

Abstract
Coalescent theory provides a powerful framework for estimating the evolutionary, demographic, and genetic parameters of a population from a small sample of individuals. Current coalescent models have largely focused on population genetic factors (e.g., mutation, population growth, and migration) rather than on the effects of experimental design and error. This study develops a new coalescent/mutation model that accounts for unobserved polymorphisms due to missing data, sequence errors, and multiple reads for diploid individuals. The importance of accommodating these effects of experimental design and error is illustrated with evolutionary simulations and a real data set from a population of the California sea hare. In particular, a failure to account for sequence errors can lead to overestimated mutation rates, inflated coalescent times, and inappropriate conclusions about the population. This current model can now serve as a starting point for the development of newer models with additional experimental and population genetic factors. It is currently implemented as a maximum-likelihood method, but this model may also serve as the basis for the development of Bayesian approaches that incorporate experimental design and error.
THE genealogy for a small random sample of sequences is influenced by a large number of evolutionary, demographic, and genetic factors for its population. By making a few basic assumptions, coalescent theory provides the framework to estimate the probabilities of these genealogies and their associated population parameters (Hudson 1990; Donnelly and Tavaré 1995; Hein et al. 2004). Current coalescent models continue to emphasize population genetic factors such as mutation, varying population size, migration, and divergence time. These models are implemented with both maximum-likelihood (ML) and sampling-based (e.g., Markov chain Monte Carlo, MCMC) approaches. Although exact, the former is generally practical or even possible only for the simpler models (i.e., those that account for a single factor) and smaller data sets. In turn, although approximate, the latter can usually accommodate more complex models and larger data sets. The sampling-based methods often rely on a Bayesian setting, where parameters are integrated over their ranges and expected values are obtained (rather than ML estimates).
In contrast to this emphasis on population genetic factors, the effects of experimental design and error on a coalescent study have been largely ignored (Felsenstein 2004). Most current coalescent models assume that haplotype data are available for diploids and that sequence variation is sampled in an unbiased manner. However, haplotypes are not always available, particularly for nuclear markers, and single-nucleotide polymorphisms (SNPs), for example, are often ascertained in ways that can bias their subsequent analysis. In light of these facts, new coalescent models have been introduced to account for these effects of experimental design (Kuhner and Felsenstein 2000; Kuhner et al. 2000; Nielsen 2000).
This study develops a new coalescent/mutation model that accounts for unobserved polymorphisms due to missing data; for sequence errors due to cloning, sequencing, and recording artifacts; and for multiple sequencing reads from the same diploid individuals (Figure 1). The development of this new model begins with the standard model for reproduction of Fisher (1930) and Wright (1931) for an unstructured population with discrete nonoverlapping generations and identical individual fitness. A mutation process is then introduced according to the infinite-sites model (Kimura 1969). Thereafter, the additional effects for unobserved polymorphisms, sequence errors, and multiple reads are incorporated. The utility of this new model is evaluated with evolutionary simulations and a real data set of nuclear gene sequences from a population of the California sea hare (Aplysia californica). The latter is particularly relevant, since it was the primary motivation for the development of this new model and its specific factors for experimental design and error.
Figure 1.—

Incorporating factors for experimental design and error into the standard coalescent models with population genetic parameters (e.g., for mutations). (A) Current coalescent models have focused on the effects of population genetic processes on the genealogy for a population sample of alleles. (B) The new model builds on the standard models by incorporating factors for the experimental determination of the allele sequences. Specifically, it acknowledges that the multiple sequences for the sampled alleles can include errors and missing regions and that the assignment of their different reads to the two homologs of a diploid can remain uncertain even when the individual source of each read is known. By accommodating these facts, better estimates of the population genetic parameters can be provided by accounting for unobserved polymorphisms due to missing data and sequence errors. The open circles and dashed lines refer to an allele of a sampled individual that is never sequenced.
NEW MODEL FOR EXPERIMENTAL DESIGN AND ERROR
Coalescence and mutation:
We begin with a short review of the infinitely many-sites model for calculating genealogical tree probabilities of Griffiths (1989) and Griffiths and Tavaré (1995) upon which our new model for experimental design and error is based. Under the Fisher/Wright model, ∼2N generations is the expected time to the most recent common ancestor (MRCA) for two randomly sampled alleles from a haploid population of 2N individuals. Thus, if time is scaled by a factor of 2N generations (and N is large), then 1 unit of time can be set to this expectation and time can now be measured as continuous rather than discrete. Working backward in this scaled continuous time, the waiting time for a coalescence of two alleles in a population sample of n is then exponentially distributed with an intensity of n(n − 1)/2. Scaling time in similar ways allows these expectations to be extended to a wide range of other discrete-time models, including those for a large diploid population with equal numbers of males and females (Kingman 1982).
