

Abstract
Small molecule inhibitors of protein tyrosine kinases have become both powerful chemical probes of biological processes and clinically effective therapeutics. In contrast, few small molecule inhibitors of protein tyrosine phosphatases have been identified and none are currently approved for clinical use. New cell-based high-content methods have been developed that should enable investigators to probe for selective inhibitors of diseases-relevant protein phosphatases. Details of these methods are described herein.
Keywords: Chemical complementation, mitogen-activated protein kinase phosphatase, high-throughput assay, high-content screening
1.INTRODUCTION
Mitogen-activated protein kinases (MAPK) are ubiquitous elements in the mitogenic signal transduction pathways involved in cell survival, stress response, and programmed death. MAPKs are activated by kinase cascades triggered by a variety of extracellular stimuli and inactivated by dual-specificity phosphatases termed MKPs [1]. Twelve human dual-specific MKPs have been identified, all possessing unique but overlapping substrate specificities and all having multiple activity and expression regulatory mechanisms [2]. Because of their central role in cellular homeostasis, MKPs have attracted attention as potential disease targets. For example, MKP-1 has been found overexpressed in prostate [3] gastric [4], breast [5], and pancreatic cancer [6]. MKP-1 expression correlates with decreased progression-free survival in ovarian cancer samples [7]. Even early phases of prostate, colon, and bladder carcinogenesis are characterized by elevated MKP-1 expression [8]. MKP-1 has also been shown to reduce the cellular toxicity of antineoplastic agents, including bortezomib [9], paclitaxel [10], and cisplatin [11], and ionizing radiation [12]. Thus, selective inhibitors of MKP-1 might potentially be useful to enhance the activity of existing anticancer therapies. MKP-3 induces endothelial cell apoptosis in response to tumor necrosis factor alpha [13] but is a tumor promoter target in initiated cells that express oncogenic Ras [14]. MKP-3 expression is markedly elevated in insulin-resistant obese rats and increases gluconeogenesis in rat hepatoma cells [15]. Thus, selective small molecule inhibitors of MKP-3 might be valuable leads for therapeutic agents.
2.ASSAYS TO DISCOVER MAPK PHOSPHATASE INHIBITORS
2.1. IN VITRO ASSAYS
While there is a plethora of specific small molecule inhibitors of MAPK activation, no such inhibitors exist for the MAPK phosphatases. This might in part be due to a lack of high-throughput assays to appropriately detect phosphatase activity. Currently, the most popular approach to identifying potential phosphatase inhibitors are in vitro assays using recombinant enzymes and small molecule synthetic substrates, such as para-nitrophenyl phosphate or O-methyl fluorescein phosphate. These small molecule substrates have been employed because they provide a robust output signal, are inexpensive and are more convenient to use than the phosphorylated protein substrate. Nonetheless, we now know that the activity of MKPs depends on their interaction with other proteins in the cell [16–19], and thus, in vitro screening assays may not accurately emulate the functionality of compounds within the context of the whole cell.
Efforts to simulate cellular conditions for enzymatic activity in vitro usually render biochemical assays complex and expensive. For example, an assay for activity of the cell-cycle phosphatase Cdc25B with its phosphorylated protein substrate Cdk2 requires production of no less than four recombinant proteins, the Cdc25B enzyme itself, full-length Cdk2 and the kinase (Myt1) to phosphorylate Cdk2, as well as the regulatory cyclin A protein, which requires an additional protease digestion step to enhance its stability [20,21]. Furthermore, when using full-length phosphoprotein substrates, methods of analysis often are no longer compatible with high-throughput requirements.
2.2. CELLULAR ASSAYS
The challenges associated with in vitro screens could potentially be circumvented by the use of cell-based assays that faithfully recapitulate the biological environment for target activity. Moreover, cellular assays can also identify compounds that indirectly inhibit target activity (so-called “non-catalytic inhibitors”). These types of inhibitors are being pursued for their putative ability to retain activity against cells resistant to catalytic inhibitors. For example, merbarone, fostriecin, and dexrazoxane (ICRF-187), are topoisomerase inhibitors that do not stabilize topoisomerase II – DNA complexes and consequently are effective against etoposide resistant leukemia cells [22]. Another example is the development of non-catalytic cAMP-specific phosphodiesterase 4 inhibitors as antinflammatory agents (reviewed in [23].
Cell-based assays can be classified as phenotypic or target-based. Phenotypic mammalian cell-based assays have been widely adopted to investigate and document the biological actions of compounds for which some information about target affinity and selectivity already exists. These assays are often used in compound credentialing with some successful examples having been reported [24,25]. A phenotypic screen for dual-specificity phosphatase inhibitors has been performed using the National Cancer Institute’s 1990 member “Diversity Set” [26] and Erk phosphorylation as an endpoint. The screen resulted in the discovery of several compounds that possessed moderate phosphatase inhibitory activity, including the first cell-active inhibitor of MKP-3, but also underscored a main limitation of phenotypic screens. Because the endpoint was Erk phosphorylation, an indirect measurement of target inhibition in the cell and influenced by a multitude of factors, the list of biologically active small molecules contained an equal number of compounds possessing or lacking tyrosine phosphatase inhibitory activity [27]. Of the five compounds with in antiphosphatase activity, none was selective when counterscreened against a panel of related phosphatases.
Over the past few years we have developed a target-specific, cellular assay for protein tyrosine phosphatases. The assay, which was termed “Chemical Complementation”, originated as a confirmatory assay for Cdc25A inhibitors [28,29] and employed the measurement of Erk phosphorylation in cells transfected with an epitope-tagged protein phosphatase (Figure 1). When stimulated with activators of mitogenic signaling such as EGF or phorbol ester (TPA), cells expressing the target no longer respond to activating stimuli with phosphorylation of Erk. Recently, we have exploited the power of high-content screening (HCS) to develop an HCS version of the chemical complementation assay. HCS is an analysis tool to acquire, analyze, search, and manage multi-dimensional information from cells [30]. The HCS embodiment of the chemical complementation assay is based on simultaneous measurement of both target phosphatase and Erk phosphorylation by immunofluorescence in cells grown in multiwell plates. In this assay, cells that are induced to overexpress that phosphatase of interest are refractory to activation of Erk signaling. The differential in Erk phosphorylation in MKP-expressing and non-expressing cells then serves as a measure of phosphatase activity. The HCS embodiment of the assay has been used for compound credentialing studies [31] and a small scale library screen [32]. The assay identified sanguinarine as a small molecule inhibitor of MKP-1 but not MKP-3.
