

Abstract
• Background and Aims Brassicaceae, with nearly 340 genera and more than 3350 species, anchors the low range of angiosperm genome sizes. The relatively narrow range of DNA content (0·16 pg < 1C < 1·95 pg) was maintained in spite of extensive chromosomal change. The aim of this study was to erect a cytological and molecular phylogenetic framework for a selected subset of the Brassicacae, and use this as a template to examine genome size evolution in Brassicaceae.
• Methods DNA contents were determined by flow cytometry and chromosomes were counted for 34 species of the family Brassicaceae and for ten Arabidopsis thaliana ecotypes. The amplified and sequenced ITS region for 23 taxa (plus six other taxa with known ITS sequences) were aligned and used to infer evolutionary relationship by parsimony analysis.
• Key Results DNA content in the species studied ranged over 8-fold (1C = 0·16–1·31 pg), and 4·4-fold (1C = 0·16–0·71 pg) excluding allotetraploid Brassica species. The 1C DNA contents of ten Arabidopsis thaliana ecotypes showed little variation, ranging from 0·16 pg to 0·17 pg.
• Conclusions The tree roots at an ancestral genome size of approximately 1x = 0·2 pg. Arabidopsis thaliana (1C = 0·16 pg; ∼157 Mbp) has the smallest genome size in Brassicaceae studied here and apparently represents an evolutionary decrease in genome size. Two other branches that represent probable evolutionary decreases in genome size terminate in Lepidium virginicum and Brassica rapa. Branches in the phylogenetic tree that represent probable evolutionary increases in genome size terminate in Arabidopsis halleri, A. lyrata, Arabis hirsuta, Capsella rubella, Caulanthus heterophyllus, Crucihimalaya, Lepidium sativum, Sisymbrium and Thlaspi arvense. Branches within one clade containing Brassica were identified that represent two ancient ploidy events (2x to 4x and 4x to 6x) that were predicted from published comparative mapping studies.
Keywords: Arabidopsis, Brassicaceae, ITS phylogeny, DNA content, genome size, chromosome number
INTRODUCTION
Genome size varies over 500-fold among angiosperms. The diploid species with the largest reported 1C DNA content is Fritillaria davisii, n = 12 (90 pg DNA or ∼88000 Mbp; Bennett and Smith, 1976). One of the smallest 1C DNA contents is in Arabidopsis thaliana, n = 5 (0·16 pg or ∼157 Mbp DNA; Bennett et al., 2003). The biological and evolutionary significance of the massive variation in DNA content is an unsolved puzzle of evolutionary biology. Research on the evolution of DNA content has been conducted in numerous laboratories over the last 40 years, but the lack of a clear phylogenetic framework limited the ability to resolve direction of evolutionary changes in DNA content. However, the recent use of DNA sequence data for assembling evolutionary trees has allowed variation in DNA content to be analyzed in a phylogenetic context (Bennetzen and Kellogg, 1997; Leitch, et al., 1998, 2005; Wendel et al., 2002; Soltis et al., 2003; Price et al., 2005).
Here we report a study of genome size in Brassicaceae. This family is large, consisting of about 340 genera and more than 3350 species (Al-Shehbaz, 1984). Brassicaceae is of particular interest from a genome evolution perspective because it has nuclear DNA contents that anchor the low range of angiosperm values, and has a relatively narrow range of DNA content compared to families such as Poaceae (1C DNA content = 0·3 pg to 26·0 pg), Fabaceae (1C DNA content = 0·4 pg to 27·4 pg) and Liliaceae (1C DNA content = 12·4 pg to 127·4 pg; Plant DNA C-values Database http://www.rbgkew.org.uk/cval/homepage.html). No DNA contents above 1C = 1·95 pg have been reported for Brassicaceae (Plant DNA C-values Database). Furthermore, numerous molecular comparisons among species of Brassicaceae have been conducted, particularly among crop species in the genus Brassica (see Hall, et al., 2002). In spite of the relatively conservative range of DNA content, extensive structural genome evolution has occurred involving chromosome duplication, rearrangements and fusions (Lagercrantz, 1998; Lan et al., 2000).
The relatively small genomes also make Brassicaceae excellent choices for genome evolution studies. The Arabidopsis Genome Initiative (2000) produced over 73 % of the nuclear genomic sequence of A. thaliana (see Bennett et al., 2003); and unprecedented genomic studies, which are now possible within a dicot species, are being extended to wild relatives of Arabidopsis through comparative studies (Acarkan et al., 2000; Rossberg et al., 2001) in which genome size evolution will be of additional interest.
MATERIALS AND METHODS
Plant material
The Brassicaceae species used and their sources are listed in Tables 1 and 2. Plants were grown in the University of Chicago greenhouse or in an environmental chamber at Texas A&M University on a light/dark regime of 14 h (22 °C)/10 h (17 °C).
Table 1.
