

Abstract
In addition to productive lytic infections, herpesviruses such as human cytomegalovirus (HCMV) establish a reservoir of latently infected cells that permit lifelong colonization of the host. When latency is established, the viral immediate-early (IE) genes that initiate the lytic replication cycle are not expressed. HCMV IE gene expression at the start of a lytic infection is facilitated by the viral pp71 protein, which is delivered to cells by infectious viral particles. pp71 neutralizes the Daxx-mediated cellular intrinsic immune defense that silences IE gene expression by generating a repressive chromatin structure on the viral major IE promoter (MIEP). In naturally latently infected cells and in cells latently infected in vitro, the MIEP also adopts a similar silenced chromatin structure. Here we analyze the role of Daxx in quiescent HCMV infections in vitro that mimic some, but not all, of the characteristics of natural latency. We show that in these “latent-like” infections, the Daxx-mediated defense that represses viral gene expression is not disabled because pp71 and Daxx localize to different cellular compartments. We demonstrate that Daxx is required to establish quiescent HCMV infections in vitro because in cells that would normally foster the establishment of these latent-like infections, the loss of Daxx causes the lytic replication cycle to be initiated. Importantly, the lytic cycle is inefficiently completed, which results in an abortive infection. Our work demonstrates that, in certain cell types, HCMV must silence its own gene expression to establish quiescence and prevent abortive infection and that the virus usurps a Daxx-mediated cellular intrinsic immune defense mechanism to do so. This identifies Daxx as one of the likely multiple viral and cellular determinants in the pathway of HCMV quiescence in vitro, and perhaps in natural latent infections as well.
The intrinsic, innate, and adaptive arms of our immune system protect us from the myriad of pathogens to which we are continuously exposed. Successful viruses can neutralize or evade immune surveillance to promote efficient viral replication and ultimately spread to a new host. Here we demonstrate that a prominent virus benefits not by inactivating a cellular intrinsic immune defense that silences its gene expression but by using it to establish the defining aspect of its replicative cycle, i.e., latency.
Human cytomegalovirus (HCMV) is a betaherpesvirus that infects the majority of the population, causes severe disease in immunocompromised patients, is the leading viral cause of birth defects, and likely plays a role in age-related immunosenescence, certain cardiovascular diseases, and some cancers (12, 64, 70, 77, 78). A cellular component of PML nuclear bodies, the Daxx protein, confers intrinsic immunity against HCMV (8, 72, 79, 91). Through the action of a histone deacetylase (HDAC), Daxx silences the expression of the viral immediate-early (IE) genes that are required to initiate the lytic replication program by inducing a repressive chromatin structure at the viral major IE promoter (MIEP). At the start of a lytic infection, the viral pp71 protein, which is packaged into the tegument layer of infectious virions, is delivered to cells, migrates to the nucleus, and degrades Daxx, thus preventing Daxx from silencing viral gene expression (79). pp71 also degrades the tumor suppressors of the retinoblastoma family, Rb, p107, and p130, to stimulate cell cycle progression (34-36), although any role this activity of pp71 may play during HCMV replication and pathogenesis has yet to be established. Degradation of the Rb proteins (36) and Daxx (31) by pp71 is through an uncommon, proteasome-dependent, ubiquitin-independent pathway.
Many viral genes are expressed during lytic viral replication, resulting in progeny virion production that allows the infection to spread within a host and to new hosts. During latency, viral genomes are maintained for extended periods with little or no gene expression, thus preventing detection by the immune system. Latency allows the infection to persist for the life of the host (reviewed in references 5 and 81), and the inability to clear an infected individual of the reservoir of latently infected cells is the primary reason that cures for viruses that establish latency, such as the herpesviruses, are not available. It is thought that in naturally infected people, latent HCMV genomes reside in a small population of incompletely differentiated cells of the myeloid lineage, such as CD34+ mononuclear hematopoietic progenitor cells (26, 39, 41, 58, 82, 86, 87). Induced terminal differentiation of naturally infected cells, as well as some in vitro latently infected cells results in reactivation, lytic replication, and virus production (73, 74, 82, 83, 87).
For the alpha- and betaherpesviruses, latency is established when the expression of lytic-phase genes from the infecting viral genome is prevented. During HCMV infection, the first lytic genes expressed are the IE genes, and their expression is controlled by the MIEP. At the start of lytic infection, Daxx silences IE gene expression (79) and induces a repressed chromatin structure at the MIEP (91) prior to being degraded by pp71, which then allows IE gene expression. This repressed chromatin structure at the MIEP is characterized by the association of heterochromatin protein 1 (HP-1) and the lack of acetylated histones and in these respects is identical to that found at the MIEP in latently infected CD34+ mononuclear hematopoietic progenitor cells. CD34+ cells show this repressed chromatin structure whether they are naturally infected (74) or cultured ex vivo and infected in vitro (73). Other in vitro models for HCMV latency, NT2 and THP-1 cells, do not faithfully mimic all of the aspects of natural latency but do assemble a repressed chromatin structure at the MIEP that is indistinguishable (by all of the criteria analyzed) from that found in CD34+ cells and in fibroblasts at the very start of a lytic infection (66, 91). These observations seem to indicate that a similar mechanism may be used to generate a repressive chromatin architecture at the MIEP at the start of lytic replication and during both natural latency in vivo and in model quiescent or “latent-like” infections in vitro.
The only clue to how the repressive chromatin structure around the MIEP is formed during the establishment of latency is that HDAC activity is required (55, 57, 66). Because the Daxx-mediated intrinsic immune defense silences IE gene expression through the action of an HDAC at the start of a lytic infection unless it is neutralized by pp71 (79, 91), we asked if Daxx also controls HCMV latency. Here we show that in two commonly used in vitro models of latent-like HCMV infections, the Daxx-mediated defense that silences IE gene expression is not inactivated upon HCMV infection because tegument-delivered pp71 fails to reach the nuclei of these quiescently infected cells. When the level of Daxx in these cells is reduced either by preexpression of nuclearly localized pp71 or by RNA interference, IE genes are expressed upon HCMV infection, indicating that a latent-like state is not established and that lytic replication is initiated. However, we found that the lytic replication cycle is not completed and infectious progeny virions are rarely produced from these cells. This work describes one of the likely multiple steps used by HCMV to establish latency, i.e., allowing a Daxx-mediated cellular intrinsic immune defense to silence viral IE gene expression. Our data, along with other recent findings (38, 93), argue that IE gene expression must be silenced in undifferentiated cells where latency is to be established because failure to do so results in an abortive viral infection that would prevent lifelong colonization of the host.
