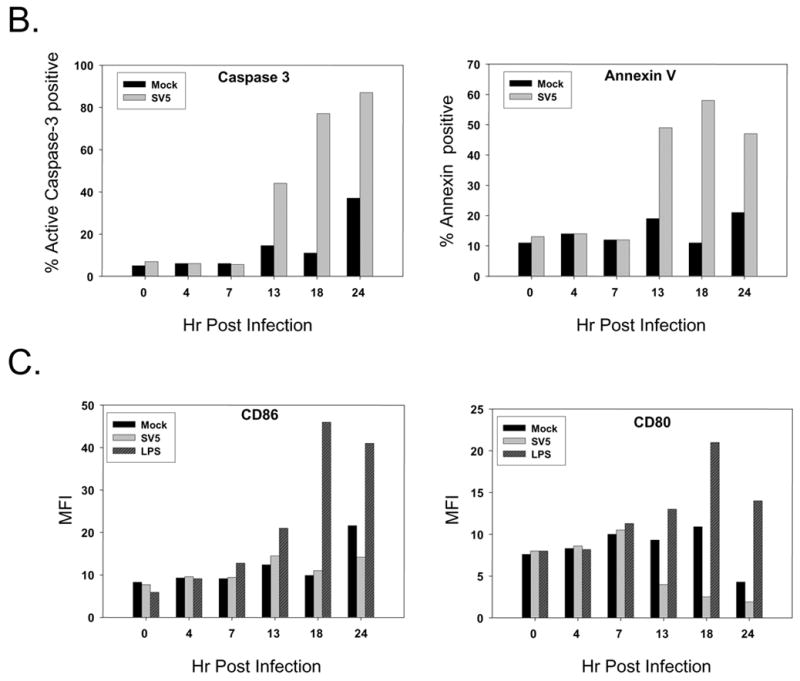
A) Immature moDC from a representative donor were mock infected (M lanes) or infected at an moi of 5 with WT rSV5 (S lanes). Cell lysates were prepared at the indicated times pi, and analyzed by western blotting for STAT1, P protein and actin. B and C) Timecourse of appearance of apoptotic markers and maturation markers. Immature DC from a representative donor were mock infected or infected at an moi of 5 with WT rSV5. At the indicated times pi, cells were analyzed by flow cytometry for intracellular levels of active caspase-3 and cell surface annexinV (panel B), or for cell surface CD86 and CD80 (panel C). In panel C, a sample of mock infected immature moDC were treated at time 0 with LPS to serve as a positive control for the timecourse of appearance of DC maturation markers.
Abundant viral protein expression and STAT1 degradation occurred before the appearance of apoptotic markers. This is evident in Fig. 3B, which shows the kinetics of appearance of cleaved caspase-3 and annexinV staining for mock infected and SV5 infected immature moDC. A large increase in the percentage of infected cells with elevated active caspase-3 was apparent between 7 and 13 h pi, and continued to increase out to 24 h pi. There was a modest increase in the percentage of mock infected cells with active caspase-3 staining at 24 h pi, but this was not reproducibly seen with all donors. Very similar kinetics were seen for the percentage of infected cells with high levels of surface annexin-V staining (right panel, Fig. 3B).
As shown in the timecourse in Fig. 3C, immature moDC treated with 200 ng/ml of LPS show elevated levels of cell surface CD80 and CD86 compare to mock treated samples beginning between 7 and 13 h pi. By contrast, WT rSV5 infected cells show either very poor induction (CD86; Fig. 3C) or decreased levels (CD80; Fig. 3C) of these maturation markers over time. Taken together, these data indicate that WT SV5 infection of immature moDC results in a very rapid reduction of STAT1 that begins prior to 6 h pi and apoptotic markers begin to increase between 7 and 13 h pi, but maturation markers do not increase during this timeperiod.
To test the hypothesis that SV5 imposes a block in DC maturation pathways, SV5-infected immature moDC were treated with compounds that activate TLR signaling, a known pathway leading to DC maturation (Iwasaki and Medzhitov, 2004). TLRs 2–6 and -8 were chosen for activation, based on the previously-established TLR expression patterns of in vitro-differentiated human moDC (Iwadaki and Medzhitov, 2004), and the role for individual TLRs in recognition of viral components (Boehme and Compton, 2004). Cells were mock infected or infected at high moi with WT rSV5. At 7 h pi, cells were then mock treated or treated with individual TLR agonists as detailed in Materials & Methods. This timepoint for agonist challenge was chosen based on the kinetics of appearance of apoptotic markers in SV5-infected DC shown in Fig. 3, with the goal of activating signaling pathways before extensive CPE had occurred. At 22 h pi, cells were then analyzed for surface maturation markers CD40, CD86, and CD80.
Fig. 4 shows the average mean fluorescence intensity (MFI) from three experiments with three donors expressed as a fold increase over that seen with immature moDC that were mock infected and mock treated. In the case of CD40, treatment of mock infected cells with each of the individual TLR agonists gave a ~2–6 fold increase in surface marker expression (Fig. 4A, black bars). The highest levels of CD40 on mock infected cells were seen after treatment with the TLR-4 agonist LPS and TLR-6 agonist FSL1. CD86 responses in TLR agonist-treated mock infected cells were similar to that seen for CD40, except that TLR-2, -3 and -6 responses were approximately equal (Fig. 4B, black bars). For CD80, TLR-4 and -6 agonists induced the highest responses, which were ~4–7 fold above mock treated cells, while the other TLR responses were much lower (e.g., TLR-2) or were not above background levels.
Figure 4. TLR-4 and -6 agonists induced cell surface maturation markers on SV5-infected immature moDC.