Genomic characteristics of species of the Brassicaceae
| Species |
Source |
Stock number |
Chromosome number (2n) |
1C Nuclear DNA Content (pg ± s.e) |
Duncan Multiple Range Grouping7 |
Genome size (1x) (Mbp) |
|---|---|---|---|---|---|---|
| Arabidopsis arenosa (L.) Lawalrée | C. Pikaard | 32 (4x) | 0·417 ± 0·001 | K | 203 | |
| Arabidopsis halleri3 (L.) O'Kane & Shehbaz ssp. gemmifera (Matsum.) O'Kane & Al-Shehbaz | SASSC1 | J020 | 164 | 0·261 ± 0·004 | Q | 255 |
| Arabidopsis lyrata (L.) O'Kane & Al-Shehbaz | S. O'Kane | 324 (4x) | 0·468 ± 0·003 | J | 230 | |
| Arabidopsis suecica3 (Fries) Norrlin | SASSC | JS6 | 26 (4x) | 0·356 ± 0·001 | M | 174 |
| Arabidopsis thaliana (L.) Heynh. ecotype Columbia | NASSC2 | 104 | 0·160 ± 0·001 | T | 157 | |
| Arabis hirsuta (L.) Scop. | SASSC | J023 | 326 (4x) | 0·686 ± 0·005 | E | 335 |
| Brassica carinata L. | T. Osborn | 34 (4x) | 1·308 ± 0·012 | A | 642 | |
| Brassica juncea (L.) Czern. | T. Osborn | 36 (4x) | 1·092 ± 0·001 | C | 534 | |
| Brassica napus L. | T. Osborn | 38 (4x) | 1·154 ± 0·006 | B | 566 | |
| Brassica nigra (L.) Koch | T. Osborn | 16 | 0·647 ± 0·009 | F | 632 | |
| Brassica oleracea L. | T. Osborn | 18 | 0·710 ± 0·002 | D | 696 | |
| Brassica rapa L. | T. Osborn | 20 | 0·539 ± 0·018 | I | 529 | |
| Capsella bursa-pastoris3 (L.) Medik | SASSC | J022 | 324 (4x) | 0·414 ± 0·002 | K | 203 |
| Capsella rubella3 Reuter | Italy | 164,6 | 0·257 ± 0·005 | Q | 250 | |
| Cardamine amara | L. Luczaj | 166 | 0·225 ± 0·001 | R S | 221 | |
| Cardamine hirsuta3 L. | SASSC | J027 | 164 | 0·229 ± 0·001 | R S | 225 |
| Cardamine impatiens3 L. | SASSC | J010 | 164 | 0·212 ± 0·009 | S | 206 |
| Caulanthus amplexicaulis var. barbarae | A. Pepper | 288 | 0·377 ± 0·001 | L | 372 | |
| Caulanthus heterophyllus (Nutt.) Payson var. heterophyllus | A. Pepper | 288 | 0·701 ± 0·008 | D E | 686 | |
| Caulanthus heterophyllus (Nutt.) Payson var. pseudosimulans R. Buck | A. Pepper | 288 | 0·687 ± 0·003 | E | 671 | |
| Cleome hassleriana Chodat | B&T Seeds | 204 | 0·307 ± 0·003 | P | 299 | |
| Crucihimalaya himalaica (Edgeworth) Al-Shehbaz, O'Kane & R. A. Price | SASSC | J018 | 164 | 0·323 ± 0·004 | N O P | 319 |
| Crucihimalaya wallichii3 (J.D. Hooker & Thompson) Al-Shehbaz, O'Kane & R. A. Price | SASSC | J012 | 164 | 0·330 ± 0·011 | N O | 323 |
| Draba nemorosa3 L. | SASSC | J021 | 164 | 0·242 ± 0·002 | Q R | 235 |
| Guillenia lasiophyllum | A. Pepper | 288 | 0·384 ± 0·001 | L | 377 | |
| Lepidium sativum3 L. | B&T Seeds | 2309 | 244 (3x) | 0·582 ± 0·001 | G | 380 |
| Lepidium virginicum3 L. | SASSC | J26 | 324 (4x) | 0·333 ± 0·001 | N | 164 |
| Olimarabidopsis cabulica3 (J.D. Hooker and Thompson) Al-Shehbaz, O'Kane & R. A. Price | SASSC | JS4 | 485 (4x?) | 0·420 ± 0·004 | K | 206 |
| Olimarabidopsis pumila3 (Stephan) Al-Shehbaz, O'Kane & R. A. Price | SASSC | JS3 | 304 (4x?) | 0·416 ± 0·005 | K | 203 |
| Raphanus sativus L. | T. Osborn | 186 | 0·583 ± 0·006 | G | 573 | |
| Sinapis alba3 L. | B&T Seeds | 51709 | 246 | 0·566 ± 0·006 | G H | 553 |
| Sisymbrium irio3 L. | SASSC | J04 | 284 (4x) | 0·533 ± 0·002 | I | 262 |
| Sisymbrium orientale3 L. | SASSC | J19 | 144 | 0·312 ± 0·003 | O P | 304 |
| Thlaspi arvense3 L. | Chicago | 144 | 0·548 ± 0·005 | H I | 539 |
Arabidopsis Seed Stock Center, Sendai, Japan.