(Part of this work was presented in 2006 at the 25th American Society for Virology Meeting and the 31st International Herpesvirus Workshop through an American Society for Virology travel award and a Vilas Travel Fellowship awarded to R.T.S.)
MATERIALS AND METHODS
Cells and viruses.
Cells were grown in a 5% CO2 atmosphere at 37°C. Human foreskin fibroblast (HF), N-Tera2 (NT2), HEK 293, and U-2 OS cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen), and THP-1 cells were cultured in RPMI 1640 medium (Invitrogen). The medium was supplemented with 10% (vol/vol) fetal bovine serum (Gemini), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.292 mg/ml glutamine (Gibco). NT2 cells were differentiated by growth in 10 μM all-trans-retinoic acid (Sigma) for 5 days and counted and reseeded prior to infection. THP-1 cells were differentiated with 100 ng/ml tetradecanoyl phorbol acetate (Sigma) for 24 h before infection. The wild-type (AD169) and pp71-null mutant (ADsubUL82, passed once through noncomplementing HFs to remove tegument-incorporated pp71) (7) viruses have already been described. For UV inactivation, virus stocks were placed on ice in a UV Stratalinker 2400 (Stratagene) and exposed to the 254-nm light source at 0.12 J/cm2 for 3 min. For infection, the medium was removed from the cells, which were then incubated with virus at 37°C for 60 min. Medium was then added back to the original volume.
A recombinant adenovirus (rAD) expression system that expresses transgenes from the EF1α promoter was constructed. The EF1α promoter (with a mutated BglII site), was cloned as a HindIII fragment from the SIN-EF1α MCS2 plasmid (a generous gift from C. Alexander) into the pShuttle vector lacking any promoter or poly(A) signal (a generous gift from B. Vogelstein). The poly(A) sequence was cloned from the pShuttle CMV vector (a generous gift from B. Vogelstein) into the pShuttle vector containing the EF1α promoter. The desired transgene of interest was cloned into the pShuttle EF1α construct as a BamHI fragment into a BglII site. Transductions with the EF1α- and pADIC-based rADs have been previously described (34). Transductions were done at the indicated particle/cell ratios (63). Different particle/cell ratios were used to achieve approximately equal transgene expression levels.
Preexpression of pp71 and HCMV infection.
THP-1 cells were transduced with rADs lacking a transgene or expressing pp71 or Did2-3 mutant pp71 (29) driven from the EF1α or CMV promoter for ∼18 h as previously described (34). Subsequently, THP-1 cells were gently pelleted and infected with HCMV at a multiplicity of infection (MOI) of 3 for 24 h.
Inhibitors and antibodies.
Lactacystin (20 μM), E64 (50 μM) (Calbiochem), or trichostatin A (100 ng/ml) (Upstate Biotechnology) dissolved in dimethyl sulfoxide and valproic acid (VPA) (3 mM) dissolved in water were added at the time of infection with HCMV. The antibodies to the following were from commercial sources: Daxx (D7810; Sigma), tubulin (DM 1A; Sigma), Oct 3/4 (sc-5279; Santa Cruz), UL44 (1202), and pp65 (1025; Rumbaugh-Goodwin Institute). Antibodies against pp71 (2H10-9 and IE-233), IE1 (1B12), and pp28 (CMV 157) have been previously described (34, 68, 95). For Western blot analysis, secondary horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit antibodies were from Chemicon. Secondary antibodies for indirect immunofluorescence conjugated with Alexa Fluor 488 (A-11029) or Alexa Fluor 546 (A-11030) were from Molecular Probes.
Western blot assays and coimmunoprecipitation.
Equal amounts of protein from cell lysates prepared in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors were analyzed by Western blotting as previously described (34). During coimmunoprecipitation (co-IP) assays, equal amounts of protein from cell lysates harvested in RIPA buffer were precleared with goat serum and heat-inactivated, protein A-positive Staphylococcus aureus (Amersham) for 30 min each. Lysates were cleared by centrifugation, and after incubation with the proper primary antibody, immune complexes were captured on protein A-agarose beads (Calbiochem) and washed six times with RIPA buffer. Bound proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analyzed by Western blotting.
Indirect immunofluorescence.
HFs, NT2 cells, T2RA cells, and THP-1-derived macrophage cells were grown on glass coverslips and infected as described above. Coverslips were harvested and washed twice with phosphate-buffered saline (PBS; Gibco) prior to fixation as described below. THP-1 cells (which grow in suspension) were infected, collected by low-speed centrifugation, and resuspended in cold PBS, and approximately 2.5 × 105 cells were allowed to attach to water-washed coverslips (12 mm in diameter) for 60 min at room temperature. The coverslips were then fixed with 4% paraformaldehyde in PBS and then washed three times with PBST (PBS plus 0.1% Triton X-100 and 0.05% Tween 20) before blocking with PBST plus 5% goat serum and 0.5% bovine serum albumin for 25 min. Antibody incubations were for 1 h at room temperature in PBST plus 5% goat serum and 0.5% bovine serum albumin, followed by three washes with PBST for 5 min each. Coverslips were then washed twice with distilled water, incubated with Hoechst 33342 for 10 min, washed twice more with distilled water, and finally mounted with Fluoromount-G (Southern Biotech). Images were taken with a Zeiss microscope and camera (Axiovert 200 M).
Small interfering RNA (siRNA) transfection.