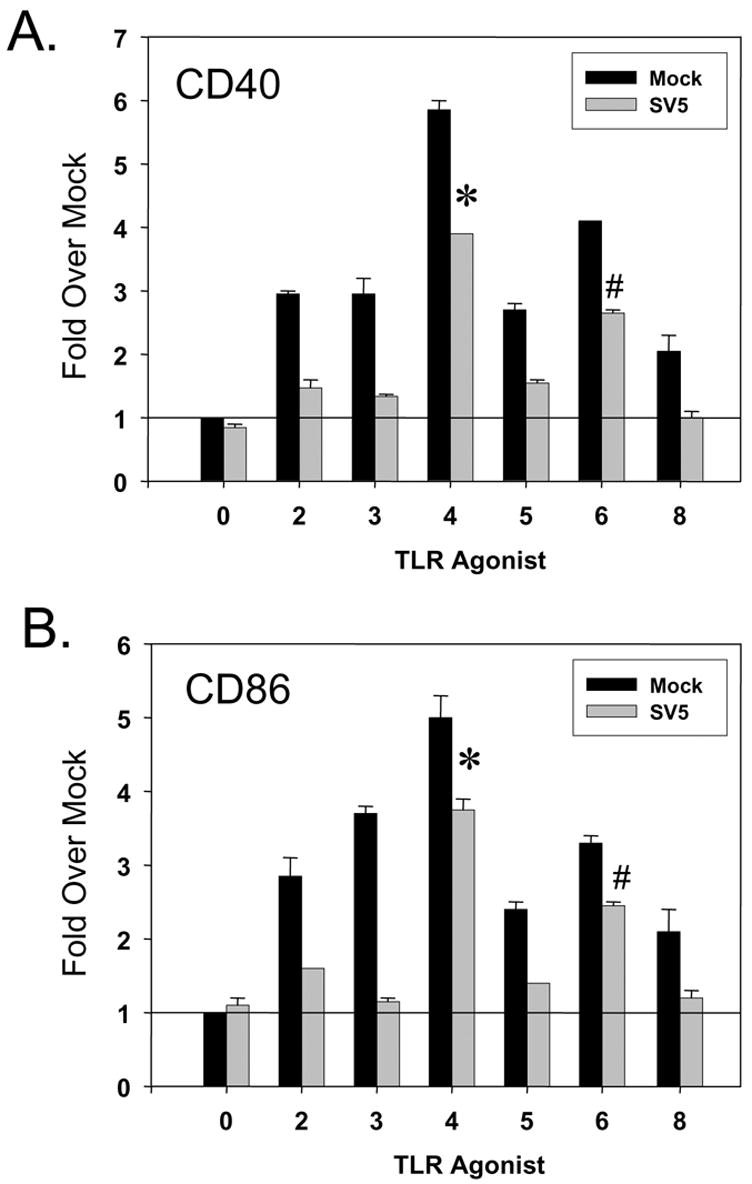
Immature moDC were mock infected or infected at an moi of 5 with WT rSV5. At 7 h pi, cells were mock treated or treated with the indicated TLR agonists as detailed in Materials and Methods. Levels of cell surface CD40 (panel A), CD86 (panel B) and CD80 (panel C) were determined by flow cytometry. Mean fluorescent intensities from three experiments using three individual donors (with standard error bars) are expressed as a fold change relative to mock infected, mock treated sample set at 1 (indicated by horizontal line). *, P values for CD40, CD86 and CD80 on TLR-4 agonist treated SV5 infected cells compared to untreated infected control cells were 0.0003, 0.002 and 0.005, respectively. #, P values for CD40, CD86 and CD80 on TLR-6 agonist treated infected cells compared to untreated SV5 infected control cells were 0.002, 0.04, and 0.007, respectively.
Treatment of infected moDC with agonists for TLR-2, -3, -5, and -8 resulted in only minimal increases in CD40, CD86 or CD80 surface expression (Fig. 4, gray bars). By contrast, treatment of infected cells with TLR-4 or -6 agonists resulted in elevated levels of all three maturation markers, with the strongest response coming from treatment with the TLR-4 activator LPS. The response of SV5 infected immature moDC was most clear in the case of CD80, where addition of TLR-4 and TLR-6 agonists gave ~5.5 and 2.5 fold increases in surface marker expression compared to the lack of response following treatment with TLR-2, -3, -5, and -8 agonists. Cell surface maturation markers on TLR-4 and -6 treated cells were always lower for infected cells compared to agonist-treated mock infected cells, but were always higher than that of untreated SV5-infected DC.
Taken together, these data indicate that SV5 infected immature moDC respond to TLR-4 and -6 agonists by expressing higher levels of cell surface maturation markers CD40, CD86, and CD80, as well as secretion of prototypic immunomodulatory cytokines. Thus, the lack of DC maturation following WT SV5 infection is not the result of a global block in maturation pathways
SV5-induced CPE in human DCs is reduced by TLR-4 and -6 agonists without elimination of virus replication
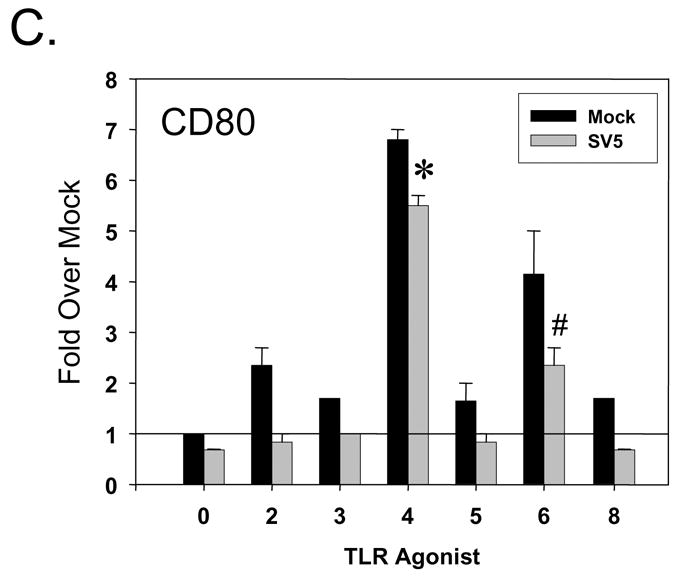
During our analysis, we observed that SV5 infected immature moDC showed reduced CPE following treatment with TLR-4 and -6 agonists (see below). To determine if treatment with TLR ligands altered SV5-induced apoptosis, immature moDC were mock infected or infected with WT SV5 at high moi. At 7 h pi, cells were treated with TLR agonists as described above, and levels of cleaved caspase-3 and surface annexin-V staining were determined by flow cytometry at 22 h pi. In the absence of TLR challenge, > 50% of WT SV5 infected immature DC expressed high levels of active caspase-3 (0 TLR samples, gray bars, Fig. 5A), and more than 40% of infected cells were positive for surface annexin-V staining (Fig. 5B). There was only a modest change in levels of cleaved caspase-3 and annexinV staining with infected cells treated with agonists for TLR-2, -3, -5, and -8. By sharp contrast, there was a substantial decrease in the percentage of infected cells with active caspase-3 staining following treatment with TLR-4 agonist LPS (<10%) or the TLR-6 agonist FSL1 (<20%). Similarly, the percentage of infected cells staining positive for Annexin-V was decreased by treatment with TLR-4 and -6 agonists (Fig. 5B), while other TLR ligands had much less of an effect.
Figure 5. Apoptotic markers in SV5 infected immature moDC are reduced by treatment with TLR-4 and -6 agonists.
Mock infected or WT rSV5 infected immature DC were treated at 7 h pi with the indicated TLR agonists as detailed in Materials and Methods. Cells were analyzed by flow cytometry at 22 h pi for levels of intracellular cleaved caspase-3 (panel A) or cell surface annexinV staining (panel B). Results are the mean values (with standard error bars) from three experiments with three individual donors. *, P values for percent active caspase-3 and annexin positive cells on TLR-4 agonist treated SV5 infected cells compared to untreated infected control cells were 0.007. #, P values for percent active caspase-3 and annexin positive cells on TLR-6 agonist treated infected cells compared to untreated infected control cells were 0.01 and 0.017, respectively.