Arabidopsis Seed Stock Centre, Nottingham, United Kingdom.
Identification confirmed by Dr. Ihsan Al-Shehbaz at the Missouri Botanical Gardens.
Chromosome numbers determined in this study.
Alpha = 0·05.
Table 2.
Genome size of ten diverse ecotypes of Arabidopsis thaliana
| Ecotype |
Source/stock number* |
Habitat |
Origin |
Elevation (m) |
Latitude |
1C DNA content (pg) |
±s.e. |
Duncan Grouping† |
|---|---|---|---|---|---|---|---|---|
| En-0 | ABRC/CS1136 | Sandy loam | Enkheim/Frankfurt, Germany | 120 | N50 | 0·172 | 0·003 | A |
| Cvi-0 | MartinKoornneef | Rocky wall with moss | Cape Verde Islands | 1200 | N16 | 0·168 | 0·001 | A |
| Berkeley-0 | Angus Murphy | Berkeley, CA, USA | 50 | N38 | 0·159 | 0·001 | B | |
| Buckhorn-0 | Angus Murphy | Non-serpentine | Trinity Mountains, CA, USA | N30 | 0·162 | 0·002 | B | |
| Yo-0 | ABRC/CS1622 | Sandy valley floor | Yosemite National Park, CA, USA | 1400 | N37 | 0·160 | 0·001 | B |
| Hod-O | ABRC/CS6178 | Hodja-Obi-Garm, Tadjikistan | 1800 | ∼N38 | 0·161 | 0·001 | B | |
| La-er | MartinKoornneef | Landsberg, Germany | 300 | N48 | 0·159 | 0·001 | B | |
| Landsberg | Jeff Chen | 0·162 | 0·001 | B | ||||
| Santa Clara-0 | Angus Murphy | Serpentine outcrop | Santa Clara, CA, USA | 100 | N37 | 0·159 | 0·001 | B |
| Tsu-1 | ABRC/CS1640 | Tsu, Japan | 0 to 100 | N34 | 0·158 | 0·001 | B |
ABRC = Arabidopsis Biological Research Center, Ohio State University, Columbus, Ohio, USA.
Alpha = 0·05.
Chromosome counts
Chromosome counts were made by the protocol of Jewell and Islam-Faridi (1994) with slight modification as described by Price et al. (2005).
Determination of DNA content
At least three plants for each species were analyzed to obtain the mean DNA content. Newly expanded leaves of the target species and of a standard species were co-chopped with a sharp razor blade and prepared using the procedures and flow cytometer as described by Price et al. (2005). Arabidopsis thaliana ecotype Columbia (1C DNA content = 0·16 pg; Bennett et al., 2003) and/or Sorghum bicolor ‘TX623’ (1C DNA content = 0·835 pg; Price et al., 2005) were used as internal calibration standards.
DNA sequence analysis
DNA was extracted from leaves of single individuals, either by the method described in Pepper and Norwood (2001) or the CTAB procedure (Doyle and Doyle, 1987). DNA of ribosomal internal transcribed spacer (ITS) fragments was amplified using primers ITS4 and ITS5 (White et al., 1990). Polymerase chain reaction (PCR) was performed using established conditions (Konieczny and Ausubel, 1993) in 50 mL reactions containing 10–20 ng of genomic DNA. The resulting PCR products were purified using the QIAquick PCR purification columns (Qiagen, Valencia, California, USA), then 20–30 ng of purified double-stranded PCR product was directly used as template for 35 cycles of sequencing using BigDye Version 3·0 terminator chemistry (Applied Biosystems, Foster City, California, USA). The primers used for direct sequencing were ITS4, ITS5, ITS11 (5′-ATCTCGGCTCTCGCATCGATG-3′) and ITS12 (5′-CAAAGACTCGATGGTTCACG-3′). Cycle-sequencing products were purified by Bio-Gel P-30 size exclusion chromatography (Bio-Rad, Richmond, California, USA) and analyzed using an ABI3100 capillary sequencer (Applied Biosystems). Double-stranded DNA sequence contigs from each taxon were assembled and edited using Sequencher 3·0 (Gene Codes, Ann Arbor, Michigan, USA). Finished sequences from the various taxa were aligned using Clustal W (Thompson et al., 1994). Alignments across insertion/deletion differences (indels) were then verified manually.