THP-1 monocytes were transfected by Amaxa transfection technology. THP-1 monocytes (1 × 106) were transfected with siRNA (3 μg) by using cell line Nucleofector kit V (VCA-1003; Amaxa Biosystems) and following the manufacturer's protocol. Synthetic, annealed 21-base oligonucleotides (siRNA) were purchased from Dharmacon. Three target sequences directed at the Daxx transcript were siDaxx-674 sense (5′-AGGCACGGTTGAAGCGTAA-3′), siDaxx-1498 sense (5′-CTACAGATCTCCAATGAAA-3′), and hDx2, in addition to an siRNA directed toward the Skp-1 sequence; both hDx2 and the Skp-1 sequence were previously described (79). Transfected cells were cultured for 48 h and then infected with HCMV at an MOI of 3 for an additional 24 h before harvesting for Western blotting.
Short hairpin RNAs (shRNAs) and retroviruses.
Sequences for shRNAs to Daxx (hDx2) and Skp-1 (79) were synthesized according to the manufacturer's specification and cloned into the pSuper.retro.puro retrovirus backbone (OligoEngine) by following the manufacturer's protocol. A retrovirus that expresses GFP was a generous gift from Bill Sugden. The retrovirus plasmids (20 μg) were transfected with calcium phosphate into 293 cells grown on 10-cm dishes along with 5 μg pMD-G (a generous gift from Y. Kawaoka), which encodes vesicular stomatitis virus glycoprotein G, and 15 μg pMD.gagpol (69), which encodes the murine leukemia virus Gag and Gag-Pol proteins. The medium was changed 16 h after transfection, and supernatants containing virus were collected at 48 and 72 h posttransfection. Supernatants were filtered through a 0.45-μm-pore-size filter. NT2 and THP-1 cells were transduced with retroviral supernatants four to six times, and cell populations were selected with 1 μg/ml puromycin (Sigma). HFs were transduced twice with the GFP-expressing retrovirus.
Real-time quantitative PCR (Q-PCR).
DNA was isolated from 1 × 106 HCMV-infected THP-1 monocytes and macrophages with the DNeasy tissue kit (69504; QIAGEN) according to the manufacturer's instructions. Nuclei were isolated as described previously (45). Briefly, infected cells were washed once with PBS, pelleted, and resuspended in RSB (10 mM NaCl, 3 mM MgCl2, 10 mM Tris-HCl [pH 7.4]) and a 20× detergent solution (10% sodium deoxycholate and 20% Tween 40 in RSB) to a 1× final concentration. Cells were vortexed gently and incubated on ice for 5 min, and the nuclei were pelleted by centrifugation at 1,000 × g for 3 min. The nuclei were washed once with RSB alone and pelleted again at 1,000 × g for 3 min. DNA was harvested from the nuclei and from total cell pellets with the DNeasy tissue kit.
Real-time Q-PCR was used to determine the efficiency of viral genome delivery to the infected nuclei of THP-1 monocytes or macrophages. Viral genomes were quantified with a primer pair (pp549s and pp812as) and a probe to UL83 as previously described (18). The number of viral genomes was normalized to cellular copies of beta-actin with a previously described primer-probe set (27). Unknown sample values were determined on the basis of a standard curve of known copy numbers of UL83 (pSG5-pp65) and beta-actin (pAB1-bactinPCRscript, a generous gift from T. Compton). PCRs contained 5 μl of 200 μl extracted DNA, 50 nM primers, 50 nM probe, 12.5 μl Taq-Man Universal PCR Master Mix (4304437; Applied Biosystems), and nuclease-free water (9937; Ambion) to 25 μl. Real-time PCR was run on an ABI 7900HT, and data were analyzed by the SDS 2.2.1 program.
RESULTS
Tegument-delivered pp71 is unable to bind and degrade Daxx in cells where HCMV latency is modeled in vitro, but newly synthesized pp71 binds and degrades Daxx in these cells.
Because the cell type that represents the true in vivo latent reservoir of HCMV is not known and because of the inability to efficiently prevent the differentiation of ex vivo-cultured cells, we and others use two surrogate in vitro model cell types to study HCMV latency. NT2 embryonal carcinoma cells and THP-1 monocytes are used to study silencing of the MIEP during the establishment of HCMV latency because they display the same differentiation-dependent IE gene expression observed in cells isolated from healthy donors and maintained for short periods ex vivo (6, 16, 20, 32, 48, 51, 55, 56, 66, 67, 80, 88). These cells fail to express IE genes upon infection with HCMV; however, they become fully permissive for both IE gene expression and productive replication if they are differentiated prior to infection. Furthermore, the repressed chromatin structure at the MIEP in in vitro-infected NT2, THP-1, and CD34+ progenitor cells is identical to that observed during natural latent infections (66, 73, 74, 81). Because HCMV infection of NT2 and THP-1 cells mimics many, but not all, of the characteristics of natural latency, we describe these infections as quiescent, or latent-like, infections.
We discovered that pp71 relieves repression of the MIEP at the start of lytic infections by inducing the degradation of Daxx (79). Therefore, we asked if Daxx degradation correlated with IE gene expression in undifferentiated NT2 and THP-1 cells, as well as their differentiated counterparts. We found that during infection of both NT2 and THP-1 cells, Daxx is not degraded and confirmed (55, 66) that IE genes are not expressed unless HDAC activity is inhibited (Fig. 1A and B). However, when these cells were differentiated before HCMV infection (NT2 cells with retinoic acid into T2RA cells or THP-1 monocytes with the phorbol ester tetradecanoyl phorbol acetate into macrophages), we observed Daxx degradation and confirmed (20, 88) IE gene expression (Fig. 1C and D). Control experiments established that the drop in the level of Daxx after HCMV infection of T2RA cells was due to pp71-mediated proteasomal degradation, as it is in lytically infected cells (79). We found that in T2RA cells, Daxx was degraded after infection with UV-inactivated virus that cannot express any viral genes (Fig. 1E), Daxx was stabilized in HCMV-infected cells by the proteasome inhibitor lactacystin but not the cysteine protease inhibitor E64 (Fig. 1F), and that Daxx was not degraded after infection with a pp71-null virus (Fig. 1G).
FIG. 1.

