

Abstract
Protein tyrosine phosphatases (PTPs) catalyze the dephosphorylation of phosphotyrosine, a central control element in mammalian signal transduction. Small-molecule inhibitors that are specific for each cellular PTP would be valuable tools in dissecting phosphorylation networks and for validating PTPs as therapeutic targets. However, the common architecture of PTP active sites impedes the discovery of selective PTP inhibitors. Our laboratory has recently used enzyme/inhibitor-interface engineering to generate selective PTP inhibitors. The crux of the strategy resides in the design of “inhibitor-sensitized” PTPs through protein engineering of a novel binding pocket in the target PTP. “Allele-specific” inhibitors that selectively target the sensitized PTP can be synthesized by modifying broad-specificity inhibitors with bulky chemical groups that are incompatible with wild-type PTP active sites; alternatively, specific inhibitors that serendipitously recognize the sensitized PTP’s non-natural pocket may be discovered from panels of “non-rationally” designed compounds. In this review, we describe the current state of the PTP-sensitization strategy, with emphases on the methodology of identifying PTP-sensitizing mutations and synthesizing the compounds that have been found to target PTPs in an allele-specific manner. Moreover, we discuss the scope of PTP sensitization in regard to the potential application of the approach across the family of classical PTPs.
Keywords: Protein tyrosine phosphatases (PTPs), allele-specific inhibitors, inhibitor sensitization, protein engineering, PTP1B, T-cell PTP (TCPTP), PTPH1, hematopoietic PTP (HePTP)
1. Introduction
The protein tyrosine phosphatases (PTPs), an enzyme superfamily that includes about 100 human proteins, catalyze the dephosphorylation of phosphotyrosine in protein substrates [1–3]. Phosphotyrosine is a critical cell-signaling control element, and PTP activity is essential both for cellular homeostasis and for appropriate responses to extracellular signals. In contrast to the early hypothesis that PTP activity largely represents a basal (“housekeeping”) counterforce to highly controlled protein-phosphorylation events (i.e., tyrosine kinase activity), it is now clear that PTP activity is also specific and tightly regulated, and that PTPs can exert either positive or negative effects on a signaling pathway [4–6]. Moreover, it is now beyond dispute that PTPs represent significant drug targets for a wide variety of clinically important pathologies [7–10]. Small-molecule inhibitors that can act specifically on individual PTPs would thus be important tools for both of these “worlds”: understanding the basic-science roles of individual PTPs in complex signaling pathways, and validating PTPs as viable therapeutic targets [11,12].
Unfortunately, due to the size and homology of the PTP superfamily, the identification of inhibitors that are specific for each of the ∼100 PTPs through the methods of conventional medicinal chemistry is not a practical prospect in the foreseeable future. The search for selective PTP inhibitors has intensified in recent years; however, these efforts are generally only pursued after a PTP has been unambiguously identified as a clinical target. For example, the overwhelming majority of PTP-inhibitor development has been focused on a single enzyme: PTP1B, a leading type-II-diabetes target. While the search for PTP1B inhibitors has yielded notable successes [13–18], the labor-intensive efforts that have led to the discovery of potent and selective PTP1B inhibitors highlight the difficulties inherent in such endeavors.
Our laboratory has recently attempted to develop a general method for targeting individual PTPs with small-molecule inhibitors, a method that does not rely on serendipitously exploiting the small atomic-level differences in the binding sites of homologous PTPs [19–21]. To circumvent these specificity problems, we have used engineering of PTP active sites to generate “inhibitor-sensitized” PTPs—enzymatically competent PTPs that contain active-site mutations, which allow them to be competitively inhibited by compounds that do not effectively inhibit wild-type PTPs (Figure 1). These inhibitors are generally small, organic molecules that have been designed to target a non-natural binding site (“hole”) in the sensitized PTP. In principle, since the “allele-specific” inhibitors target the sensitized PTP–and not wild-type PTPs–these compounds can be used to specifically inhibit engineered PTPs in a model cellular system (or organism, or lysate) that contains the sensitized PTP. The ability to observe the phenotype of cells after selective inhibition of a target PTP could provide a rapid method for determining the unique roles of individual PTPs in signal-transduction pathways.
Fig. 1.
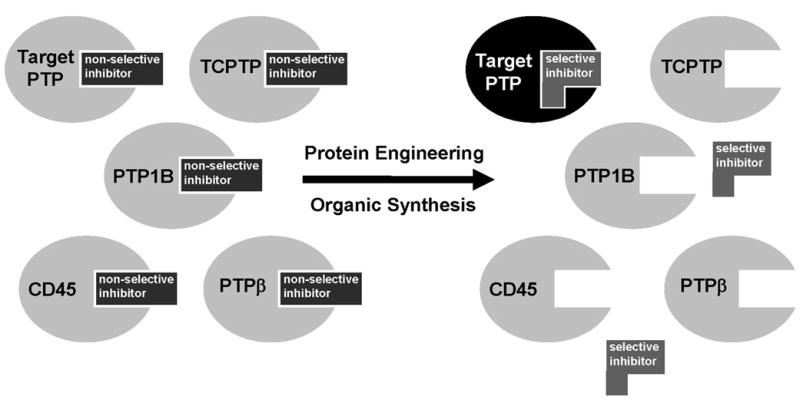
Schematic representation of an active-site-directed inhibitor-sensitization approach for PTPs. The problem of structural redundancy in PTP active sites is alleviated by artificially introducing diversity in the target PTP with a functionally silent mutation. The conversion of a large amino acid to a small amino acid creates a novel binding pocket that is not present in wild-type PTPs. A specific inhibitor of the engineered PTP is synthesized by modifying a known PTP inhibitor with a chemical group designed to fit the novel active-site pocket.
It has been previously shown in a number of systems that the introduction of chemical diversity into a target protein (through mutagenesis), coupled with small-molecule diversification (through organic synthesis), can lead to the rapid identification of specific ligand/receptor pairs [22–24]. To cite the most relevant examples, protein/small-molecule interface engineering has been used to design cell-specific calcineurin inhibitors [25], and to generate inhibitor-sensitized protein methyltransferases [26] and protein kinases [27–29]. Inhibition of sensitized protein kinases has been of particular importance in demonstrating the utility of chemical approaches in cell-signaling studies: information gathered from chemical kinase-inhibition experiments is often distinct from that obtained by genetically knocking out a kinase, or suppressing its expression through RNAi [30].
Building on these studies, our laboratory’s attempts at designing inhibitor-sensitive PTPs started with the recognition that all classical PTPs adopt a conserved fold in their respective catalytic domains [31]. Therefore, any classical PTP could, in principle, be used as a prototype for the design of inhibitor-sensitized PTP mutants. Moreover, due to the conserved nature of the PTP active site, once a sensitizing mutation is discovered in a prototype PTP, it is likely that corresponding mutations in other PTPs would also be sensitizing [27,32]. As a prototype for a first generation of sensitized PTPs we used PTP1B. This enzyme can be expressed in E. coli [33] and readily purified as a GST-fusion protein [17]. Importantly, many crystal structures of PTP1B have been solved [31] making it an ideal PTP on which to perform the initial enzyme engineering. ]
Our PTP1B-sensitization was guided by the following criteria. (i.) An amino acid that is chosen for mutagenesis must be large enough such that substitution by a small amino acid will create a novel binding pocket. (ii.) The corresponding residue in PTPs other than PTP1B, according to primary sequence alignments, should generally not be occupied by small aminoacid residues (Figure 2). (iii.) The mutant PTP1B must retain enzymatic activity that is comparable to that of the wild-type. (iv.) The amino acid used for sensitization should be present in other PTPs, eliminating the need to redesign the PTP/inhibitor interface for each target.