The infinite-sites model can now be added to this standard coalescent model to incorporate a mutation process, whereby only a single mutation can occur at any homologous position (Kimura 1969). Thus, a maximum of only two nucleotides can occur at any site, thereby allowing for the recoding of each sequence as a vector of 0's and 1's (with the former designating the ancestral state). Furthermore, our mutation process assumes that all mutations are neutral (Kimura 1983). With time scaled as above, the total expected divergence time between two sequences is 4N generations given that the expected time to their MRCA is 2N generations. Thus, the scaled mutation rate is equal to the expected number of mutations between two sequences (θ = 4Nμ, where μ is the mutation rate per gene per generation).
Incorporating missing data and unobserved polymorphisms:
Let S refer to the ordered set of n sequences, s1, s2,…, sn, at a particular time in the genealogy. Each sequence is now defined as a triplet, s = (a, g, σ), with its three entries representing its allele configuration (vector of 0's and 1's), pattern of missing data, and total number of singletons (unique derived variants or 1's due either to mutations or to sequence errors), respectively. As one works backward in time, S will change as mutations are accounted for and identical alleles coalesce (Figure 2). In particular, this means that a will be redefined for alleles with the most recent derived mutations and σ will be updated since these mutations are counted as singletons as one works backward toward the MRCA.
Figure 2.—

Illustration of how singletons are transferred from a to σ for the n sequences of S as mutation and coalescent events are accounted for, working backward in time. At the top, the history for four sampled sequences (bars at the bottom of the tree, with bars further up the tree representing their common ancestors) is shown with asterisks and solid circles highlighting their derived mutations at four polymorphic sites (v1–v4). Three different times in this history are highlighted with A corresponding to the present. At time A, two matrices are shown for the four extant sequences, before and after the transfer of their singletons to σ. In these two matrices, rows correspond to a for the four sampled sequences (as listed in the same order from top to bottom as presented from left to right in their genealogy), columns refer to polymorphic sites v1–v4, and “0” and “1” distinguish between the ancestral and derived states at these variable positions. As illustrated here, all singletons in the left matrix are transferred to their corresponding σ in the right matrix, with the latter now tracking these unique mutations. Time B then highlights an older point in the genealogy prior to the mutation events at polymorphic sites v2 and v3. Thus, at this time, only two polymorphic sites (v1 and v4) occur among the four sequences of S. The derived mutation at v1, which represents a singleton of the fourth extant sequence, continues to be tracked by its σ = 1. Finally, time C is highlighted, because it represents a point prior to the first coalescent event in the genealogy. As a result of this coalescent event, n = 3 with the first two rows for the first two sampled sequences in matrix B now replaced by the single first row for their common ancestor in the two C matrices. Correspondingly, the shared derived mutation for the first two extant sequences in matrix B now constitutes a singleton of their ancestral sequence. Thus, as before, this singleton is transferred from a to σ for their common ancestor to further track this now unique mutation. This process of accounting for mutations, coalescences, and singletons as one works backward in time continues until the MRCA of the four sampled sequences is reached.
In turn, let g refer to a series of closed intervals that summarizes what regions are known for each sampled sequence relative to their full-length alignment (Figure 3). For a complete sequence, g = (0:1). For any partial sequence (i.e., one with missing data), g is represented by narrower closed intervals on this 0:1 scale. As one works backward in time and coalescent events occur, g is then calculated for each common ancestor as the union of g for its two coalescing sequences. Finally, let 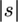 represent the total available length for an extant or ancestral sequence, which is calculated as the sum of its closed intervals in g. This tracking of total available lengths allows for the later introduction of a correction factor for mutations that are overlooked because they occur within a missing region of the sequences (see below). In these ways, the known and missing regions of sequences, along with the potential to observe mutations, are accounted for as one works backward in time to the MRCA for the sampled sequences.
represent the total available length for an extant or ancestral sequence, which is calculated as the sum of its closed intervals in g. This tracking of total available lengths allows for the later introduction of a correction factor for mutations that are overlooked because they occur within a missing region of the sequences (see below). In these ways, the known and missing regions of sequences, along with the potential to observe mutations, are accounted for as one works backward in time to the MRCA for the sampled sequences.
