

Abstract
Cerebral microvascular amyloid β protein (Aβ) deposition and associated neuroinflammation are increasingly recognized as an important component leading to cognitive impairment in Alzheimer’s disease and related cerebral amyloid angiopathy (CAA) disorders. Transgenic mice expressing the vasculotropic Dutch/Iowa (E693Q/D694N) mutant human Aβ precursor protein in brain (Tg-SwDI) accumulate abundant cerebral microvascular fibrillar amyloid deposits exhibiting robust neuroinflammation. In the present study, we sought to determine if the unique amyloid pathology of Tg-SwDI mice was associated with deficits in behavioral performance. Behavioral performance tests that assessed a variety of psychological functions, including overall activity, motor ability, balance and strength, anxiety, impulsivity, and learning were conducted on homozygous Tg-SwDI mice and similarly aged wild-type C57Bl/6 mice. Our results indicate that Tg-SwDI mice were impaired in the performance of the Barnes maze learning and memory task at 3, 9, and 12 months of age. While more widespread cerebral microvascular Aβ pathology was evident in older animals, the evaluation of the Aβ pathology in the 3 months old transgenic animals revealed specific accumulation of microvascular amyloid and markedly elevated numbers of reactive astrocytes and activated microglia restricted to the subiculum. These findings indicate that early-onset accumulation of subicular microvascular amyloid and accompanying neuroinflammation correlates with impaired performance in the learning and memory task in Tg-SwDI mice.
Keywords: cerebral microvascular amyloid, cognitive impairment, neuroinflammation, subiculum, transgenic mice
One of the primary pathological features of Alzheimer’s disease (AD), as well as other related disorders, is the accumulation of amyloid β-protein (Aβ) in the parenchyma and cerebral vasculature of the brain (Selkoe, 2001). Aβ is formed by the sequential cleavage of amyloid β-protein precursor (APP) by β- and γ-secretases. Parenchymal Aβ deposits come in two forms: diffuse plaques and fibrillar plaques. Although both types of plaque deposits have been associated with dementia, fibrillar deposits show dystrophic neurites and evidence of inflammation including activated microglia (Selkoe, 2001; Walsh and Selkoe, 2004). Cerebral vascular Aβ deposits are largely of fibrillar form and are associated with a localized neuroinflammatory response and cognitive impairment (Vinters, 2001; Bailey et al., 2004; Atterns and Jellinger, 2004; Greenberg et al., 2004). Furthermore, recent studies suggested that dementia shows a better correlation with microvascular Aβ accumulation than parenchymal Aβ accumulation (Thal et al., 2003; Atterns and Jellinger, 2004). However, direct demonstration of microvascular amyloid-induced cognitive impairment remains lacking.
Two mutations within the Aβ region of the APP gene that result in severe, early onset cerebral amyloid angiopathy (CAA) are the Dutch E22Q and Iowa D23N mutations (Levy et al., 1990; Van Broeckhoven et al., 1990; Grabowski et al., 2001). Recently, we created a line of transgenic mice (Tg-SwDI) that express human Swedish/Dutch/Iowa mutant APP in the brain and consequently develops early-onset, robust accumulation of fibrillar Aβ in the cerebral microvasculature (Davis et al., 2004). This accumulation occurs despite the mice expressing low levels of human APP and producing low levels of human mutant Aβ (Davis et al., 2004; Deane et al. 2004). The insufficient removal of the Dutch/Iowa mutant Aβ across the blood-brain barrier into circulation appears to contribute to this extensive accumulation (Deane et al., 2004; Davis et al., 2006). These mice were further shown to exhibit vascular degeneration and neuroinflammation specifically around the sites of microvascular amyloid deposits (Miao et al., 2005a; 2005b). Nevertheless, the relationship between cerebral microvascular amyloid-related pathological changes and behavioral performance in this unique model was unknown.
In the present study, we characterized the behavioral performance of Tg-SwDI mice as compared to non transgenic C57Bl/6 wild-type mice. Since the impact of cerebral microvascular amyloid pathology on behavior has not been assessed, we tested the mice on several inter-supporting tests that examined a range of psychological functions, including overall activity, motor ability, balance and strength, anxiety, impulsivity, and learning. Our results show that Tg-SwDI mice exhibit a specific deficit in the performance of a learning and memory test. This behavioral deficit was detected in Tg-SwDI mice as early as three months of age. Analysis of the Aβ pathology at this early age revealed accumulation of microvascular amyloid and highly elevated numbers of reactive astrocytes and activated microglia restricted to the subiculum. These findings indicate that early-onset accumulation of subicular microvascular amyloid and neuroinflammation is associated with impaired performance in a spatial learning and memory task in Tg-SwDI mice.
Experimental Procedures
Tg-SwDI mice
All work was carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23, revised 1996) and was approved by the Stony Brook University Institutional Animal Care and Use Committee. The generation of Tg-SwDI mice on a pure C57Bl/6 background was as described (Davis et al., 2004). For the behavioral studies and subsequent pathological analyses, all Tg-SwDI were homozygous for the human Swedish/Dutch/Iowa APP transgene. Homozygous Tg-SwDI mice developed more extensive pathology earlier but remained highly fertile and exhibited no early lethality. There were three age cohorts of behavioral testing at ages 3, 9, and 12 months. Each cohort consisted of 12–14 Tg-SwDI and wild-type C57Bl/6 mice. Each group consisted of an even mix of male and female animals. Mice were weighed at the beginning of each week of behavioral testing.
Tissue preparation
Mice were euthanized with an overdose of 2.5% avertin, the brains were immediately removed and bisected in the mid-sagittal plane. One hemisphere was snap-frozen and the other hemisphere was placed in 70% ethanol, followed by xylene treatment and embedding in paraffin for immunohistochemical and histological analyses.