Fig. 2.

Partial sequence alignment of PTPs discussed in this review, in addition to two further examples (SHP1 and CD45). Numbering is according to human PTP1B.
Inhibitor-sensitizing mutations identified by the above constraints have a potential advantage over medicinal-chemistry approaches to PTP-inhibitor discovery: the mutations are identified based on the common features of PTP active sites, not the small differences between PTPs. That is, due to the conserved nature of classical PTP domains, sensitizing mutations that are discovered using PTP1B may be applicable across the family of classical PTPs, by simple inspection of primary-sequence alignments (e.g., Ile219 in PTP1B corresponds to Ile846 of PTPH1, see Figure 2). (Our approach is based on the primary-sequence homology of classical PTPs. Since, outside of the PTP consensus sequence, other families of PTPs share little homology with classical PTPs, alternative engineering strategies will be presumably be needed to sensitize other PTP families.) In principle, a sensitizing mutation discovered on PTP1B could be used to generate an inhibitor-sensitive version of a classical PTP that has never been crystallized—or even one that has never been expressed in vitro. Ultimately, a series of highly selective and allele-specific inhibitors could be used to systematically delineate the precise functions of many PTPs in signaling cascades and to validate PTPs as therapeutic targets.
Our laboratory’s progress in identifying PTP-sensitizing mutations, synthesizing the attendant allele-specific inhibitors, and analyzing the scope of the PTP-sensitization strategy across the family of classical PTPs will comprise the focus of this review.
2. Materials and reagents
All chemicals for organic synthesis were purchased from Sigma-Aldrich (www.sigmaaldrich.com) or Acros (www.fishersci.com) and used without further purification, unless otherwise noted. All E. coli cultures were grown in LB Broth, obtained from Fisher Scientific (www.fishersci.com). Ampicillin (sodium salt) was obtained from EM Science (www.vgdusa.com) and used at 100 μg/mL. All plasmid preparations were performed with Wizard Plus Minipreps DNA Purification System from Promega (www.promega.com). Bacterial Protein Extraction Reagent (BPER), SwellGel Nickel Chelated Discs, and the SwellGel Bulk Immobilized Glutathione Discs were obtained from Pierce (www.piercenet.com). Dpn I restriction enzyme (20000 U/mL) was purchased from New England Biolabs, Inc. (www.neb.com). The Centriprep Centrifugal Filter Devices (Ultracel YM-30) are products of Amicon (www.amicon.com). QuikChange Site-Directed Mutagenesis Kit was obtained from Stratagene (www.stratagene.com). Mutagenic primers (desalted) were purchased from Invitrogen (www.invitrogen.com). All DNA sequencing was performed by the Cornell Biotechnology Resource Center.
3. Identification and evaluation of potentially inhibitor-sensitizing PTP mutations
3.1. Introduction
For a first-generation of potentially PTP1B-sentizing mutation sites we selected valine 49 (V49) and isoleucine 219 (I219). These active-site amino-acid residues possess hydrophobic side chains that are not thought to play critical roles in the PTP mechanism [34], leading us to hypothesize that mutation of these amino acids to the smaller residues, alanine and glycine, could sensitize PTP1B without disrupting the enzyme’s catalytic function. Additionally, primary-sequence alignments suggest that mutation of the corresponding residues could, if successful in PTP1B, similarly confer novel inhibitor-sensitivity in other PTPs (Figure 2). A sequence alignment of all 37 classical PTP catalytic domains in the human genome shows that the position corresponding to Val49 of human PTP1B is occupied by isoleucine or valine in 35 of the 37 PTPs [2]. Likewise, the position corresponding to Ile219 of human PTP1B contains either valine or isoleucine in 28 of 37 human classical PTP domains [2]. Based on the analysis above, we generated the following PTP1B mutants: V49G, I219G, V49A, I219A, and V49A/I219A.
Evaluation of these mutants for in vitro catalytic activity showed that the glycine mutants (V49G and I219G) and the double-alanine mutant (V49A/I219A) were severely catalytically compromised [21]. Apparently, the PTP1B active site is sensitive to significant losses of hydrophobic surface area in this phosphotyrosine-binding site. Additionally, drastic diminution in the hydrophobic components of the active site may also destabilize the protein, impairing the catalytic activity. Regardless of the reasons for their inactivity, V49G, I219G, V49A/I219A PTP1B were not investigated further. By contrast, V49A and I219A PTP1B turn over substrate at rates that are essentially indistinguishable from wild-type PTP1B, while their KM values are approximately 7- and 4-fold, respectively, higher than the wild-type value (Table 1). (The slight increase in the kcat value of I219A PTP1B relative to wild-type may not indicate an actual increase in turnover rate; error in determining concentrations of active enzyme could be responsible for modest differences in kcat values.) The catalytic efficiencies of V49A and I219A PTP1B are both within an order of magnitude (8-fold and 2-fold lower, respectively) of the wild-type value, leading us to surmise that these enzymes are suitable for an inhibitor design strategy that requires a silent mutation [27]. (V49A PTP1B had been prepared and characterized previously by Zhang and coworkers [35]; these authors observed an even smaller reduction of catalytic efficiency, less than 2-fold, when V49A PTP1B was assayed at pH 7 and 30°C.)
Table 1.
Kinetic constants for inhibitor-sensitized PTP1B mutants, assayed with pNPP as substrate at pH 5.2
| PTP | kcat (s−1) | KM (mM) | kcat/KM (s−1 mM−1) |
|---|---|---|---|
| wild-type PTP1B | 12 ± 1.8 | 0.32 ± 0.067 | 40 |
| V49A PTP1B | 11 ± 3.7 | 2.2 ± 0.057 | 5.5 |
| I219A PTP1B | 24 ± 1.3 | 1.4 ± 0.35 | 17 |
It is impossible to predict a priori what level of change in catalytic efficiency is tolerable for a mutant to complement the function of a wild-type enzyme. However, it seems unlikely that a mutation that leads to a modest increase in the mutant’s KM for pNPP would significantly affect the biological function of the PTP. The true physiological substrates of PTP1B, tyrosine-phosphorylated proteins, are far more structurally complex than pNPP, and PTP1B makes an array of important binding contacts with phosphoproteins outside of the phosphotyrosine-recognition site [36,37]. Therefore, the small increase in KM for pNPP for the alanine-mutant PTP1Bs, relative to wild-type, would most likely not be reflected in a change in biological substrate recognition. Future cellular experiments, however, are needed to confirm this working hypothesis.
3.2. Identification of V49 and I219 analogs in other PTPs
Primary-sequence alignments can be used to identify positions corresponding to V49 or I219 PTP1B in any mammalian classical PTP (see Figure 2). Alignment of other target PTPs not described in this review may be generated using any standard web-based alignment application (ClustalW, NCBI’s Blast 2 Sequences, etc.). Alternatively, Andersen et al’s very helpful alignment of all human PTP domains may be consulted [2].
3.3. Introduction of inhibitor-sensitizing PTP mutations
Once a suitable overexpression vector for a target PTP is obtained, inhibitor-sensitizing mutations can be introduced. The QuikChange Site-Directed Mutagenesis Kit, which provides detailed instructions for mutagenic-primer design, was used according to the manufacturer’s instructions with some minor modifications, which are detailed below.