Figure 1. A single-cell chemical complementation assay for MPK-3 inhibition. The assay is based on the differential response of cells that do or do not overexpress a target of interest, namely MKP-3.
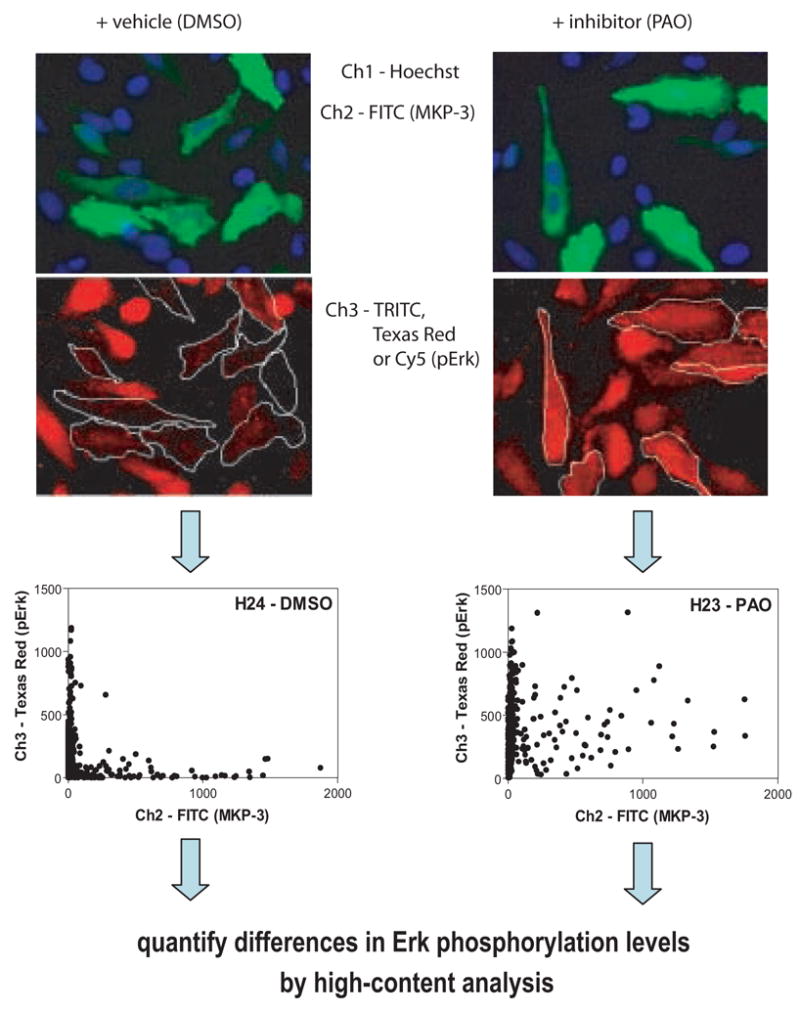
HeLa cells were transfected with c-myc-tagged MKP-3, stimulated with phorbol ester (TPA), treated with vehicle (DMSO) or phenylarsine oxide (PAO) and immunostained simultaneously with anti-c-myc (green) or anti-phospho-Erk (red) antibodies. Plates are analyzed on an ArrayScan II using a triple-band pass filter set. Cellular data from randomly chosen wells are shown as two-dimensional scatter plots with target expression (c-myc-MKP-3) depicted on the x-axis and Erk phosphorylation on the y-axis. In both vehicle-treated and PAO-treated wells, cells with low levels of MKP-3 show a heterogeneous distribution of phospho-Erk that is similar for both conditions. In cells expressing higher levels of MKP-3, phospho-Erk intensities were reduced. Inclusion of the control inhibitor, PAO, elevates phospho-Erk levels in MKP-3 expressing cells to those seen in cells not expressing MKP-3. High-content analysis procedures were developed to quantify differences in Erk phosphorylation between vehicle treated and PAO-treated cells.
Here we present the reader with a review of the process of implementing the chemical complementation assay as a high-throughput screening tool for inhibitors of MKP-3. We provide a validated assay protocol and discuss in detail the identification and optimization of several critical parameters that collectively permitted the assay to fulfill the stringent requirements for large scale library screening. Data supporting advancement of the assay to the primary screening stage and automation protocols are presented.
3.HTS IMPLEMENTATION OF THE MKP-3 CHEMICAL COMPLEMENTATION ASSAY
Based on our past experience, we first attempted to assess assay variability and performance using our previously described protocol [33] but soon discovered that several critical areas that, collectively, appeared to impair HTS compatibility of the Chemical Complementation assay. Although the following discussion contains specifics unique to the MKP-3 assay, the critical elements that we identified during the implementation process represent general findings that, in principle, should be applicable to any HCS assay. The three specific areas that were targeted for improvement were: (1) Single-cell analysis procedures, (2) Methods to analyze multiparameter distribution functions and (3) Use of alternative positive controls for maximum signal.
3.1. CELL SUBPOPULATION ANALYSIS
We made two major modifications of our previous data analysis procedure to reduce variability and to improve HTS validation. Figure 2 illustrates the effect that these combined changes had on assay window. The first change encompassed measurement of Erk phosphorylation specifically in the MKP-3 expressing cells. When comparing phospho-Erk intensities in MKP-3 transfected cells, the relative frequency distributions between vehicle-treated and inhibitor-treated cells were similar (Figure 2A) and differences in well average phospho-Erk levels were low (65 relative fluorescence units (RFU). This represented an increase in signal of only 30%, or a signal-to-background (S:B) ratio of 1.3. In contrast, the difference in phospho-Erk levels between the two treatments increased to 260 RFU (170% increase; S:B = 2.7) when the HCS data set was subsetted into MKP-3 positive cells by gating in the green channel and Erk phosphorylation was measured only in the MKP-3 expressing cells. This was illustrated by a more pronounced shift of phospho-Erk frequency distributions to higher values after treatment with the nonspecific protein tyrosine phosphatase inhibitor phenylarsine oxide (PAO) (Figure 2B). The main reason for the larger profile shift was that in transient transfection assays, only a proportion of all cells in a well overexpressed the target, and the remaining non-expressing cells muted the overall response. The results illustrated the power of HCS, as its ability to analyze cell subpopulations eliminated the contribution of the non-expressing cells’ lack of response to the desired readout.