Daxx degradation correlates with IE gene expression. NT2 cells (A) and THP-1 cells (B) were mock infected (M) or infected with HCMV (V) in the absence or presence of trichostatin A (T) at an MOI of 3. Lysates were harvested at the indicated times (hours postinfection [hpi]) and analyzed by Western blotting. Tubulin (Tub) was used as an internal loading control. (C) NT2 and T2RA cells were mock infected (M) or infected with HCMV (V) at an MOI of 3 or 1, respectively. Lysates were harvested at 12 hpi and analyzed by Western blotting. (D) THP-1 monocytes (mono) or THP-1-derived macrophages (macroφ) were mock infected (M) or infected with HCMV (V) at an MOI of 3. Lysates were harvested at 12 hpi and analyzed by Western blotting. (E) T2RA cells were mock infected (M) or infected with HCMV (V) or UV-inactivated HCMV (UV) at an MOI of 3. Lysates were harvested at 12 hpi and analyzed by Western blotting. UV inactivation was confirmed by lack of IE1 gene expression (data not shown). (F) T2RA cells were mock infected (M) or infected with HCMV (V) in the presence of lactacystin (L) or E64 at an MOI of 3. Lysates were harvested at 12 hpi and analyzed by Western blotting. (G) T2RA cells were mock infected (M) or infected with HCMV (V) or the HCMV mutant lacking pp71 (Δ71) at an MOI of either 1 or 0.1, respectively. Lysates were harvested at 12 hpi and analyzed by Western blotting. The tegument protein pp65 served as a marker for viral entry.
Surprisingly, we found that while tegument-delivered pp71 fails to degrade Daxx in HCMV-infected NT2 and THP-1 cells (Fig. 1A to D), pp71 produced after transduction with an rAD was perfectly capable of degrading Daxx in these cells (Fig. 2A and B), as well as in their differentiated counterparts (Fig. 2C and D). Daxx degradation was not a result of rAD transduction because a pp71 mutant (termed D2) unable to bind Daxx (29) and expressed in these cells after rAD transduction was unable to degrade Daxx (Fig. 2A to D). Wild-type pp71, but not the Daxx-binding mutant, bound to Daxx when expressed from an rAD in both NT2 and T2RA cells (Fig. 3A). In contrast, HCMV tegument-delivered wild-type pp71 bound to Daxx in T2RA cells but not in undifferentiated NT2 cells (Fig. 3B). Although only assayed once, tegument-delivered pp71 likewise only bound Daxx in THP-1-derived macrophages and not in THP-1 monocytes (Fig. 3C). Thus, HCMV infection fails to induce Daxx degradation in both NT2 and THP-1 cells because tegument-delivered pp71 fails to bind to Daxx, even though ectopically expressed, newly synthesized pp71 is able to bind and degrade Daxx in these cells.
FIG. 2.

Newly expressed pp71 induces Daxx degradation in undifferentiated and differentiated cells. (A) NT2 cells were mock transduced (lane M) or transduced with an rAD that expresses wild-type pp71 (lane 71) or a mutant pp71 protein (lane D2) which fails to bind Daxx at 2,000 or 4,000 particles per cell (ppc), respectively. Lysates were collected at 24 h posttransduction (hpt) and analyzed by Western blotting. (B) THP-1 monocytes (mono) were mock transduced (lane M) or transduced with the rADs that express pp71 (lane 71) or the pp71 Daxx-binding mutant (lane D2) at 30,000 ppc. Lysates were harvested at 24 hpt and analyzed by Western blotting. (C) Differentiated T2RA cells were mock transduced (lane M) or transduced with an rAD that expresses pp71 (lane 71) or the pp71 Daxx-binding mutant (lane D2) at 300 or 6,000 ppc, respectively. Lysates were harvested at 18 hpt and analyzed by Western blotting. (D) THP-1-derived macrophages (macroφ) were mock transduced (lane M) or transduced with an rAD expressing pp71 (lane 71) or the pp71 Daxx-binding mutant (lane D2) at 5,000 or 30,000 ppc, respectively. Lysates were harvested at 24 hpt and analyzed by Western blotting. Tub, tubulin.
FIG. 3.

Tegument-delivered pp71 fails to bind Daxx in undifferentiated NT2 and THP-1 cells. (A) NT2 cells were mock transduced (lane M) or transduced with an rAD expressing either pp71 (lane 71) or the pp71 Daxx-binding mutant (lane D2) at 2,000 or 6,000 particles per cell (ppc), respectively, for 24 h, while differentiated T2RA cells were transduced with pp71 (lane 71) or the pp71 Daxx-binding mutant (lane D2) at 500 or 1,000 ppc, respectively, for 12 h. Lysates were harvested and used in a co-IP/Western blot assay. Lysates were immunoprecipitated with an antibody to Daxx. (B) NT2 or differentiated T2RA cells were mock infected (lane M) or infected with HCMV (lane V) at an MOI of 3 or 1, respectively. Lysates were harvested at 6 hpi and used in a co-IP/Western blot assay. Lysates were immunoprecipitated with an antibody to Daxx. (C) THP-1 monocytes (mono) or THP-1-derived macrophages (macroφ) were mock infected (lane M) or infected with HCMV (lane V) at an MOI of 3 for 12 h. Lysates were harvested and used in a co-IP/Western blot assay. Lysates were immunoprecipitated with an antibody to Daxx. Result from one experiment is shown. Tub, tubulin.
Viral genomes efficiently enter nuclei, but tegument-delivered pp71 remains in the cytoplasm when in vitro latent-like HCMV infections are established.