Plasmid DNA from a miniprep (1 μL), cloned Pfu 10× reaction buffer (5 μL), cloned Pfu DNA polymerase (1 μL, 2.5 U), 2 mM dNTP mix (5 μL), and water (35 μL) were combined with appropriate primers (1.5 μL of each at a concentration of 100 ng/μL), and placed in a temperature cycler. The reaction mixtures were subjected to one cycle of 95 ºC for 30 seconds, then 12 cycles of 95 ºC for 30 seconds, 55 ºC for 1 minute, and 68 ºC for 16 minutes.
After cooling to room temperature, 1 μL of Dpn I (20 U) was added and the reaction mixtures were incubated at 37 °C for 1 hour.
DNA in the reaction mixtures was precipitated by adding 40 μL of water, 10 μL of 3M sodium acetate (pH 5.2), and 250 μL of ice-cold ethanol, incubating at −20 °C for 20 minutes, and centrifuging at 13000 rpm at 4 °C for 15 minutes. The supernatants were discarded and the DNA pellets were dissolved in 3 μL of sterile water. The entirety of the resulting DNA solutions was transformed into DH5α E. coli competent cells, and the transformed cultures were plated on LB/ampicillin by standard methods.
Ampicillin-resistant colonies were picked and grown overnight in 3 mL of liquid LB/ampicillin. Plasmid DNA from the resulting cultures was extracted and sequenced. Confirmed mutant plasmids were transformed into BL21(DE3) E. coli competent cells for protein expression (for PTP-encoding genes harbored in pET vectors).
3.4. Expression of inhibitor-sensitized PTPs
Expression and purification conditions vary based on the expression vector of choice, the expression level of the particular PTP, and the desired amount of purified PTP. Expression-optimization experiments should be carried out for every PTP target. Thus, the following directions are generalized, and should be modified as needed.
A culture of a BL21(DE3) strain containing the appropriate mutant plasmid was grown in liquid LB/ampicillin until an absorbance (600 nm) of 0.5 was reached.
Isopropyl-1-thio-β-D-galactopyranoside (IPTG) was added to a final concentration of 0.2 mM. The cell suspension was shaken at 37 °C for additional 5–6 hours and was centrifuged at 10,000×g for 15 minutes. The supernatant was discarded and the pellet was stored at −80 °C.
Immediately before purification, the cell pellet was lysed in Bacterial Protein Extraction Reagent (BPER) according to the manufacturer’s instructions.
3.5. Purification of inhibitor-sensitized PTPs
All of the inhibitor-sensitized PTPs purified in our laboratory to date have contained a purification tag: glutathione-S-transferase (GST) for PTP1B; and 6-histidine (His6) for TCPTP, PTPH1, and HePTP. GST-tagged proteins were purified with the SwellGel Bulk Immobilized Glutathione Discs and His6-tagged proteins were purified with SwellGel Nickel Chelated Discs according to the manufacturer’s instructions. After PTP purification, regardless of the purification method of choice:
PTP-activity-containing eluents were pooled, concentrated with a Centriprep-30 filtration unit, and exchanged into pH 7.0 buffer containing 50 mM 3,3-dimethylglutarate, 1 mM EDTA, and 1 mM dithiothreitol.
Glycerol was added to the purified protein to a final concentration of 30%, and the resulting solution was aliquoted and stored at −20 °C. Note: We have recently started flash-freezing PTP aliquots (no glycerol) in liquid nitrogen and storing them at −80°C; we have found this storage method to be preferable.
3.6. Kinetic characterization of inhibitor-sensitized PTPs
Note
All PTP assays should be performed in low-protein-binding microcentrifuge tubes (e.g., Eppendorf LoBind, Fisher Scientific catalog #13–698–793).
The kinetic competence of sensitized PTPs was determined using the artificial PTP substrate para-nitrophenyl phosphate (pNPP). Activity assays were carried out at 22 °C in a total reaction volume of 200 μL containing pNPP (0.5–10 mM) and the appropriate enzyme (20–150 nM) in the appropriate 1×PTP Buffer (pH 7.0: 50 mM 3,3-dimethylglutarate pH 7.0, 1 mM EDTA, 50 mM NaCl; pH 5.2: 100 mM NaOAc pH 5.2, 50 mM NaCl). Reactions were quenched after 8 min by the addition of 40 μL of 5 M NaOH. The reaction mixtures (200 μL) were loaded onto a 96-well plate, and the absorbance at 405 nm was measured with a Molecular Devices Versamax plate-reader. The kinetic constants for each enzyme were determined by fitting the data to the Michaelis-Menten equation using SigmaPlot. Values given in Tables 1 and 2 represent the averages and standard deviations of at least three independent experiments.
Table 2.
Kinetic constants for inhibitor-sensitized HePTP mutants, assayed with pNPP as substrate at pH 7.0
| PTP | kcat (s−1) | KM (mM) | kcat/KM (s−1 mM−1) |
|---|---|---|---|
| wild-type HePTP | 1.3 ± 0.39 | 6.9 ± 3.0 | 0.19 |
| I107A HePTP | 1.7 ± 0.49 | 6.2 ± 2.4 | 0.27 |
| I274A HePTP | 1.3 ± 0.33 | 7.3 ± 2.7 | 0.18 |
4. Design and synthesis of allele-specific PTP inhibitors
4.1. Introduction
Compared to the design criteria for PTP-sensitizing mutations, the criteria for the small-molecule inhibitors are more straightforward. In fact, compounds that target the novel binding pocket of sensitized PTPs can either be rationally designed or identified from screens of non-directed compound libraries.
For rationally designed allele-specific inhibitors, the parent inhibitor, or scaffold, should be capable of targeting multiple wild-type PTPs (Figure 1). Also, the binding orientation of the inhibitor in the enzyme active site should be known or readily predictable; and the molecule should bind in a manner in which the position pointing toward the novel binding pocket can be synthetically modified. As a starting point for a panel of rationally designed allele-specific inhibitors that meet these criteria, we chose 6-(oxalyl-amino)-1H-indole-5-carboxylic acid (compound 1, Figure 3) as a scaffold. This general PTP inhibitor, which was discovered by workers at Novo Nordisk, inhibits a variety of classical PTPs at micromolar concentrations (PTP1B, PTPα, PTPβ, PTPε, CD45, SHP-1) [38]. Also, the crystal structure of 1 bound to PTP1B has been reported [38]. Importantly, the indole nitrogen of 1 binds close in space to V49 and I219. This nitrogen atom thus represents a “hook” onto which chemical “bumps” can be synthetically appended. These bumps, which are targeted toward the engineered pockets of V49A and I219A PTP1B, are designed to be sterically incompatible with the active sites of wild-type PTPs (Figure 1).
Fig. 3.
