

Abstract
To better understand determinants of susceptibility/resistance of uveal melanomas to herpes simplex virus type 1 (HSV-1) oncolytic therapy, uveal melanoma cell lines of low (OCM1a) and of high (M619, MUM2B) invasive potential were infected with HSV-1 either in the presence or absence of a laminin-rich extracellular matrix (Matrigel). OCM1a cultures were destroyed faster by HSV-1 than M619 and MUM2B cultures. In the presence of Matrigel, all melanoma cultures demonstrated delayed destruction by HSV-1 relative to Matrigel-free cultures. As sequestration of chromatin is a characteristic feature of highly invasive uveal melanomas that is further increased by exposure to laminin, we explored whether chromatin sequestration could be reversed by HSV-1 infection. HSV-1 infection induced a global reversal of chromatin sequestration in highly invasive uveal melanoma cells. However, this viral effect was first observed only 2 hours following virus infection and required novel protein synthesis from input viral DNA. These findings suggest that tumor invasiveness, the spatial relationship of tumor cells to laminin and chromatin sequestration are determinants of susceptibility/resistance of melanomas to HSV-1 oncolytic therapy. Furthermore, these findings indicate for the first time that HSV-1 infection is associated with global exposure of normally highly sequestered cellular DNA in malignant cells.
Keywords: uveal melanoma, herpes simplex virus, extracellular matrix, laminin, oncolytic therapy, chromatin sequestration, chromatin exposure
Introduction
Melanomas involving the skin and other organs, including uveal melanomas remain a significant cause of morbidity and mortality (Namkoong et al, 2006; Woll et al, 1999). Therefore, much work has been focused on finding novel approaches for the treatment of these tumors.
Oncolytic therapy with herpes simplex virus type 1 (HSV-1) is a promising novel approach to treat several human malignancies including melanomas (Latchman, 1997; Randazzo et al, 1995, 1997;Shen and Nemuniatis, 2006; Varghese and Rabkin, 2002). This treatment involves the inoculation of genetically modified, replication-competent HSV-1 strains into melanomas leading to HSV-1 infection and destruction of melanoma cells without significant virus replication in and destruction of normal cells (Latchman, 2005; MacKie et al. 2001; Randazzo et al, 1995, 1997; Shen and Nemunaitis 2006; Varghese and Rabkin 2002). Studies also indicate that the effectiveness of oncolytic HSV-1 therapy is dependent upon virus replication in tumor cells and is augmented by host antiviral and infection-induced anti-tumor immune responses (Miller and Fraser, 2003; Randazzo et al, 1997; Toda et al. 1999). Treatment efficacy can be compromised by incomplete delivery of virus to all tumor cells and can be improved by degradation of fibrillar collagen in tumor (McKie et al. 2006).
In spite of significant progress, several aspects of HSV-1 melanoma treatment are incompletely understood. For example, it is not known whether melanomas of different invasive potential differ in their susceptibility to HSV-1-mediated destruction. Although HSV-1 infects and replicates in most cell types, there are striking differences in the extent of virus replication among cell types (Roizman and Knipe, 2001). These differences in susceptibility are often not completely understood but are thought to be due to differences in the expression of cellular proteins that can positively or negatively influence the viral replication process including among others cellular receptors for viral entry and cellular transcription factors that can regulate the expression of viral genes. As significant differences in gene expression exist between uveal melanoma cells of low and high invasive potential (Folberg et al, 2006, Maniotis et al, 2005), it is possible that uveal melanomas of different invasiveness differ in their susceptibility to HSV-1 replication.
Studies also indicate that the extracellular matrix (ECM) environment is a critically important determinant of the gene expression profile and morphology of uveal melanomas in vivo and of uveal melanoma cultures in vitro (Folberg et al, 2000, 2006; Maniotis et al, 1999, 2005). Association of highly invasive uveal melanoma cells with a laminin-rich ECM environment in vivo and in vitro and the consequent formation of vasculogenic mimicry patterns is paradoxically associated both with increased mortality and with the expression of phenotypic and biochemical features of indolence among cells in direct contact with the laminin-rich vasculogenic mimicry patterns (Folberg et al, 2006). These observations raise the possibility that the susceptibility of uveal melanomas to HSV-1 replication is dependent on the presence of a laminin-rich ECM.
Highly invasive cancer cells feature increased sequestration of cellular chromatin and this may affect their susceptibility to HSV-1 replication. DNA in cancer cells is tightly packaged in protein complexes that contain disulfide bonds, making it highly resistant to digestion by restriction enzymes (Maniotis et al, 2005). Detergent extraction assays, cell smear assays, flow cytometry and touch preparations, and isolated chromosome sets all demonstrate that the more invasive the cancer, the greater its DNA is protected from restriction enzyme cleavage (Maniotis et al, 2005). The sequestration of DNA as a specific physical marker for malignancy, as opposed to other disease or normal states, has been proposed as a novel method for cancer detection (Maniotis et al, 2005; Stein, 2005). Sequestered genes in cancer may represent specific differentiation genes (Kryostek and Puck, 1990).
HSV-1 infection of cells is associated with dramatic changes in cellular physiology and morphology and many of these changes are required for the progression and completion of the HSV-1 replication cycle (reviewed in Roizman and Pellet, 2001; Roizman and Knipe, 2001; Valyi-Nagy et al, 2006). It is possible that sequestration of cellular chromatin represents a cellular condition unfavorable for the progression of viral replication. Currently it is not known whether HSV-1 infection alters the pattern of cellular chromatin sequestration in uveal melanomas.
Identification of cellular determinants of susceptibility/resistance of melanomas to HSV-1 replication is important for the development of effective HSV-1 oncolytic melanoma therapy. Therefore, in the experiments reported here were designed to find answers to three interrelated questions: (1) Does the susceptibility of uveal melanoma cultures to HSV-1-mediated destruction vary with differences in tumor cell invasiveness, (2) Does the susceptibility of tumor cells to HSV-1 vary between microenvironments deficient or enhanced with laminin, and (3) What is the effect of HSV-1 on chromatin sequestration in tumor cells? We found that uveal melanoma cells of low invasive potential are more susceptible to HSV-1-mediated destruction than uveal melanoma cells of high invasive potential. Furthermore, we show that in the presence of laminin-rich ECM, uveal melanoma cultures demonstrate delayed destruction by HSV-1 relative to cultures deficient in laminin. We also report that HSV-1 infection induces a reversal of cellular chromatin sequestration in highly invasive MUM2B cells.