Phylogenetic inference
Phylogeny reconstructions were performed using PAUP* 4·0·1b3a (Swofford, 1999). For parsimony analysis, indels of one nucleotide or longer were defined as a new (5th) character state. Ambiguous nucleotides (e.g. divergent paralogs) were defined as polymorphisms. Branch and bound searches using unweighted parsimony and accelerated transformation (ACCTRAN) of character state optimization were performed to identify the most-parsimonious trees. Branches of zero length were collapsed. For neighbour-joining analysis (Saitou and Nei, 1987), Kimura two-parameter distances (Kimura, 1980) and a minimum evolution objective function using the branch-and-bound method were employed, and ambiguous and ‘missing’ data (e.g. indels and divergent paralogs) were ignored. Cleome hassleriana (Brassicaceae) was used as an outgroup. Relative support for various clades was determined by bootstrap analysis (Felsenstein, 1985) employing 1000 replicates using a ‘branch and bound’ search strategy.
RESULTS
DNA content
DNA content and chromosome numbers of the 32 species of Brassicaceae are listed in Table 1. DNA content among the species of this study ranged over 8-fold from 1C = 0·16 pg (A. thaliana) to 1·31 pg (B. carinata). Excluding the allotetraploid Brassica species, 1C DNA content ranges 4·4-fold from 0·16 pg in A. thaliana to 0·71 pg in B. oleracea.
Arabidopsis suecica (2n = 4x = 26) is a tetraploid with genomes contributed from A. arenosa (2n = 16) and A. thaliana (2n = 10) (O'Kane, et al., 1996). The A. arenosa used in the current study is a tetraploid with 2n = 4x = 32 chromosomes and, therefore, the diploid 1C DNA content extrapolates to 0·21 pg. The 1C DNA content determined for A. suecica (0·355 pg) is proportionately less (96 %) than the sum of 0·21 pg and the 1C value of 0·16 for A. thaliana.
Three species, B. napus, B. juncea and B. carinata, are allotetraploids derived from B. rapa × B. oleracea, B. rapa × B. nigra and B. nigra × B. oleracea, respectively (U, 1935). The DNA content of these three amphidiploids is proportionately less (92 %, 92 % and 97 %, respectively) than the sum of the DNA content of their putative parental species.
The genome sizes of ten diverse ecotypes of A. thaliana are presented in Table 2. The 1C DNA content of all these biotypes is 0·16 pg, except for the ecotypes En-0 and Cvi-0, which were 0·17 pg. Although these differences are statistically significant, they are none-the-less small, and based upon three replicates may not be real.
Phylogenetic reconstruction based on ribosomal DNA ITS sequences
ITS sequences for 22 taxa of Brassicaceae were determined (GenBank accession numbers AY662277–AY662298). Additional ITS sequences of Caulanthus heterophyllus var. heterophyllus, C. heterophyllus var. pseudosimulans, C. amplexicaulis var. amplexicaulis and Guillenia lasiophylla were determined previously (Pepper and Norwood, 2001), and ITS sequences of A. thaliana and L. rapa were obtained from Yang et al. (1999).
Analysis using ClustalW yielded an alignment of ±599 nucleotides containing ITS1, the 5·8S ribosomal RNA gene, and ITS2. Nucleotide ambiguities consisted of a G/A dimorphism in ITS1 of C. himalaica, a G/A dimorphism in ITS2 of Draba nemorosa, and a C/T dimorphism in the 5·8S ribosomal RNA gene and a G/A dimorphism in ITS2 of C. wallichii. No other ambiguous nucleotides were observed.
A 20-nucleotide region (located in ITS1), in which alignments across several indel polymorphisms could not be confidently resolved, was excluded from further analyses. The modified alignment had 579 characters, of which 211 were parsimony-informative. Parsimony analysis yielded a single most-parsimonious tree of 910 steps. The most-parsimonious tree (Fig. 1) had a consistency index of 0·614 (0·543 excluding uninformative characters), retention index of 0·673, and homoplasy index of 0·393 (0·455 excluding uninformative characters). Neighbour-joining yielded a tree that differed from the parsimony tree only in the placement of Guillenia lasiophylla (this taxon was grouped with Caulanthus heterophyllus in the parsimony tree, while it was a sister group to the genus Caulanthus in the parsimony tree), and in the placement of a clade containing Thlaspi and Lepidium (sister to the clade containing Brassica and Cardamine in neighbour-joining analysis, but sister to the clade containing Arabidopsis and Capsella in the parsimony tree). Neither pattern was strongly supported by bootstrap analysis.
Fig. 1.