To confirm these results, TLR agonist-treated immature moDC were analyzed by microscopy. As shown in Fig. 6A, the majority of immature mock-infected and untreated (UT) DC were found as flat cells with a small percentage of rounded but attached cells. This contrasts with the large CPE seen following rSV5-EGFP infection (Fig. 6A), including cell rounding, detachment from the dish, and increased cellular debris. Importantly, treatment of infected immature DC with the TLR-4 agonists LPS or TLR-6 agonist FSL1 resulted in a decrease in CPE and cellular debris, and a large fraction of cells were found in an elongated form as well as rounded cells still attached to the dish (Fig. 6A). Similar morphological changes were induced by agonist treatment of mock infected cells. Immature DC infected with both WT rSV5 or rSV5-EGFP showed reduced CPE following agonist treatment with was indistinguishable (not shown).
Figure 6. TLR-4 and -6 agonist treatment of SV5 infected immature moDC does not reduce virus replication.
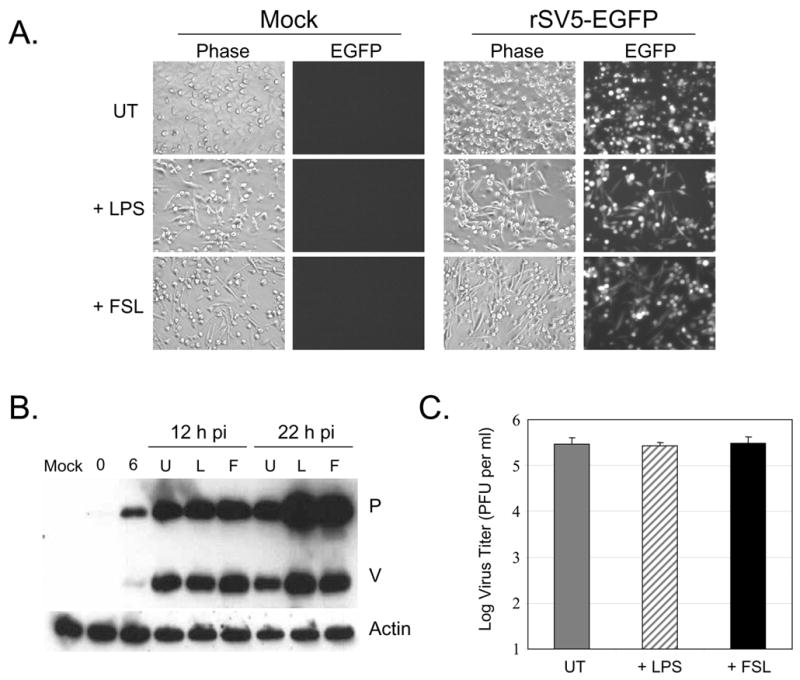
A) Immature moDC from a representative donor were mock infected or infected with rSV5-EGFP at an moi of 5. At 6 h pi, samples were left untreated (UT) or treated with the TLR-4 agonist LPS (200 ng/ml) or TLR-6 agonist FSL1 (100 ng/ml). Phase contrast pictures and EGFP expression were assayed at 22 h pi. Representative of three experiments. B) Immature moDC were infected as described for panel A, except that cell lysates were prepared at 0 and 6 h pi. Parallel samples were then left untreated (U lanes) or treated with LPS (L lanes) or FSL1 (F lanes). Cell lysates were prepared at 12 and 22 h pi, and levels of SV5 P and V proteins or cellular actin were assayed by western blotting. Representative of two experiments. C) At 6 h pi, immature moDC infected with rSV5-EGFP were left untreated (UT) or treated with LPS or FSL1. Virus titers were determined at 22 h pi. Values are the average of three experiments using three donors with standard deviations indicated by bars.
As shown in Fig. 6A, LPS or FSL1 treatment of SV5-EGFP infected DC did not reduce EGFP expression, suggesting that TLR activation of maturation pathways did not abort viral replication. To directly test this, rSV5-EGFP infected immature moDC were treated with LPS or FSL1 at 6 h pi, and the accumulation of viral proteins over time was analyzed by western blotting. As shown in Fig. 6B, viral P and V protein levels in infected immature moDC increased between 6 and 12 h pi, but importantly, the increased levels of P and V did not differ significantly between control untreated cells and cells treated with LPS or FSL1 (Fig. 6B, U lane versus L and F lanes). By 22 h pi, the levels of P and V proteins were slightly higher in cells treated with the TLR-4 and TLR-6 agonists. In addition, treatment of WT rSV5 infected immature moDC with LPS or FSL1 did not decrease virus yield compared to untreated cells (Fig 6C). Taken together, these data indicate that treatment of SV5 infected immature moDC with TLR-4 or TLR-6 ligands leads to enhanced DC maturation and reduced apoptosis and CPE, but does not reduce virus replication.
Reduced CPE and enhanced maturation following treatment of SV5-infected immature moDC with the non-TLR activator IL-1β
The above data indicate that activation of TLR-4 and -6 pathways in SV5 infected immature moDC leads to decreased apoptosis and maturation. Since TLR activation leads to induction of NFkB activity (Pasare and Medzhitov), we hypothesized that other non-TLR ligands that similarly activate NFkB could have a similar effect on infected immature DC. To test this hypothesis, mock infected or WT rSV5 infected immature moDC were treated at 6 h pi with or without IL-1beta (10 ng/ml). At 24 h pi, DC maturation and apoptotic markers were analyzed by flow cytometry. As shown in Fig. 7A and B, IL-1beta treatment increased surface expression of CD40 and CD80 on mock infected as well as SV5-infected cells. The levels of maturation marker expression induced by IL-1beta were lower than that seen after LPS treatment, but were similar to that induced by the TLR-6 agonist FSL1. Importantly, IL-1beta treatment of infected immature DC also reduced the percentage of cells with high levels of activated caspase-3 (Fig. 7C) and overall CPE in the culture (not shown). Together, these data indicate that the ability of exogenous ligands to increase maturation and decrease CPE and apoptosis of SV5-infected immature DC is not limited to responses initiated by the TLR agonists LPS and FSL1.
Figure 7. Enhanced maturation and reduced CPE in SV5 infected moDC treated with the non-TLR agonist IL-1beta.