To determine why HCMV tegument-delivered pp71 fails to bind and degrade Daxx in undifferentiated NT2 and THP-1 cells, we asked whether these proteins localize to the same subcellular compartment. When pp71 is expressed from an rAD in fibroblasts (data not shown), NT2 cells, T2RA cells, THP-1 monocytes, or THP-1-derived macrophages, it induces the degradation of Daxx and thus, not surprisingly, accumulates in the nucleus, where Daxx is localized (Fig. 4A). Unlike lytically infected fibroblasts, where tegument-delivered pp71 migrates to the nucleus, we found that tegument-delivered pp71 remains in the cytoplasm of quiescently infected NT2 and THP-1 cells as punctate speckles (Fig. 4B). When GFP-expressing fibroblasts are cocultured with NT2 cells, the difference between the localization of tegument-delivered pp71 in lytically versus quiescently infected cells is readily apparent (Fig. 4C). In fibroblasts, most of the pp71 enters the nucleus, while in NT2 cells, it remains in the cytoplasm. Interestingly, some cytoplasmic punctate pp71 was observed in fibroblast cells. It is possible that these cytoplasmic speckles represent an intermediate in the ill-defined process of viral tegument disassembly and that this process occurs efficiently in lytically infected cells but is interrupted in cells where a latent-like infection is established. In agreement with the Daxx degradation and IE gene expression observed in the differentiated forms of these cells (Fig. 1C and D), we found that, upon infection, HCMV tegument-delivered pp71 enters the nucleus in differentiated T2RA cells and THP-1-derived macrophages (Fig. 4D). The ability of tegument-delivered pp71 to localize to the nuclei of differentiated T2RA cells is not dependent on viral gene expression because UV-inactivated HCMV successfully delivers pp71 to the nucleus (data not shown), where pp71 induces the degradation of Daxx (Fig. 1E).
FIG. 4.

Recombinant adenovirus-expressed pp71, but not HCMV tegument-delivered pp71, localizes to the nuclei of both undifferentiated and differentiated NT2 and THP-1 cells. (A) NT2, differentiated T2RA cells, THP-1 monocytes (mono), and THP-1-derived macrophages (macroφ) were transduced with the rAD expressing pp71 at 2,000, 1,000, 10,000, or 5,000 particles per cell, respectively. pp71 subcellular localization was visualized by indirect immunofluorescence as described in Materials and Methods. Nuclei were counterstained with Hoechst. pp71 is green, and cellular DNA is blue. (B) HFs, NT2 cells, and THP-1 monocytes (mono) were infected with HCMV at an MOI of 3 for 6 h. Cells on coverslips were stained with the appropriate antibody. Nuclei were counterstained with Hoechst. pp71 is red in HFs and NT2 cells and green in THP-1 monocytes. In all of the panels, cellular DNA is blue. (C) HFs transduced with a retrovirus that expresses GFP were cocultured with NT2 cells grown on coverslips and then infected with HCMV at an MOI of 1. Coverslips were harvested at 10 hpi and assayed by indirect immunofluorescence. GFP is green, pp71 is red, and cellular DNA is blue. (D) Differentiated T2RA cells and THP-1-derived macrophages (macroφ) grown on coverslips were infected with HCMV at an MOI of 1 or 3, respectively. Coverslips were harvested at 8 hpi and assayed by indirect immunofluorescence. pp71 is green, and cellular DNA is blue.
While HCMV tegument-delivered pp71 remains in the cytoplasm of quiescently infected NT2 and THP-1 cells, the viral genomes presumably enter the nucleus because we (Fig. 1A and B) and others (55, 57, 66) have shown that viral IE genes are expressed in the presence of HDAC inhibitors. Thus, there appear to be differences in the entry and/or uncoating pathways in quiescently and lytically infected cells. To compare the overall efficiency of genome delivery to the nucleus in undifferentiated THP-1 monocytes and differentiated THP-1-derived macrophages, we used real-time Q-PCR to determine the percentage of viral genomes that traffic to the nucleus upon infection of these cell populations. THP-1 monocytes and macrophages were infected with HCMV, and cellular and viral DNAs were isolated from either total cell extract or isolated nuclei. To demonstrate the efficiency of our cytoplasmic and nuclear fractionation, total cell lysates were compared to the cytoplasmic and nuclear fractions by Western blotting. As expected, the cellular lamin A/C proteins were localized to the nuclear fraction while tubulin was found in the cytoplasmic fraction (Fig. 5A). To quantitate the number of viral genomes which localize to the nucleus, primers to the UL83 gene were used to determine viral genome copies and normalized to the number of cellular copies of the gene for beta-actin. In undifferentiated THP-1 cells, 69% of the viral genomes were detected in the nucleus, while in THP-1-derived macrophages, 83% localized to the nucleus (Fig. 5B). Furthermore, we infected THP-1 monocytes and THP-1-derived macrophages alone or in the presence of another HDAC inhibitor which activates HCMV gene expression, VPA (46, 59, 60), and determined the percentage of IE1-positive nuclei. As expected, there were essentially no IE1-positive cells in undifferentiated monocytes. However, in the presence of VPA or after infection of differentiated macrophages in the absence or presence of VPA, roughly 30% of the cells were positive for IE1 expression (Fig. 5C). This second conformation further demonstrates that viral genomes are being delivered to the nuclei of THP-1 cells, irrespective of the ability of pp71 to transit to the nucleus. These experiments indicate that although there is a qualitative difference in the entry pathway between quiescently and lytically infected cells (as shown by the different localizations of tegument-delivered pp71), there does not appear to be a significant quantitative difference in the ability of viral genomes to localize to the nucleus.
FIG. 5.

Viral genomes localize to the nucleus as efficiently in undifferentiated THP-1 cells as in their derivatives. (A) THP-1 monocytes (mono) and macrophages (macroφ) were infected with HCMV at an MOI of 3 for 12 h. Total cell extracts (T) or cytoplasmic (C) and nuclear (N) fractions were analyzed by Western blotting. Lamin (Lam) A/C represents nuclear proteins, while tubulin (Tub) represents cytoplasmic proteins. (B) DNA was harvested from total cell extracts or from isolated nuclei of HCMV-infected THP-1 monocytes (mono) or macrophages (macroφ) and analyzed by real-time Q-PCR. The number of viral genomes detected in the nuclear fraction was divided by the number of viral genomes detected in the total cell extracts. The data represent the average from three independent experiments. (C) THP-1 monocytes (mono) or macrophages (macroφ) were infected with HCMV at an MOI of 3 for 24 h in the absence (V) or presence of VPA. At least 300 nuclei per sample from randomly selected fields were counted, and the data represent an average of the percentage of IE1-positive cells from three independent experiments.
Daxx silences viral gene expression when latent-like HCMV infections are established in vitro.