Figure 3.—

Illustration of how g is determined for both extant and ancestral sequences as one works backward in time toward their MRCA. The history for four sampled sequences, each with missing data, is shown with their known and unknown regions represented as solid and open bars. The full-length alignment for these four extant sequences is 1000 bases. The first number for each sampled sequence marks the relative alignment position at the end of its unknown region (if any) to its left. For example, the leftmost sampled sequence is missing the first 200 bases of the full-length alignment. In turn, the following interval in parentheses corresponds to its g [i.e., its summary of known regions as scored over the closed interval of (0:1) for the full alignment]. Thus, g = (0.2:1.0) for the first extant sequence. As one works backward in time and coalescent events are accounted for, g is then calculated for each ancestral sequence as the union of g for its two coalescing sequences [e.g., g = (0.2:1.0) for the common ancestor of the two leftmost sampled sequences]. In these ways, known and unknown regions of both extant and ancestral sequences are tracked back to the MRCA of the population sample.
For now, we assume that all singletons are due to mutations (those reflecting sequence errors are accounted for in the next section). We furthermore assume that the allele configurations are known, even for those missing regions of the sequences. This assumption is allowed since we can sum over all possible states at the variable sites in the missing regions of the partial sequences (see below). If ai = aj and σi = σj = 0, then si and sj are combinable (si ∼ sj) since their known regions are identical. In contrast, incombinable sequences differ by one or more mutations that must have occurred since their common ancestor. Thus, only combinable sequences can coalesce given that the next event (as always, working backward in time) is not a mutation. If si and sj do indeed coalesce, then the sequence for their common ancestor (sij) must be identical to si and sj. As sij should now have missing regions only where both si and sj lack information, sij = (a, gi ∪ gj, 0), an initial condition that is defined only when si ∼ sj.
Under the infinite-sites model, if the next event is a mutation then the mutation must also be a singleton, even if it is shared among multiple extant sequences (Figure 2). If shared among the extant sequences, then under the infinite-sites model, this mutation must have arisen in the branch leading to their common ancestor. If si of the current set of sequences is selected for this next mutation and σi > 0, then its immediate ancestor before the mutation can be denoted as 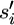 = (ai, gi, σi − 1). Furthermore, define αi(S) = (S\{si}) ∪
= (ai, gi, σi − 1). Furthermore, define αi(S) = (S\{si}) ∪ 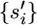 and βij(S) = (S\{si, sj}) ∪ {sij} as the current set of sequences before this mutation and before the split of si and sj, respectively. As before, the βij(S) definition requires that any unique mutations (i.e., 1's) for sij be removed from its a and accounted for instead by its σ and that any regions known for only one sequence of the current set be removed from g (Figures 2 and 3).
and βij(S) = (S\{si, sj}) ∪ {sij} as the current set of sequences before this mutation and before the split of si and sj, respectively. As before, the βij(S) definition requires that any unique mutations (i.e., 1's) for sij be removed from its a and accounted for instead by its σ and that any regions known for only one sequence of the current set be removed from g (Figures 2 and 3).
We now find the probability of observing S by direct calculation from the recursion:
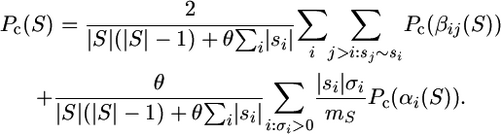 |
(1) |
The final step is Pc({(0, g, 0)}) = 1 for any g. Working backward in time, the expected waiting times for both a coalescence and an observed mutation are exponential, with their scaled rates equal to 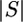 (
(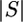 − 1)/2 and
− 1)/2 and 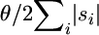 , respectively (given no allele information). The use of
, respectively (given no allele information). The use of 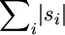 in the latter factor reflects the fact that mutations are observed only when they occur within the known regions of the sequences.
in the latter factor reflects the fact that mutations are observed only when they occur within the known regions of the sequences.
The probability that the next (observed) event is a specific coalescence is
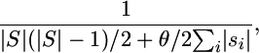 |
(2) |
which is the first factor of the first term in Equation 1. If indeed a coalescent event occurs, then it can happen only between combinable sequences as indicated by the double sum.