Immunochemical analysis of cerebral Aβ peptides
Soluble pools of Aβ 40 and Aβ 42 were determined by using an specific enzyme-linked immunosorbent assay (ELISA) of carbonate extracted mouse forebrain tissue and subsequently the insoluble Aβ 40 and Aβ 42 levels were determined by ELISA of guanidine lysates of the insoluble pellets resulting from the carbonate extracted brain tissue (Johnson-Wood et al., 1997; DeMattos et al., 2002). Total Aβ 40 and Aβ 42 levels were determined by combining the soluble and insoluble levels of each form.
Soluble Aβ oligomers were analyzed in Tris buffered saline (TBS)-soluble forebrain fractions using dot blot analysis. Briefly, brain fractions were loaded to nitrocellulose membranes (Schleicher and Schuell, Hertogen-Bosch, The Netherlands). Blots were washed for 15 min in PBS containing 0.05% Tween 20 (PBST), preincubated with blocking solution (1% milk powder in PBST), washed three times with PBST, and subsequently incubated with primary polyclonal anti-oligomer antibody OC11 and peroxidase-labeled secondary antibodies (Amersham, Arlington Heights, IL). Detection was performed by chemiluminescence according to the description of the manufacturer (Biorad Laboratories, Hercules, CA) and quantitated using BioRad VersaDoc system and the Quantity One software.
Immunohistochemical analysis
Immunohistochemistry and histology were performed as previously described (Davis et al., 2004, Miao et al., 2005a). Briefly, sections were cut in the sagittal plane at 10 Jm thickness using a microtome, deparaffinated and rehydrated. Antigen retrieval was performed by treatment with proteinase K (0.2 mg/ml) for 10 min at 22°C for Aβ and collagen staining, and by 10 mM sodium citrate solution (pH 9.0) for 30 min at 90°C in a water-bath for activated microglia staining. Primary antibodies were detected with horseradish peroxidase-conjugated or alkaline phosphatase-conjugated secondary antibodies and visualized either with a stable diaminobenzidine solution (Invitrogen, CA) or with the fast red substrate system (Spring Bioscience, Fremont, CA), respectively, as substrate. Sections were counterstained with hematoxylin. Cerebral microvascular amyloid burden in the regions of the subiculum, thalamus and fronto-temporal cortex was respectively quantified on the same set of systematically sampled Aβ-immunostained sections using stereological principles as described (Long et al., 1998). The following antibodies were used for immunohistochemical analysis: mouse monoclonal antibody 66.1, which recognizes residues 1–5 of human Aβ (Deane et al., 2003) (1:200), rabbit polyclonal antibody to collagen type IV for identification of microvessel (1:100, Research Diagnostics Inc., Flanders, NJ), mouse monoclonal antibody to glial fibrillary acidic protein (GFAP) for the detection of astrocytes (1:300, Chemicon, Temecula, CA), mouse monoclonal anti-keratan sulfate antibody for the detection of activated microglia (clone: 5D4, 1:200, Seikagaku Corporation, Japan).
Quantitative analysis of reactive astrocyte and activated microglia cell densities
Total numbers of astrocytes and activated microglia in the subiculum, dentate gyrus, thalamus, and fronto-temporal cortex regions were estimated using a computerized stereology system (Stereologer, Systems Planning and Analysis, Alexandria, VA). Every tenth section was selected and generated 10–15 sections per reference space in a systematic-random manner. Immuno-positive cells were counted using the optical fractionator method with the dissector principle and unbiased counting rules (Long et al., 1998).
Behavioral testing
The following battery of behavioral tasks was utilized in the analysis of Tg-SwDI and wild-type mice. The measurements on all tests were either mechanically scored or objectively scored by the experimenters and each task was conducted by multiple two person teams.
Digiscan
On test Day 1, an assessment of overall activity was performed using a commercial Digiscan activity monitor (Columbus Instruments, Columbus, OH). Each mouse was placed in a plastic tub cage measuring 43.2 cm long by 21.6 cm wide by 20.3 cm deep for five minutes. Horizontal movements were recorded automatically. An experimenter blind to treatment condition counted unsupported rearings and wall-supported rearings from videotape records of the sessions.
Open field
Day 2 of testing assessed open field activity. The open field apparatus was 54 × 54 cm and is enclosed by a 19 cm high wall marked with lines forming thirty six 9 cm2 squares along the floor of the apparatus. The mice were placed in the open field for two minutes each and the number of lines the mouse crossed and number of times the mouse traversed the center four squares was recorded from videotape. Day 3 of testing assessed 2 min of open field activity again except that a novel object, e.g. a clear 6 cm3 plastic box filled with pennies, was present in the center of the field. Time to approach novel object in addition to total lines crossed is recorded from videotape records of the sessions.
Wirehang
Day 4 of testing assessed muscle endurance using an apparatus consisting of a 43 cm wire stretched between two wooden poles attached to a base where the wire was 48 cm above the base. Foam padding is placed on the platform to cushion a fall. The mouse’s two front paws were placed on the wire and the mouse allowed to hang allowed until falling off for a maximum of one minute. The latency to fall was recorded.
Inverted crawl
Day 5 of testing assessed strength and agility using an apparatus in which a section of metal grid hardware cloth 82.5 cm long by 9 cm wide and 56 cm above the base was stretched between two sealed, vertical wooden 2.5 × 10 cm planks attached to a base. All four paws of the mouse were placed on the bottom side of the hardware cloth so that they were hanging upside down and allowed to traverse for a maximum of one minute. The distanced traversed as well as total time hanging were recorded.
Light/dark box
Day 6 of testing employed a light/dark box apparatus (Crawley, 2000) to assess anxiety. The light/dark box was a 43 × 21.5 × 20 cm clear tub cage with 16.5 × 21.5 × 20 cm of the volume subdivided into a dark compartment with a 4 × 4 cm aperture that allowed the mouse to move freely between the dark side and the light side. The mouse was initially placed in the dark side of the box and then allowed to explore for five minutes. The number of entries into the light and total time spent in the light were recorded.