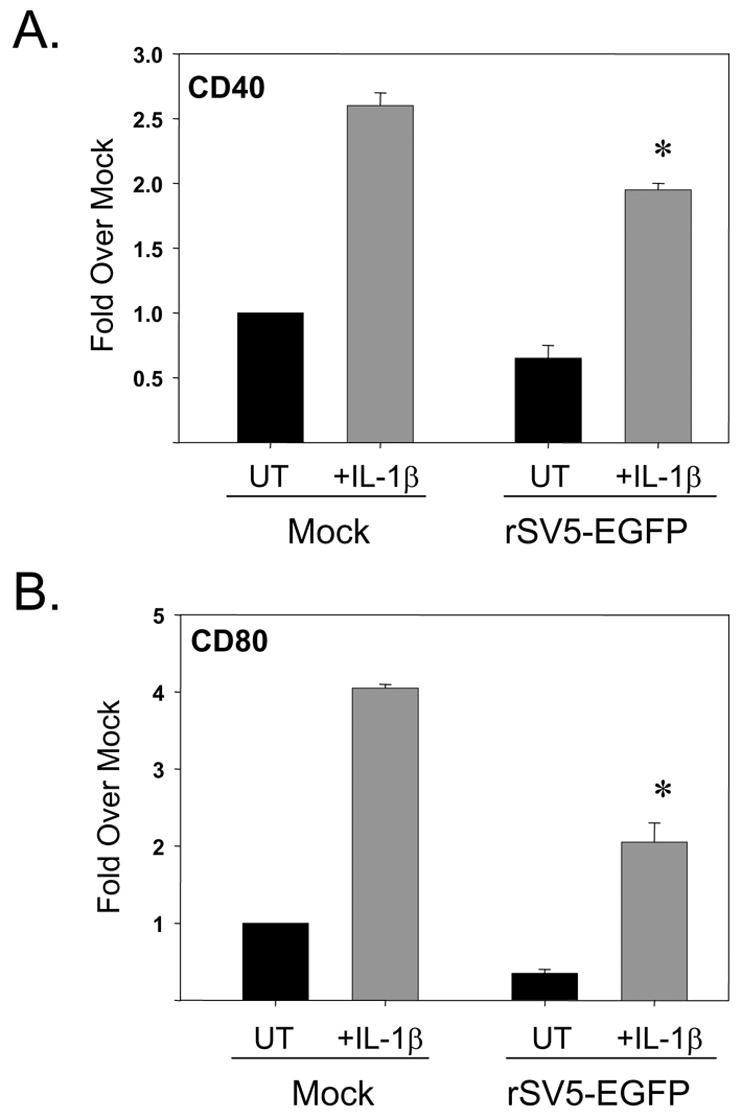
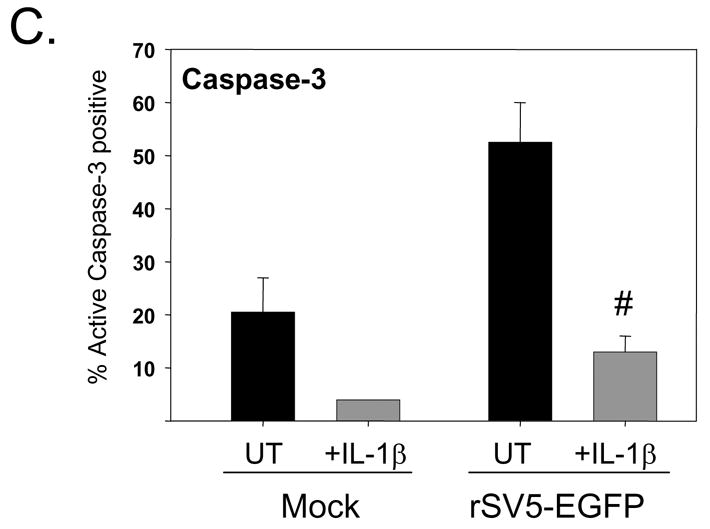
Immature moDC were mock infected or infected at high moi with WT rSV5. At 6 h pi, cells were left untreated (UT samples) or treated with 10 ng/ml IL-1beta. Levels of cell surface CD40 (panel A), CD80 (panel B), and intracellular cleaved caspase-3 (panel C) were determined by flow cytometry. Results are the average of mean fluorescent intensities from three experiments using three individual donors (with standard error bars). CD40 and CD80 results are expressed as a fold change relative to mock infected, mock treated sample set at 1. *, P values for CD40 and CD80 on IL-1beta treated infected cells compared to untreated infected control cells were 0.007 and 0.02, respectively. #, P values for percent active caspase-3 on IL-1beta treated infected cells compared to untreated infected control cells was 0.05.
TLR-4 mediated maturation and reduced apoptosis of SV5 infected immature moDC is blocked by NFkB inhibitors
The above data demonstrate an inverse relationship between the maturation of SV5 infected immature moDC by exogenous stimuli and reduced levels of CPE and apoptosis. To test the hypothesis that maturation and reduced apoptosis were linked through common pathways, infected immature moDC were treated with inhibitors of NFkB, a transcription factor that is essential for TLR-mediated induction of CD40 and CD80 (Ardeshna et al. 2000). Immature DC were mock infected or infected at high moi with WT rSV5 and cells were then cultured with or without the NFkB inhibitor PDTC (Schreck et al., 1992). At 6 h pi, samples were left unchallenged or challenged with LPS to induce maturation.
As shown in Fig. 8, addition of PDTC to both mock infected and WT rSV5 infected cells had little effect on the levels of maturation markers CD40 and CD80 (compare UT and P bars, panels A and B), as well as the percentage of infected cells with high apoptotic markers (compare UT and P bars, panels C and D). LPS treatment enhanced maturation marker expression and decreased apoptotic markers on both mock infected and infected immature moDC, as shown in prior figures. Most importantly, in the presence of the NFkB inhibitor, LPS treatment did not increase maturation of infected or control immature moDC, and the percentage of infected cells with high apoptotic markers was similar to that of untreated (UT) or PDTC treated cultures (P). Very similar results were found using the TLR-6 agonist FSL1 and the non-TLR ligand IL-1beta as inducers of maturation and Bay11943 as an alternative NFkB inhibitor (not shown). These data indicate that NFkB is a common factor playing a key role in the ability of exogenous ligands to enhanced maturation and override apoptosis in SV5 infected immature moDC.
Figure 8. TLR-4 mediated maturation and reduced apoptosis of SV5 infected immature moDC is blocked by NFkB inhibitors.