The mutation process is modeled by the second term in Equation 1. The first factor is for the probability that the latest event is a mutation in a specific sequence, whereas the sum is over all sequences with at least one singleton. The σi factor counts the number of singletons and thereby possible sites for this mutation. Given that the segregating sites are ordered, the probability is then divided by mS, which denotes the number of variable positions in S. As indicated above, the missing regions of each sequence can be dealt with by summing over all possible states for it, at those sites where a mutation is observed among the other sequences. However, unlike other coalescent/infinite-sites-mutation models, ours also accounts for those mutations that go unobserved since they occur in the missing regions of these partial sequences. This class of unobserved mutations is once again accounted for by 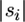 , which represents the probability of a singleton occurring within an observed part of si.
, which represents the probability of a singleton occurring within an observed part of si.
Apart from some combinatorial terms for labeling and grouping, if 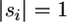 for all i, we note that our starting Equation 1 is identical to Equation 1.4 in Griffiths (1989) (see also Griffiths and Tavaré 1995). That is, both equations use recursion to calculate the exact probability of a population sample of alleles under the Wright/Fisher model for reproduction and the infinite-sites model for mutation. This recursion is guaranteed to converge, because each step leads to fewer polymorphic sites or sequences, thereby leading to the MRCA at some point. However, if the mutation process does not obey the infinite-sites model, then this recursion may converge onto an incorrect probability.
for all i, we note that our starting Equation 1 is identical to Equation 1.4 in Griffiths (1989) (see also Griffiths and Tavaré 1995). That is, both equations use recursion to calculate the exact probability of a population sample of alleles under the Wright/Fisher model for reproduction and the infinite-sites model for mutation. This recursion is guaranteed to converge, because each step leads to fewer polymorphic sites or sequences, thereby leading to the MRCA at some point. However, if the mutation process does not obey the infinite-sites model, then this recursion may converge onto an incorrect probability.
Incorporating sequence errors and multiple reads:
The infinite-sites process is now extended to include sequence errors as well as mutations (i.e., only one error or mutation can occur at any homologous position). A uniform distribution of errors along the sequences is assumed, with ε denoting the expected number for a full sequence. Thus, the total number of observed errors is Poisson distributed with an intensity of  and the probability of observing i errors is
and the probability of observing i errors is
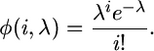 |
(3) |
Let v = (v1, v2,…, vn) be an n-dimensional vector of errors per sequence, let 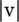 be its sum of entries, and let S − v be the set of sequences with vi singletons removed from si. In calculating the probability of observing S, sequence errors can now be accommodated as
be its sum of entries, and let S − v be the set of sequences with vi singletons removed from si. In calculating the probability of observing S, sequence errors can now be accommodated as
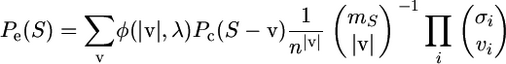 |
(4) |
The first factor is for the probability of observing 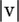 errors, whereas the second is for the coalescent/mutation probability of the sequences without the errors (Equation 1). The last factors are for the probability that
errors, whereas the second is for the coalescent/mutation probability of the sequences without the errors (Equation 1). The last factors are for the probability that 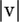 errors are distributed among the sequences as indicated by v. The sum is over all nonnegative integers of the vi's where no sequence in S − v can have a negative number of singletons.
errors are distributed among the sequences as indicated by v. The sum is over all nonnegative integers of the vi's where no sequence in S − v can have a negative number of singletons.
As multiple reads of the same allele or haplotype should at least be combinable, any discrepant nucleotides among them can be attributed to sequence errors rather than polymorphism. Thus, sequence errors can be readily distinguished among the multiple sequencing reads for a haploid individual, but not for a diploid or a polyploid one. The reason is that the different multiple reads for a diploid or a polyploid individual can vary due to allelic variation as well as sequencing errors. This ambiguity is distinct from the problem of identifying haplotypes from the direct sequencing of PCR products from potential heterozygotes (Clark 1990). Instead, the problem here is concerned with assigning the multiple individual reads of the haplotypes for a diploid or a polyploid, as obtained from the direct sequencing of its cloned DNA inserts, e.g., to its two or more homologs.