Materials and Methods
Cells
Uveal melanoma cells of low (OCM1a) and high (M619 and MUM2B) invasive potential were maintained in Eagle’s Minimal Essential Medium (EMEM, BioWhittaker Inc., Walkersville, MD) supplemented with heat inactivated 15% fetal bovine serum (Fisher, Ontario, Canada) without the addition of exogenous extracellular matrix molecules or growth factors. The characteristics of these cell lines have been described in detail previously (Maniotis et al, 1999, 2005). Melanoma cells used for HSV-related experiments were grown on six well plates in EMEM medium either in the presence or in the absence of extracellular matrix rich in laminin (Matrigel, BD Biosciences, Bedford, MA). Matrigel was poured onto tissue culture plates to a depth of approximately 0.2 mm followed by polymerization for 1 hour at 37° C before addition of melanoma cells.
Viruses
HSV-1 strain F was a generous gift of Dr. B. He (University of Illinois at Chicago). HSV-2 strain 333 was obtained from Dr. Deepak Shukla (University of Illinois at Chicago). Virus inactivation by heat and by ultraviolet (UV) light was performed as previously described (Valyi-Nagy et al, 1991).
Determination of susceptibility of uveal melanoma cultures to HSV-1-mediated destruction
Uveal melanoma cells were grown on 6-well tissue culture plates in the presence or absence of Matrigel. Cell numbers per well were then counted. Cells in some wells were left untreated and were further incubated (untreated controls) while the tissue culture medium was removed from other wells and the cells were exposed at 37° C to one of the following inocula: (i) 0.5 ml of sterile PBS (mock infection); (ii) HSV-1 (strain F) with a calculated multiplicity of infection (MOI) ranging from 0.00001 plaque forming units (PFU) per cell to 10 PFU/cell diluted in PBS to a final volume of 0.5 ml. After incubation for 2 hours, the original inocula were removed and fresh tissue culture medium (3 ml) was added to each well and further incubated in repeatedly refreshed culture medium for up to 2 weeks. During this 2-week period, cultures were observed daily under an inverted light microscope (Leica, Bannockburn, IL) for evidence of viral cytopathic effects and the day when at least 95% of the melanoma cells were destroyed was noted. Cell death was confirmed by the uptake of the charged cationic dye Trypan blue (0.2%) by more than 95% of residual cells following incubation of cultures with Trypan blue (0.2%) for 10 minutes at 37 °C. 0.5 ml of medium from virus inoculated cultures demonstrating at least 95% destruction was removed and was used to infect Vero cell cultures with known susceptibility to productive HSV-1 infection to confirm the presence of infectious virus in the cultures. Photographs were taken of the cultures before HSV-1 inoculation and at selected times after HSV-1 inoculation. Experiments were performed in duplicates and were repeated at least three times.
Cell smear assay to determine the effect of HSV-1 infection on chromatin sequestration in uveal melanoma cells
Uveal melanoma cells of low (OCM1a) and high (MUM2B) invasive potential were grown to approximately 70% confluency on 6-well tissue culture plates in the absence of Matrigel. Cell numbers per well were then counted. OCM1a and MUM2B cells in some wells were then left untreated while the tissue culture medium was removed from OCM1a and MUM2B cells in other wells and the cells were exposed at 37 °C to one of the following inocula: (i) 0.5 ml of sterile PBS (mock infection); (ii) HSV-1 (strain F) with a calculated MOI ranging from 0.1 PFU/cell to 10 PFU/cell diluted in PBS to a final volume of 0.5 ml (HSV-1 infection); (iii) HSV-2 (strain 333) with a calculated MOI ranging from 0.1 PFU/cell to 10 PFU/cell diluted in PBS to a final volume of 0.5 ml (HSV-2 infection). Untreated OCM1a and MUM2B cells and OCM1a and MUM2B cells exposed to HSV-1, HSV-2 or mock infection for selected times (5 minutes, 1, 2, or 3 hours), were washed twice in PBS and were then mechanically dislodged from the wells, pelleted and resuspended in 1X PBS. A drop containing 15 ul of the cell suspension was then placed on a glass slide. The drops were allowed to evaporate over 30 minutes to 1 hour at room temperature. Alu I restriction enzyme (Promega) (0.5 ul in 40 ul of DMEM) was applied to the dried cells, and the preparation was placed in a humidified 37° C chamber to optimize enzyme activity and minimize enzyme evaporation. Endonuclease digestions were terminated at pre-selected time points (30 minutes and hourly increments thereafter up to 24 hours) with ethidium bromide (Sigma; 100 ng/ml) and chromatin digestion was visualized using an inverted fluorescence microscope (Leica, Bannockburn, IL). The preparations were photographed immediately and scored qualitatively as follows: (i) nuclei in which no fluorescence was detected except for nucleoli were scored as digested nuclei; (ii) nuclei in which non-nucleolar nuclear and nucleolar fluorescence was detected was scored as undigested.