If the inability of HCMV infection to induce Daxx degradation and initiate IE gene expression in undifferentiated cells stems from the inability of tegument-delivered pp71 to enter the nucleus, then preexpression of nuclearly localized pp71 prior to HCMV infection should activate IE gene expression in these cells. Because pp71 expressed after transduction with rADs localizes to the nucleus, binds Daxx, and induces its degradation (Fig. 2, 3, and 4), we transduced THP-1 cells with an rAD that expresses pp71 from the EF1α promoter and then infected them with HCMV. We found that prior expression of pp71 after transduction induced the degradation of Daxx and subsequent infection with HCMV resulted in the expression of IE1 (Fig. 6). Transduction with an rAD expressing Did2-3 mutant pp71 from the CMV promoter, or ones lacking a transgene (driven by either the EF1α or the CMV promoter), failed to induce the degradation of Daxx and failed to rescue IE1 gene expression (Fig. 6). Thus, nuclear localization of pp71 prior to HCMV infection allows the expression of viral IE genes. Note that we used different promoters to achieve approximately equal levels expression of the two proteins (Fig. 6). When wild-type pp71 and Did2-3 mutant pp71 were both expressed from the MIEP, the wild-type protein accumulated to a much higher level (data not shown), probably because it degrades Daxx and stimulates its own production, whereas the Did2-3 mutant protein is unable to degrade Daxx and thus cannot induce its own expression. Interestingly, high-level overexpression of wild-type pp71 (from the CMV promoter) was, for unknown reasons, not compatible with IE gene expression upon HCMV infection (data not shown).
FIG. 6.

Preexpression of nuclear pp71 rescues IE1 gene expression. THP-1 monocytes were mock transduced (lane M) or transduced with rAD-EF1α pp71 (lane 71), rAD-EF1α without a transgene (lane E), rAD-CMV pp71 Did2-3 (lane D2), or rAD-CMV without a transgene (lane C) at 2,000 particles per cell for 18 h. Subsequently, all of the cells were infected with HCMV at an MOI of 3 for 24 h. Lysates were harvested and analyzed by Western blotting. Tub, tubulin.
All of these experiments establish a correlation between Daxx degradation and lytic gene expression and between Daxx stability and the gene silencing that occurs when a latent-like infection is established. To determine specifically if Daxx silences IE gene expression in undifferentiated cells, we used RNA silencing to knock down the level of either Daxx or Skp-1 in THP-1 cells prior to infection with HCMV and then assayed for IE gene expression by Western blotting. Skp-1 is a component of cellular ubiquitin ligase complexes (14) that is not required for pp71-mediated Daxx degradation (79) and served as a negative control. We found that three independent siRNA molecules directed to the Daxx transcript were able to enhance IE gene expression in HCMV-infected THP-1 cells compared to the siRNA against Skp-1 (Fig. 7A). This indicates that the Daxx protein represses viral IE gene expression in HCMV-infected undifferentiated THP-1 cells.
FIG. 7.

IE1 is expressed after HCMV infection of undifferentiated cells with reduced Daxx levels, but the infection is abortive. (A) THP-1 monocytes were transfected with siRNA directed at Daxx (674, 1498, or hDx2) or Skp-1 (Skp) for 48 h and then infected with HCMV at an MOI of 3. Lysates were harvested at 24 hpi and analyzed by Western blotting. (B) NT2 or differentiated T2RA cells expressing either shRNA to Daxx or Skp-1 were infected with HCMV at an MOI of 0.5. Lysates were harvested at 12 hpi and analyzed by Western blotting. Oct-4 was used as a marker for the differentiation status of the NT2 cells. (C) THP-1 monocytes expressing either shRNA to Daxx or Skp-1 were mock infected (M) or infected with HCMV (V) at an MOI of 3. Lysates were harvested at 12 hpi and analyzed by Western blotting. (D) HFs, NT2 cells expressing shRNA to Daxx, differentiated T2RA cells expressing shRNA to Daxx, and U-2 OS cells were infected with HCMV at an MOI of 3. Lysates were harvested on the indicated days postinfection (dpi) and analyzed by Western blotting. UL44 served as a marker for early gene expression, and pp28 served as a marker for late gene expression. (E) Supernatants collected at 5 dpi from panel D were used in a plaque assay to determine viral titers. Data collected from two independent experiments are shown with standard deviations. (F) U-2 OS cells were plated on coverslips and infected with HCMV at an MOI of 3. Coverslips were harvested at 8 hpi, and pp71 subcellular localization was visualized by indirect immunofluorescence staining. Nuclei were counterstained with Hoechst. Tub, tubulin.
We also generated NT2 and THP-1 cell populations in which the levels of Daxx or Skp-1 were knocked down by transducing them with retroviruses that make shRNAs directed to either the Daxx (with the hDx2 sequence) or the Skp-1 transcript (79). Confirming and extending the results of the transient assays described above, we found that in the absence of Daxx, IE genes are expressed in NT2 and THP-1 cells upon HCMV infection, indicating that Daxx silences viral IE gene expression in both of these undifferentiated cell types (Fig. 7B and C). Control experiments showed that the retroviral transduction and selection did not induce cellular differentiation. The shDaxx-NT2 cells still express Oct-4 (Fig. 7B), a stem cell marker that is lost upon differentiation into T2RA cells (15), and the shDaxx-THP-1 cells are nonadherent, unlike THP-1-derived macrophages that attach to culture plates (data not shown). Furthermore, we have observed IE gene expression in shDaxx-NT2 cells, where tegument-delivered pp71 localized only to the cytoplasm (data not shown), another independent confirmation of their undifferentiated state.
Thus, in cells where IE genes are normally silenced and in vitro latent-like infections are established, IE genes are expressed and lytic replication is initiated in the absence of Daxx, indicating that Daxx is required for HCMV to establish quiescent infections in vitro and thus perhaps natural latent infections as well. We next asked if the lytic replication initiated in these shDaxx cells was productive. In addition to the expression of the IE1 gene, infection of shDaxx-NT2 cells also resulted in the expression of the early UL44 gene (Fig. 7D). However, the late pp28 gene was not expressed to detectable levels (Fig. 7D) and release of progeny virions was a rare event (Fig. 7E). Differentiation of these cells into shDaxx-T2RA cells prior to infection allows the productive lytic replication cycle to be completed (Fig. 7D and E), indicating that we have not selected for a nonpermissive cell line.