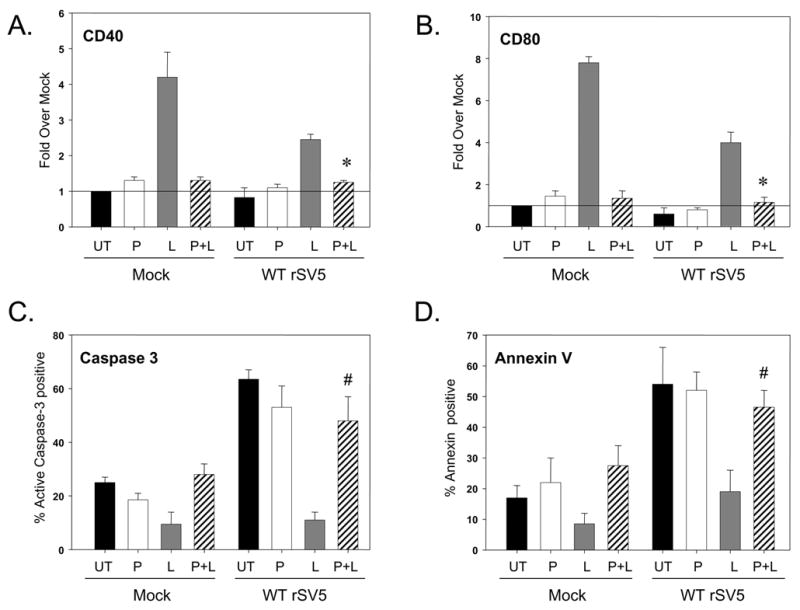
Immature moDC were mock infected or infected at high moi with WT rSV5 and left untreated (UT; black bars) or cultured in the presence of 10 uM PDTC (P, white bars). At 6 h pi, 200 ng/ml LPS was added to samples lacking PDTC (L, gray bars) or samples treated with PDTC (P + L, striped bars). At 22 h pi, samples were analyzed as detailed in the legend to Fig. 7. *, P values for CD40 and CD80 on P + L treated infected cells compared to L treated infected control cells were 0.006 and 0.018, respectively. #, P values for percent active caspase-3 and annexin positive cells on P + L treated infected cells compared to L treated infected control cells were 0.1 and 0.034, respectively.
DISCUSSION
The paramyxovirus SV5 is unusual among this family of viruses since it is a poor inducer of apoptosis in human epithelial- and fibroblast-derived cells (Choppin, 1964; Lin et al, 2003; He et al., 2002; Wansley and Parks, 2002; Wansley et al., 2003). Remarkably, the ability of SV5 to limit the activation of host cell death pathways is not evident in primary cultures of human moDC (Arimilli et al, 2006), since WT SV5 infection results in high CPE and activation of apoptosis. In the case of the paramyxoviruses respiratory syncytial virus (RSV) and human parainfluenza virus type 3 (HPIV-3), infection of human immature DC is highly cytopathic, but infected cells still upregulate maturation markers (Bartz et al., 2003; Guerrero-Plata et al, 2006; Plotnicky-Gilquin et al., 2001). This contrasts with the cytopathic SV5 infection of immature DC that does not result in maturation. The goal of the work described here was to determine the relationship between activation of cell death and the lack of maturation in SV5 infected immature moDC. We have shown that under conditions of reduced CPE and apoptosis, infected immature DC did not show a substantial increase in cell surface maturation markers. This result demonstrates that the lack of maturation is not the result of apoptosis per se, and suggests that SV5 infection either blocks maturation or does not produce viral components that are sufficient for inducing maturation.
Virus induced DC maturation can be viewed in part as an anti-viral response to infection, since many of the signaling pathways and cellular sensors are shared between these two host cell responses (Lopez et al., 2003; 2006). Thus, viruses that specifically target innate immune responses may also target DC maturation pathways. A number of negative strand RNA viruses have been shown to impose a block in the ability of DC to mature. For example, influenza virus attenuates maturation of immature moDC through the action of the NS1 protein (Fernandez-Sesma et al., 2006). RSV infection of human moDC and pDC can result in a block in dsRNA-mediated activation of maturation pathways and IFN production (Guerrero-Plata et al, 2006; Schlender et al., 2005). In a mouse transgenic system using immature bone marrow-derived DC, measles virus infection results in a block in the IL-12 response to TLR-4 agonists by a mechanism that is independent of the viral V or C proteins (Hahm et al., 2006).
Based on these examples and the known effect of SV5 on suppressing antiviral signaling pathways in epithelial and fibroblast cells (Didcock et al., 1999a; 1999b; Poole et al., 2002), we tested the hypothesis that SV5 also imposed a global block in maturation pathways. Infected immature moDC showed a rapid loss of STAT1, consistent with SV5 imposing a block in IFN signaling pathways (Didcock et al. 1999a), and this occurred before detectable CPE and the appearance of apoptotic markers (Fig. 2). However, at this time pi, SV5 infected immature moDC were still capable of responding strongly to TLR-4 and TLR-6 agonists by showing upregulation of maturation markers CD40, CD80 and CD86. Thus, SV5 selectively targets the inhibition of some innate response pathways in moDC (e.g., IFN induction and signaling), but other innate pathways remain functional. The ability of SV5 infected immature moDC to respond to exogenous ligands was not limited to agonists 4 and 6, since IL-1beta also enhanced moDC maturation. This is consistent with previous results with epithelial cells showing that while SV5 is a poor inducer of cytokines, infection does not result in a block in the ability of infected cells to respond to exogenous cytokines such as TNF-alpha or IL-1beta (Young et al., 2003). Together, these data demonstrate that the lack of maturation of SV5 infected immature moDC is not due to a global inhibition of signaling pathways initiated by exogenous ligands.
When TLR agonists were tested, maturation of SV5 infected moDC was clearly apparent with TLR-4 and -6 ligands, indicating that SV5 did not impose a global block on DC maturation. The lack of response to TLR-2, -3, -5 and -8 agonists could reflect specific blocks in these individual signaling pathways, or a nonspecific CPE that precludes responses to treatment. The ability of infected cells to specifically respond to TLR-4 and -6 ligands raises the question of what unique features allow these signaling pathways to override SV5-induced apoptosis and activate DC maturation. TLR-6 functions as an oligomer with TLR-2, but the TLR-6/2 complex recognizes distinct microbial products and activates cells through different pathways than TLR-2 homo-oligomers (Iwasaki and Medzhitov, 2004). Importantly, TLR-4 and TLR-6/2 signaling pathways have been shown to differ from other TLR pathways (Takeda and Akira, 2004) by their dependence on MyD88 in conjunction with a cytoplasmic adaptor protein called TIRAP (TIR domain-containing adaptor protein) or Mal (MyD88-adaptor-like). Thus, the ability of moDC to signal through TIRAP-dependent pathways may be responsible for the striking ability of TLR-4 and TLR-6/2 agonists to reverse apoptosis and promote maturation of SV5 infected DC compared to other TLR agonists.