Figure 2. Enhancing assay window by measuring cellular response in target- overexpressing cell subpopulations.
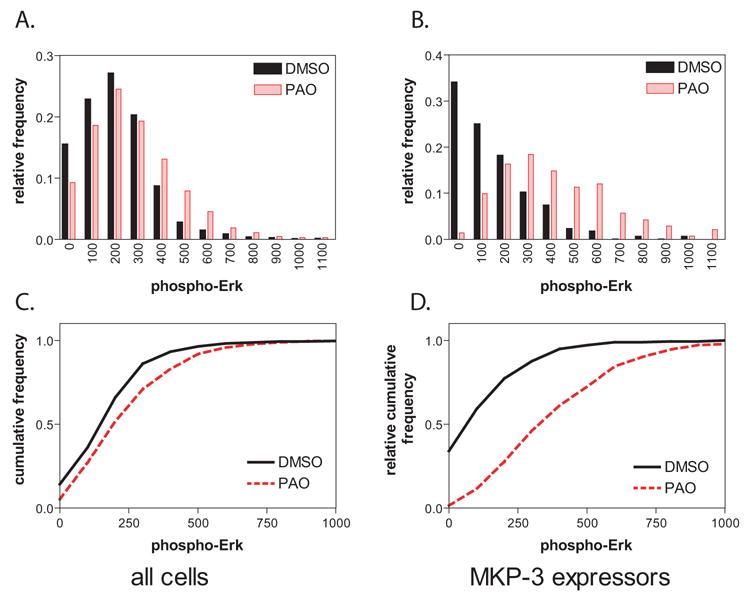
Using the data presented in Figure 1, relative phospho-Erk frequency distributions were established using (A) all cells in the well or (B) only those cells that were classified as MKP-3 positive on the basis of AlexaFluor 488 fluorescence. When using all cells, phospho-Erk distributions are almost identical regardless of treatment. In MKP-3 expressing cells, there is a shift in Erk distribution to higher levels after PAO treatment, although substantial overlap remains. Representing pErk levels as cumulative distributions (C, D) magnifies differences and prepares data for quantification by Kolmogorov-Smirnov (KS) statistics. Solid line, DMSO; dotted line, 5 μM PAO. See text for details on quantitative evaluation.
3.2. METHODS TO ANALYZE DIFFERENCES BETWEEN FREQUENCY DISTRIBUTIONS
When comparing the average response from histograms of cell populations, one often finds that average differences between treatments are small. This reflects the inherent sample heterogeneity, resulting in a distribution of signal across the cell population. Measurements of response then are influenced not only by the magnitude of response but also by the shape and width of the distribution function. This effect can be further magnified when only a small proportion of cells contribute to changes in signal (see Section 3.1. above).
A method that takes into account the shape of the distribution function is to compare samples by Kolmogorov-Smirnov (KS) statistics. KS statistics have been described as a method to quantitatively compare the distributions of individual cell populations [34–37], and were shown to be a valuable tool for the quantification of HCS data [38]. The resulting KS values can assume a value between 0 (identical distributions) and 1 (completely different distributions). In previous reports, we had experimented with a two-dimensional implementation of the KS statistic and shown it dramatically reduced assay variability and the number of false positives compared with measurements based on well averages [33,39]. At the time, an appealing feature of the two-dimensional variation of the KS method was that it condensed all of the information content in each well into a single numerical value by ranging over data in an (x,y) plane in search of a maximum cumulative difference between two two-dimensional data distributions [40,41]. Application of this algorithm to a 720 compound HCS data set, however, revealed two shortcomings that limited its use to the analysis of small compound collections. First, the 2D KS value was influenced not only by Erk phosphorylation but also by changes in target expression. Therefore, a high KS value could potentially be obtained when compounds were autofluorescent in the channel used to detect target expression. Second, histogram comparisons were performed on the entire cell population rather than the target-expressing cells, hence not taking full advantage of the power of single-cell analyses (vide supra). As a result, the assay, while occasionally yielding Z-factors in excess of 0.5, did not reproducibly perform with the same level of statistical significance. We therefore implemented an algorithm that performed a single-channel KS analysis for Erk phosphorylation in MKP-3 expressing cells. The algorithm, which was implemented in the S-Plus statistical program, generated a data subset containing only MKP-3 expressing cells, followed by establishment of a reference distribution from 16 wells transfected with MKP-3 and treated with vehicle. Sixteen wells of cells treated with vehicle or PAO-treated were then compared to this reference distribution. KS values in MKP-3 expressing cell subpopulations were 0.11 and 0.70, respectively, corresponding to a 550% increase in signal or a S:B ratio of 6.5. In contrast, when cells were not subsetted into MKP-3 expressors, the average KS values for the 16 vehicle controls and inhibitor controls were 0.10 and 0.30, respectively (a S:B of only 3.0). Thus, the combination of subpopulation gating and KS statistics resulted in a five-fold increase in assay window. A preliminary Z-factor [42] calculated from these 32 wells of positive and negative controls was 0.49.