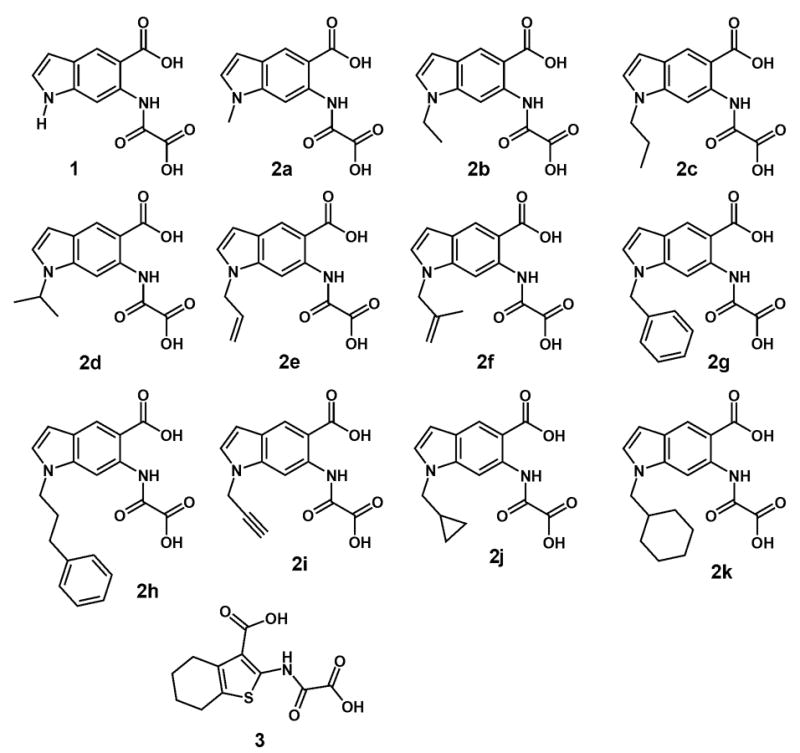
Chemical structures of compounds 1, 2a–2k, and 3.
In addition to rationally designed allele-specific inhibitors, we have found that the novel pockets of sensitized PTPs can also preferentially bind compounds that are not co-engineered with the enzyme active site. In principle, any compound that possesses some PTP-inhibitory ability could serendipitously exploit the sensitized-PTP active site. Along these lines, we have found that a compound that was previously identified as a wild-type PTP inhibitor (compound 3, Figure 3), can act in an allele-specific manner, inhibiting I219A PTP1B much more potently than it does wild-type PTP1B [19].
4.2. Synthesis of indole-based inhibitors 2a-2k
N-substituted analogs of 1 (compound series 2a-2k) were prepared according to a seven-step synthesis, the last three steps of which are depicted in Scheme 1.The indole scaffold 4 was synthesized as described by Showalter and coworkers [39]. Although we found no literature precedent for a high yield N-1 alkylation of either an aminoindole [40] or an oxalylaminoindole, we were able to achieve the synthesis of compounds 2a-2k by two distinct routes, without employing protective group chemistry. In an effort to minimize repetitive post-alkylation chemistry, we initially attempted to alkylate the oxalylated compound 5.We hypothesized that, in an excess of sodium hydride, the doubly deprotonated conjugate base of 5 would be formed. This dianion would presumably alkylate at N-1, owing to the greater basicity (hence, nucleophilicity) of this position. This strategy was carried out with limited success: excellent SN2 electrophiles, such as allyl bromide and benzyl bromide (yielding compounds 7e and 7g, respectively), were compatible with this approach. However, compound 5 is inherently unstable under the alkylation conditions, and longer incubation times (or elevated temperatures) were not possible when less reactive electrophiles were used. Alternatively, we found that compound 4 could be directly alkylated with poor to quantitative yields (17–98%), furnishing compound series 6.Although this scheme requires a larger amount of post-derivatization chemistry, it proved to be the more robust approach, allowing for the use of a broad range of alkyl-halide electrophiles. The two synthetic routes converge at compound series 7, which was subjected to standard ester hydrolysis conditions, affording a panel of putative inhibitors, compounds 2a–2k. The following protocols were used (Scheme 1):
Representative procedure for the alkylation of compound 4 (synthesis of 6a): 10.7 mg (0.45mmol, 1.3 equivalents) of dry, 95% sodium hydride were weighed into a flame-dried round bottom flask containing a magnetic stir bar. The flask was sealed and purged with argon. 2.5 mL of anhydrous DMF were introduced via syringe. The resulting suspension of sodium hydride in DMF was cooled to 0 °C. While the suspension was cooling, two separate solutions were prepared at room temperature: 70.9 mg (0.35 mmol) of 4 dissolved in 1 mL anhydrous DMF, and 49.7 mg (0.35 mmol) of methyl iodide in 1 mL anhydrous DMF. The solution of 4 in DMF was added via syringe to the cooled suspension of sodium hydride. Once visible evolution of hydrogen gas had ceased, the methyl-iodide solution was added. The reaction was monitored by TLC (40% ethyl acetate/60% hexanes) until consumption of 4 was complete. The reaction mixture was quenched with 4 mL saturated aqueous ammonium chloride, poured into water, and extracted twice with ethyl acetate. The organic layers were pooled, washed 5–10 times with water to remove trace DMF, and dried over magnesium sulfate. Ethyl acetate was removed under reduced pressure to yield 6a (90% yield). [When necessary, other alkylation products were purified by flash column chromatography in ethyl acetate/hexanes. Alkylations of compound 5 (conditions á, Scheme 1) to furnish 7e and 7g were carried out under identical conditions, with the exception that three equivalents of NaH were used.]
Representative oxalylation procedure (synthesis of 7a): A solution of ethyl oxalyl chloride (51.4 mg, 0.38 mmol) in 6 mL of THF was added to 60.6 mg (0.28 mmol) of 6a, and the reaction was stirred at room temperature for 30 minutes. The reaction mixture was poured into water and extracted. The organic layers were pooled and solvent was removed under reduced pressure to yield compound 7a in quantitative yield.
Representative procedure for the ester hydrolysis of compounds 7a–7k (synthesis of 2a): Compound 7a (85.7 mg, 0.27 mmol) was dissolved in 6 mL of a 1:1 EtOH:H2O solution. 5N NaOH (1 mL, 5 mmol, 19 equivalents) was added and the reaction mixture was stirred for 16 hours at room temperature. Ethanol was removed by evaporation under reduced pressure and the resulting aqueous mixture was acidified with 1M HCl over ice. The precipitate was collected by filtration, washed with cold water, and dried under reduced pressure to yield dicarboxylic acid product 2a (80% yield).
4.3. Synthesis of thiophene-based inhibitor 3
Compound 3 was prepared as described by others [41].
5. Evaluation of inhibitor selectivity for mutant PTPs
5.1. Introduction
We have identified compounds that display significant selectivity for V49A and I219A PTP1B over wild-type PTP1B, by screening compound series 2 for PTP inhibition [21]. Nine members of series 2 exhibited varying degrees of selectivity for at least one of the alanine mutants [21]. As a rule, selectivity was greater for I219A PTP1B than for V49A. This preference for I219A mutants over V49A mutants has been true of almost every putative inhibitor analyzed to date; thus, V49A PTP1B will not be discussed further here. The most selective mutant/inhibitor pair consisted of I219A PTP1B and the N-methyl derivative of 1 (2a, Figures 2 and 4A), which demonstrated 10-fold selectivity for I219A PTP1B [KI (pH 5.2) = 1.1 μM)] over wild-type PTP1B [KI (pH 5.2) = 11 μM)] [21].
Fig. 4.