Why does SV5 fail to induce maturation of human immature moDC, despite growing to high titers and producing large quantities of viral gene products? Our results are similar to those reported for Lassa virus (Baize et al., 2006), where infection of immature moDC is highly productive but DC do not express maturation markers unless they are challenged with exogenous TLR agonists. Baize et al. (2006) proposed that infected DC do not mature because Lassa virus replication does not produce high levels of key viral components that trigger intracellular signaling pathways. DC maturation through intracellular pathways may occur by dsRNA-activated cellular proteins such as PKR, MDA-5 or RIG-I (Lopez et al., 2006), and may require a threshold level of dsRNA to activate signaling pathways. It has recently been shown using transfected cell lines that the paramyxovirus V protein can inhibit MDA-5 but not RIG-I pathways (Childs et al., 2007). RIG-I has been shown to play an important role in IFN and proinflammatory cytokine responses in paramyxovirus infected murine cDC (Kato et al., 2005), raising the question of why the high levels of SV5 replication in immature moDC do not activate RIG-I dependent maturation pathways. Virus preparations that are rich in defective interfering (DI) particles are potent inducers of DC maturation (Yount et al., 2006), which presumably reflects the large amounts of dsRNA produced by copy-back DI particles during replication. Thus, SV5 may limit TLR-independent DC maturation by limiting the production of dsRNA or other intracellular signals. Taken together, our data are consistent with the proposal of Baize et al. (2006) that the lack of maturation of infected immature moDC is not due to a global block in signaling, but rather, results from virus replication that fails to produce significant levels or types of key components such as dsRNA that trigger intracellular maturation pathways (Lopez et al., 2006).
Treatment of SV5 infected immature moDC with exogenous ligands revealed an inverse relationship between the induction of maturation and decreased apoptosis. It is well established that in DC the upregulation of maturation markers (Ardeshna et al. 2000) and proinflammatory cytokine synthesis are dependent on NFkB activation. NFkB can also play important roles in expression of either proapoptotic or antiapoptotic genes, depending on the host cell type and the type of stimulation (Dutta et al., 2006). In this regard, immature moDC have been shown to be dependent on NFkB for both maturation and survival following LPS treatment (Ardeshna et al, 2000). Consistent with this, results with NFkB inhibitors showed that in SV5 infected immature moDC, the ability of TLR-4 and TLR-6 agonists to both decreased apoptosis and to activate maturation are blocked by NFkB inhibitors. Thus, NFkB may serve as a common transcription factor for two distinct pathways that lead to maturation of SV5 infected DC as well as overriding apoptosis signals by inducing prosurvival genes (Ardeshna et al., 2000).
Our results suggest a model whereby SV5 infection of immature moDC activates apoptosis through pathways that are independent of NFkB, and this is consistent with our finding that NFkB inhibitors do not prevent SV5-induced apoptosis (Fig. 8). In our model, DC maturation pathways and NFkB function are not activated during virus replication because SV5 does not encode the prototypic TLR ligands described for other viruses (Boehme and Compton, 2004) and the production of intracellular signals such as dsRNA is limited. Addition of exogenous ligands to SV5 infected DC activates NFkB signaling, which leads to induction of genes encoding maturation markers and cytokines, as well as expression of pro-survival gene products which override virus-induced apoptosis.
Due to their critical role in activation of naïve T cells, there is intense interest in developing methods to promote maturation and antigen presentation in virus infected DC. In the case of viruses that are highly cytopathic to DC, cell killing could contribute to poor activation of T cell responses and adaptive immunity as proposed previously for a number of viruses (e.g., Muller et al. 2004; Wahid et al., 2005). It may be possible to develop vaccine strategies such that TLR-4 or -6/2 pathways are activated in virus infected DC, which could lead to decreased DC death and increased capacity of infected DC to initiate antiviral immune responses.
MATERIALS AND METHODS
Cells, viruses and reagents
Immature peripheral blood mononuclear cell (PBMC)-derived human DC were prepared as described previously (Arimilli et al, 2006). Briefly, PBMC were isolated from randomly selected healthy donors by standard density gradient centrifugation on Ficoll-Hypaque. CD14+ cells were isolated by positive selection using CD14 microbeads. Typically, the purity of monocytes isolated by this procedure was ≥95%. Enriched CD14+ cells (106 cells/ml in 5 ml) were cultured for six days in culture medium containing 10 ng/ml human interleukin-4 and 10 ng/ml human granulocyte-macrophage colony-stimulating factor. On day 3, half of the medium was replaced with fresh medium supplemented with cytokines to the above final concentrations. Greater than 95% of this immature DC population was CD11c+ (not shown).
Recombinant WT SV5 (WT rSV5) was recovered as described previously (He et al. 1997) from cDNA plasmid kindly provided by Robert Lamb and Biao He (Northwestern University). The rSV5-GFP infectious clone kindly provided by Robert Lamb and Biao He was used as starting material to generate an rSV5 that encoded the gene for enhanced green fluorescence protein (EGFP) between HN and L (details available on request). The EGFP open reading frame also contained an 18 residue C-terminal extension encoding the immunodominant p60 epitope from Lysteria monocytogenes (single letter amino acid code: ALSVKYGVSVQDIMSWNN). Viruses were grown in MDBK cells, and purified as described previously (Arimilli et al., 2006). SV5 infections were carried out as described previously (Arimilli et al., 2006). In experiments with TLR agonists, DC were infected and then treated at the indicated time post infection with the following agonists according to the manufacturers recommendations (Invivogen; San Diego, CA): heat killed Listeria monocytoenes for TLR2 (108 cells per ml); Poly(I:C) for TLR3 (2.5 ug/ml); E. coli K12 LPS for TLR4 (200 ng/ml); S. typhimurium flagellin for TLR5 (200 ng/ml); Pam2CGDPKHPKSF (FSL1), a synthetic lipoprotein from Mycoplasma salivarium, for TLR6 (100 ng/ml); Imiquimod R837 for TLR7 (1 ug/ml); ssRNA40 for TLR8 (1 ug/ml). Cells were analyzed for maturation markers or cytokine secretion at 22–24 h pi. Pyrrolidine dithiocarbamate (PDTC) and z-VAD-FMK were from Sigma and Promega, respectively.
Cytokine production and cell surface staining
Cytokines IL-1β, IL-6, IL-8, IL-10, IL-12p70 and TNF-alpha, were quantified by an inflammatory response cytometric bead array assay (BD Pharmingen, San Diego, CA). For detection of surface markers, DCs were stained with the following conjugated monoclonal antibodies according to the manufacturer’s recommendations, CD11c (B-ly6), CD14 (M5E2), CD40 (5C3), CD80 (L307.4) and CD86 (FUN-1) all from BD Pharmingen (San Diego, CA). Samples were analyzed on a FACScan instrument using Cell Quest software (Becton Dickinson, San Diego, CA). A Student’s t test was used to determine significance between samples.
Western blotting, microscopy and cytometric analyses of apoptotic cells
Levels of viral and cellular protein were analyzed by western blotting as described previously (Arimilli et al, 2006). Equivalent amounts of protein were analyzed by western blotting using polyclonal rabbit antibodies specific for SV5 P (Parks et al., 2001), a monoclonal antibody V5 specific for an epitope in the N-terminal region of the SV5 P and V (Invitrogen), rabbit polyclonal antibodies against cellular STAT1 protein (clone 554, Santa Cruz Biotechnology), or a monoclonal antibody specific for actin (Sigma) followed by HRP-conjugated secondary antibodies and ECL.