While the lytic replication cycle is initiated in undifferentiated cells in the absence of Daxx, it is not completed and resembles abortive infections described recently in infected NT2 cells treated with forskolin (38), THP-1 cells that ectopically express HCMV IE genes (93), and osteosarcoma cells (49), such as U-2 OS cells. Interestingly, tegument-delivered pp71 enters the nuclei of U-2 OS cells infected with HCMV (Fig. 7F), strengthening the correlations between the nuclear entry of tegument-delivered pp71 and the initiation of lytic replication and between the establishment of quiescent or latent-like infections and the nuclear exclusion of tegument-delivered pp71.
Our data demonstrate that Daxx represses viral gene expression in HCMV-infected undifferentiated cells because pp71 is sequestered in the cytoplasm and thus is prevented from binding to and degrading Daxx. However, differentiation prior to HCMV infection permits nuclear localization of pp71 and subsequent degradation of Daxx, allowing viral gene expression (Fig. 8A). We have shown that Daxx degradation is required for IE gene expression at the start of a low-multiplicity lytic infection (79), and our data now support a model in which Daxx also plays a role in repressing viral gene expression in two in vitro models used to study latent-like HCMV infections. Furthermore, our data show not only that Daxx silences viral gene expression in undifferentiated cells when latent-like infections are established in vitro but also that Daxx degradation must be prevented when the virus infects an undifferentiated cell during the establishment of a latent-like infection. In the absence of Daxx, viral IE genes are not silenced, leading to the initiation of a lytic replication cycle which is not efficiently completed in these cells, resulting in an abortive replication cycle that would terminate the infection (Fig. 8B).
FIG. 8.
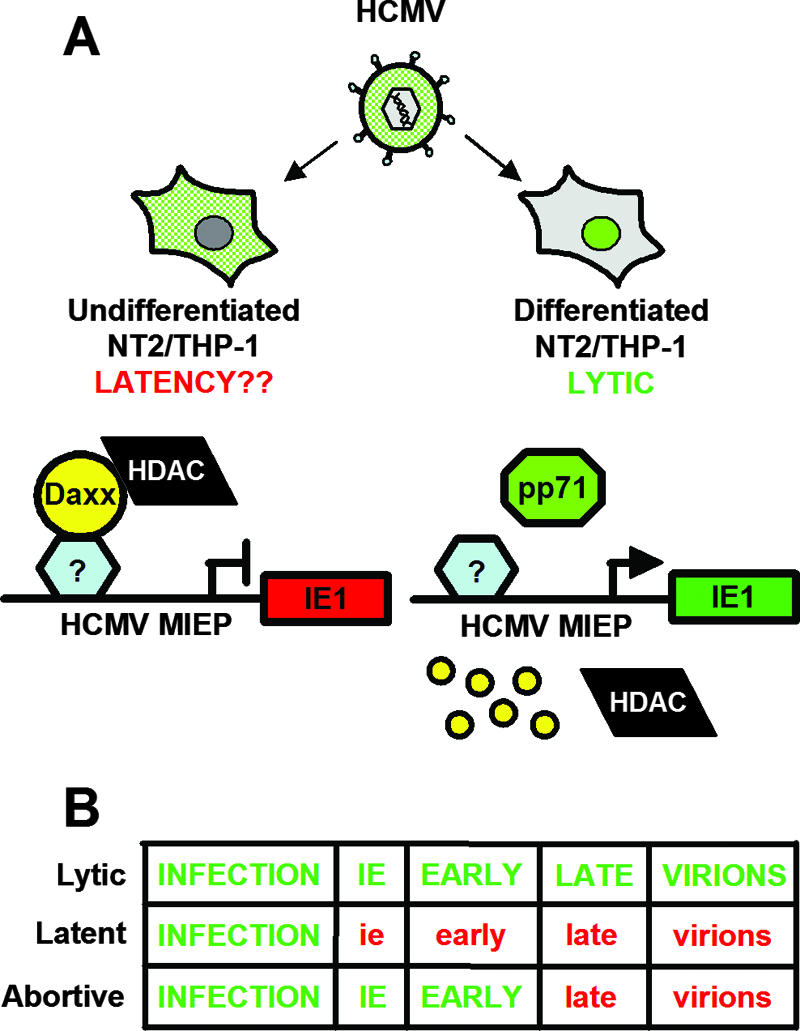
Model of differentiation-dependent regulation of pp71 localization, Daxx degradation, and IE gene expression. (A) In undifferentiated NT2/THP-1 monocytes, tegument-delivered pp71 (green) is sequestered in the cytoplasm upon HCMV infection, which allows Daxx to repress IE gene expression through the action of an HDAC. This may mimic what happens when HCMV establishes latency in vivo. However, prior differentiation allows pp71 to localize to the nucleus, induce Daxx degradation, activate viral IE gene expression, and initiate a lytic infection. The small yellow circles represent Daxx degradation. (B) Schematic of the regulation of viral gene expression during lytic, latent, and abortive infections (e.g., when Daxx is absent from cells that would normally establish a latent infection). Green capital letters indicate completion of the particular stage, while red lowercase text indicates failure to complete the phase.
DISCUSSION
Modeling of HCMV latency in vitro.
On the basis of the paradigms of the gammaherpesviruses Epstein-Barr virus (40) and Kaposi's sarcoma-associated herpesvirus (13), efforts to describe HCMV latency have focused on identifying viral transcripts either in naturally infected cells harboring latent HCMV genomes or in ex vivo-cultured cells latently infected in vitro. About 84 viral latency-associated transcripts have been identified with these methods, and although one or more of the transcripts likely play a role during some stage of latent HCMV infections, none as yet have been found to be required for any facet of latency (4, 11, 21, 33, 42, 43, 89). Indeed, deciphering the molecular mechanisms of HCMV latency has been hindered by the lack of an animal model system for viral pathogenesis, by the inability to effectively use genetic approaches in model systems to investigate roles that cellular proteins may play during latency, and because the cell type that represents the in vivo latent reservoir has not been fully defined. As a complement to others’ powerful but descriptive methods, we have combined a functional assay with a genetic approach that together identified a cellular determinant of the establishment of HCMV latency. We found that Daxx plays a role in the silencing of viral IE gene expression that occurs when latent-like infections are established in vitro. We propose that pp71 and Daxx play similar roles in natural latent infections.