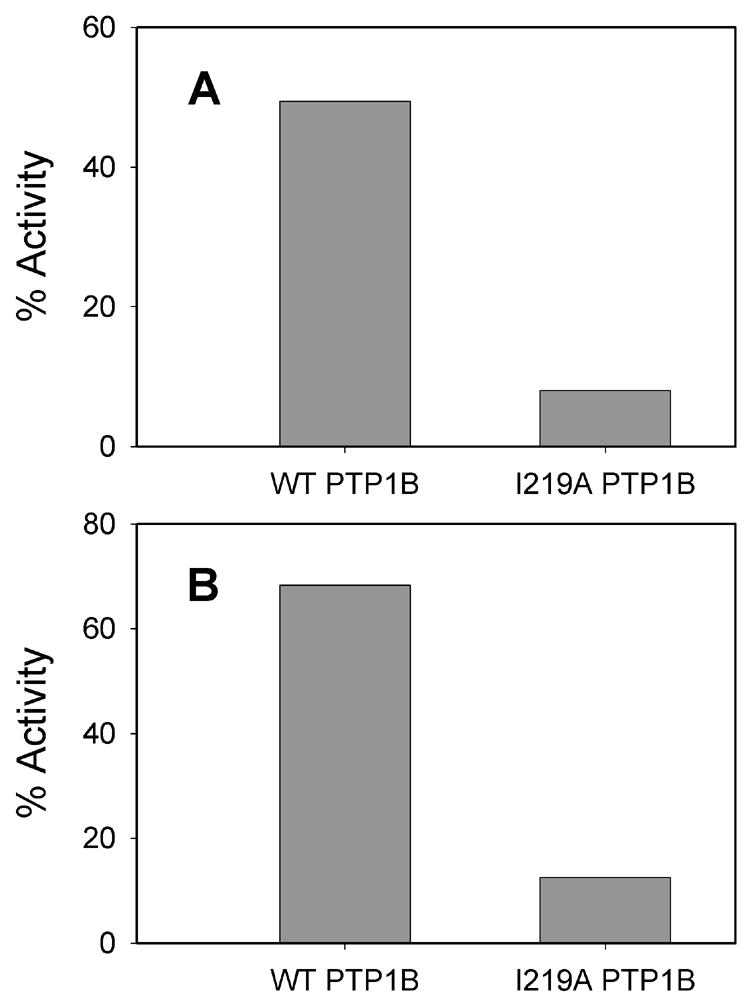
(A) Selective inhibition of I219A PTP1B by compound 2a. Compound 2a (25 μM) was incubated at 22 °C with 100 mM NaOAc pH 5.2; 50 mM NaCl; pNPP (concentration corresponding to the Km value for the particular enzyme); and the indicated PTP. Percent PTP1B activities in the presence of 2a (normalized to a no-inhibitor control) are shown as bars. (B) Selective inhibition of I219A PTP1B by compound 3.Compound 3 (20 μM) was incubated at 22 °C with 50 mM 3,3-dimethylglutarate pH 7.0; 1 mM EDTA; 50 mM NaCl; pNPP (concentration corresponding to the KM for the particular enzyme); and the indicated PTP. Percent PTP1B activities in the presence of 3 (normalized to a no-inhibitor control) are shown as bars.
In further experiments, we have found that previously described PTP inhibitors may also act in an allele-specific manner. Compound 3 (Figure 3) belongs to a family of well known fused-thiophene-based PTP inhibitors [41–44]. Surprisingly, we found that this ostensibly wild-type inhibitor targets I219A much more effectively than it does wild-type PTP1B (Figure 4B). Compounds 3’s selectivity for I219A PTP1B is even greater than that of the rationally designed 2a: 3 is a 30-fold more potent inhibitor of I219A PTP1B than wild-type PTP1B at pH 7.0 (wild-type PTP1B: KI = 34 μM; I219A PTP1B: KI = 1.0 μM) [19]. We hypothesize that the I219 side chain of wild-type PTP1B precludes the optimal binding orientation of compound 3’s cyclohexene moiety. More generally, these results show I219 can control inhibitor access to a PTP active site, even if the relevant compound is not concertedly designed as allele-specific. This analysis highlights the possibility that even more selective allele-specific PTP inhibitors may be identified by subjecting large compound libraries to inhibition screens with sensitized PTPs.
Regardless of the target PTP, or the set of compounds being evaluated, the same basic sequence can be carried out to identify allele-specific PTP inhibitors: first, screen sets of putative inhibitors at a fixed concentration to look for “hits” that unambiguously inhibit a sensitized PTP more potently than the corresponding wild-type; second, perform more thorough experiments to determine the relevant inhibition constants (KI) for the hit compounds.
5.2. Percent-activity screens
Percent-activity screens (see Figures 4 and 5) were performed with pNPP-based PTP assays essentially as described in section 3.6. (Characterization of inhibitor-sensitized PTPs), with the following notes:
Fig. 5.
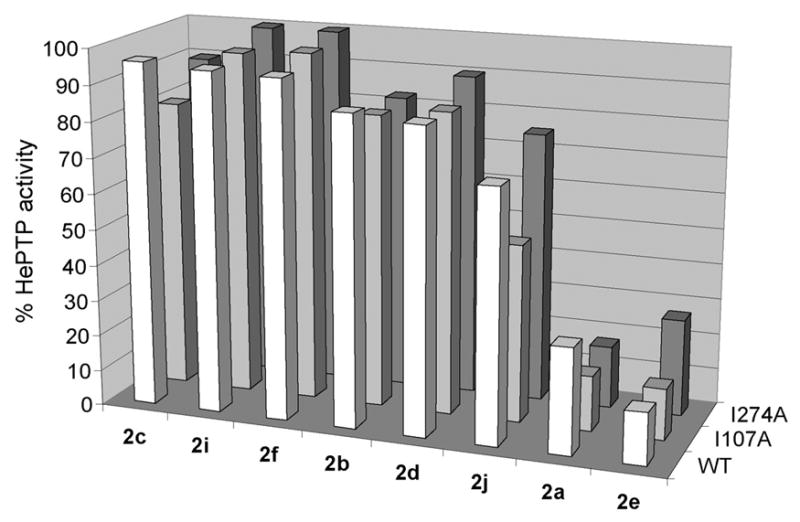
Screen of compound panel 2 for selective inhibition of engineered HePTP mutants. The indicated compounds (100 μM) were incubated at 22 °C with 50 mM 3,3-dimethylglutarate pH 7.0; 1 mM EDTA; 50 mM NaCl; pNPP (concentration corresponding to the KM for the particular enzyme); and wild-type (foreground), I107A (middle), or I274A (background) HePTP. Percent HePTP activities in the presence of the inhibitors (normalized to a no-inhibitor control) are shown as bars, which represent the mean values from at least three experiments.
Either 4 μL DMSO (vehicle) or 4 μL of a 50× stock solution of the appropriate inhibitor dissolved in DMSO (e.g., 1 mM stock for a final inhibitor concentration of 20 μM) was added to the reaction mixture.
The percent activity in the presence of an inhibitor was calculated by dividing the average A405 of that enzyme with inhibitor by the average A405 of the controls (DMSO only).
The pNPP concentration used in percent-activity screens should correspond to the KM of the enzyme being analyzed; this is because the inhibitors being analyzed are competitive, and the inhibitor-sensitized PTPs may have altered substrate KM values (see Table 1). When all enzymes are assayed at substrate concentrations corresponding to their respective KM values, wild-type and mutant percent-activity data can be directly compared.
5.3. KI measurement
KI determinations were performed with pNPP-based PTP assays essentially as described in section 3.6. (Characterization of inhibitor-sensitized PTPs), with the following notes:
Either 4 μL DMSO (vehicle) or 4 μL of a 50× stock of the inhibitor being assayed (dissolved in DMSO) was added to the reaction mixture.
At various fixed concentrations of inhibitor, the initial rate of pNPP hydrolysis was measured at five different pNPP concentrations. Inhibition constants were determined by fitting the data to classical Michaelis-Menten kinetic models for competitive inhibition (vo = Vmax[S]/αKM+[S], where α = 1 + [I]/KI).