Microscopy was carried out as described previously (Arimilli et al, 2006) using a Nikon Eclipse fluorescence microscope using a 20X lens. Images were captured using a QImaging digital camera and processed using QCapture software. Exposure times were manually set to be constant between samples.
Apoptosis was measured as described previously (Arimilli et al 2006) by intracellular staining for active caspase−3 using PE-conjugated polyclonal anti-caspase3 antibody (Pharmingen, San Diego, CA) along with CD11c-APC, or by measuring cell surface staining with PE-conjugated AnnexinV. Samples were analyzed by flow cytometry. A Student’s t test was used to determine significance between samples.
Acknowledgments
We thank members of the Parks and Alexander-Miller labs for helpful comments on the manuscript. We are grateful to Drs. Biao He and Robert A. Lamb (Northwestern University) for the gift of the rSV5 infectious clones. This work was supported by NIH grants HL-071985 (MAM) and AI-060642 (GDP, SBM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexopoulou LA, Holt C, Medzhitov R, Flavell Ra. Recognition of double-stranded RNA and activation of NF-kB by toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Ardeshna KM, Pizzey AR, Devereux S, Khwaja A. The PI3 kinase, p38 SAP kinase, and NF-kB signal transduction pathways are involved in the survival and maturation of lipopolysaccharide-stimulated human monocyte-derived dendritic cells. Blood. 2000;96:1039–1046. [PubMed] [Google Scholar]
- Arimilli S, Alexander-Miller MA, Parks GD. A simian virus 5 (SV5) P/V mutant is less cytopathic than wild-type SV5 in human dedritic and is a more effective activator of dendritic cell maturation and function. J Virol. 2006;80:3416–3427. doi: 10.1128/JVI.80.7.3416-3427.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baize S, Kaplon J, Faure C, Pannetier D, Georges-Courbot MC, Deubel V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J Immunol. 2004;172:2861–2869. doi: 10.4049/jimmunol.172.5.2861. [DOI] [PubMed] [Google Scholar]
- Baize S, Pannetier D, Faure C, Marianneau P, Marendat L, Georges-Courbot MC, Deubel V. Role of interferons in the control of Lassa virus replication in human dendritic cells and macrophages. Microbes and Infection. 2006;8:1194–1202. doi: 10.1016/j.micinf.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- Barchet W, Cella M, Colonna M. Plasmacytoid dendritic cells- virus experts of innate immunity. Semin Immunol. 2005;17:253–261. doi: 10.1016/j.smim.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Bartz H, Turkel O, Hoffjan S, Rothoeft T, Gonschorek A, Schauer U. Respiratory syncytial virus decreases the capacity of myeloid dendritic cells to induce interferon-gamma in naïve T cells. Immunobiology. 2003;109:49–57. doi: 10.1046/j.1365-2567.2003.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron CA. Initial and innate responses to viral infections--pattern setting in immunity or disease. Curr Opin Microbiol. 1999;2:374–381. doi: 10.1016/s1369-5274(99)80066-6. [DOI] [PubMed] [Google Scholar]
- Boehme KW, Compton T. Innate sensing of viruses by toll-like receptors. J Virol. 2004;78:7867–7873. doi: 10.1128/JVI.78.15.7867-7873.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosio CM, Aman MJ, Grogan C, Hogan R, Ruthel G, Negley D, Mohamadzadeh M, Bavari S, Schmaljohn A. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J Infec Dis. 2003;188:1630–1638. doi: 10.1086/379199. [DOI] [PubMed] [Google Scholar]
- Childs K, Stock N, Craig R, Andrejeva J, Hilton L, Skinner M, Randall R, Goodbourn S. Mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007 doi: 10.1016/j.virol.2006.09.023. in press. [DOI] [PubMed] [Google Scholar]
- Choppin PW. Multiplication of a myxovirus (SV5) with minimal cytopathic effects and without interference. Virology. 1964;23:224–233. doi: 10.1016/0042-6822(64)90286-7. [DOI] [PubMed] [Google Scholar]
- Didcock L, Young DF, Goodbourn S, Randall RE. Sendai virus and SV5 block activation of IFN-responsive genes: importance of virus pathogenesis. J Virol. 1999a;73:3125–3133. doi: 10.1128/jvi.73.4.3125-3133.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didcock L, Young DF, Goodbourn S, Randall RE. The V protein of SV5 inhibits interferon signaling by targeting STAT1 for proteasome-mediated degradation. J Virol. 1999b;73:9928–9933. doi: 10.1128/jvi.73.12.9928-9933.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold S, Kaisho S, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR-7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–15431. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. Current insights into the regulation of program cell death by NFkB. Oncogene. 2006;25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- Engelmayer J, Larsson M, Subklewe M, Chahorudi A, Cox WI, Steinman RM, Bhardwaj N. Vaccinia virus inhibits the maturation of human dendritic cells: a novel mechanism of immune evasion. J Immunol. 1999;163:6762–6768. [PubMed] [Google Scholar]
- Fernandez-Sesma A, Marukian S, Ebersole BJ, Kaminski D, Park M-S, Yuen T, Sealfon SC, Garcia-Sastre A, Moran TM. Influenza virus evades innate an dadaptive immunity via the NS1 protein. J Virol. 2006;80:6295–6304. doi: 10.1128/JVI.02381-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean I, Caux C, Bella C, Berger I, Wild F, Banchereau J, Kaiserlian D. Measles virus infects human dendritic cells and blocks their allostimulatory properties for CD4+ T cells. J Exp Med. 1997;186:801–812. doi: 10.1084/jem.186.6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Plata A, Casola A, Suearez G, Yu X, Spetch L, Peeples ME, Garofalo RP. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respr Cell Mol Biol. 2006;34:320–329. doi: 10.1165/rcmb.2005-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm B, Cho JH, Oldstone MBA. Measles virus-dendritic cell interactcion via SLAM inhibits innate immunity: Selective signaling through TLR4 but not other TLRs mediates suppression of IL-12 synthesis. Virology. 2007;358:251–257. doi: 10.1016/j.virol.2006.10.004. [DOI] [PubMed] [Google Scholar]
- He B, Paterson RG, Ward CD, Lamb RA. Recovery of infectious SV5 from cloned DNA and expression of a foreign gene. Virology. 1997;237:249–260. doi: 10.1006/viro.1997.8801. [DOI] [PubMed] [Google Scholar]