3.3. CHOICE OF POSITIVE CONTROL
Because readily available, selective inhibitors of MKP-3 are lacking, we had used the PAO as a positive control in assay development. We soon discovered, however, that PAO had too many liabilities to be useful to validate the assay for HTS, most notably cellular toxicity. PAO inhibited MKP-3 with an IC50 of 4.3 μM, which was only about three fold less than the cytotoxic concentration. Furthermore, PAO toxicity was cell density dependent, and often potentiated in the presence of transfection agent. The effects of PAO on transfection toxicity were partly but not completely alleviated by replacing Lipofectamine 2000 (Invitrogen) with Fugene 6 (Roche Biosciences). In some cases PAO also reduced transfection efficiency, and occasionally reduced overall phospho-Erk levels, presumably by inhibiting tyrosine phosphatases involved in Erk activation. Because the cellular effects of PAO were extremely difficult to control and standardize, we investigated the use of untransfected control wells for maximum signal, and found this was a more stable and reliable control. To accommodate this change, we altered our S-Plus analysis scripts to include in the analysis all cells from untransfected controls and MKP-3 subsetted cells from unknowns and vehicle-treated controls (see also Section 4.3.). With these combined modifications, the assay was subjected to a formal variability assessment procedure. The following is a detailed assay protocol that was used to generate the HTS implementation and validation data presented in Figures 3 and 4.
Figure 3. Three-day variability assessment.
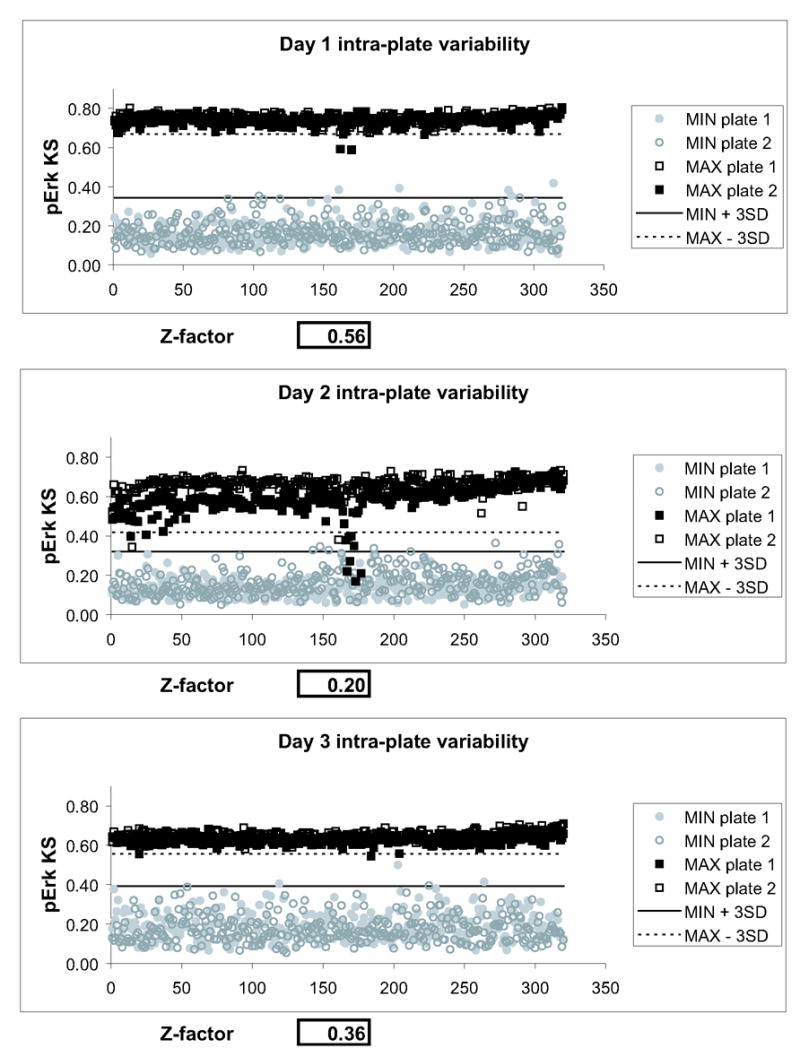
Two plates of minimum (MKP-3 transfected and FITC gated) and maximum (untransfected, ungated) signal were processed on three consecutive days. For each day, minimum and maximum signals from both plates were averaged and Z-factors were calculated. On all three days, the assay showed positive separation bands, as indicated by the solid and dotted lines, which indicate three standard deviations from the means of minimum and maximum signal, respectively. The assay gave Z-factors above 0.2 on all three days. Visual examination of plate graphs demonstrated a systematic error in one of the center rows of plate #1 on day 2, which was due to a clogged dispense nozzle on one of the liquid handlers.
Figure 4. HTS validation.