Results
Tumor invasiveness and the presence of laminin-rich ECM (Matrigel) determine the susceptibility uveal melanoma cultures to HSV-1-mediated destruction
In our first series of experiments, we compared the HSV-1-mediated destruction of uveal melanoma cells of low (OCM1a) and high (M619, MUM2B) invasive potential in the presence and absence of laminin-rich ECM (Matrigel). In the absence of Matrigel, all cell lines (OCM1a, M619, MUM2B) grew in monolayers (Fig. 1.A, C, E). In the presence of Matrigel, all cell lines (OCM1a, M619, MUM2B) developed three dimensional structures: highly invasive (M619, MUM2B) melanoma cells formed vasculogenic mimicry patterns (Fig. 1. D, F) while OCM1a cells of low invasive potential grew in aggregates (Fig. 1. B). The morphologic appearance of vasculogenic mimicry patterns formed by highly invasive (M619, MUM2B) melanoma cells in culture upon exposure to Matrigel was similar to the vasculogenic mimicry patterns that can be observed in histologic sections of invasive human uveal melanomas (Folberg et al, 2000; Maniotis et al, 1999). No such patterns are present in histologic sections of non-invasive human uveal melanomas (Folberg et al, 2000; Maniotis et al, 1999). HSV-1 inoculation was performed once the formation of vasculogenic mimicry patterns and cell aggregates was observed. Cells in some wells were left untreated and were further incubated in the original media (untreated controls) while the tissue culture medium was removed from other wells and the cells were exposed to one of the following inocula: (i) 0.5 ml of sterile PBS (mock infection); (ii) HSV-1 (strain F) with a calculated multiplicity of infection (MOI) of 10 PFU/cell diluted in PBS to a final volume of 0.5 ml; and (iii) HSV-2 (strain 333) with a calculated MOI of 10 PFU/cell.
Fig. 1.
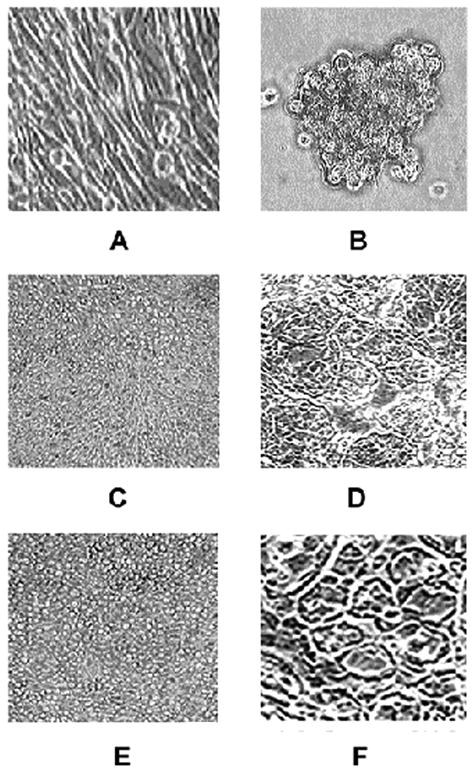
Morphologic appearance of non-invasive and invasive uveal melanoma cells cultured in the presence or absence of laminin-rich extracellular matrix (Matrigel). OCM1a uveal melanoma cell line of low invasive potential cultured in the absence (A) or in the presence (B) of Matrigel. Highly invasive M619 uveal melanoma cells cultured in the absence (C) or in the presence (D) of Matrigel. Highly invasive MUM2B uveal melanoma cells cultured in the absence (E) or in the presence (F) of Matrigel. Note the presence of prominent vasculogenic mimicry patterns in panels D and F.
We found that OCM1a cultures were destroyed by HSV-1 faster than M619 and MUM2B cultures (Table 1.). In the absence of Matrigel, OCM1a cultures were destroyed by 2 days, while MUM2B and M619 cultures were destroyed by 4 days (Table 1.). In the presence of Matrigel, OCM1a cultures were destroyed by 3 days, while MUM2B and M619 cultures were destroyed by 6 days (Table 1). Identical results were achieved with HSV-2 (data not shown).
Table 1.
Elapsed time (days) from inoculation of HSV-1 (MOI=10 PFU per cell) to at least 95% destruction of uveal melanoma cultures of low (OCM1a) and high (M619, MUM2B) invasive potential in the presence or absence of Matrigel
| Uveal melanoma cell line | Matrigel used | Day of 95% destruction |
|---|---|---|
| OCM1a | No | 2 |
| Yes | 3 | |
| M619 | No | 4 |
| Yes | 6 | |
| MUM2B | No | 4 |
| Yes | 6 |
Untreated and mock infected cultures showed no cytopathic effects and demonstrated normal growth and morphology for 14 days. Portions of 0.5 ml of medium were removed from HSV-1 or HSV-2 inoculated cultures demonstrating at least 95% destruction and were used to infect Vero cell cultures with known susceptibility to productive HSV infection. These studies confirmed the production of infectious HSV-1 and HSV-2 in uveal melanoma cultures.
To determine whether the susceptibility of uveal melanoma cells to HSV-1 is dependant of viral MOI, we compared the destruction of melanoma cell cultures of low (OCM1a) and high (MUM2B) invasive potential following infection with a wide range of HSV-1 doses. OCM1a and MUM2B cells for these experiments were cultured without Matrigel. Cells in some wells were left untreated and were further incubated (untreated controls) while the tissue culture medium was removed from other wells and the cells were exposed either HSV-1 strain F at a calculated MOI ranging from 0.00001 PFU/per cell to 10 PFU/cell or to sterile PBS (mock infection). After incubation for 2 hours, the original inocula were removed and fresh tissue culture medium (3 ml) was added to each well and further incubated in repeatedly refreshed culture medium for up to 2 weeks. We again noted that highly invasive MUM2B cells were more resistant to HSV-1-mediated destruction than OCM1a cells that are of low invasive potential (Table 2.). At the highest used HSV-1 inoculum at MOI=10, OCM1a cultures were destroyed by 2 days, while MUM2B cultures were destroyed by 4 days (Table 2.). At MOI=1, OCM1a cultures were destroyed by 3 days, while MUM2B cultures were destroyed by 5 days (Table 2., Fig. 2.). At lower HSV-1 inocula, the detected differences were larger. At the lowest virus dose used (MOI=0.00001), OCM1a cultures were destroyed by 7 days, while MUM2B cultures were destroyed by 14 days (Table 2.). Untreated and mock infected cultures showed no cytopathic effects and demonstrated normal growth and morphology for 14 days. Portions of 0.5 of medium were removed from HSV-1 inoculated cultures demonstrating at least 95% destruction and were used to infect Vero cell cultures with known susceptibility to productive HSV-1 infection. These studies confirmed the production of infectious HSV-1 in uveal melanoma cultures.