Rotarod
Days 7, 8, and 9 of testing use a standard Rotarod (Med Associates, Inc., St. Albans, VT) to assess motor performance and motor learning. The rod rotation increased over the course of the five min from 3 rotations per min (rpm) to 30 rpm on day one of testing, from 3.5 rpm to 35 rpm on day 2, and from 4 rpm to 40 rpm on day 3. Each mouse was given three trials per day, with a thirty-minute intertrial interval on a given day to reduce fatigue effects. Each trial was five min long or until the mouse fell off. The latency to fall was recorded automatically by the apparatus.
Barnes maze
Days 10 through 14 of testing utilize the Barnes maze apparatus (Barnes, 1979) to assess ability to learn the location of an escape box over the course of five days. The Barnes maze used is a circular table with a diameter of 91 cm, where eight, 5 cm diameter holes were located equidistantly around the perimeter. Each hole is 35.5 cm from the center of the table. The Barnes maze was positioned in the center of the test room and 71 cm from the ground. During a trial, the escape box, measuring 11 × 7.5 × 7.5 cm, is placed under the escape hole. The escape hole was in a different position for each mouse, but constant for each mouse over the five training days. Each mouse was tested twice per day for five days, with a fifteen-minute intertrial interval separating each trial. Each trial began when the mouse was released from a start box, measuring 16.5 × 9 × 5 cm, attached to a onemeter pole. Each trial lasted up to two min or until the mouse entered the escape box. The mice who did not find the escape hole within two minutes were guided by the experimenter to the correct hole. Once the mouse entered the escape box, it was allowed to remain in the box for one minute.
Statistical Analysis
Measures for the Barnes maze and the Rotarod were analyzed using a repeated measures ANOVA. One-way ANCOVAs, with body weight as the covariate, were run for the Wire Hang and Inverted Crawl. All other data were analyzed using one-way ANOVAs.
Results
Homozygous Tg-SwDI mice exhibit more extensive cerebral Aβ accumulation than heterozygous animals
Earlier we described the generation of Tg-SwDI mice, a novel transgenic mouse line expressing human Swedish/Dutch/Iowa vasculotropic mutant APP in neurons (Davis et al., 2004). Heterozygous Tg-SwDI mice were shown to develop early-onset and robust accumulation of fibrillar amyloid in the cerebral microvasculature with only diffuse Aβ deposits in the brain parenchyma (Davis et al., 2004; Miao et al., 2005a; 2005b). The fibrillar cerebral microvascular amyloid was associated with a strong localized neuroinflammatory response involving activated microglia and elevated levels of inflammatory cytokines (Miao et al., 2005a; 2005b). Homozygous Tg-SwDI mice exhibited more extensive accumulation of cerebral Aβ as compared with identically aged heterozygous mice (Fig. 1A,B). As expected, wild-type mice showed no human Aβ accumulation as they lack the human APP transgene (data not shown). At one year of age the levels of total Aβ 40 and Aβ 42 were markedly higher in the homozygous Tg-SwDI mice (Fig. 1C). The increase in total Aβ levels in homozygous Tg-SwDI mice resulted in corresponding increase in the levels of soluble and insoluble Aβ compared with the heterozygous animals (Fig. 1D). Despite the large increase in cerebral Aβ pools in homozygous compared with heterozygous Tg-SwDI mice, the ratios of insoluble to soluble Aβ remained essentially the same with values of 45 ± 6 and 42 ± 4, respectively. In light of the abundant accrual fibrillar microvascular Aβ in Tg-SwDI mice, we sought to determine if this unique pattern of Aβ accumulation causes behavioral deficits in the mice. We chose to perform our analyses on homozygous Tg-SwDI mice since they accumulated Aβ earlier and higher levels than the heterozygous animals.
Figure 1.
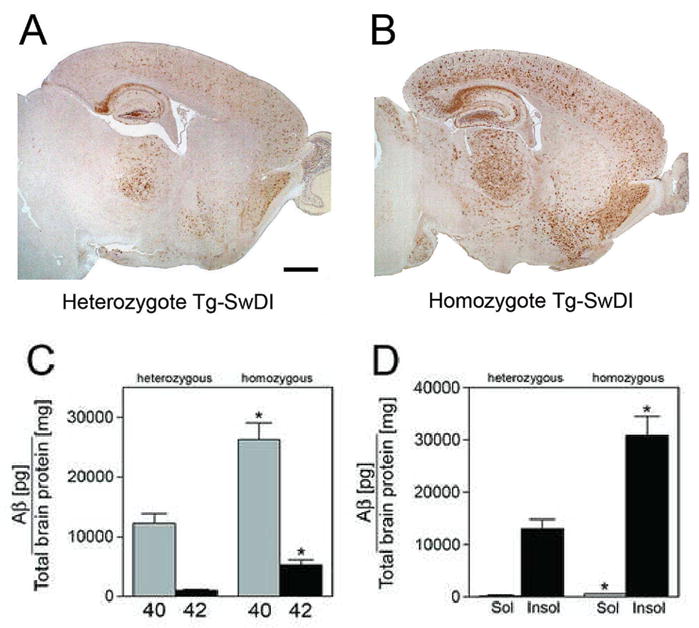
Homozygous Tg-SwDI mice accumulate markedly more Aβ than heterozygous animals. Aβ immunostaining in twelve months old heterozygous (A) homozygous (B) Tg-SwDI reveals more extensive deposition in the latter. Scale bar = 1 mm. (C) Total Aβ 40 and Aβ 42 loads in homozygous Tg-SwDI mice is more than double compared to heterozygous animals. *P < 0.001. (D) The levels of soluble Aβ and insoluble Aβ are similarly increased in homozygous Tg-SwDI mice compared with heterozygous animals. Data presented are the mean ± S.D. (n = 10 per group). *P < 0.0005.