To address this ambiguity due to the uncertain mapping of known multiple reads to homologs, let us assume that each read was obtained from a cloned DNA insert or a single haploid gamete and that each can be matched to a specific individual but not to one of its p alleles (with p = 1 or 2 for haploid and diploid, respectively). Now, order the pk alleles for k p-ploid individuals such that those for individual i are labeled {p(i − 1) + 1, p(i − 1) + 2,…, pi} and then map the n reads to this arrangement. Of the pnk alternative mappings, let F define the final set of valid f 's where only combinable reads are assigned to the same allele. In the case of haploid individuals (p = 1), F contains only a single mapping of the various reads for each individual to a single sequence.
In the absence of sequence errors, the probability of observing S can now be calculated as
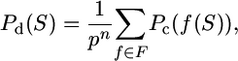 |
(5) |
where f (S) refers to a valid transformation of S reads to the alleles of the p-ploid individuals. Multiple reads that map to the same allele are then joined as in a coalescent event. In contrast, when sequence errors can be present, Pc(f (S)) in the above equation is replaced by Pe(f (S)) from Equation 4.
Full algorithm:
The complete algorithm for the new model uses expectation maximization for parameter optimization (Durbin et al. 1998). Nevertheless, it remains computationally intensive, since it requires sums over all missing data, error configurations, allele mappings, and ancestral states. Fortunately, the algorithm also benefits from its infinite-sites assumption that allows for only certain allele matches, missing configurations, and ancestral states and for some numbers of singletons as sequence errors for particular mappings. A further acceleration is gained by the reuse of partial results in computer memory and by the reduction of the ML optimization for θ and ε from a two- to a one-dimensional search in effect. The latter is achieved by rewriting the total probability of the data as a sum over the probabilities for different numbers of sequence errors:
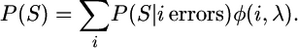 |
(6) |
Since P(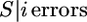 ) is independent of ε, P(S) can now be more efficiently calculated for a given θ in the ML optimization of ε. (An implementation of the algorithm is available upon request from B.K.)
) is independent of ε, P(S) can now be more efficiently calculated for a given θ in the ML optimization of ε. (An implementation of the algorithm is available upon request from B.K.)
EVOLUTIONARY SIMULATIONS AND A. CALIFORNICA DATA
To evaluate the new model, evolutionary simulations were conducted according to standard methods (Hein et al. 2004). Two hundred data sets apiece were simulated for either 8 or 16 sequences of length 500 from a single population. The baseline conditions for these simulations were those of the standard Fisher/Wright and infinite-sites models with θ = 1 or 2. To this baseline, sequence errors and/or missing data were incorporated as four or eight randomly placed errors among the 8 and 16 sequences, respectively (for an expected ε value of 0.5), and by the removal of the starting 150 sites for 4 or 8 of the 8 and 16 total sequences, respectively. In addition, in the trials with 8 sequences, missing data were also simulated by removing the first 200 sites from 2 sequences and the last 200 from 2 others. Estimates of θ and ε (when appropriate) were then obtained for the 200 data sets of each tested combination with the standard coalescent/mutation model and the new model that accounts for experimental design and error. Simulating the sequences as independent samples of the population (rather than as multiple reads of diploid individuals) allowed for more direct tests of the effects of unobserved polymorphisms due to missing data and sequence errors.
The standard model underestimated θ in all of the simulated cases with missing data but no sequence errors (Table 1). These underestimates were significant in all of these cases, except for the nearly significant result with 8 sequences, θ = 1, and missing data (I) (simulations A3, B2, B3, C2, and D2 vs. A2, respectively). The standard model ignores unobserved polymorphisms due to missing data, which thereby leads to these underestimates of θ. As expected of a systematic bias due to model failure, this problem was more pronounced for the larger samples (cf. the significant underestimate of θ = 1 for simulation C2 with 16 sequences vs. the nearly significant outcome for its counterpart A2 with 8 sequences).
TABLE 1.