Table 2.
Elapsed time (days) from inoculation of various doses of HSV-1 to at least 95% destruction of uveal melanoma cultures of low (OCM1a) and high (MUM2B) invasive potential in the absence of Matrigel
| HSV-1 inoculum dose (MOI) | OCM1a (day of 95% destruction) | MUM2B (day of 95% destruction) |
|---|---|---|
| 10 | 2 | 4 |
| 1 | 3 | 5 |
| 0.1 | 3 | 7 |
| 0.01 | 5 | 7 |
| 0.001 | 5 | 14 |
| 0.0001 | 5 | 14 |
| 0.00001 | 7 | 14 |
Fig. 2.
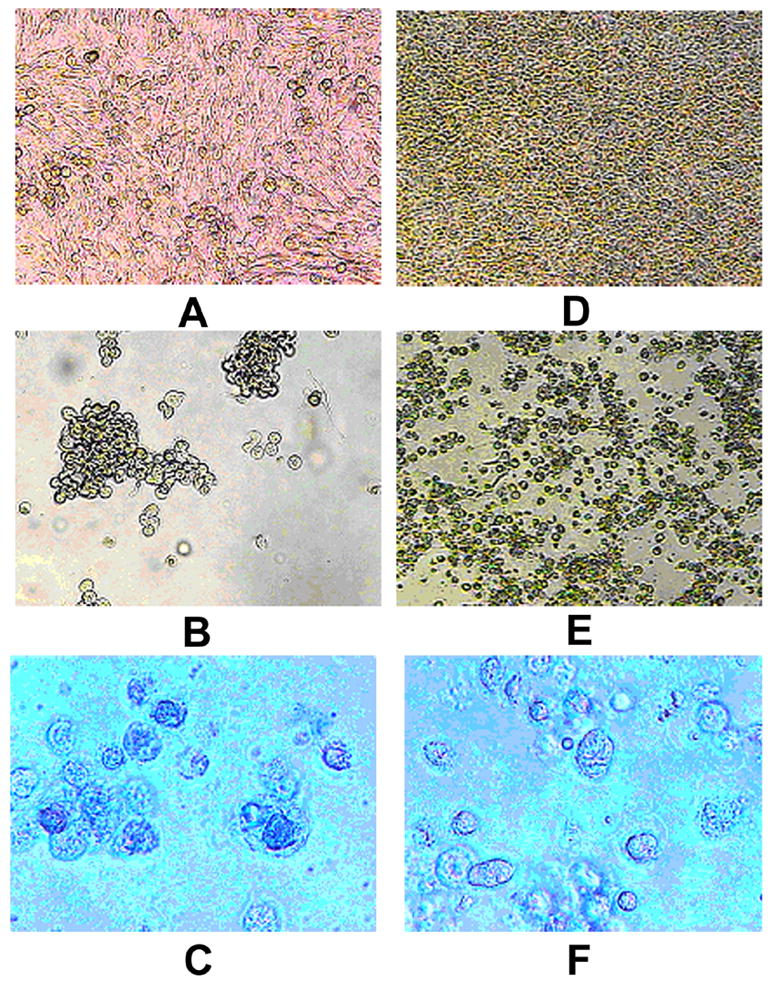
Morphology of mock infected and HSV-1 infected OCM1a and MUM2B melanoma cells. OCM1a and MUM2B cells were grown to approximately 70% confluency on 6-well tissue culture plates and were either exposed to 0.5 ml of sterile PBS (mock infection) or to HSV-1 diluted in PBS at a MOI=1. After incubation for 2 hours, the original inocula were removed and fresh tissue culture medium was added to each well and cultures were further incubated for times as indicated below when photographs were taken using an inverted light microscope. A. OCM1a culture 3 days after mock infection, B. OCM1a culture 3 days after HSV-1 infection, C. OCM1a culture 3 days after HSV-1 infection and following incubation of cultures with Trypan blue (0.2%) for 10 minutes, D. MUM2B culture 5 days after mock infection, E. MUM2B culture 5 days after HSV-1 infection, F. MUM2B culture 5 days after HSV-1 infection and following incubation of cultures with Trypan blue (0.2%) for 10 minutes.
These experiments suggest that uveal melanoma cells of low invasive potential are more susceptible to HSV-1-mediated destruction than uveal melanoma cells of high invasive potential. Differences in susceptibility are most pronounced at low MOI. Furthermore, these observations indicate that in the presence of Matrigel, uveal melanoma cultures demonstrate delayed destruction by HSV-1 relative to Matrigel-free cultures with vasculogenic mimicry-forming highly invasive melanoma cultures showing particularly decreased susceptibility to HSV-1.
Reversal (exposure) of cellular chromatin sequestration in highly invasive MUM2B cells following HSV-1and HSV-2 infection
To determine whether HSV-1 and/or HSV-2 infection affects cellular chromatin organization in highly invasive uveal melanoma cells, cell smears were prepared from cultures of uninfected uveal melanoma cells of low (OCM1a) and high (MUM2B) invasive potential and from MUM2B cells that were previously exposed for various times to either HSV-1 or HSV-2 (at a MOI ranging from 0.1 to 10 PFU/cell) or to sterile PBS (mock infection). Cell smears were then exposed to Alu I as described previously (Maniotis et al, 2005).