Tg-SwDI mice do not show changes in mobility, strength, and coordination
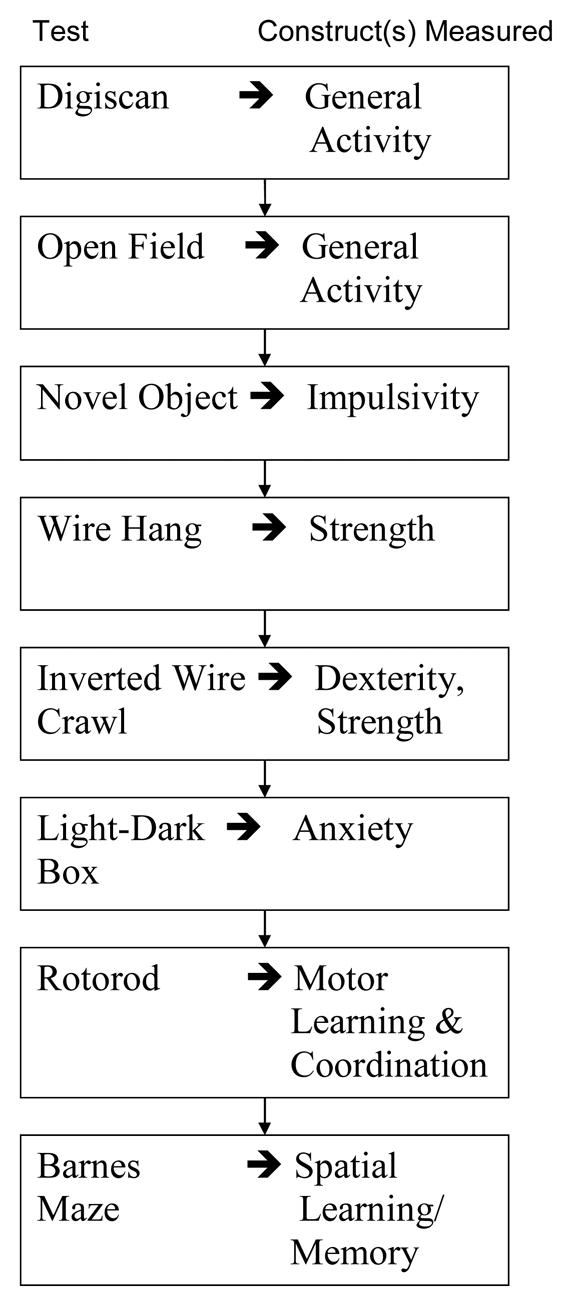
Homozygous Tg-SwDI mice were assessed for deficits in behavioral performance using the tasks outlined in Table 1. Digiscan analysis was used to determine if twelve months old Tg-SwDI mice showed differences in exploratory movements compared to age-matched wild-type mice. There were no significant differences in horizontal movements, free rearings, or wall supported rearings. Similarly, in open field testing the twelve month old homozygous Tg-SwDI mice showed no significant differences in center line crosses but they did approach the novel object significantly more quickly on day 3 (F(1,26)=5.7, p<.02). Using the light/dark box as a measure of anxiety we found no significant difference in the amount of time spent in the light between the wild-type and Tg-SwDI mice. Also, wire hang and inverted crawl tasks, measures of strength, did not show differences between the two groups of mice. Finally, in rotarod analysis to measure motor learning and coordination a significant main effect for Trial 1 (F(1,2)=18.9, p<.0001), Trial 2 (F(1,2)=7.2, p<.002), and Trial 3 (F(1,2)=4.4, p<.02), showed that all of the mice, whether wild-type or Tg-SwDI, improved performance over the course of testing. However, there were no significant group X trial interaction effects for Trial 1 (F(1,2)=.67, n.s.), Trial 2 (F(1,2)=.85, n.s.), or Trial 3 (F(1,2)=.74, n.s.) that would indicate a difference between the wild-type and Tg-SwDI in rate of motor learning. A significant weight difference between wild-type and Tg-SwDI mice was evident at 12 months (F(1,26)=43.20, p<.0001; Tg-SwDI mean = 27.8, s.e.m = 0.99; WT Mean = 37.5, s.e.m. = 1.1).
Table 1.
Behavioral testing scheme for analysis of Tg-SwDI mice.
Tg-SwDI mice exhibit impaired learning and memory task performance
We next used the Barnes maze to determine if Tg-SwDI showed an impairment of learning and memory. Fig. 2A shows the results for latency to find the escape hole of the Barnes maze for twelve month old animals, showing that Tg-SwDI mice, in general, took significantly longer to find the escape hole than the age-matched wild-type mice across sessions (main effect F(1,26)=4.4, p < 0.05; group × trial interaction F(1,9)=0.5, n.s.). The results of the fitting of exponential functions by least-squares regression to the data points for each group revealed a more rapid rate of change (Tg-SwDI e = −0.087 vs. WT e = −0.03) though comparable y-intercepts (Tg-SwDI = 94.1 vs. WT = 93.9). The body weights of the Tg-SwDI were not predictive of averaged latency to find the escape platform in the Barnes Maze, (F1,12) = 0.23, p. = 0.63, R2 = 0.1. A very similar deficit in Barnes maze performance was observed in homozygous twelve months old Tg-SwDI/line A mice, an independently generated Tg-SwDI mouse line (data not shown). This additional Tg-SwDI mouse line similarly expresses low levels of human APP and shows essentially the same type of accumulative cerebral microvascular Aβ pathology (Davis et al., 2004). Importantly, this indicates that the observed impairment of learning and memory is consistent across independent Tg-SwDI mouse lines and reflects the behavioral effect of the Aβ accumulation and associated pathology that are unique to these transgenic lines. A similar Barnes maze impairment was also observed in nine months old Tg-SwDI homozygotes as compared to aged matched wild-type controls (data not shown).
Figure 2.