6. Scope of PTP-sensitization strategy across the classical-PTP family
6.1. PTP1B and TCPTP
As discussed in detail above, PTP1B has served as the prototype for our laboratory’s PTP-sensitization endeavor. Since a general PTP-sensitization approach aims to exploit the commonalities between PTP active sites, one would expect that, the more homologous an enzyme is to PTP1B, the more amenable to homology-based sensitization the target PTP would be. To date, this supposition has been borne out by our experiments with PTP1B’s closest homolog, TCPTP [2]. Owing to the almost identical active sites of PTP1B and TCPTP, the vast majority of literature PTP inhibitors demonstrate similar potencies against both phosphatases [13–16]. Consistent with this, we have found that our allele-specific inhibitors target I219A PTP1B and I220A TCPTP with roughly equivalent KI and selectivity (compared to wild-type PTPs) values [19,21]. Thus, it seems safe to assume that, with future discoveries of inhibitor-sensitizing mutations or allele-specific inhibitors, the data obtained from a PTP1B prototype will be readily transferable to engineering unique TCPTP/inhibitor pairs (and vice versa, if the engineering is performed on TCPTP).
6.2. PTPH1
For an active-site-directed PTP sensitization strategy to be truly general it must work for PTPs that have not been subjected to inhibitor studies, whose crystal structures are not known, and/or have not been biochemically characterized. As our first test of these criteria, we attempted to engineer novel inhibitor sensitivity in PTPH1 [20], a biologically important PTP—somatic PTPH1 mutations have been discovered in colorectal cancers [45]—that is less closely related to PTP1B than TCPTP (36% catalytic-domain identity between PTP1B and PTPH1). When we performed our sensitization on PTPH1, it had not been subjected to targeted inhibitor-discovery studies, and a PTPH1 crystal structure was not known. [A structure of the PTPH1 catalytic domain has since been deposited in the Protein Data Bank (PDB ID: 2B49), but it has not yet been described in a publication.]
We found that primary-sequence analysis could, indeed, be used to impart novel inhibitor sensitivity to a PTP whose structure had not been solved. The analog of I219A PTP1B, I846A PTPH1 (Figure 2), was made and screened against compound series 2.As with I219A PTP1B, all but two of the compounds tested exhibited some degree of selectivity for I846A PTPH1 with respect to the wild-type PTPH1 [20]. This high hit rate of compound series 2 is notable, particularly considering that the inhibitor design for these compounds was performed on PTP1B, which is not a particularly close homolog of PTPH1. Additionally, the observation that compound 2a, the most selective inhibitor of I219A PTP1B and I220A TCPTP, was also the most selective for I846A PTPH1 lent further support to our use of the PTP1B structure as a prototype [20]. That said, the overall I846A selectivity of compound 2a (with respect to wild-type PTPH1) was only 4-fold [20], suggesting that further inhibitor discovery will be required to identify a compound that targets the sensitized PTPH1 with high selectivity.
6.3. HePTP
In more recent work on a PTP from a different subclass, our laboratory has learned that some PTPs may not be readily sensitized to compound panel 2 through introduction of alanine at positions 49 and 219.Hematopoietic PTP (HePTP) is a member of the kinase-interaction-motif (KIM)-containing PTP family, members of which interact with and modulate the substrate specificity of the ubiquitous signaling enzymes, mitogen-activated-specific kinases (MAPKs) [46]. HePTP, in particular, is a negative regulator of T-cell activation [47] and is often overexpressed in preleukemic myeloproliferative diseases [48]. In addition to its biological importance, several other factors made HePTP an attractive candidate for sensitization: no selective HePTP inhibitors have been described; HePTP contains isoleucine residues at the amino-acid positions corresponding to 49 and 219 (I107 and I274, respectively; see Figure 2); and HePTP is evolutionarily more diverged from PTP1B than either TCPTP or PTPH1 [46], providing a further test for the limits of the PTP-sensitization strategy.
We expressed and purified the HePTP analogs of V49A and I219A PTP1B, I107A and I274A HePTP, and measured their catalytic activities with pNPP. As we have observed with other alanine-mutant PTPs, I107A and I274A HePTP retain catalytic efficiencies comparable to that of wild-type HePTP (Table 2). Unlike previously sensitized PTPs, however, the HePTP mutants demonstrate essentially no measurable difference in activity (within error) when compared to wild-type HePTP. (Compare Table 2 to Table 1: V49A and I219A PTP1B have modestly increased KM values for pNPP, presumably due to a decrease in substrate-binding affinity caused by the “hole.”) These kinetic data show that these HePTP mutants meet a central criterion of an inhibitor-sensitization strategy: that the putatively sensitized enzymes retain the catalytic activity of the wild-type.
When screened against compound panel 2 for selective PTP inhibition, I107A and I274A HePTP proved not to be inhibitor-sensitized, at least in regard to the indole-based compound panel 2.As shown in Figure 5, none of the inhibitors demonstrated substantially heightened potency for I107A or I274A over wild-type HePTP in a fixed-concentration inhibitor screen. (Compounds 2a and 2j show only a marginal preference for I107A over wild-type HePTP.) These results, particularly those for the I219A analog, I274A HePTP, stand in marked contrast to those we have obtained for PTP1B, TCPTP, and PTPH1, all of which could be readily sensitized with an alanine mutation at position 219. (Data for compounds 2d, 2g, 2h, and 2k are not shown, as our supply of these compounds was exhausted prior to the HePTP work.)
The recently solved HePTP crystal does provide some clues as to why sensitization has, to date, not worked for HePTP [49]. The HePTP catalytic domain, like those of the other KIM PTPs, has a structure that is somewhat distinct from other classical PTPs [46,49]. The KIM motif, which is not present in canonical PTPs, such as PTP1B, forms an α-helix (α0) at the N-terminal portion of the KIM PTP catalytic domain; also, there are substantial backbone-structure deviations between the KIM PTP and PTP1B catalytic domains [50]. More relevantly, the positioning of I219 in the PTP1B active site differs substantially from that of I274 in HePTP (Figure 6): PTP1B’s I219 side chain comprises a large part of the “tunnel” surface leading to the enzyme’s active-site cysteine (Figure 6A), whereas HePTP’s I274 side chain is mostly buried and does not make up a significant part of the active site’s surface (Figure 6B). These structures may help to explain why the I274A HePTP mutant demonstrated both kinetic activity and inhibitor sensitivity that is unchanged with respect to wild-type HePTP. Intriguingly, I107 (the analog of V49 in PTP1B) appears to make up a significant part of the active-site path in HePTP (Figure 6B). Thus, if future classes of allele-specific inhibitors can be designed to effectively target the 49/107 interface, HePTP may yet prove to be amenable to sensitization.
Fig. 6.