- He B, Paterson RG, Stock N, Durbin JE, Durbin RK, Goodbourn S, Randall RE, Lamb RA. Recovery of paramyxovirus simian virus 5 with a V protein lacking the conserved cysteine-rich domain: the multifunctional V protein blocks both interferon-beta induction and interferon signaling. Virology. 2002;303:15–32. doi: 10.1006/viro.2002.1738. [DOI] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Liford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1481–1482. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Hengel H, Koszinowski UH, Conzelmann KK. Viruses know it all: new insights into IFN networks. Trends Immunol. 2005;26:396–401. doi: 10.1016/j.it.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immun. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Klagge IM, Schneider-Schaulies S. Virus infections with dendritic cells. J Gen Virol. 1999;80:823–833. doi: 10.1099/0022-1317-80-4-823. [DOI] [PubMed] [Google Scholar]
- Lin Y, Bright AC, Rothermel TA, He B. Induction of apoptosis by paramyxovirus simian virus 5 lacking a small hydrophobic protein. J Virol. 2003;77:3371–383. doi: 10.1128/JVI.77.6.3371-3383.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CB, Garcia-Sastre A, Williams BR, Moran TM. Type I interferon induction pathways, but not released interferon, participates in the maturation of dendritic cells induced by negative strand RNA viruses. J Infect Dis. 2003;187:1126–1136. doi: 10.1086/368381. [DOI] [PubMed] [Google Scholar]
- Lopez CB, Yount JS, Moran TM. Toll-like receptor-independent triggering of dendritic cell maturation by viruses. J Virol. 2006;80:3128–3134. doi: 10.1128/JVI.80.7.3128-3134.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund J, Alexopolou ML, Sato A, Karow M, Adams NC, Gale MW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by toll-like receptor 7. Proc Natl Acad Sci USA. 2004;101:5598–5603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mckenna K, Beingnon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol. 2005;79:17–27. doi: 10.1128/JVI.79.1.17-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutaftsi M, Mehl AM, Borysiewicz LK, Tabi S. Human cytomegalovirus inhibits maturation and impairs function of monocyte-derived dendritic cells. Blood. 2002;99:2913–21. doi: 10.1182/blood.v99.8.2913. [DOI] [PubMed] [Google Scholar]
- Muller DB, Raftery MJ, Kather A, Giese T, Schonrich G. Frontline: Induction of apoptosis and modulation of c-FLIPL and p53 in immature dendritic cells infected with herpes simplex virus. Eur J Immunol. 2004;34:941–951. doi: 10.1002/eji.200324509. [DOI] [PubMed] [Google Scholar]
- Parks GD, Ward KR, Rassa JC. Increased readthrough transcription across the simian virus 5 M-F gene junction leads to growth defects and a global inhibition of viral mRNA synthesis. J Virol. 2001;75:2213–2223. doi: 10.1128/JVI.75.5.2213-2223.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 2004;6:1382–1387. doi: 10.1016/j.micinf.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Plotnicky-Gilquin H, Cyblat D, Aubry JP, Delneste Y, Blaecke A, Bonnefoy JY, Corvaia N, Jeannin P. Differential effects of parainfluenza virus type 3 on human monocytes and dendritic cells. Virology. 2001;285:82–90. doi: 10.1006/viro.2001.0933. [DOI] [PubMed] [Google Scholar]
- Poole E, He B, Lamb RA, Randall RE, Goodbourn S. The V proteins of simian virus 5 and other paramyxoviruses inhibit induction of interferon-β. Virology. 2002;303:33–46. doi: 10.1006/viro.2002.1737. [DOI] [PubMed] [Google Scholar]
- Raftery MJ, Kraus AA, Ulrich R, Kruger DH, Schonrich G. Hantavirus infection of dendritic cells. J Virol. 2002;76:10724–33. doi: 10.1128/JVI.76.21.10724-10733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis e Sousa C. Dendritic cells as sensors of infection. Immunity. 2001;14:495. doi: 10.1016/s1074-7613(01)00136-4. [DOI] [PubMed] [Google Scholar]
- Salio M, Cella M, Suter M, Lanzavecchia A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur J Immunol. 1999;29:3245–53. doi: 10.1002/(SICI)1521-4141(199910)29:10<3245::AID-IMMU3245>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Schlender J, Hornung V, Finke S, Gunthner-Biller M, Marozin S, Brzozka K, Moghim S, Endres S, Hartmann G, Conzelmann KK. Inhibition of toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J Virol. 2005;79:5507–5515. doi: 10.1128/JVI.79.9.5507-5515.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Schaulies S, Klagge IM, ter Meulen V. Dendritic cells and measles virus infection. Curr Top Microbiol Immunol. 2003;276:77–101. doi: 10.1007/978-3-662-06508-2_4. [DOI] [PubMed] [Google Scholar]
- Schreck R, Meier B, Mannel DN, Droger W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor kB activation in intact cells. J Exp Med. 1992;175:1181–1194. doi: 10.1084/jem.175.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Akira S. Microbial recognition by toll-like receptors. J Derm Sci. 2004;34:73–82. doi: 10.1016/j.jdermsci.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Wahid R, Cannon MJ, Chow M. Dendritic cells and macrophages are productively infected by poliovirus. J Virol. 2005;79:401–409. doi: 10.1128/JVI.79.1.401-409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansley EK, Parks GD. Naturally occurring substitutions in the P/V gene convert the noncytopathic paramyxovirus simian virus 5 into a virus that induces alpha/beta interferon synthesis and cell death. J Virol. 2002;76:10109–21. doi: 10.1128/JVI.76.20.10109-10121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansley EK, Grayson JM, Parks GD. Apoptosis induction and interferon signaling but not IFN-beta promoter induction by an SV5 P/V mutant are rescued by coinfection with wild-type SV5. Virology. 2003;316:41–54. doi: 10.1016/s0042-6822(03)00584-1. [DOI] [PubMed] [Google Scholar]
- Wansley EK, Dillon PJ, Gainey MD, Tam J, Cramer SD, Parks GD. Growth sensitivity of a recombinant simian virus 5 P/V mutant to type I interferon differs between tumor cell lines and normal primary cells. Virology. 2005;335:131–144. doi: 10.1016/j.virol.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Young VA, Parks GD. Simian virus 5 is a poor inducer of chemokine secretion from human lung epithelial cells: identification of viral mutants that activate interleukin-8 secretion by distinct mechanisms. J Virol. 2003;77:7124–7130. doi: 10.1128/JVI.77.12.7124-7130.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young VA, Dillon PJ, Parks GD. Variants of the paramyxovirus simian virus 5 with accelerated or delayed viral gene expression activate proinflammatory cytokines. Virology. 2006;350:90–102. doi: 10.1016/j.virol.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Yount JS, Krause TA, Horvath CM, Moran TM, Lopez CB. A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. J Immunol. 2006;177:4503–4513. doi: 10.4049/jimmunol.177.7.4503. [DOI] [PubMed] [Google Scholar]