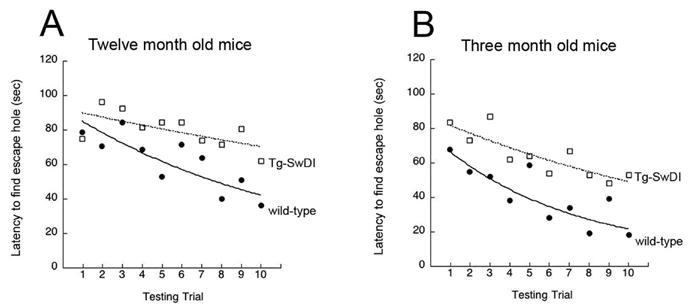
Twelve months old (A) and three months old (B) Tg-SwDI mice exhibited slower rates of decrease in the latencies to find the escape hole in the Barnes Maze spatial learning and memory test compared with age-matched wild type mice. Shown are the best fitting exponential decay curves for each group (12 months old WT y = 93.8e−.09; 12 months old Tg-SwDI y = 94.1e−.03; 3 months old WT y = 77.4e−.15; 3 months old Tg-SwDI y = 88.8e−.06).
We next sought to determine if the deficit in performance on the Barnes maze was evident in younger Tg-SwDI mice. At the younger age of three months, the Tg-SwDI mice took significantly more time to find the hole over the course of five days compared to wild-type mice as tested by a repeated measures (Fig. 2B ANOVA, F(1,26)=8.7, p < 0.01; group × trial interaction F(1,9)=0.79, n.s.). The results of the fitting of exponential functions by least-squares regression to the data points for each group again revealed a more rapid rate of change (Tg-SwDI e = −0.15 vs. WT e = −0.06) though less comparable y-intercepts (Tg-SwDI = 88.8 vs. WT = 77.4). Similar to the twelve months old Tg-SwDI mice, the younger three months old mice also showed no deficits in mobility, strength, and coordination. Also, there was no significant difference in weights between the wild-type and Tg-SwDI mice at 3 months (F(1,26)=.14, n.s; Tg-SwDI mean = 22.7, s.e.m = 0.82; WT Mean = 23.2, s.e.m. = 0.7).
Tg-SwDI deficits in Barnes maze performance do not strictly correlate with cerebral Aβ levels
Since a similar impairment in learning and memory task performance of Tg-SwDI was evident at three months as observed at twelve months we sought to determine if there was a link in the extent and location of cerebral Aβ accumulation and pathology at these different ages. ELISA measurements expectedly showed that the twelve months old Tg-SwDI mice had markedly higher levels of Aβ 40 and Aβ 42 peptides in brain compared with the younger animals (Fig. 3A). Similarly, the older animals had much higher levels of both soluble and insoluble pools of Aβ in brain compared with the three months old Tg-SwDI mice (Fig. 3B). Since the soluble pool of Aβ peptides was significantly higher in the older animals we evaluated the levels of soluble Aβ oligomers in the two age groups of Tg-SwDI mice. Quantitative dot blot analysis, using a polyclonal antibody designated OC11 that is specific for oligomer forms of Aβ, revealed that twelve months old Tg-SwDI mice had nearly three times higher levels of soluble Aβ oligomers compared with the younger animals (Fig. 3C,D). Similarly, using a sandwich ELISA composed of mAb 3D6 to capture Aβ and biotinylated mAb 3D6 to detect any size oligomeric Aβ species as described by DeMattos et al. (2002) revealed the higher amounts of soluble Aβ oligomers in twelve months old Tg-SwDI mice (data not shown). Together, these data indicate that twelve months old Tg-SwDI mice have markedly higher levels of all Aβ peptide pools in brain compared to the younger animals and does not correlate with the similar behavioral learning deficit observed in the young and older animals.
Figure 3.

Accumulation of cerebral Aβ in young and older Tg-SwDI mice. (A) ELISA measurements of total Aβ 40 (gray bars) and Aβ 42 (black bars) in mouse forebrain tissue. (B) ELISA measurements of soluble Aβ (gray bars) and insoluble Aβ (black bars) in mouse forebrain tissue. ELISA data shown are mean ± S.D. (n = 6 mice per age group). *P < 0.000001, **P < 0.0001. (C) Representative dot blot analysis of Aβ oligomers in soluble mouse forebrain extracts. (D) Quantitation of soluble Aβ oligomers from dot blots of young and older Tg-SwDI mice. Data shown are mean ± S.D. (n = 6 mice per age group). *P < 0.0002.
Tg-SwDI deficits in Barnes maze performance are associated with early-onset subicular microvascular amyloid and neuroinflammation
We next compared the regional accumulation of cerebral microvascular amyloid, the prominent pathological feature of this model, in the different aged Tg-SwDI mice. At twelve months of age the Tg-SwDI mice showed high amounts of cerebral microvascular amyloid in subiculum and thalamus, with much lower levels in the fronto-temporal cortex as previously reported (Davis et al., 2004, Miao et al., 2005a, 2005b) (Fig. 4A–D). Analysis of the three months old mice showed that only the subiculum exhibited high amounts of cerebral microvascular amyloid whereas the thalamus and cortex only showed occasional diffuse parenchymal deposits at this young age (Fig. 4E–H). Wild-type mice at either ages show no cerebral microvascular amyloid (data not shown).
Figure 4.
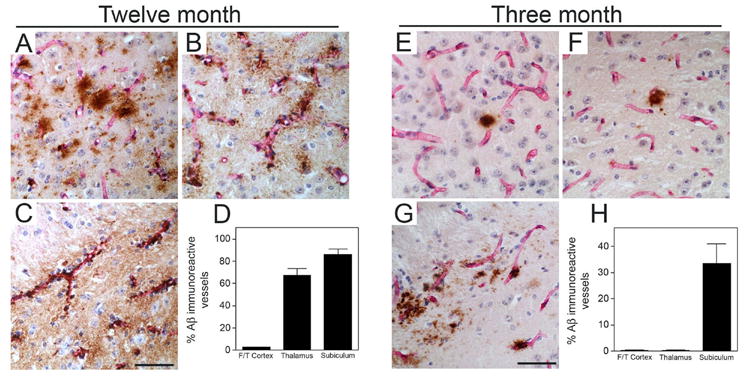
Cerebral microvascular amyloid accumulation in young and older Tg-SwDI mice. Microvascular amyloid deposition in Tg-SwDI mouse brain revealed by immunostaining for Aβ (brown) and collagen type IV for identification of microvessels (red) in the fronto-temporal cortex (A,E), thalamus (B,F), and subiculum (C,G) from twelve months old and three months old Tg-SwDI mice, respectively. Scale bars = 50 μm. Quantitative stereological measurement of cerebral microvascular Aβ deposition in different brain regions of twelve months old (D) and three months old (H) Tg-SwDI mice. Data shown are the mean ± S.D. (n = 6 mice per age group).