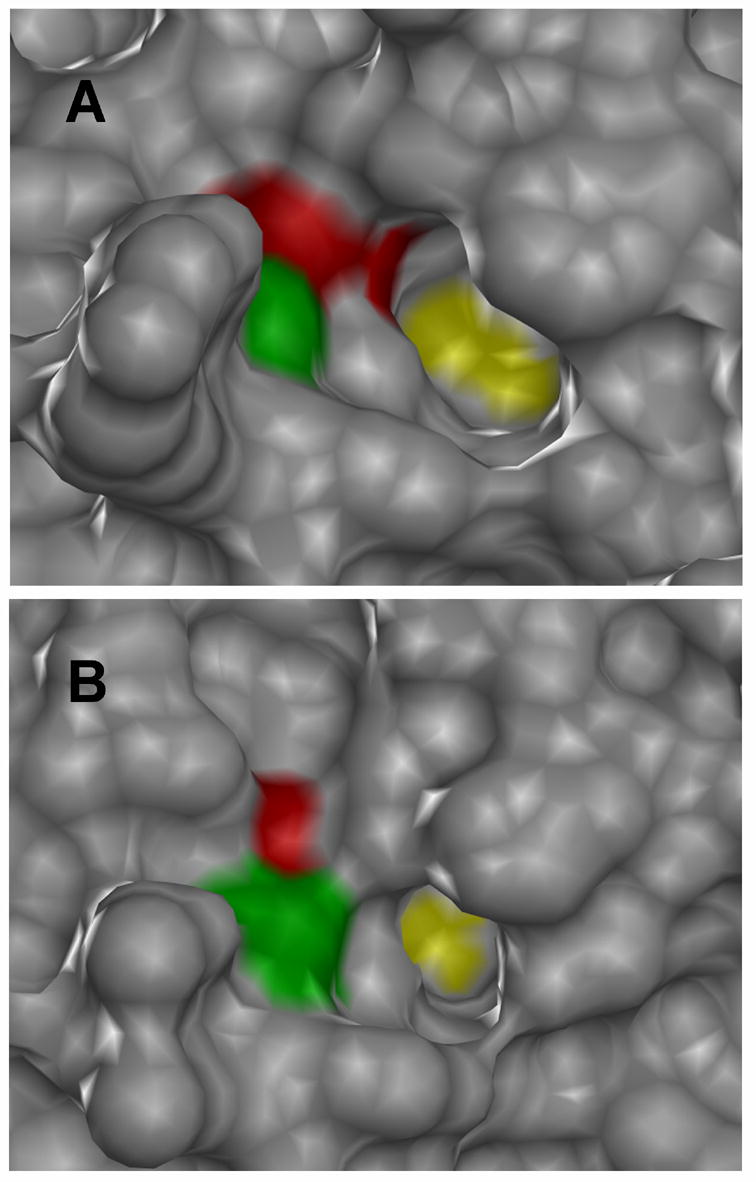
Active-site structural comparison of PTP1B and HePTP. The protein surfaces of PTP1B (A, PDB: 1C83 [38]) and HePTP (B, PDB: 1ZC0 [49]) are shown in gray, with the portions of the surface comprising V49/I107 shown in green and the portions comprising I219/I274 shown in red. For perspective, the active-site catalytic cysteine residues of PTP1B and HePTP are shown in yellow. Both enzymes are crystallized in the closed conformation: an inhibitor (compound 1) bound to the PTP1B active site and a phosphate ion bound to the HePTP active site have been removed for clarity. Images were generated using the Chimera software package (http://www.cgl.ucsf.edu/chimera).
6.4. Prospects for other classical PTPs
The negative results for HePTP described above do somewhat complicate the prospects for application of PTP sensitization across PTPs; it is apparent that there won’t be one “answer” (i.e., one mutation and one molecule) for sensitizing all classical PTPs. The generalized “bump-hole” picture depicted in Figure 1 is, of course, highly simplistic, eliding the obvious fact that PTP active sites do possess some level of structural diversity. Most sub-families of classical PTPs, however, are more closely related to PTP1B than is the KIM PTP family [46]. So, it still quite possible that the prototype engineering performed on PTP1B—and the I219A mutation in particular—will be applicable to a significant percentage of classical PTPs. The lesson to date: the more closely related a PTP is to a prototype, the more reasonable the assumption of active-site similarity.
For PTPs, more general solutions to the active-site-sensitization problem will likely come in the form of new inhibitors, not new mutations. Since the constraints for sensitizing active-site mutations are quite narrow (see Section 1.), the number of amino-acid residues that constitute plausible sensitization candidates is quite small. Additionally, introduction of alanine mutations at positions 49 and 219 has proven to be remarkably robust: four PTPs from three different families have been mutated, and, in each case, active and stable enzymes could be purified and analyzed. It seems reasonable, therefore, to retain positions 49 and 219 as the starting points in any future sensitization endeavors on new PTPs. By contrast, there is no limit to the number of putative inhibitors that can be synthesized and screened. Since we have found that non-targeted compounds can inhibit sensitized mutants in an allele-specific manner, there appears to be no reason to limit future screens to rationally designed compounds. New libraries of potential inhibitors, along with discoveries of new PTP-targeting pharmacophores, will most likely provide the next generation of allele-specific PTP inhibitors.
Scheme 1.
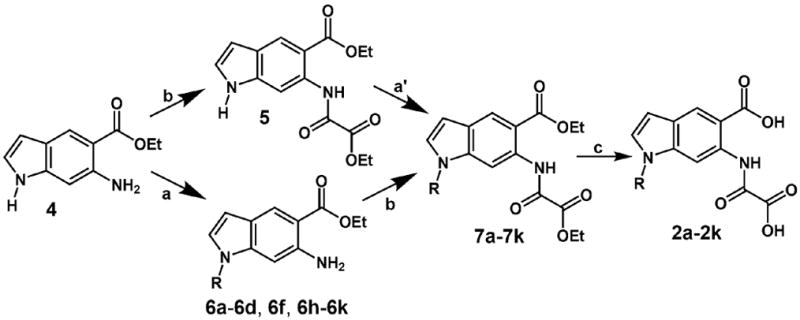
Synthesis of the N-derivatized oxalylaminoindole inhibitors, compound panel 2a-2k. Compound 4 was synthesized in five steps essentially as described [39]. Reaction conditions: (a) NaH (1.3 equiv), DMF, RX; (á) NaH (3.0 equiv), DMF, RX; (b) ethyl oxalyl chloride, THF; (c) 1.NaOH, H2O, EtOH; 2. HCl.