Simulated results, summarized as the averages plus or minus twice the standard deviations for 200 data sets each
| Standard coalescent/mutation model: | New model
|
|||
|---|---|---|---|---|
| Evolutionary simulations | θ | θ | ε | |
| A1 | θ = 1, 8 sequences | 1.016 ± 0.107 | 0.747 ± 0.107 | 0.072 ± 0.015 |
| A2 | With missing data (I) | 0.918 ± 0.101 | 0.682 ± 0.105 | 0.094 ± 0.019 |
| A3 | With missing data (II) | 0.892 ± 0.102 | 0.692 ± 0.111 | 0.091 ± 0.021 |
| A4 | With ε = 0.5 | 3.280 ± 0.145 | 0.908 ± 0.137 | 0.534 ± 0.028 |
| A5 | With missing data (I) and ε = 0.5 | 2.733 ± 0.147 | 0.822 ± 0.131 | 0.550 ± 0.033 |
| A6 | With missing data (II) and ε = 0.5 | 2.519 ± 0.144 | 0.868 ± 0.135 | 0.526 ± 0.037 |
| B1 | θ = 2, 8 sequences | 2.064 ± 0.177 | 1.602 ± 0.167 | 0.108 ± 0.027 |
| B2 | With missing data (I) | 1.830 ± 0.156 | 1.555 ± 0.164 | 0.116 ± 0.030 |
| B3 | With missing data (II) | 1.828 ± 0.160 | 1.555 ± 0.172 | 0.132 ± 0.030 |
| B4 | With ε = 0.5 | 4.510 ± 0.208 | 1.901 ± 0.213 | 0.542 ± 0.037 |
| B5 | With missing data (I) and ε = 0.5 | 3.808 ± 0.187 | 1.869 ± 0.216 | 0.540 ± 0.047 |
| B6 | With missing data (II) and ε = 0.5 | 3.635 ± 0.194 | 1.833 ± 0.221 | 0.561 ± 0.047 |
| C1 | θ = 1, 16 sequences | 0.952 ± 0.093 | 0.775 ± 0.088 | 0.029 ± 0.007 |
| C2 | With missing data (I) | 0.862 ± 0.084 | 0.719 ± 0.085 | 0.038 ± 0.008 |
| C3 | With ε = 0.5 | 4.891 ± 0.132 | 0.934 ± 0.109 | 0.503 ± 0.012 |
| D1 | θ = 2, 16 sequences | 1.865 ± 0.138 | 1.623 ± 0.140 | 0.037 ± 0.009 |
| D2 | With missing data (I) | 1.711 ± 0.127 | 1.565 ± 0.139 | 0.046 ± 0.012 |
| D3 | With ε = 0.5 | 6.048 ± 0.179 | 1.866 ± 0.171 | 0.501 ± 0.016 |
Missing data (I) refer to the simulations where the first 150 bases were removed from half of the total sequences in each data set. Missing data (II) correspond to the simulations with 8 sequences where the first 200 sites of 2 sequences and the last 200 positions of 2 others were removed. The simulations with 16 sequences and both missing data and errors remain unavailable, since they proved too computationally intensive (see text).
Conversely, the standard model overestimated θ when sequence errors occurred (simulations A4–A6, B4–B6, C3, and D3 in Table 1). These highly significant overestimates were evident even when opposed by the underestimates of θ due to missing data and unobserved polymorphisms (simulations A5, A6, B5, and B6). The standard model erroneously attributes singletons due to sequence errors to mutations, which thereby results in these highly significant overestimates of θ.
In contrast, the new model significantly underestimated θ when the sequences were error free (simulations A1–A3, B1–B3, C1, C2, D1, and D2 in Table 1). For a related reason, the new model also significantly overestimated ε as greater than zero in these simulations. The related reason for these significant under- and overestimations is that the mutations of error-free sequences may at times be attributed to sequence errors by the new model. To avoid this problem of overparameterization, a statistical test of the null hypothesis that ε = 0 can first be performed (e.g., with a likelihood-ratio test; Huelsenbeck and Rannala 1997). If this null hypothesis cannot be rejected, then Equation 1 can be used instead of Equation 4 in the full algorithm to eliminate sequence errors as an explanation for the singletons.
Furthermore, there was also a slight tendency for the new model to underestimate θ and overestimate ε when ε = 0.5 (simulations A4–A6, B4–B6, C3, and D3 in Table 1). In particular, there were four cases using 8 sequences where such under- and/or overestimations were significant (simulations A4, A5, B4, and B6). However, in contrast to the systematic bias for the standard model (see above), these under- and overestimations were less pronounced for the cases with the larger samples (cf. the insignificant outcomes for simulations C3 and D3 using 16 sequences vs. the significant results for their counterparts A4 and B4 using 8 sequences). The reason here is that these under- and overestimations are due to sampling errors (rather than systematic bias), whereby the larger samples provide extra information for the resolution of both sequence errors and mutations.