We next measured the numbers of microvascular amyloid-associated neuroinflammatory cells in the different brain regions of the young and older Tg-SwDI mice. Immunohistochemical staining and quantitative stereological counting revealed that compared with similarly aged wild-type mice the GFAP-positive reactive astrocytes were significantly elevated in all brain regions of twelve months old Tg-SwDI mice with higher numbers observed in the subiculum and thalamus, regions with more extensive cerebral microvascular amyloid (Fig. 5A–D). On the other hand, astrocyte numbers were only elevated in the subiculum of the younger animals, the only region showing appreciable cerebral microvascular amyloid at this age (Fig. 5E–H). Similarly, activated microglia were significantly elevated in all brain regions of twelve months old Tg-SwDI mice with higher numbers again observed in the cerebral microvascular amyloid-rich subicular and thalamic regions (Fig. 6A–D). Again, activated microglia were only elevated in the subiculum of the younger animals, the only region showing appreciable cerebral microvascular amyloid at this age (Fig. 6E–H). Few, if any, activated microglia were found in any brain region of young or older wild-type mice. These results suggest that accumulation of cerebral microvascular Aβ and promotion of localized neuroinflammation in the subiculum is associated with impairment of learning and memory in young Tg-SwDI mice.
Figure 5.
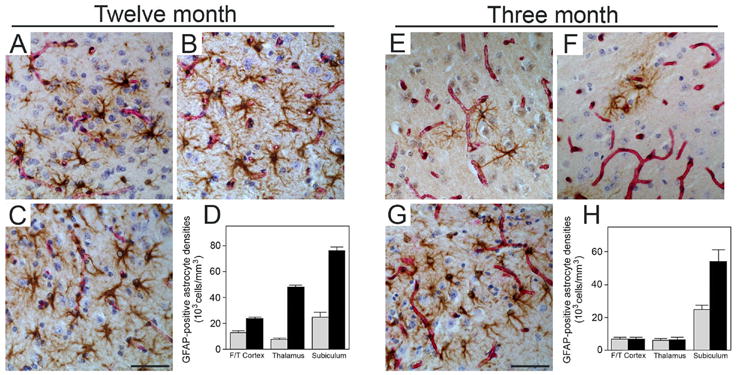
Elevated numbers of reactive astrocytes in the subiculum of young Tg-SwDI mice. Microvascular-associated reactive astrocytes revealed by GFAP-positive immunostaining (brown) and collagen type IV for identification of microvessels (red) in the fronto-temporal cortex (A,E), thalamus (B,F), and subiculum (C,G) from twelve months old and three months old Tg-SwDI mice, respectively. Scale bars = 50 μm. Quantitative stereological measurement of reactive astrocyte densities in different brain regions of twelve months old (D) and three months old (H) wild-type (gray bars) or Tg-SwDI mice (black bars). Data shown are the mean ± S.D. (n = 6 mice per age group).
Figure 6.
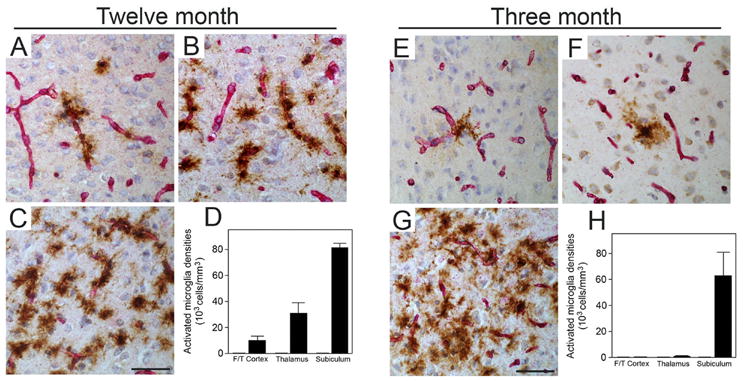
Elevated numbers of activated microglia in the subiculum of young Tg-SwDI mice. Microvascular-associated activated microglia revealed by 5D4-positive immunostaining (brown) and collagen type IV for identification of microvessels (red) in the fronto-temporal cortex (A,E), thalamus (B,F), and subiculum (C,G) from twelve months old and three months old Tg-SwDI mice, respectively. Scale bars = 50 μm. Quantitative stereological measurement of activated microglia densities in different brain regions of twelve months old (D) and three months old (H) wild-type (gray bars) or Tg-SwDI mice (black bars). Data shown are the mean ± S.D. (n = 6 mice per age group).