As expected based on our previous observations (Maniotis et al, 2005), nuclei of MUM2B cells in the uninfected control cultures demonstrated increased resistance to Alu I digestion relative to untreated OCM1a cells. Specifically, digested nuclei were observed in OCM1a smears after as short as 2 hours of Alu I digestion with essentially complete digestion of all nuclei by 12 hours of Alu I exposure (data not shown). In contrast, MUM2B cell smears essentially did not show any digestion until 12 hours and Alu I digestion was typically not complete even after 24 hours of Alu I exposure (data not shown). We also found that Alu I-sensitive sites remained sequestered in MUM2B cells exposed to HSV-1 or HSV-2 for 5 minutes or to 1 hour similarly to mock infected and uninfected, untreated cells. In sharp contrast, 2 hours exposure to Alu I was sufficient to cause complete digestion of many cells in MUM2B cultures that were previously exposed to HSV-1 or HSV-2 for 2 or 3 hours (Figs 3 and 4). This virus effect was particularly striking in MUM2B cultures inoculated with HSV-1 or HSV-2 at a MOI of 1 or 10 as infection in these cultures lead to chromatin exposure in nearly all cells present (Figs. 3. and 4.). The number of MUM2B cells demonstrating exposure of Alu I-sensitive sites following HSV-1 or HSV-2 inoculation at 0.1 MOI was significantly lower and involved less than 10% of cells (Fig. 3). Exposure to sterile PBS and exposure of cells to heat- or UV-inactivated HSV-1 for 2 or 3 hours did not lead to exposure of AluI-sensitive sites relative to untreated cultures (Fig. 5) indicating that viral protein synthesis from input viral DNA is required for the reversal of chromatin sequestration and that virion-cell interactions are not sufficient for this effect.
Fig. 3.
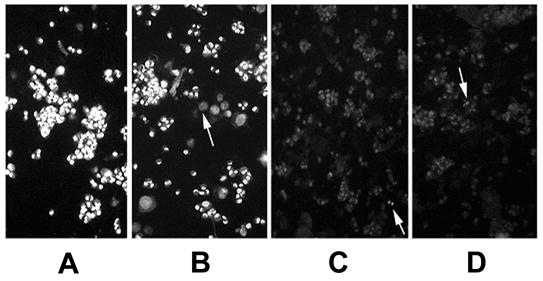
Reversal of chromatin sequestration in MUM2B melanoma cells following HSV-1 inoculation. MUM-2 cells were grown to approximately 70% confluency on 6-well tissue culture plates. The tissue culture medium was then removed from the wells and the cells were exposed at 37° C to either 0.5 ml of sterile PBS (mock infection)(A) or to HSV-1 diluted in PBS to a final volume of 0.5 ml and a calculated MOI of 0.1 (B), 1 (C), or 10 (D) PFU/cell. After incubation for 2 hours, cells were washed twice in PBS and were then mechanically dislodged from the wells, pelleted and resuspended in 1X PBS. A drop containing 15 ul of the cell suspension was then placed on a glass slide. The drops were allowed to evaporate over a 1-hour period at room temperature. Alu I restriction enzyme (Promega) (0.5 ul in 40 ul of DMEM) was applied to the dried cells, and the preparation was placed in a humidified 37° C chamber for 2 hours when ethidium bromide was added to terminate the digestion and the preparation was photographed immediately using an inverted fluorescence microscope. Arrows in 0.1 MOI picture (B) point to the nuclei of cells that demonstrate chromatin exposure, whereas, the arrows in the 1.0 MOI (C) and the 10 MOI (D) cultures point to residual cells whose chromatin demonstrates chromatin sequestration.
Fig. 4.
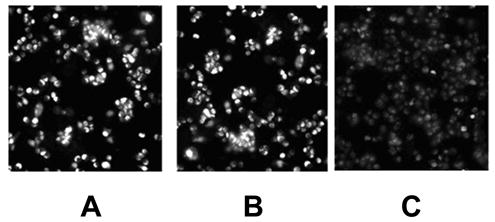
Reversal of chromatin sequestration in MUM2B melanoma cells following HSV-2 inoculation. MUM-2 cells were grown to approximately 70% confluency on 6-well tissue culture plates. The tissue culture medium was then removed from the wells and the cells were exposed at 37° C to either 0.5 ml of sterile PBS (mock infection, B) or to HSV-2 diluted in PBS to a final volume of 0.5 ml and a MOI of 1 PFU/cell (C). After incubation for 2 hours, cells were washed twice in PBS and were then mechanically dislodged from the wells, pelleted and resuspended in 1X PBS. A drop containing 15 ul of the cell suspension was then placed on a glass slide. The drops were allowed to evaporate over a 1-hour period at room temperature. Alu I restriction enzyme (Promega) (0.5 ul in 40 ul of DMEM) was applied to the dried cells, and the preparation was placed in a humidified 37° C chamber for 2 hours when ethidium bromide was added to terminate the digestion and the preparation was photographed immediately using an inverted fluorescence microscope. Panel A shows the appearance of tumor cells without treatment (mock or HSV-2 infection) and without Alu I digestion.
Fig. 5.
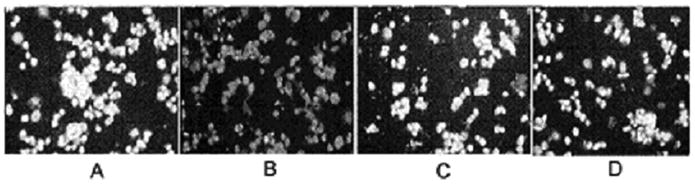
Heat- or UV inactivated HSV-1 fails to affect chromatin exposure to Alu I digestion in highly invasive uveal melanoma cells. MUM2B cells were grown to approximately 70% confluency on 6-well tissue culture plates. The tissue culture medium was then removed from the wells and the cells were exposed at 37° C to either 0.5 ml of sterile PBS (mock infection)(A) or to HSV-1 diluted in PBS to a final volume of 0.5 ml and a MOI of 10 (B) or to UV (C) or heat-inactivated (D) HSV-1. After incubation for 3 hours, cells were washed twice in PBS and were then mechanically dislodged from the wells, pelleted and resuspended in 1X PBS. A drop containing 15 ul of the cell suspension was then placed on a glass slide. The drops were allowed to evaporate over a one-hour period at room temperature. Alu I restriction enzyme (0.5 ul in 40 ul of DMEM) was applied to the dried cells, and the preparation was placed in a humidified 37° C chamber for 2 hours when ethidium bromide was added to terminate the digestion and the preparation was photographed immediately using an inverted fluorescence microscope.
These findings indicate that HSV-1 and HSV-2 infection lead to reversal of chromatin sequestration in highly invasive uveal melanoma cells and that this reversal requires at least 2 hours following virus inoculation.