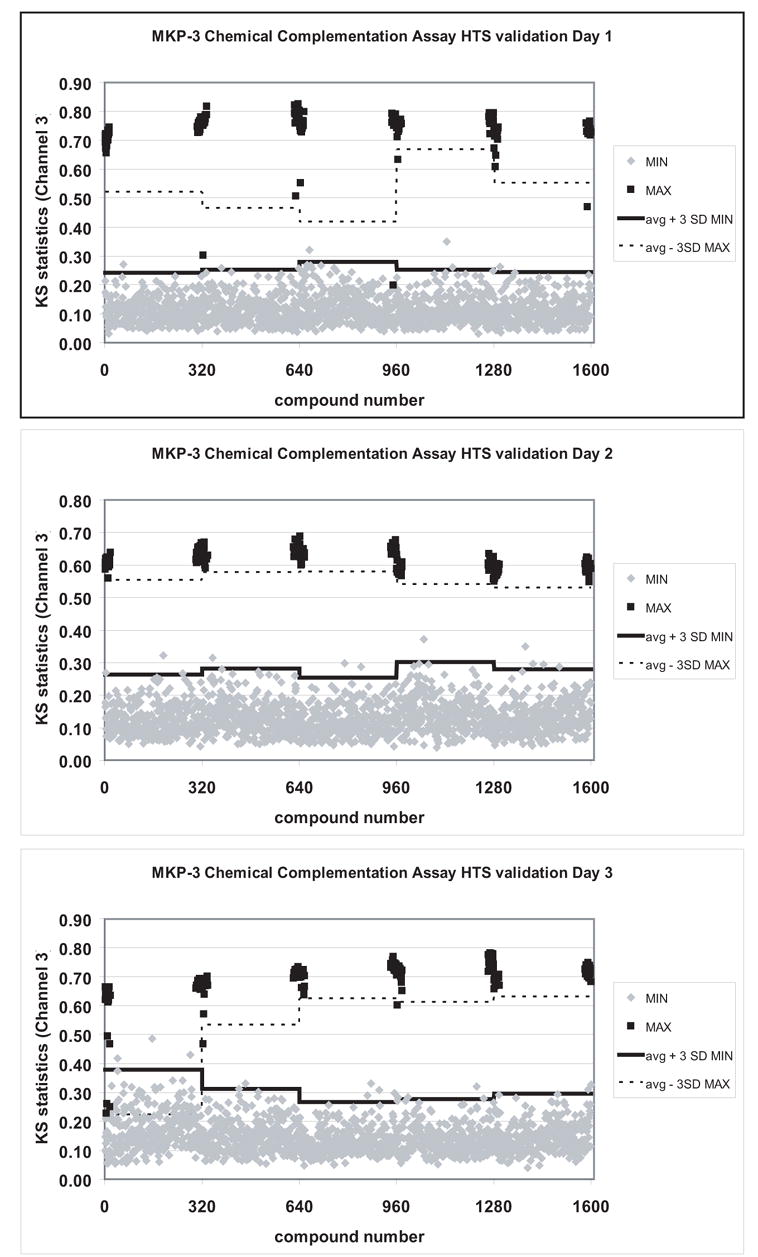
Five 384 well microplates each containing 320 vehicle-controls and 16 positive and negative control wells were run on three separate days. Graphs represent aggregate plate data from each day with positive controls shown as solid black squares. Every plate except one had a positive assay window, with 10/15 plates showing Z-factors above 0.5. Plate failures were due to inconsistencies in positive controls. All outliers were traced to liquid handling inconsistencies, which could be addressed by modification of instrument maintenance routines. None of the plates showed any false positives. Solid line, mean + 3SD of minimum signal, dotted line, mean – 3SD of maximum signal.
4.METHODS
4.1 Materials (for one 384 well microplate)
HeLa cells (ATCC)
384 well flat bottom microplates, collagen-coated (Falcon Biocoat)
384 well Greiner polystyrene storage plates (for treatments)
Fugene 6 transfection reagent (Roche Biosciences)
plasmid encoding c-myc-tagged MKP-3 in mammalian expression vector pSG5 [43,44]
DMEM growth medium, supplemented with 10% Fetal bovine serum and antibiotics
Serum-free DMEM
0.2% Triton-X100 (Sigma cat# T-9284)
Blocking solution: 25.1 ml 1xPBS, 900 μl BSA (Sigma cat# A-0336). Filter through 0.22 μm syringe filter.
Primary antibody cocktail: 35 μl anti-cMyc (Santa Cruz cat# sc-40, 1:200 dilution), 35 μl anti-pErk (Cell Signaling cat# 9101, 1:200 dilution) in 7 ml blocking buffer
Secondary antibody cocktail : 14 μl 1:500 dilution of AlexaFluor 488 goat anti-mouse IgG (FITC equivalent), 14 μl AlexaFluor 594 goat anti-rabbit IgG (Texas Red), 7 μl 10 mg/ml Hoechst 33342 (Molecular Probes cat# A11029, A11037 and H-1399, respectively) in 7 ml blocking solution.
Formaldehyde: Ultra-Pure formaldehyde, 16% (Polysciences Inc. cat# 18814–20).
12-O-tetradecanoylphorbol 13-acetate (TPA) (Sigma cat# T-1585), frozen in 50 μl aliquots at 1 mg/ml
Test compounds as 100X DMSO stocks
4.2. Sample preparation
4.2.1. Plating and transfection
Calculate total amount of cell suspension needed based on a final volume of 30 μl per well.
Calculate total amount of Fugene6 assuming a ratio of 5 μl Fugene6 per μg of DNA in serum-free medium.
Preincubate Fugene for 5 min. in serum-free medium. Use 1/15th the final cell suspension volume and a polystyrene tube (Falcon 2059).
Dilute plasmid DNA into serum-free medium. Use 1/15th the final cell suspension volume and a polystyrene tube (Falcon 2059).
Combine DNA and Fugene and incubate 20 min at RT.
Prepare a cell suspension of 2,000 HeLa cells in 30 μl complete DMEM.
Remove calculated amount of cell suspension for transfection and combine with DNA-Fugene complexes.
Save remainder and use to plate untransfected control wells.
Plate 30 μl of cell suspension into wells according to plate layout.
Incubate cells for 2 days at 37C and 5% CO2.
4.2.2. Stimulation and automated immunostaining
Prepare 384 well treatment plates (Greiner clear polystyrene) containing 50–100 μl of compounds at 3X the desired screening concentration in complete growth medium containing 1.5% DMSO. Load columns 1, 2, 23, and 24 with 1.5% DMSO in DMEM.
Prepare 2 μg/ml TPA in HBSS from 1 mg/ml DMSO stock of phorbol ester (TPA) in DMSO.
Prepare all staining solutions as described under “Materials”.
Transfer 15 μl of compound treatment solution from reconstituted compound plates to assay plates without removing growth medium. Use a liquid handler with 384 well transfer for this step (Velocity 11 VPrep).
Return plates to incubator and incubate for 15–30 min. at 37°C.
Transfer plates to a Titertek MAP-C2 plate processor and add 15 μl 2 μg/ml TPA directly to drug-treated wells.
Return plates to incubator and incubate for 15 min. at 37°C.
-
Transfer plates to a Titertek MAP-C2 plate processor for automated immunostaining.
-
Add 15 μl 16% formaldehyde directly to each well without removing medium and incubate at room temperature for 10 min.
Remove fixative and wash with PBS (3 cycles, 50 μl, fast wash speed, 1 sec. aspirate delay).
Permeabilize for 10 min with 15 μl Triton-X100.
Wash with PBS and incubate in 15 μl blocking solution at room temperature for 1 h.
Remove blocking solution and incubate with 15 μl primary antibody cocktail at room temperature for 1 h.
Wash with PBS.
Incubate with 15 μl secondary antibody cocktail for 1 h at room temperature.