Discussion
The present study suggests that early-onset accumulation of cerebral microvascular amyloid in the subiculum and localized activation of astrocytes and microglia are associated with impaired performance in Tg-SwDI mice of the Barnes Maze test of spatial learning and memory. The Tg-SwDI mouse is a unique model in that it develops fibrillar amyloid deposits primarily in the cerebral microvasculature whereas Aβ deposits in the brain parenchyma are essentially diffuse. In heterozygous Tg-SwDI mice cerebral microvascular amyloid starts to deposit at about 4–5 months and extensively accumulates as the mice age (Davis et al. 2004). Here we show that homozygous Tg-SwDI mice accumulate more than double the amount of cerebral Aβ as heterozygous animals at one year of age. Despite this large increase in accumulated Aβ the ratio of insoluble Aβ to soluble Aβ remained essentially the same. This strong accumulation of Aβ in brain likely reflects the markedly deficient ability to clear Dutch/Iowa mutant Aβ peptides from brain across the blood-brain barrier into the circulation (Davis et al., 2004; Deane et al., 2004; Davis et al., 2006). The exclusive collection of fibrillar amyloid in the cerebral vasculature suggests that specific factors in this compartment may enhance CAA mutant Aβ fibril assembly and deposition. For example, we recently showed that GM3 ganglioside, which is abundantly found in cultured cerebrovascular cells and in isolated cerebral vessels but not in the brain parenchyma, selectively promotes fibrillar assembly of CAA mutant forms of Aβ (Yamamoto et al, 2005). In any case, the restriction of fibrillar amyloid predominantly to the cerebral vasculature accurately mirrors the site of fibrillar Aβ deposition in patients with the Dutch-type or Iowa-type familial CAA disorders (Grabowski et al., 2001; Maat-Schieman et al., 2004).
The results of the behavioral battery demonstrate that Tg-SwDI mice exhibit phenotypic differences when compared to wild-type mice beginning as early as three months of age. These deficits were apparent in the Barnes maze test for spatial learning and memory. In particular, both the three and twelve months old animals exhibited slower rate of change in the rate, although at three months a difference in the average starting level was also indicated by the non-linear regression analysis. These differences are consistent with the performance change expected if spatial learning ability was impaired. Gross impairments in motor competence, general activity and well-being were not apparent, pointing to the higher order nature of the differences. The lack of difference in the rate of motor learning between the Tg-SwDI and wild-type animals in the rotorod task suggests that spatial learning and memory may be specifically affected.
The present findings of behavioral impairment in three months old Tg-SwDI mice are significant for several reasons. Firstly, the primary site of microvascular Aβ accumulation in these young Tg-SwDI mice is within the subiculum (Fig. 4). The subiculum is a major output structure for the hippocampal formation. Postcommissural fornix fibres originate largely in the subicular region and project through the hypothalamus on their way to the mammillary bodies, anterior and laterodorsal thalamus, and lateral septal regions (Swanson and Cowan, 1977, 1979; Canteras and Swanson, 1992). The subicular region also has important cortical outputs, especially to prefrontal, anterior cingulate and parahippocampal areas, as well as to the midbrain tegmental region (Amaral and Witter, 1989; Jay and Witter, 1991; Van Hoesen, 1995). Given the well-established role for the hippocampal formation and these associated cortical and limbic regions in regulating learning/memory, it is highly plausible that the pathology in this region could account for the presently observed deficit.
Secondly, at three months of age appreciable amounts of cerebral microvascular fibrillar amyloid is present in the subiculum region of the Tg-SwDI mice. Previously we reported that in the Tg-SwDI model cell degeneration and neuroinflammation were restricted to the sites of cerebral microvascular amyloid and were unrelated to either diffuse Aβ deposits or soluble Aβ oligomers in the brain parenchyma (Miao et al., 2005a, 2005b). Here we show that the learning deficits observed in young and older Tg-SwDI mice did not correlate with either the amount of accumulated Aβ peptides nor with the levels of soluble Aβ oligomers in brain (Fig. 3). Importantly, this suggests that behavioral deficits in Tg-SwDI mice correlate with the presence of subicular cerebral microvascular amyloid. However, it remains possible that the behavioral deficits are not the result of this accumulation. Stronger evidence for a causal link would come from the demonstration that the degree of behavioral impairment was correlated with the levels of cerebrovascular amyloid, especially if the analyses also including heterozygote animals, which would presumably show lower levels of accumulation. Additionally, while the behavioral deficits observed were reliable across time points and mouse line, the generality of the present conclusions may also be limited by the present use of only one spatial learning/memory task and the lack of complete blinding of investigators to genotype. These limitations should be addressed by future work that employs additional learning and memory tests and uniform blinding to mouse genotype.
Lastly, numerous reactive astrocytes and activated microglia were found intimately associated with the microvascular amyloid deposits in the subiculum of the young Tg-SwDI mice (Figs. 5 and 6, respectively). In contrast, other regions of the brain without microvascular amyloid showed no elevation in astrocytes and few or no activated microglia. This suggests that the early onset of spatial learning and memory deficits correlates with the initial cerebral microvascular amyloid deposition and localized neuroinflammation evidenced by evidenced by elevated astrocytes and activated microglia. Recently, the activation of microglia in response to parenchymal amyloid deposition has been directly linked to behavioral deficits in a human mutant β APP transgenic mouse model of parenchymal fibrillar Aβ plaques (Seabrook et al., 2006). These findings together suggest that microglial activation and neuroinflammation in response to either cerebral microvascular fibrillar amyloid or parenchymal plaque fibrillar amyloid can lead to behavioral impairments in mice.
The emerging concept of cerebral microvascular amyloid and associated neuroinflammation in promoting cognitive impairment has been supported by recent studies showing that treatments aimed at reducing CAA-induced neuroinflammation in afflicted individuals have improved the cognitive deficits associated with this particular pathology (Eng et al., 2004; Harkness et al., 2004). In light of our findings that glial activation in response to cerebral microvascular amyloid correlates with poor behavioral performance in Tg-SwDI mice, specifically targeting cerebral microvascular amyloid-induced neuroinflammation may provide relief of cognitive impairment that is specifically associated with this particular amyloid pathology.
Acknowledgments
This work was supported in part by National Institute of Neurological Disorders and Stroke grants RO1-NS55118 and RO1-NS36645. Antibody reagents for the Aβ ELISA and detection of soluble Aβ oligomers were generously provided by Lilly Research Laboratories, Indianapolis, IN and Dr. Charles Glabe, University of California, Irvine, respectively. The authors thank Ms. Tiffany Buono for assistance with the behavioral testing.