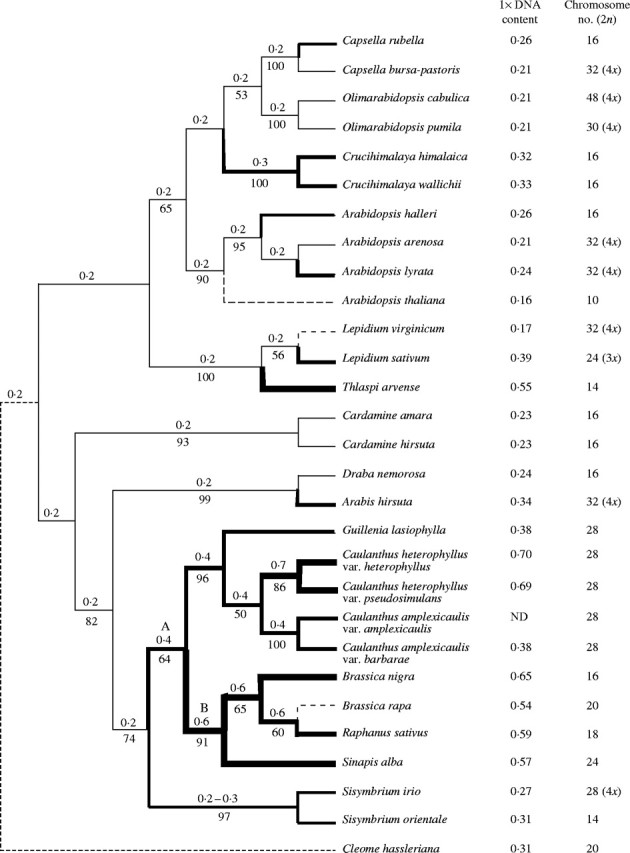
Phylogenetic analysis of the evolution of genome size in representative members of Brassicaceae based on a single most-parsimonious ITS tree. Bootstrap support (%) for various nodes (1000 replicates) is indicated beneath the corresponding node. Nodes without an indicated bootstrap support had bootstrap values of less than 50 %. Genome size (1× DNA content) expressed in pg, and chromosome numbers for each taxon are indicated. Numbers above each node indicate a hypothetical ancestral genome size (pg). Thickness of the lines shown in the tree is used to illustrate hypothetical changes in genome size in the Brassicaceae family. A dashed line is used to indicate a decrease in genome size. Cleome hassleriana (Capperaceae) merely provides an outgroup for the phylogenetic analysis, and was not used to estimate the ancestral genome size of Brassicaceae. A and B indicate nodes discussed in the text.
Where there were overlapping taxa, the topological arrangements reported here were largely in agreement with those previously obtained through analysis of nucleotide sequences from the ITS (Koch et al., 1999; Yang et al., 1999; Pepper and Norwood, 2001; Hong et al., 2003), coding and non-coding plastid DNA (Koch et al., 2001; Pepper and Norwood, 2001; Yang et al., 2002), chalcone synthase and alcohol dehydrogenase (Koch et al., 2000; Koch et al., 2001), and the S-locus related gene SLR1 (Inaba and Nishio, 2002). Specifically, there were notable differences between our topological arrangements and that of Koch et al. (2001) in the placement of Arabis hirsuta, Cardamine amara and Thlaspi arvense. These differences most likely arise from overlapping but non-congruent sets of taxa sampled and different genes used (ITS vs. CHS and matK). For example, Koch et al. (2001) place T. arvense in a sister group to Sisymbrium and Sinapus, when using the combined CHS and matK dataset. These differences in taxonomic arrangement would not alter our interpretation of patterns of genome change in the taxa examined. Although not strongly supported throughout, our parsimony tree (which closely matched the neighbour-joining tree) was used as a framework for a preliminary phylogenetic analysis of genome size in Brassicaceae (Fig. 1).
The genome size and 2n chromosome number are listed to the right of each species in the phylogeny (Fig. 1). The 1C value for known tetraploids, or species that are very likely tetraploids, was halved to give the 1x genome size. At each dichotomy, the presumed ancestral genome size was indicated.
Because the species sampled were a very small subset of the total Brassicaceae, and because the mode of genome size evolution is not known, a very conservative approach was used to calculate ancestral genome size. Assuming the genome size changes rarely and change is associated with speciation events, the assumed ancestral genome is the 1C value (rounded to one decimal place) common to members at each branch of the clade.
DISCUSSION
Bennetzen and Kellogg (1997) proposed that plants may have a ‘one-way ticket to genomic obesity’ as a consequence of retroelement accumulation and polyploidy. Although this may be a general trend among the angiosperms as a whole (Leitch et al., 1998; Soltis et al., 2003), apparent decreases in genome size accompanying angiosperm phylogeny are also apparent (Soltis et al., 2003). Evolutionary decreases in genome size have been detected in the evolution of cotton relatives (Wendel et al., 2002) and in the evolution of species of the genus Sorghum (Price et al., 2005). Although the evolution of DNA content in Brassicaceae is generally from low to high, the data support the concept of a dynamic nature of genome size evolution involving both increases and decreases.