Wash with PBS, leaving last wash on cells.
Seal and barcode plate and image on ArrayScan or store at 4°C until needed for imaging.
-
Notes
The objective is to obtain reasonable transfection efficiencies while retaining cell monolayers suitable for imaging without compromising cell health. Because MKP-3 expressing cells are gated out, it is not necessary to have all cells express the target.
Key parameters that required optimization were cell plating density, transfection reagents and time of expression. We found that transfecting cells directly after trypsinization with 20 ng of DNA and a 1:5 ratio of DNA to Fugene 6 gave the most reliable results.
For automated immunostaining, it is essential that all solutions are free of particles. Solutions should be filtered through a 0.2 μm membrane.
Formaldehyde vapors generated during processing are a health hazard and should be trapped according to institutional guidelines.
4.3. Data analysis
To quantify cellular responses, our strategy to measure Erk phosphorylation based on KS statistics [45] was modified (Scheme 1). The new procedure quantifies differences in Erk phosphorylation levels in MKP-3 expressing cells compared with phospho-Erk levels in untransfected cells.
Scheme 1. Data analysis of phospho-Erk levels
Acquire blue, green, and red fluorescence intensities from 1,000 cells per well on an ArrayScan II using a three-channel target activation algorithm. (Channel 1, Hoechst; Channel 2, FITC; Channel 3, Texas Red.)
Extract individual cell data (average fluorescence intensities in Channel 2 and Channel 3) from Cellomics Store database file.
Isolate data from untransfected control wells and store in a temporary data file.
-
Threshold all data for FITC (=c-myc-MKP-3) positive cells based on distribution of green signal in untransfected wells.
Pool FITC distributions of untransfected wells
Set threshold as average + SD of vector-transfected cells. Alternatively, visually/manually determine FITC threshold from histograms or (x,y) scatter plots and subset all plates using the same value. This is faster and less affected by artifacts in the untransfected wells.
Select from the FITC subsetted data file only those wells that were transfected with MKP-3 and store in a temporary data file.
Combine data from untransfected wells with those from MKP-3 transfected and FITC subsetted wells.
-
Perform a KS analysis
Pool phospho-Erk distributions in MKP-3 transfected and vehicle-treated wells
Compare phospho-Erk distributions in all wells against this reference distribution.
Output is a matrix of KS values that are low for MKP-3 transfected and vehicle-treated cells, and high for untransfected (or inhibitor-treated) cells.
Notes
To perform the analysis, it is necessary to extract single-cell data from a full microplate, which can be as many as 1 million objects depending on cells acquired per field. Numbers this large are not manageable by Microsoft Excel-based programs. We use S-Plus (Insightful, Inc.) for statistical analysis and Spotfire Decision Site for graphing of individual cell data.
Extraction of single–cell data from HCS platform requires custom data analysis scripts The ArrayScan vHCS interface permits export of individual objects but is limited to 65,000 objects because it is Microsoft Excel-based. We have, therefore, developed two methods of extracting individual cell data: (1) Querying the Microsoft Access database file that the system generates for archiving (see [33] , or (2) By direct SQL query of the Cellomics Store database. In both cases, the extraction of a full microplate takes about 15 minutes. One needs to have knowledge about database structure and parameter names.
5.VARIABILITY ASSESSMENT AND HTS VALIDATION
5.1. Three day variability
Scaling up assays to a point where they can be considered for large scale screening necessitates formal assessment of assay variability. Such studies are routinely performed as part of HTS assay implementation and constitute a critical decision point in the assay development process. Many assays that perform well on a small scale fail at this stage because they are not robust enough for large sample sizes. Data scatter and numbers of outliers increase with increasing numbers of data points; automation adds additional variability. To ensure that an assay can perform reliably and robustly on a large scale, most HTS organizations perform a three-day variability assessment similar to that reported by Ghosh [46]. The general principle is that multiple plates of minimum and maximum control are generated on three consecutive days. From these data, signal-to-background (S : B), assay window (at least 1 standard deviation (SD) between the mean maximum value ± 3 SD and the minimum value - 3 SD) , Z-factors [42], intra-plate variability, inter-plate variability, and day-to-day variability were calculated. Table 1 lists the requirements for progression of an ideal assay through validation and screening. While many biochemical assays routinely meet these stringent requirements, complex cell-based assays often fail to meet one or more of the related performance standards. These assays may still be screenable if the source of variability can be identified and a positive assay window is routinely obtained, or if Z-factors are consistently above 0.2, a level of statistical significance suitable for screening with relaxed hit selection criteria [42].
Table 1.
Performance requirements for ideal HTS assays
| Parameter | Performance standard |
|---|---|
| S : B | > 3 |
| separation band or assay window | 1 SD between the mean maximum value - 3 SD and the minimum value + 3 SD |
| Z-factors | greater than 0.5 |
| intra-plate variability | < 20% COV |
| inter-plate variability | each plate within 20% of the mean of the 2 plates |
| day-to-day variability | < 20% COV between plate pairs |
SD, standard deviation
COV, coefficient of variance
For the MKP-3 chemical complementation assay, two plates of minimum (MKP-3 transfected and FITC gated) and maximum (untransfected, ungated) signal were processed on three consecutive days. Each plate contained 16 untransfected wells and 16 wells transfected with MKP-3, placed on the left and right side to control for edge effects. For minimum (MIN) and maximum (MAX) signal, the 320 remaining wells in the center of the plate were untransfected or transfected with MKP-3, respectively. All wells were treated with DMSO, stimulated with TPA, and immunostained for c-myc (MKP-3) and phospho-Erk. KS values were calculated by comparing each well on the microplate to a reference phospho-Erk distribution function derived from the 16 minimum signal controls. For each day, minimum and maximum signals from both plates were averaged and Z-factors were calculated according to [42]. Figure 3 shows that on all three days, the assay showed a positive separation bands, as indicated by the solid and dotted lines indicating three standard deviations from the means of minimum and maximum signal, respectively. Visual examination of plate graphs demonstrate a systematic error in one of the center rows of plate #1 on day 2, which was due to a clogged dispense nozzle on one of the liquid handlers. Although Z-factors exceeded 0.5 only once, the assay gave Z-factors above 0.2 on all three days, which has been described as suitable for screening [42]. Thus, while the assay did not reach the statistical significance seen with many biochemical assays, it performed on a level comparable to many commercially available cellular assays.