List of Abbreviations
- Aβ
amyloid β protein
- APP
amyloid β-protein precursor
- ANOVA
analysis of variance
- ANCOVA
analysis of covariance
- CAA
cerebral amyloid angiopathy
- ELISA
enzyme-linked immunosorbent assay
- Tg-SwDI
transgenic mice expressing neuronal vasculotropic Dutch/Iowa (E693Q/D694N) mutant human Aβ precursor protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31:571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Atterns J, Jellinger KA. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—a pilot study. Acta Neuropathol. 2004;107:83–90. doi: 10.1007/s00401-003-0796-9. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Rivara CB, Rocher AB, Hof PR. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res. 2004;26:573–578. doi: 10.1179/016164104225016272. [DOI] [PubMed] [Google Scholar]
- Barnes CA. Memory deficits associated with senescence: A neuropsychological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- Canteras NS, Swanson LW. Projections of the ventral subiculum to the amygdala, septum, and hypothalamus: a PHAL anterograde tract-tracing study in the rat. J Comp Neurol. 1992;324:180–194. doi: 10.1002/cne.903240204. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: A disorder of cortical cholinergic innervation. Science. 1983;219:1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- Crawley JN. What’s wrong with my mouse?: Behavioral phenotyping of transgenic and knockout mice. New York: Wiley-Liss; 2000. [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Ziegler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid β-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid β-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- Davis J, Xu F, Miao J, Previti ML, Romanov G, Ziegler K, Van Nostrand WE. Deficient cerebral clearance of vasculotropic Dutch/Iowa double mutant Aβ in human Aβ PP transgenic mice. Neurobiol Aging. 2006;26:946–954. doi: 10.1016/j.neurobiolaging.2005.05.031. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, O’dell MA, Parsadanian M, Taylor JW, Harmony JA, Bales KR, Paul SM, Aronow BJ, Holtzman DM. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2002;99:10843–10848. doi: 10.1073/pnas.162228299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol. 2004;55:250–256. doi: 10.1002/ana.10810. [DOI] [PubMed] [Google Scholar]
- Grabowski TJ, Cho HS, Vonsattel JPG, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35:2616–2619. doi: 10.1161/01.STR.0000143224.36527.44. [DOI] [PubMed] [Google Scholar]
- Harkness KA, Coles A, Pohl U, Xuereb JH, Baron JC, Lennox GG. Rapidly reversible dementia in cerebral amyloid inflammatory vasculopathy. Eur J Neurol. 2004;11:59–62. doi: 10.1046/j.1351-5101.2003.00707.x. [DOI] [PubMed] [Google Scholar]
- Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontalcortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313:574–86. doi: 10.1002/cne.903130404. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: An update. J Neural Trans. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ 42 deposition in a transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GTAM, Lyunendijk W, Frangoine B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- Long JM, Kalehua AN, Muth NJ, Calhoun ME, Jucker M, Hengemihle JM, Ingram DK, Mouton PR. Stereological estimation of total microglia number in mouse hippocampus. J Neurosci Meth. 1998;84:101–108. doi: 10.1016/s0165-0270(98)00100-9. [DOI] [PubMed] [Google Scholar]
- Maat-Schieman MLC, Yamaguchi H, Hegeman-Kleinn I, Welling-Graafland C, Natte R, Roos RAC, van Duinen SG. Glial reactions and the clearance of amyloid β protein in the brains of patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type. Acta Neuropathol. 2004;107:389–398. doi: 10.1007/s00401-004-0824-4. [DOI] [PubMed] [Google Scholar]
- Miao J, Vitek MP, Xu F, Previti ML, Davis J, Van Nostrand WE. Reducing cerebral microvascular amyloid-β protein deposition diminishes regional neuroinflammation in vasculotropic mutant amyloid precursor protein transgenic mice. J Neurosci. 2005a;25:6271–6277. doi: 10.1523/JNEUROSCI.1306-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid β-protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid β-protein precursor. Am J Pathol. 2005b;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensink AA, de Waal RM, Kremer B, Verbeek MM. Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev. 2003;43:207–223. doi: 10.1016/j.brainresrev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Seabrook TJ, Jiang L, Maier M, Lemere CA. Minocycline affects microglia activation, Aβ deposition, and behavior in APP-tg mice. Glia. 2006;53:776–782. doi: 10.1002/glia.20338. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: Genes, proteins, and therapy. Physiol Rev. 2001;8:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Cowan WM. An autoradiographic study of the organization of the efferent connections of the hippocampal formation in the rat. J Comp Neurol. 1977;172:49–84. doi: 10.1002/cne.901720104. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Cowan WM. The connections of the septal region in the rat. J Comp Neurol. 1979;186:621–55. doi: 10.1002/cne.901860408. [DOI] [PubMed] [Google Scholar]
- Thal DR, Ghebremedhin E, Orantes M, Wiestler OD. Vascular pathology in Alzheimer’s disease: Correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol. 2003;62:1287–1301. doi: 10.1093/jnen/62.12.1287. [DOI] [PubMed] [Google Scholar]
- Van Broekhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter Van der Vlis M, Roos RA. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch) Science. 1990;248:1120–1122. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW. Anatomy of the medial temporal lobe. Magn Reson Imaging. 1995;13:1047–55. doi: 10.1016/0730-725x(95)02012-i. [DOI] [PubMed] [Google Scholar]
- Vinters HV. Cerebral amyloid angiopathy: A microvascular link between parenchymal and vascular dementia? Ann Neurol. 2001;49:691–692. doi: 10.1002/ana.1055. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Hirabayashi Y, Amari M, Yamaguchi H, Romanov G, Van Nostrand WE, Yanagisawa K. Assembly of hereditary amyloid β-protein variants in the presence of favorable gangliosides. FEBS Lett. 2005;579:2185–2190. doi: 10.1016/j.febslet.2005.03.013. [DOI] [PubMed] [Google Scholar]