The generally conservative nature of the evolution of genome size in Brassicaceae allows for the detection of changes in genome size, including polyploidization events, in a phylogenetic context. The ITS sequence phylogeny for Brassicaceae presents a tree (Fig. 1) that can be rooted with a genome size of about 0·2 pg (∼200 Mbp). This is a tentative value. The number of species is small relative to the entire family, and the single outgroup has 1C = 0·3 pg. Further, the absence of a suitable model of the mechanism involved in genome change means no best method exists to estimate the base value. And yet, the value 0·2 pg, while tentative, is common to most branches in the phylogeny (Fig. 1) and, as such, serves as a good starting point to study genome size evolution in Brassicacae. Assuming that the root 1C genome size is 0·2 pg, then the radiation of Brassicaceae has involved both increases and decreases in genome size. Since the family is rooted by small hypothetical genomes, we would expect that the general evolutionary trend should be to larger genomes. This was in fact observed; genome size decreased in one diploid species, A. thaliana, while genome size increased in eight other diploid species including A. halleri, A. lyrata, C. rubella, C. himalaica, L. sativum, S. irio, S. orientale and T. arvense. Two additional increases and one decrease that occurred concurrent with doubling and tripling of DNA content in diploidized species in the clade that includes Brassica and Raphanus are addressed below.
Studies of comparative linkage maps (Lagercrantz and Lydiate, 1996; Lagercrantz, 1998; Lan et al., 2000) indicate extensive duplication and triplication in the genomes of the ‘diploid’ Brassica species relative to A. thaliana, thus suggesting that modern Brassica species are descendants of a hexaploid ancestor. Evolution of extant diploid Brassica species involved extensive chromosomal rearrangement, including chromosome fusion events resulting in a reduction of chromosome number (Lagercrantz, 1998). In the phylogeny presented, branches where the putative polyploidization events occurred can be identified (Fig. 1, nodes A and B). Node A represents a hypothetical 2-fold increase (0·2 to 0·4 pg) in DNA content that would be expected in proceeding from the diploid to the tetraploid condition. Node B represents an increase from 0·4 to 0·6 pg, which would be expected going from tetraploid to hexaploid. When these putative polyploid events are assumed to be true, and the genome size of the putative polyploids is reduced to 1x to permit direct comparisons to the other 1x values in Fig. 1, the extent of genome size evolution in Brassicaceae is further apparent.
Arabidopsis thaliana has the smallest genome detected in this study and may represent the smallest genome in Brassicaceae. Cardamine amara has a reported genome size of 1C = 0·06 pg (Angiosperm DNA C-values Database) and until recently had been considered the smallest genome in Brassicaceae. However, we report a genome size of 1C = 0·23 pg (Table 1). Similar 1C values for C. amara was determined by L. Hanson and M. Bennett (see Bennett and Leitch, 2005) and J. Greilhuber (pers. com.).
The Arabidopsis thaliana genome of 1C = 0·16 pg (∼157 Mbp) apparently represents an evolutionary decrease in genome size. Other branches representing probable evolutionary decreases in genome size terminate in L. virginicum and B. rapa. On the other hand, Capsella rubella, Crucihimalaya, A. halleri, A. lyrata, L. sativum, Thlaspi arvense, Arabis hirsuta, Caulanthus heterophyllus and Sisymbrium represent branches in the phylogenetic tree leading to increases in genome size.
Further decreases in DNA content have apparently occurred in the extant allopolyploids, B. napus, B. juncea and B. carinata formed from hybridization between B. rapa × B. oleracea, B. rapa × B. nigra, and B. nigra × B. oleracea. The DNA content of B. napus, B. juncea and B. carinata, respectively, is proportionally less (0·92, 0·92 and ·97 pg, respectively) than the sum of the DNA content of their putative parental species. Similar reductions were seen for the tetraploid A. suecica, which has a genome size ∼4 % less than the sum of its diploid ancestors. Narayan (1998) also observed that the genomes of these present-day allopolyploid Brassica species average >6 % less than the expected value. Genomic instability in artificially synthesized allopolyploids from crosses of B. rapa × B. nigra and of B. rapa × B. oleracea was detected as unexpected changes in restriction fragment profiles (Song et al., 1995). Song et al. (1995) interpreted these observations as demonstrating rapid genomic changes following polyploid formation in Brassicaceae. If hybrid instability also influences DNA content, then the DNA content of polyploids is not necessarily expected to be strictly additive with respect to their progenitors.
Among angiosperms in general, DNA content variation greater than 2- to 3-fold is common among congeneric species. In well-studied genera such as Microseris (Price and Bachmann, 1975), Lathyrus, Nicotiana, Clarkia and Allium (Narayan, 1998) the DNA content variation is not continuous, but rather falls into discontinuous groupings. Although DNA content may vary greatly among congeneric species (Price, 1976) it is typically very constant within species, seldom exceeding the approximate 2–5 % resolution of microspectrophotometric and flow cytometric methods commonly used for determining nuclear DNA amount (see Bennett et al., 2000). Relative constancy of C-value was observed among the ten geographically diverse ecotypes of A. thaliana included in the current study (Table 2). Only two of the biotypes were significantly different from the others and these differences were small.