Discussion
We report here for the first time that uveal melanoma cells of low invasive potential are more susceptible to HSV-1-mediated destruction in vitro than uveal melanoma cells of high invasive potential. Furthermore, we show that in the presence of laminin-rich ECM (Matrigel), uveal melanoma cultures demonstrate delayed destruction by HSV-1 relative to Matrigel-free cultures. We also report novel observations indicating that HSV-1 and HSV-2 infection induce a reversal of cellular chromatin sequestration in highly invasive MUM2B cells.
These findings suggest that the efficacy of oncolytic HSV-1 melanoma therapy depends on tumor invasiveness and thus suggest that viral doses used for melanoma therapy need to be adjusted according to the invasiveness of the treated tumor. Our findings also suggest that the efficacy of oncolytic HSV-1 melanoma therapy depends on the relationship of tumor cells with laminin. A previous study has found that the effectiveness of oncolytic HSV-1 therapy is dependant on the ECM environment (McKie et al 2006). This study reported that the efficacy of HSV-1 therapy can be improved by degradation of fibrillar collagen in tumors and the improved therapeutic efficacy was attributed to improved delivery of virus to tumor cells (McKie et al 2006). Our current study suggests that the ECM environment can affect the efficacy of HSV-1 oncolytic therapy not only by this indirect way of blocking virus access to tumor cells but also directly through modulating tumor cell susceptibility to virus replication. Our findings also suggest that anti-laminin treatment prior to HSV-1 therapy may increase the efficiency of HSV-1 oncolytic melanoma therapy.
Highly invasive uveal melanoma cell lines demonstrate increased chromatin sequestration relative to uveal melanoma cell lines of low invasive potential (Maniotis et al, 2005) raising the possibility that increased chromatin sequestration is a cause of the decreased susceptibility of highly invasive uveal melanoma cells to HSV-1-mediated destruction. Our findings indicate that exposure of highly invasive MUM2B cells to HSV-1 leads to a complete reversal of cellular chromatin sequestration. However, this viral effect requires at least 2 hours and is dependent on protein synthesis from input viral DNA. These findings suggest that increased chromatin sequestration may indeed contribute to the decreased susceptibility of highly invasive uveal melanoma cells to HSV-1. Our findings also indicate that a reversal of chromatin sequestration in invasive melanoma cells also occurs after HSV-2 infection and thus this viral effect likely represents a general cytopathic effect of HSV infection in malignant tumor cells. In addition to the potential significance of these observations to a better understanding of mechanisms of tumor resistance to HSV oncolytic therapy, the observed effect of HSV on chromatin organization in malignant tumor cells is novel and very interesting.
It has been long known that active genes in normal cells are preferentially digested by DNase-I (Weintraub and Goudine, 1976). DNA in malignant neoplastic cells is tightly packaged (sequestered), making it highly resistant to digestion by restriction enzymes (Maniotis et al, 2005). It is also known that during the process of reverse transformation when malignant cells are restored to a normal phenotype by exposure to cyclic AMP derivates, a succession of metabolic changes occurs varying from early alterations in calcium dynamics and changes in cytoskeletal structures to subsequent chromatin exposure associated with an increase in DNA sensitivity to enzyme digestion (Hsie and Puck, 1971; Kryostek and Puck, 1990; Puck et al, 1990). In malignant uveal melanoma cells, Alu I-sensitive sites become profoundly sequestered when cells are incubated with laminin (Matrigel) or a circular RGD peptide (RGD-C), but become exposed when cells are exposed to serum or collagen I (Maniotis et al, 2005).
The mechanisms by which extracellular signals induce changes in Alu I binding site sequestration and exposure appear to depend on the cytoskeleton. For instance, disruption of actin leads to exposure, while disruption of microtubules results in increased sequestration of Alu I binding sites (Maniotis et al, 2005). The mechanism(s) by which HSV-1 infection leads to the reversal of chromatin sequestration is unknown at this point. Our finding that inoculation of melanoma cells with heat- or UV-inactivated virus did not lead to exposure of Alu I-sensitive sites relative to untreated cultures indicate that (i) the detected chromatin changes were caused by virus and not by media components, and (ii) viral protein synthesis from input viral DNA is required for reversal of chromatin sequestration and that virion-cell interactions are not sufficient for this effect.
HSV-1 infection-induced nuclear changes are well documented (reviewed in Simpson-Holey et al, 2005). Following HSV-1 infection, replication compartments form and host chromatin is eventually marginalized. Chromatin is later dispersed, and virus replication compartments reach the nuclear edge. The nuclear lamina is disrupted and there is nuclear expansion detectable from 8 to 24 hours after virus inoculation (Simpson-Holey et al, 2005). It is notable that these nuclear changes occur later than 2 hours after virus inoculation and thus later than chromatin exposure reported in this study. HSV-1 and HSV-2 infection can induce chromatid and chromosome breaks and uncoiling of the centromeres of some chromosomes as early as 2–3 hours post infection (reviewed by Fortunato and Spector, 2003), however, global reversal of chromatin sequestration following HSV infection observed in our current study has not been reported.
There are multiple known mechanisms by which HSV-1 infection interferes with cellular metabolism and alters cellular including nuclear morphology (reviewed in Roizman and Pellet, 2001; Roizman and Knipe, 2001; Valyi-Nagy et al, 2006). Known effects of HSV infection include among others shutoff of cellular macromolecular synthesis, degradation of cellular RNA, inhibition of splicing of messenger RNA, selective degradation or stabilization of cellular proteins and interference with the cell cycle machinery with induction of cell cycle block in either G1 or G2 (reviewed in Roizman and Pellet, 2001; Roizman and Knipe, 2001; Valyi-Nagy et al, 2006). HSV infection leads to an up-regulation of various cellular transcription factors, stress response genes, cell cycle regulatory genes and genes involved in apoptotic pathways (Hobbs and DeLuca, 1999; Khodarev et al, 1999; Stingley et al, 2000; Taddeo et al, 2002; Valyi-Nagy et al, 1991). Interaction of HSV with the cellular apoptotic machinery is complex and involves both proapoptotic and anti-apoptotic viral effects. HSV has multiple antiapoptotic genes and the inhibition of apoptosis is thought to be beneficial for HSV because it requires the environment of a living cell to replicate (reviewed in Roizman and Knipe, 2001).