5.2. HTS VALIDATION
Based on the data obtained using the optimized procedure, the assay progressed into HTS validation. This is an important stage of HTS development as it mimics conditions of drug screening. Typically, independent test runs of multiple plates treated with vehicle control only are performed on three separate days, followed by a single run at the maximum number of plates on a single day. This design provides a tool to estimate the rate of false positives that can be expected during a screen. For the MKP-3 chemical complementation assay, five plates of DMSO controls were run on three separate days. Because of assay complexity and cost, the maximum batch size was set to 5 plates. Figure 4 shows that results from the HTS validation run. Ten out of 15 plates had Z-factors > 0.5. In the case of the two worst plates, low Z-factors were traced to transient malfunctions of the liquid dispenser, which was addressed by modifying instrument maintenance and QC procedures. All of the outliers came from positive control wells, and the rate of false positives was very low. With the exception of a single microplate, few data points exceeded a threshold of three standard deviations above the mean minimum value. None was greater than three standard deviation below the mean of the maximum controls.
6.CONCLUSIONS AND FUTURE DIRECTIONS
In this article, we have reviewed methodology to adapt a challenging, complex cellular assay to the stringent requirements for HTS. We selected the MKP-3 chemical complementation assay as an example of a challenging, multistep assay whose performance is affected by a multitude of factors. The assay’s complexity, however, provided an opportunity to generate general knowledge about the critical issues in rendering high-content screening assays HTS compatible. As a result, we were able to develop tools of general utility during the implementation process. Many of the aspects we identified as critical and that we discussed in detail will be applicable to any single-cell immunofluorescece-based assay.
Acknowledgments
This work was supported by NIH grants MH074411 and CA52995, and MH76330. We thank Peter McPherson for excellent technical assistance, Paul Johnston for providing technical advice and Tongying Shun for help with data analysis.
Abbreviations
- HCS
high-content screening
- HTS
high-throughput screening
- MAPK
mitogen-activated protein kinase
- MKP
mitogen-activated protein kinase phosphatase
- PAO
phenylarsine oxide
- TPA
12-O-tetradecanoylphorbol 13-acetate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Keyse SM. Curr Opin Cell Biol. 2000;12:186–192. doi: 10.1016/s0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 2.Farooq A, Zhou MM. Cellular Signalling. 2004;16:769–779. doi: 10.1016/j.cellsig.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 3.Magi-Galluzzi C, Mishra R, Fiorentino M, Montironi R, Yao H, Capodieci P, Wishnow K, Kaplan I, Stork PJ, Loda M. Lab Invest. 1997;76:37–51. [PubMed] [Google Scholar]
- 4.Bang YJ, Kwon JH, Kang SH, Kim JW, Yang YC. Biochem Biophys Res Commun. 1998;250:43–47. doi: 10.1006/bbrc.1998.9256. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Cheng Z, Malbon CC. Cancer Lett. 2003;191:229–237. doi: 10.1016/s0304-3835(02)00612-2. [DOI] [PubMed] [Google Scholar]
- 6.Liao Q, Guo J, Kleeff J, Zimmermann A, Buchler MW, Korc M, Friess H. Gastroenterology. 2003;124:1830–1845. doi: 10.1016/s0016-5085(03)00398-6. [DOI] [PubMed] [Google Scholar]
- 7.Denkert C, Schmitt WD, Berger S, Reles A, Pest S, Siegert A, Lichtenegger W, Dietel M, Hauptmann S. Int J Cancer. 2002;102:507–513. doi: 10.1002/ijc.10746. [DOI] [PubMed] [Google Scholar]
- 8.Loda M, Capodieci P, Mishra R, Yao H, Corless C, Grigioni W, Wang Y, Magi-Galluzzi C, Stork PJ. Am J Pathol. 1996;149:1553–1564. [PMC free article] [PubMed] [Google Scholar]
- 9.Small GW, Shi YY, Edmund NA, Somasundaram S, Moore DT, Orlowski RZ. Mol Pharmacol. 2004;66:1478–1490. doi: 10.1124/mol.104.003400. [DOI] [PubMed] [Google Scholar]
- 10.Wu W, Pew T, Zou M, Pang D, Conzen SD. J Biol Chem. 2004 doi: 10.1074/jbc.M411200200. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Xu J, Zhou JY, Liu Y, Wu GS. Cancer Res. 2006;66:8870–8877. doi: 10.1158/0008-5472.CAN-06-1280. [DOI] [PubMed] [Google Scholar]
- 12.Nyati MK, Feng FY, Maheshwari D, Varambally S, Zielske SP, Ahsan A, Chun PY, Arora VA, Davis MA, Jung M, Ljungman M, Canman CE, Chinnaiyan AM, Lawrence TS. Cancer Res. 2006;66:11554–11559. doi: 10.1158/0008-5472.CAN-06-1935. [DOI] [PubMed] [Google Scholar]
- 13.Rossig L, Haendeler J, Hermann C, Malchow P, Urbich C, Zeiher AM, Dimmeler S. J Biol Chem. 2000;275:25502–25507. doi: 10.1074/jbc.M002283200. [DOI] [PubMed] [Google Scholar]