The new model was next evaluated with the real sequences and multiple reads for a population of A. californica under investigation at The Whitney Laboratory for Marine Bioscience, University of Florida (L. L. Moroz and A. B. Kohn, unpublished data). Three different clones, each carrying the FMRF gene, were selected from the plasmid genomic libraries for six individuals of this population. Each insert was then sequenced as a pair of single passes starting from both ends of an internal segment of 1731 bp for the protein-coding region of this nuclear gene (Figure 4). These pairs of passes overlapped in the middle for nine sequences, but at most by only 58 bases.
Figure 4.—

Summary of the available data for the 18 sequencing reads from six individuals (ind.) of A. californica. Forty-four singletons and 10 shared polymorphisms occur among these 18 sequences (the tick marks and x's, respectively). At position 736, three nucleotides (C, G, and T) are found in violation of the infinite-sites model and its maximum of 2 bases per site. Thus, in the analysis of these 18 sequences, this position was divided into two separate ones for its G vs. T singletons (as marked).
The relatively high ratio of singletons (44) to shared polymorphisms (10) for the 18 reads of the six individuals suggested that sequence errors are a major source of variation in this data set (Figure 4). Correspondingly, the joint estimation of θ and ε by the new model proved too time consuming for these 18 sequences, primarily because of their interval of 0–44 possible errors to sum over in their likelihood calculations (see below). Thus, a two-step procedure was instead adopted, whereby the number of errors was first ML estimated, followed by the determination of θ for this specific ML value. In turn, ε was calculated as the ML number of errors divided by 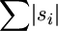 for the original sequences. In this way, ML estimates of ε = 2.52 (or 42 errors for the 18 complete and partial sequences) and θ = 6.32 were obtained for this data set with the new model. Given these ML estimates for 1731 positions, nucleotide diversity, or π, and the error rate were calculated as 0.0037 mutations/site and 0.0015 errors/site, respectively. As these 18 reads were based on single sequencing passes, their error rate of 0.0015 was ∼15 times greater than the accepted cutoff of 0.0001 for finished sequences (Richterich 1998).
for the original sequences. In this way, ML estimates of ε = 2.52 (or 42 errors for the 18 complete and partial sequences) and θ = 6.32 were obtained for this data set with the new model. Given these ML estimates for 1731 positions, nucleotide diversity, or π, and the error rate were calculated as 0.0037 mutations/site and 0.0015 errors/site, respectively. As these 18 reads were based on single sequencing passes, their error rate of 0.0015 was ∼15 times greater than the accepted cutoff of 0.0001 for finished sequences (Richterich 1998).
The computational complexity for a particular data set is strongly influenced by its number of segregating sites, with more variable positions involving more choices as one works back through the coalescent/mutation recursion (Equation 1). The coalescent/mutation process introduces a large variance in the number of segregating sites due to the heterogeneity in both the total branch length of the genealogy and the Poisson mutation process (Hudson 1990; Felsenstein 2004). In the evolutionary simulations with 16 sequences, θ = 2, and missing data (D2 in Table 1), the full-likelihood calculations had a median computation time of 29 CPU sec on a computer using a single core of an Intel Quad-Core Xeon X3210 CPU at 2.13 GHz. In contrast, 53 CPU hr were required for the most complex of these data sets, and indeed, the full-likelihood calculations for 16 sequences with both missing data and sequence errors proved too time consuming. In turn, the A. californica data set was even more complex given the need to sum across its 44 singletons and ambiguous haplotype assignments, uncertain error specifications, ancestral states, and missing data (Equations 4 and 5). Thus, its likelihood evaluations necessitated the two-step procedure that still took several CPU hours to complete.
DISCUSSION
In this study, sequence errors are modeled as unique events of single reads that are primarily due to cloning, sequencing, and recording inaccuracies. Thus, when sequence errors are not accounted for, they are misinterpreted as unique mutations in the terminal branches of the genealogy (Felsenstein 2004). Correspondingly, the coalescent times for the sequences are artificially extended as their genealogy becomes more like that for an expanding population (Harpending et al. 1998). In conclusion, when sequences are error prone (e.g., as for expressed sequence tags and other single-read data), a failure to account for sequence errors can lead to overestimates of θ and artificially older coalescent times (Table 1). As a result, this failure can also lead to inaccurate conclusions about the biology of the population.
The new model is presented as a starting point for the further development of coalescent/mutation models that account for experimental design and error. It is presently designed to accommodate three specific experimental factors that are of particular interest to the study of the A. californica data set. As for other starting models, the new model currently overlooks other experimental and population genetic factors that are regarded as less pertinent to its targeted goal (e.g., SNP ascertainment bias, migration, and varying population size for this laboratory population; see also below).