Perhaps most relevant to the observed virus-induced reversal of cellular chromatin sequestration are previously described HSV-induced changes in the cellular cytoskeleton and viral mechanisms that block silencing of HSV-1 genes in infected cells. Shortly after HSV infection, microtubules at the junction with the plasma membrane become disrupted, and more extensive rearrangements of the microtubular network occur later during the infection (Roizman and Knipe, 2001; Ward et al, 1998). HSV-1 infection is also associated with the reorganization of microfilaments, actin and myosin (Bedows et al, 1983; Winkler et al, 1982). The US3 gene product of HSV is a serine/threonine protein kinase that during HSV-2 infection mediates the disruption of actin filaments (Maruta et al, 2000).
Cellular reactions to infection that HSV needs to overcome include silencing of viral DNA by the complex formed by CoRest/REST and histone deacetylases (HDAC) 1 and 2. Early in infection, viral DNA and the immediate early (IE) viral protein ICP0 colocalize with nuclear structures known as nuclear domain (ND) 10 (Everett and Maul, 1994; Maul and Everett, 1994; Maul et al, 1993). This colocalization appears to serve two objectives (Gu et al, 2005; Roizman et al, 2005). The first objective, particularly important during infections at low multiplicities, is to dissociate HDACs 1 and 2 from the CoRest/REST repressor complex and thereby to block silencing of post-IE HSV gene expression (24). The second objective is to disperse ND10 components (Everett and Maul, 1994; Everett and Murray, 2005; Maul and Everett, 1994; Maul et al, 1993) and degrade the promyelocytic leukemia protein (PML) that is responsible for the organization of ND10 and thereby to block the IFN-mediated host response to infection.
The mechanism(s) by which HSV-1 infection leads to a reversal of cellular chromatin sequestration and how this viral effect is related to already described virus-induced changes are not known and clearly further detailed studies are indicated to define these mechanisms. There are numerous genetically defined HSV-1 strains available deficient in specific virus genes and the use of these mutants will help in future studies to define the molecular mechanisms of HSV-1-induced cellular chromatin desequestration (exposure). These future studies may provide important information on the molecular level about changes in chromatin structure in neoplastic and viral diseases.
In summary, findings reported here provide potentially useful practical information concerning HSV-1-based melanoma therapy. Furthermore, these findings represent the first report of global reversion of cellular chromatin sequestration in malignant cells following virus infection.
Acknowledgments
This work was supported by Public Health Service grant EY10457 to R.F. and a TRECC Accelerator Award by the University of Illinois to R.F., A.J.M, K.V-N, and T.V-N.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Bedows E, Rao KM, Welsh MJ. Fate of microfilaments in Vero cells infected with measles virus and herpes simplex virus type 1. Mol Cell Biol. 1983;3:712–719. doi: 10.1128/mcb.3.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Maul GG. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994;13:5062–5069. doi: 10.1002/j.1460-2075.1994.tb06835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Murray J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol. 2005;79:5078–5089. doi: 10.1128/JVI.79.8.5078-5089.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folberg R, Hendrix MJ, AJ Maniotis AJ. Vasculogenic mimicry and tumor angiogenesis. Am J Pathol. 2000;156:361–381. doi: 10.1016/S0002-9440(10)64739-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folberg R, Arbieva Z, Moses J, Hayee A, Sandal T, Kadkol S, Lin AK, Valyi-Nagy K, Setty S, Leach L, Chavez-Barrios P, Larsen P, Mujamdar D, Pe’er J, Maniotis AJ. Tumor cell plasticity in uveal melanoma: microenvironment directed dampening of the invasive and metastatic genotype and phenotype accompanies the generation of vasculogenic mimicry patterns. Am J Pathol. 2006;169:1376–1389. doi: 10.2353/ajpath.2006.060223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunato A, Spector DH. Viral induction of site-specific chromosome damage. Rev Med Virol. 2003;13:21–37. doi: 10.1002/rmv.368. [DOI] [PubMed] [Google Scholar]
- Gu H, Liang Y, Mandel G, Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. PNAS USA. 2005;102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsie AW, Puck TT. Morphologic transformation of Chinese hamster cells by dibutyryl adenosine cyclic 3’5’-monophosphate and testosterone. PNAS USA. 1971;68:358–361. doi: 10.1073/pnas.68.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs WE, 2nd, DeLuca NA. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J Virol. 1999;73(10):8245–8255. doi: 10.1128/jvi.73.10.8245-8255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodarev NN, Advani SJ, Gupta N, Roizman B, Weichselbaum RR. Accumulation of specific RNAs encoding transcriptional factors and stress response proteins against a background of severe depletion of cellular RNAs in cells infected with herpes simplex virus 1. PNAS U S A. 1999;96 (21):12062–12067. doi: 10.1073/pnas.96.21.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystosek A, Puck TT. The special distribution of exposed nuclear DNA in normal, cancer, and reverse-transformed cells. PNAS USA. 1990;87:6560–6564. doi: 10.1073/pnas.87.17.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman DS. Herpes simplex virus-based vectors for the treatment of cancer and neurodegenerative disease. Curr Opin Mol Ther. 2005;7:415–418. [PubMed] [Google Scholar]
- Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LMG, Pe’er J, Trent JM, Meltzer PS, Hendrix MJC. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155:739–752. doi: 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniotis AJ, Valyi-Nagy K, Karavitis J, Moses J, Boddipali JV, Wang Y, Nuñez R, Setty S, Bissell MJ, Folberg R. Chromatin organization measured by Alu I restriction enzyme changes with malignancy and is regulated by the extracellular matrix and the cytoskeleton. Am J Pathol. 2005;166:1187–1203. doi: 10.1016/S0002-9440(10)62338-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul GG, Everett RD. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J Gen Virol. 1994;75:1223–1233. doi: 10.1099/0022-1317-75-6-1223. [DOI] [PubMed] [Google Scholar]
- Maul GG, Guldner HH, Spivack JG. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene product. J Gen Virol. 1993;74:2679–2690. doi: 10.1099/0022-1317-74-12-2679. [DOI] [PubMed] [Google Scholar]
- MacKie RM, Stewart B, Brown SM. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet. 2001;357:525–526. doi: 10.1016/S0140-6736(00)04048-4. [DOI] [PubMed] [Google Scholar]
- McKie TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP, Bawaendi MG, Boucher Y, Breakefield XO, Jain RK. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66:2509–2513. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- Miller CG, Fraser NW. Requirement of an integrated immune response for successful neuroattenuated HSV-1 therapy in an intracranial metastatic melanoma model. Mol Ther. 2003;7:741–747. doi: 10.1016/S1525-0016(03)00120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Goshima F, Daikoku T, Takakuwa H, Nishiyama Y. Expression of herpes simplex virus type 2 US3 affects the Cdc42/Rac pathway and attenuates c-Jun N-terminal kinase activation. Genes Cells. 2000;5:1017–1027. doi: 10.1046/j.1365-2443.2000.00383.x. [DOI] [PubMed] [Google Scholar]
- Namkoong J, Martino JJ, Chen S. From existing therapies to novel targets: a current view of melanoma. Front Biosci. 2006;11:2081–2092. doi: 10.2741/1951. [DOI] [PubMed] [Google Scholar]
- Puck TT, Krystosek A, Chan DC. Genome regulation in mammalian cells. Somat Cell Mol Genet. 1990;16:257–265. doi: 10.1007/BF01233362. [DOI] [PubMed] [Google Scholar]
- Randazzo BP, Kesari S, Gesser RM, Alsop D, Ford JC, Brown SM, McLean A, Fraser NW. Treatment of experimental intracranial murine melanoma with a neuroattenuated herpes simplex virus 1 mutant. Virology. 1995;211:94–101. doi: 10.1006/viro.1995.1382. [DOI] [PubMed] [Google Scholar]
- Randazzo BP, Bhat MG, Kesari S, Fraser NW, Brown SM. Treatment of experimental subcutaneous melanoma with a replication-restricted herpes simplex virus mutant. J Invest Dermatol. 1997;108:933–937. doi: 10.1111/1523-1747.ep12295238. [DOI] [PubMed] [Google Scholar]
- Roizman B, Gu H, Mandel G. The first 30 minutes in the life of a virus: unrest in the nucleus. Cell Cycle. 2005;4:8. doi: 10.4161/cc.4.8.1902. [DOI] [PubMed] [Google Scholar]
- Roizman B, Knipe DM. Herpes simplex viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 2399–2459. [Google Scholar]
- Roizman B, Pellet PE. The family of Herpesviridae: a brief introduction. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 2381–2397. [Google Scholar]
- Shen Y, Nemunaitis J. Herpes simplex virus 1 (HSV-1) for cancer treatment. Cancer Gene Ther Apr. 2006 doi: 10.1038/sj.cgt.7700946. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Simpson-Holey M, Colgrove RC, Nalepa G, Harper JG, Knipe DM. Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J Virol. 2005;79:12840–12851. doi: 10.1128/JVI.79.20.12840-12851.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein GS. Mechanogenomic control of DNA exposure and sequestration. Am J Pathol. 2005;166(4):959–62. doi: 10.1016/S0002-9440(10)62317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingley SW, Ramirez JJ, Aguilar SA, Simmen K, Sandi-Goldin RM, Ghazal P, Wagner EK. Global analysis of herpes simplex virus type 1 transcription using an oligonucleotide-based DNA microarray. J Virol. 2000;74(21):9916–9927. doi: 10.1128/jvi.74.21.9916-9927.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddeo B, Esclatine A, Roizman B. The patterns of accumulation of cellular RNAs in cells infected with a wild-type and a mutant herpes simplex virus 1 lacking the virion host shutoff gene. PNAS U S A. 2002;99(26):17031–17036. doi: 10.1073/pnas.252588599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda M, Rabkin SD, Kojima H, Martuza RL. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther. 1999;10:385–393. doi: 10.1089/10430349950018832. [DOI] [PubMed] [Google Scholar]
- Valyi-Nagy T, Deshmane SL, Dillner AJ, Fraser NW. Induction of cellular transcription factors in trigeminal ganglia of mice by corneal scarification, HSV-1 infection and explantation of trigeminal ganglia. J Virol. 1991;65:4142–4152. doi: 10.1128/jvi.65.8.4142-4152.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valyi-Nagy T, Shukla D, Engelhard HH, Kavouras J, Scanlan P. Latency strategies of alphaherpesviruses: herpes simplex virus and varicella-zoster virus latency in neurons. In: Minarovits J, Gonczol E, Valyi-Nagy T, editors. Latency strategies of herpesviruses. Springer; New York: 2006. In press. [Google Scholar]
- Varghese S, Rabkin SD. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002;9:967–978. doi: 10.1038/sj.cgt.7700537. [DOI] [PubMed] [Google Scholar]
- Ward PL, Avitabile E, Campadelli-Fiume G, Roizman B. Conservation of the architecture of the Golgi apparatus related to a differential organization of microtubules in polykaryocytes induced by syn-mutants of herpes virus type 1. Virology. 1998;241:189–199. doi: 10.1006/viro.1997.8972. [DOI] [PubMed] [Google Scholar]
- Weintraub H, Goudine M. Chromosomal subunits in active genes have an altered conformation. Science. 1976;193:848–856. doi: 10.1126/science.948749. [DOI] [PubMed] [Google Scholar]
- Winkler M, Dawson GJ, Elizan TS, Berl S. Distribution of actin and myosin in a rat neuronal cell line infected with herpes simplex virus. Arch Virol. 1982;72:95–103. doi: 10.1007/BF01314454. [DOI] [PubMed] [Google Scholar]
- Woll E, Bedikian A, Legha SS. Uveal melanoma: natural history and treatment options for metastatic disease. Melanoma Res. 1999;9:575–581. [PubMed] [Google Scholar]