- 14.Warmka JK, Mauro LJ, Wattenberg EV. J Biol Chem. 2004 doi: 10.1074/jbc.M403120200. [DOI] [PubMed] [Google Scholar]
- 15.Xu H, Yang Q, Shen M, Huang X, Dembski M, Gimeno R, Tartaglia LA, Kapeller R, Wu Z. J Biol Chem. 2005;280:36013–36018. doi: 10.1074/jbc.M508027200. [DOI] [PubMed] [Google Scholar]
- 16.Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, Boschert U, Arkinstall S. Science. 1998;280:1262–1265. doi: 10.1126/science.280.5367.1262. [DOI] [PubMed] [Google Scholar]
- 17.Hutter D, Chen P, Barnes J, Liu Y. Biochem J. 2000;352(Pt 1):155–163. [PMC free article] [PubMed] [Google Scholar]
- 18.Slack DN, Seternes OM, Gabrielsen M, Keyse SM. J Biol Chem. 2001;276:16491–16500. doi: 10.1074/jbc.M010966200. [DOI] [PubMed] [Google Scholar]
- 19.Chen P, Hutter D, Liu P, Liu Y. Protein Expr Purif. 2002;24:481–488. doi: 10.1006/prep.2001.1599. [DOI] [PubMed] [Google Scholar]
- 20.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Nat Struct Biol. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 21.Rudolph J, Epstein DM, Parker L, Eckstein J. Anal Biochem. 2001;289:43–51. doi: 10.1006/abio.2000.4906. [DOI] [PubMed] [Google Scholar]
- 22.Fattman CL, Allan WP, Hasinoff BB, Yalowich JC. Biochem Pharmacol. 1996;52:635–642. doi: 10.1016/0006-2952(96)00338-3. [DOI] [PubMed] [Google Scholar]
- 23.Burnouf C, Pruniaux MP. Curr Pharm Des. 2002;8:1255–1296. doi: 10.2174/1381612023394665. [DOI] [PubMed] [Google Scholar]
- 24.Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- 25.Kau TR, Schroeder F, Ramaswamy S, Wojciechowski CL, Zhao JJ, Roberts TM, Clardy J, Sellers WR, Silver PA. Cancer Cell. 2003;4:463–476. doi: 10.1016/s1535-6108(03)00303-9. [DOI] [PubMed] [Google Scholar]
- 26.Vogt A, Cooley KA, Brisson M, Tarpley MG, Wipf P, Lazo JS. Chem Biol. 2003;10:733–742. doi: 10.1016/s1074-5521(03)00170-4. [DOI] [PubMed] [Google Scholar]
- 27.Vogt A, Cooley KA, Brisson M, Tarpley MG, Wipf P, Lazo JS. Chem Biol. 2003;10:733–742. doi: 10.1016/s1074-5521(03)00170-4. [DOI] [PubMed] [Google Scholar]
- 28.Vogt A, Takahito A, Ducruet AP, Chesebrough J, Nemoto K, Carr BI, Lazo JS. J Biol Chem. 2001;276:20544–20550. doi: 10.1074/jbc.M100078200. [DOI] [PubMed] [Google Scholar]
- 29.Lazo JS, Aslan DC, Southwick EC, Cooley KA, Ducruet AP, Joo B, Vogt A, Wipf P. J Med Chem. 2001;44:4042–4049. doi: 10.1021/jm0102046. [DOI] [PubMed] [Google Scholar]
- 30.Giuliano KA, DeBiasio RL, Dunlay T, Gough A, Volosky JM, Zock J, Pavlakis GN, Taylor DL. J Biomol Screen. 1997;2:249–259. [Google Scholar]
- 31.Vogt A, Cooley KA, Brisson M, Tarpley MG, Wipf P, Lazo JS. Chem Biol. 2003;10:733–742. doi: 10.1016/s1074-5521(03)00170-4. [DOI] [PubMed] [Google Scholar]
- 32.Vogt A, Tamewitz A, Skoko J, Sikorski RP, Giuliano KA, Lazo JS. J Biol Chem. 2005;280:19078–19086. doi: 10.1074/jbc.M501467200. [DOI] [PubMed] [Google Scholar]
- 33.Vogt A, Lazo JS. Methods Mol Biol. 2006;356:389–400. doi: 10.1385/1-59745-217-3:389. [DOI] [PubMed] [Google Scholar]
- 34.Young IT. J Histochem Cytochem. 1977;25:935–941. doi: 10.1177/25.7.894009. [DOI] [PubMed] [Google Scholar]
- 35.Lampariello F. Cytometry. 2000;39:179–188. doi: 10.1002/(SICI)1097-0320(20000301)39:3<179::AID-CYTO2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 36.Watson JV. Cytometry. 2001;43:55–68. doi: 10.1002/1097-0320(20010101)43:1<55::aid-cyto1019>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 37.Cox C, Reeder JE, Robinson RD, Suppes SB, Wheeless LL. Cytometry. 1988;9:291–298. doi: 10.1002/cyto.990090404. [DOI] [PubMed] [Google Scholar]
- 38.Giuliano KA, Chen YT, Taylor DL. J Biomol Screen. 2004;9:557–568. doi: 10.1177/1087057104265387. [DOI] [PubMed] [Google Scholar]
- 39.Vogt A, Tamewitz A, Skoko J, Sikorski RP, Giuliano KA, Lazo JS. J Biol Chem. 2005;280:19078–19086. doi: 10.1074/jbc.M501467200. [DOI] [PubMed] [Google Scholar]
- 40.Peacock JA. Monthly Notices of the Royal Astronomical Society. 1983;202:615–627. [Google Scholar]
- 41.Fasano G, Francescini A. Monthly Notices of the Royal Astronomical Society. 1987;225:155–170. doi: 10.1093/mnras/225.3.491. [DOI] [PubMed] [Google Scholar]
- 42.Zhang JH, Chung TDY, Oldenburg KR. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 43.Dowd S, Sneddon AA, Keyse SM. J Cell Sci. 1998;111(Pt 22):3389–3399. doi: 10.1242/jcs.111.22.3389. [DOI] [PubMed] [Google Scholar]
- 44.Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM. EMBO J. 1996;15:3621–3632. [PMC free article] [PubMed] [Google Scholar]
- 45.Vogt A, Tamewitz A, Skoko J, Sikorski RP, Giuliano KA, Lazo JS. J Biol Chem. 2005;280:19078–19086. doi: 10.1074/jbc.M501467200. [DOI] [PubMed] [Google Scholar]
- 46.Ghosh RN, Chen YT, DeBiasio R, DeBiasio RL, Conway BR, Minor LK, Demarest KT. Biotechniques. 2000;29:170–175. doi: 10.2144/00291pf01. [DOI] [PubMed] [Google Scholar]