Acknowledgments
This research was supported by the National Institutes of Health (1 R15 GM071388–01A1), Research Corporation (CC6372), and Amherst College. The plasmids encoding wild-type PTP1B, TCPTP, and HePTP were generous gifts from Professor Zhong-Yin Zhang (Indiana University School of Medicine), Professor Harry Charbonneau (Purdue University), and Professor Rebecca Page (Brown University), respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain
References
- 1.Tonks NK. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 2.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 4.Tonks NK, Neel BG. Curr Opin Cell Biol. 2001;13:182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 5.Van Vactor D, O'Reilly AM, Neel BG. Curr Opin Genet Dev. 1998;8:112–126. doi: 10.1016/s0959-437x(98)80070-1. [DOI] [PubMed] [Google Scholar]
- 6.Mackeigan JP, Murphy LO, Blenis J. Nat Cell Biol. 2004;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- 7.Zhang ZY. Curr Opin Chem Biol. 2001;5:416–423. doi: 10.1016/s1367-5931(00)00223-4. [DOI] [PubMed] [Google Scholar]
- 8.Easty D, Gallagher W, Bennett DC. Curr Cancer Drug Targets. 2006;6:519–532. doi: 10.2174/156800906778194603. [DOI] [PubMed] [Google Scholar]
- 9.Mustelin T. Adv Exp Med Biol. 2006;584:53–72. doi: 10.1007/0-387-34132-3_5. [DOI] [PubMed] [Google Scholar]
- 10.Tautz L, Pellecchia M, Mustelin T. Expert Opin Ther Targets. 2006;10:157–177. doi: 10.1517/14728222.10.1.157. [DOI] [PubMed] [Google Scholar]
- 11.Kumar S, Liang F, Lawrence DS, Zhang ZY. Methods. 2005;35:9–21. doi: 10.1016/j.ymeth.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Zhang ZY. Biochim Biophys Acta. 2005;1754:100–107. doi: 10.1016/j.bbapap.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Zhang ZY, Lee SY. Expert Opin Investig Drugs. 2003;12:222–233. doi: 10.1517/13543784.12.2.223. [DOI] [PubMed] [Google Scholar]
- 14.Taylor SD. Curr Top Med Chem. 2003;3:759–782. doi: 10.2174/1568026033452311. [DOI] [PubMed] [Google Scholar]
- 15.Taylor SD, Hill B. Expert Opin Investig Drugs. 2004;13:199–214. doi: 10.1517/13543784.13.3.199. [DOI] [PubMed] [Google Scholar]
- 16.Bialy L, Waldmann H. Angew Chem Int Ed. 2005;44:3814–3839. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 17.Shen K, Keng YF, Wu L, Guo XL, Lawrence DS, Zhang ZY. J Biol Chem. 2001;276:47311–47319. doi: 10.1074/jbc.M106568200. [DOI] [PubMed] [Google Scholar]
- 18.Xie L, Lee SY, Andersen JN, Waters S, Shen K, Guo XL, Moller NP, Olefsky JM, Lawrence DS, Zhang ZY. Biochemistry. 2003;42:12792–12804. doi: 10.1021/bi035238p. [DOI] [PubMed] [Google Scholar]
- 19.Bishop AC, Blair ER. Bioorg Med Chem Lett. 2006;16:4002–4006. doi: 10.1016/j.bmcl.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Blair ER, Hoffman HE, Bishop AC. Bioorg Med Chem. 2006;14:464–471. doi: 10.1016/j.bmc.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 21.Hoffman HE, Blair ER, Johndrow JE, Bishop AC. J Am Chem Soc. 2005;127:2824–2825. doi: 10.1021/ja043378w. [DOI] [PubMed] [Google Scholar]
- 22.Clackson T. Curr Opin Struct Biol. 1998;8:451–458. doi: 10.1016/s0959-440x(98)80122-x. [DOI] [PubMed] [Google Scholar]
- 23.Koh JT. Chem Biol. 2002;9:17–23. doi: 10.1016/s1074-5521(02)00087-x. [DOI] [PubMed] [Google Scholar]
- 24.Bishop A, Buzko O, Heyeck–Dumas S, Jung I, Kraybill B, Liu Y, Shah K, Ulrich S, Witucki L, Yang F, Zhang C, Shokat KM. Annu Rev Biophys Biomol Struct. 2000;29:577–606. doi: 10.1146/annurev.biophys.29.1.577. [DOI] [PubMed] [Google Scholar]
- 25.Belshaw PJ, Schreiber SL. J Am Chem Soc. 1997;119:1805–1806. [Google Scholar]
- 26.Lin Q, Jiang F, Schultz PG, Gray NS. J Am Chem Soc. 2001;123:11608–11613. doi: 10.1021/ja011423j. [DOI] [PubMed] [Google Scholar]
- 27.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 28.Bishop AC, Kung CY, Shah K, Witucki L, Shokat KM, Liu Y. J Am Chem Soc. 1999;121:627–631. [Google Scholar]
- 29.Bishop AC, Shah K, Liu Y, Witucki L, Kung C, Shokat KM. Curr Biol. 1998;8:257–266. doi: 10.1016/s0960-9822(98)70198-8. [DOI] [PubMed] [Google Scholar]
- 30.Knight ZA, Shokat KM. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 31.Zhang ZY. Annu Rev Pharmacol Toxicol. 2002;42:209–234. doi: 10.1146/annurev.pharmtox.42.083001.144616. [DOI] [PubMed] [Google Scholar]
- 32.Bishop AC, Shokat KM. Pharmacol Ther. 1999;82:337–346. doi: 10.1016/s0163-7258(98)00060-6. [DOI] [PubMed] [Google Scholar]
- 33.Taing M, Keng YF, Shen K, Wu L, Lawrence DS, Zhang ZY. Biochemistry. 1999;38:3793–3803. doi: 10.1021/bi9813781. [DOI] [PubMed] [Google Scholar]
- 34.Zhang ZY. Acc Chem Res. 2003;36:385–392. doi: 10.1021/ar020122r. [DOI] [PubMed] [Google Scholar]
- 35.Guo XL, Shen K, Wang F, Lawrence DS, Zhang ZY. J Biol Chem. 2002;277:41014–41022. doi: 10.1074/jbc.M207347200. [DOI] [PubMed] [Google Scholar]
- 36.Jia Z, Barford D, Flint AJ, Tonks NK. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 37.Sarmiento M, Puius YA, Vetter SW, Keng YF, Wu L, Zhao Y, Lawrence DS, Almo SC, Zhang ZY. Biochemistry. 2000;39:8171–8179. doi: 10.1021/bi000319w. [DOI] [PubMed] [Google Scholar]
- 38.Andersen HS, Iversen LF, Jeppesen CB, Branner S, Norris K, Rasmussen HB, Moller KB, Moller NP. J Biol Chem. 2000;275:7101–7108. doi: 10.1074/jbc.275.10.7101. [DOI] [PubMed] [Google Scholar]
- 39.Showalter HDH, Sun L, Sercel AD, Winters RT, Denny WA, Palmer BD. J Org Chem. 1996;61:1155–1158. [Google Scholar]
- 40.Glennon RA, Van Strandtmann M. J Het Chem. 1975;12:135–138. [Google Scholar]
- 41.Andersen HS, Olsen OH, Iversen LF, Sorensen AL, Mortensen SB, Christensen MS, Branner S, Hansen TK, Lau JF, Jeppesen L, Moran EJ, Su J, Bakir F, Judge L, Shahbaz M, Collins T, Vo T, Newman M, Ripka WC, Moller NP. J Med Chem. 2002;45:4443–4459. doi: 10.1021/jm0209026. [DOI] [PubMed] [Google Scholar]
- 42.Drake PG, Peters GH, Andersen HS, Hendriks W, Moller NP. Biochem J. 2003;373:393–401. doi: 10.1042/BJ20021851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iversen LF, Andersen HS, Branner S, Mortensen SB, Peters GH, Norris K, Olsen OH, Jeppesen CB, Lundt BF, Ripka W, Moller KB, Moller NP. J Biol Chem. 2000;275:10300–10307. doi: 10.1074/jbc.275.14.10300. [DOI] [PubMed] [Google Scholar]
- 44.Pedersen AK, Guo XL, Moller KB, Peters GH, Andersen HS, Kastrup JS, Mortensen SB, Iversen LF, Zhang ZY, Moller NP. Biochem J. 2004;378:421–433. doi: 10.1042/BJ20030565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, Parmigiani G, Yan H, Wang TL, Riggins G, Powell SM, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. Science. 2004;304:1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 46.Barr AJ, Knapp S. Trends Pharmacol Sci. 2006;27:525–530. doi: 10.1016/j.tips.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 47.Saxena M, Williams S, Gilman J, Mustelin T. J Biol Chem. 1998;273:15340–15344. doi: 10.1074/jbc.273.25.15340. [DOI] [PubMed] [Google Scholar]
- 48.Zanke B, Squire J, Griesser H, Henry M, Suzuki H, Patterson B, Minden M, Mak TW. Leukemia. 1994;8:236–244. [PubMed] [Google Scholar]
- 49.Mustelin T, Tautz L, Page R. J Mol Biol. 2005;354:150–163. doi: 10.1016/j.jmb.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 50.Szedlacsek SE, Aricescu AR, Fulga TA, Renault L, Scheidig AJ. J Mol Biol. 2001;311:557–568. doi: 10.1006/jmbi.2001.4890. [DOI] [PubMed] [Google Scholar]