Several observations suggest that genome size evolution is more than a slow equilibrium process resulting from accumulation of small insertions and deletions as hypothesized by Petrov (2002): (1) differences of 0·05 pg and 0·04 pg, respectively, were observed between the 1x genomes of sister species within the genera Capsella and Sisymbrium; while there are no differences between taxa separated for much greater periods of time such as Capsella bursa-pastoris, Olimarabidopsis cabulica and O. pumila. (2) DNA content variation among congeneric species is often discontinuous (Narayan, 1998), and (3) DNA content is normally controlled within narrow limits within species (Bennett et al., 2000). Bennett et al. (2000) proposed that a ‘counting’ mechanism must exist that regulates genome size within tightly defined or preselected limits. It is likely that such canalization of genome size results from some ubiquitous property of DNA replication and/or repair that only occasionally allows jumps to higher or lower DNA amounts. Their primary role does not have to be regulation of genome size, as long as the consequences of intrinsic checkpoints generally ensure constancy of DNA amount within a species.
Acknowledgments
This research was supported in part by the Texas Agricultural Experiment Station and a Plant Genome Training Grant from the TAMU Life Science Task Force. Research work in the Z. J. Chen laboratory was supported in part by a grant 0077774 from the National Science Foundation Plant Genome Research Program and a grant GM67015 from the National Institutes of Health. The opinions expressed are those of the authors and do not reflect the official policy of the National Institutes of Health, National Science Foundation or the US government. We thank Dr. Ihsan Al-Shehbaz for species identification, and Dr. T. C. Osborn and C. S. Pikaard for providing seeds of Brassica species and A. arenosa.
LITERATURE CITED
- Acarkan A, Rossberg M, Koch M, Schmidt R. 2000. Comparative genome analysis reveals conservation of genome organization for A. thaliana and Capsella rubella Plant Journal 23: 55–62. [DOI] [PubMed] [Google Scholar]
- Al-Shehbaz IA. 1984. The tribes of cruciferae (Brassicaceae) in the southeastern United States. Journal of the Arnold Arboretum 65: 343–373. [Google Scholar]
- Arabidopsis Genome Initiative. 2000. Analysis of the genome of the flowering plant Arabidopsis thaliana Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Bennett MD, Johnston JS, Hodnett GL, Price HJ. 2000.Allium cepa L. cultivars from four continents compared by flow cytometry show nuclear DNA constancy. Annals of Botany 85: 351–357. [Google Scholar]
- Bennett MD, Leitch IJ. 2005. Nuclear DNA amounts in angiosperms— progress, problems and prospects. Annals of Botany 95: 45–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MD, Leitch IJ, Price HJ, Johnston JS. 2003. Comparisons with Caenorhabditis (∼100 Mb) and Drosophila (∼175Mb) using flow cytometry show genome size in arabidopsis to be ∼157 Mb and thus ∼25 % larger than the Arabidopsis Genome Initiative of ∼125 Mb. Annals of Botany 91: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MD, Smith JB, 1976. Nuclear DNA amounts in angiosperms. Philosophical Transactions of the Royal Society of London Series B, Biological Sciences 274: 227–274. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Kellogg EA. 1997. Do plants have a one-way ticket to genomic obesity? Plant Cell 9: 1901–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–11. [Google Scholar]
- Felsenstein J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39: 783–791. [DOI] [PubMed] [Google Scholar]
- Hall AE, Fiebig A, Preuss D. 2002. Beyond the Arabidopsis genome: Opportunities for comparative genomics. Plant Physiology 129: 1439–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong RL, Hamaguchi L, Busch MA, Weigel D. 2003. Regulatory elements of the floral homeotic gene AGAMOUS identified by phylogenetic footprinting and shadowing. Plant Cell 15: 1296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba R, Nishio T. 2002. Phylogenetic analysis of Brassicaceae based on the nucleotide sequences of the S-locus related gene, SLR1 Theoretical and Applied Genetics 105: 1159–1165. [DOI] [PubMed] [Google Scholar]
- Jewell DC, Islam-Faridi MN. 1994. Details of a technique for somatic chromosome preparation and C-banding of maize. pp 484–493 in The Maize Handbook, M. Freeling and V. Walbot (eds.), NY: Springer-Verlag. [Google Scholar]
- Kimura M. 1980. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution 16: 111–120. [DOI] [PubMed] [Google Scholar]
- Koch MA, Bishop J, Mitchell-Olds T. 1999. Molecular systematics and evolution of Arabidpsis and Arabis Plant Biology 1: 529–537. [Google Scholar]
- Koch MA, Haubold B, Mitchell-Olds T. 2000. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Molecular Biology and Evolution 17: 1483–1498. [DOI] [PubMed] [Google Scholar]