One obvious future direction for the new model is to implement it as an MCMC or other sampling-based procedure that will allow for the incorporation of additional experimental and population genetic factors. This implementation of sampling-based procedures will facilitate the study of ancestral recombination graphs for the accommodation of missing data too and the development of a Bayesian counterpart for posterior probability testing (Griffiths 2001; Hein et al. 2004). In particular, these advances will allow for predictions about which of the three distinct reads for an individual A. californica map to the same allele and which of its singletons therefore represent errors (Figure 4). They will also account for recombination, which is likely the most important factor overlooked in our present analysis of the nuclear FMRF gene for this laboratory population. Along these lines, this study also encourages the further development of coalescent/mutation models that account for experimental design and error, but under a finite-sites process (Kuhner and Felsenstein 2000; Kuhner et al. 2000; Nielsen 2000). Here, these complementary finite-sites models will benefit from the availability of existing phylogenetic procedures for their accommodation of missing data and sequence errors as well as site-to-site variation in rates (Felsenstein 2004).
Acknowledgments
We thank Leonid L. Moroz and Andrea B. Kohn for the use of their unpublished A. californica data; Jan Gorodkin for providing computer time for the initial work at The Royal Veterinary and Agricultural University, Denmark; the Intel Corporation for lending us a computer with two quad-core CPUs used in the final analyses; the Carlsberg Foundation for its fellowship to B.K.; and the Department of Zoology, University of Florida for its support.
References
- Clark, A. G., 1990. Inference of haplotypes from PCR-amplified samples of diploid populations. Mol. Biol. Evol. 7: 111–122. [DOI] [PubMed] [Google Scholar]
- Donnelly, P., and S. Tavaré, 1995. Coalescents and genealogical structure under neutrality. Annu. Rev. Genet. 29: 401–421. [DOI] [PubMed] [Google Scholar]
- Durbin, R., S. Eddy, A. Krogh and G. Mitchison, 1998. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press, Cambridge, UK.
- Felsenstein, J., 2004. Inferring Phylogenies. Sinauer Associates, Sunderland, MA.
- Fisher, R. A., 1930. The Genetical Theory of Natural Selection. Clarendon Press, Oxford.
- Griffiths, R. C., 1989. Genealogical-tree probabilities in the infinitely-many-sites model. J. Math. Biol. 27: 667–680. [DOI] [PubMed] [Google Scholar]
- Griffiths, R. C., 2001. Ancestral inference from gene trees, pp. 137–172 in Genes, Fossils, and Behaviour: An Integrated Approach to Human Evolution, edited by P. Donnelly and R. Foley. IOS Press, Amsterdam.
- Griffiths, R. C., and S. Tavaré, 1995. Unrooted genealogical tree probabilities in the infinitely-many-sites model. Math. Biosci. 127: 77–98. [DOI] [PubMed] [Google Scholar]
- Harpending, H. C., M. A. Batzer, M. Gurven, L. B. Jorde, A. R. Rogers et al., 1998. Genetic traces of ancient demography. Proc. Natl. Acad. Sci. USA 95: 1961–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein, J., M. Schierup and C. Wiuf, 2004. Sequence Variation, Genealogies and Evolution. Oxford University Press, New York.
- Hudson, R. R., 1990. Gene genealogies and the coalescent process, pp. 1–44 in Oxford Surveys in Evolutionary Biology, Vol 7, edited by J. Antonovics and D. Futuyma. Oxford University Press, Oxford.
- Huelsenbeck, J. P., and B. Rannala, 1997. Phylogenetic methods come of age: testing hypotheses in an evolutionary context. Science 276: 227–232. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61: 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1983. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK.
- Kingman, J. F. C., 1982. The coalescent. Stoch. Proc. Appl. 13: 235–248. [Google Scholar]
- Kuhner, M. K., and J. Felsenstein, 2000. Sampling among haplotype resolutions in a coalescent-based genealogy sampler. Genet. Epidemiol. 19: S15–S21. [DOI] [PubMed] [Google Scholar]
- Kuhner, M. K., P. Beerli, J. Yamato and J. Felsenstein, 2000. Usefulness of single nucleotide polymorphism data for estimating population parameters. Genetics 156: 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., 2000. Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 154: 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richterich, P., 1998. Estimation of errors in “raw” DNA sequences: a validation study. Genome Res. 8: 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]