- Koch MA, Haubold B, Mitchell-Olds T. 2001. Molecular systematics of the Brassicaceae: evidence from coding plastidic matK and nuclear Chs sequences. American Journal of Botany 88: 534–544. [PubMed] [Google Scholar]
- Konieczny A, Ausubel FM. 1993. A procedure for mapping Arabidopsis mutations using co-dominant ecotype specific PCR-markers. Plant Journal 4: 403–410. [DOI] [PubMed] [Google Scholar]
- Lagercrantz U. 1998. Comparative mapping between Arabidopsis thaliana and Brassica nigra indicates that Brassica genomes have evolved through extensive genome replication accompanied by chromosomal fusions and frequent rearrangements. Genetics 150: 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagercrantz U, Lydiate D. 1996. Comparative genome mapping in Brassica Genetics 144: 1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T-H, DelMonte TA, Reischmann K, Hyman J, Kowalski SP, McFerson J, Kresovich S, Paterson AH. 2000. An EST-enriched comparative map of Brassica oleraceae and Arabidopsis thaliana Genome Research 10: 776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch IJ, Chase MW, Bennett MD. 1998. Phylogenetic analysis of DNA C-values provides evidence for a small ancestral genome size in flowering plants. Annals of Botany 82 (Suppl. A): 85–94. [Google Scholar]
- Leitch IJ, Soltis DE, Soltis PS, Bennett MD. 2005. Evolution of DNA amounts across land plants (Embryophyta). Annals of Botany 95: 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Kane SL, Schaal BA, Al-Shehbaz IA. 1996. The origins of Arabidopsis suecica (Brassicaceae) as indicated by nuclear rDNA sequences. Systematic Botany 21: 559–566. [Google Scholar]
- Narayan RKJ. 1998. The role of genomic constraints upon evolutionary changes in genome size and chromosomal organization. Annals of Botany 82 (Supplement A): 57–66. [Google Scholar]
- Pepper AE, Norwood LE. 2001. Evolution of C. amplexicaulus var. barbarae (Brassicaceae), a rare serpentine endemic plant: a molecular phylogenetic perspective. American Journal of Botany 88: 1479–1489. [PubMed] [Google Scholar]
- Petrov DA. 2002. Mutational equilibrium model of genome size evolution. Theoretical Population Biology 61: 533–546. [DOI] [PubMed] [Google Scholar]
- Price HJ. 1976. Evolution of DNA content in higher plants. Botanical Reviews 42: 27–52. [Google Scholar]
- Price HJ, Bachmann K. 1975. DNA content and evolution in the Microseridinae. American Journal of Botany 62: 262–267. [Google Scholar]
- Price HJ, Dillon SL, Hodnett G, Rooney W, Ross L, Johnston JS. 2005. Genome Evolution in the genus Sorghum (Poaceae). Annals of Botany 95: 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins RC. 1983.The Cruciferae of continental North America: systematics of the mustard family from the arctic to Panama. Palo Alto: Stanford University Press. [Google Scholar]
- Rossberg M, Theres K, Acarkan A, Herrero R, Schmitt T, Schumacher K, Schmitz G, Schmidt R. 2001. Comparative sequence analysis reveals extensive microcolinearity in the lateral suppressor regions of the tomato, Arabidopsis and Capsella genomes. Plant Cell 13: 979–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M, Nei N. 1987. The neighbor joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4: 406–425. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Soltis PS, Bennett MD, Leitch IJ. 2003. Evolution of genome size in the angiosperms. American Journal of Botany 90: 1596–1603. [DOI] [PubMed] [Google Scholar]
- Song K, Lu P, Tang K, Osborn TC. 1995. Rapid genome change in synthetic polyploids of Brassica and its implications for polyploid evolution. Proceedings of the National Academy of Sciences USA 92: 7719–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. 1999.PAUP*: phylogenetic analysis using parsimony (*and other methods), version 4. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Research 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UN. 1935. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Japanese Journal of Botany 7: 389–452. [Google Scholar]
- Wendel JF, Cronn RC, Johnston JS, Price HJ. 2002. Feast and famine in plant genomes. Genetica 115: 37–47. [DOI] [PubMed] [Google Scholar]
- White TJ, Bruns T, Lee S, and Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In Innis M, Gelfand JJ, Sninsky JJ, and White TJ [eds.], PCR protocols, 315–322. New York: Academic Press. [Google Scholar]
- Yang Y-W, Lai K-N, Tai P-Y, Ma D-P, Li W-H. 1999. Molecular phylogenetic studies of Brassica, Rorippa, Arabidopsis, and allied genera based on the internal transcribed spacer region of 18S-25S rDNA. Molecular Phylogenetics and Evolution 13: 455–462. [DOI] [PubMed] [Google Scholar]
- Yang Y-W, Tai P-Y, Lai K-N, Chen Y, Li W-H. 2002. A study of the phylogeny of Brassica rapa, B. nigra, Raphanus sativus and their related genera using noncoding regions of chloroplast DNA. Molecular Phylogenetics and Evolution 23: 268–275. [DOI] [PubMed] [Google Scholar]