Exploring cellular determinants of HCMV latency is challenging because of the difficulty of genetically manipulating the ex vivo-cultured cells used to study HCMV latency while maintaining them in an undifferentiated state. Thus, we took advantage of two in vitro models of HCMV latency, NT2 and THP-1 cells (6, 16, 20, 32, 48, 51, 55, 56, 66, 67, 80, 88). These cells can be stably maintained in an undifferentiated state and, unlike other latency models, can be manipulated by RNA interference techniques that allow the identification of cellular genes that control HCMV latency. Furthermore, the stem cell-like nature of NT2 cells (2) and the almost terminally differentiated status of THP-1 monocytes represent the opposite extremes of the spectrum of cells in which the in vivo HCMV latent reservoir likely resides. NT2 and THP-1 cells have been previously used to study viral gene silencing when latency is established, and importantly, the silenced chromatin structure of the MIEP observed in these cells is associated with similar core histone modifications, as observed during natural latent infections (66, 73, 74). Clearly, neither of these two immortal cell lines is a true latent reservoir of HCMV. Because not all of the characteristics of true latent infections are found in these cells, we refer to infections of THP-1 and NT2 cells as quiescent, or latent-like, infections. We and others are developing methods to genetically manipulate other cell types that are likely to be more physiologically relevant models for HCMV latency. However, because NT2 and THP-1 cells currently represent the most versatile and powerful tools to study certain aspects of latency (such as the mechanisms through which viral genomes are repressed when latency is established), we use them here as an in vitro model system.
The viral IE genes that initiate a productive lytic replication cycle are not expressed upon HCMV infection of undifferentiated NT2 or THP-1 cells unless HDAC activity is inhibited (55, 66). This is in contrast to fibroblasts and differentiated T2RA cells and THP-1-derived macrophages, where IE genes are expressed after HCMV infection in the absence of drugs that inhibit HDACs (20, 88). HDAC inhibitors are not required for IE gene expression in fibroblasts because tegument-delivered pp71 degrades Daxx and thus inactivates the cellular intrinsic immune defense that, in the absence of pp71, institutes the Daxx- and HDAC-dependent repression of viral IE gene expression (79, 91). The data we present here demonstrate that this same cellular defense mechanism is not inactivated when quiescence is established in two different in vitro model systems but is in fact used by the virus as one means to silence its genome and thus perhaps to establish latency. We showed that Daxx is not degraded upon HCMV infection of undifferentiated NT2 (Fig. 1A) and THP-1 (Fig. 1B) cells but that preexpression of nuclearly localized pp71 (Fig. 6) or prior knockdown of Daxx levels in these cells permits IE gene expression in the absence of HDAC inhibitors (Fig. 7A, B, and C), indicating that these cells have not established a latent infection but have initiated the lytic replication cycle.
We should note that while IE genes are expressed in undifferentiated Daxx knockdown cells, adding an HDAC-inhibitory drug at the time of HCMV infection increases the number of cells that initiate IE gene expression and the overall level of IE1 accumulation (data not shown). The augmenting effect of HDAC inhibition may be because of an incomplete knockdown of Daxx (perhaps because loss of Daxx may induce apoptosis [10, 61, 62]), the presence of additional cellular factors that might repress viral IE gene expression during latency through the recruitment of HDACs (30, 53, 80, 92), or likely a combination of both factors. Thus, while Daxx is an important cellular determinant of HCMV latency, it is undoubtedly not the only contributing factor. Having multiple mechanisms to silence gene expression to establish latency may benefit the virus by restricting gene expression to cellular sites where production of virus progeny can ensue.
Viral latency in the face of cellular defenses: victory, defeat, or detente?
Our data show that the Daxx-mediated cellular intrinsic immune defense that is inactivated by pp71 at the start of a lytic infection is not inactivated when latent-like infections are established in two different in vitro model systems. One could envision that latency is a boon for the virus, allowing a prolonged infection with episodic reactivation (victory), the calamitous fate of a virus unable to initiate productive replication because of efficient cellular defense mechanisms (defeat), or a compromise between the cell and virus that allows persistence of the viral genome but significantly delays pathogenesis (detente). Our results lead us to propose that HCMV latency is not a compromise between the virus and its host (and certainly not a calamitous fate) but that it benefits the virus at the expense of the host.
We suggest that HCMV exploits an intrinsic immune defense designed to silence expression from infecting viral genomes to extend its cellular tropism by establishing latency in cells that cannot support lytic viral replication but can be converted through differentiation signals into cell types where latent genomes are activated to yield productive infections. This proposal is based on our finding that, in the absence of Daxx, cells that would otherwise be quiescently infected commence lytic replication but do not support the complete lytic cycle (Fig. 7D and E). IE and early genes are expressed, but the late protein pp28 is not synthesized and the release of viral progeny was a rare event. Such nonproductive HCMV infections in which IE genes are expressed are termed abortive infections (1, 19, 22, 37, 38, 93). In the absence of Daxx in vivo, abortively infected undifferentiated cells may trigger an immune response that, when combined with the lack of a latently infected population of cells, may lead to successful clearance of the viral infection. Therefore, the Daxx-mediated silencing of viral IE gene expression may help to establish a reservoir of latently infected cells that is critical to the success of an in vivo HCMV infection, and refraining from inactivating this defense when latency is established may be the result of positive selection.
Sequestering tegument-delivered activators of IE gene expression in the cytoplasm may be a conserved mechanism that allows herpesviruses to establish latency.
Daxx silences viral gene expression when latent-like infections are established because virion-delivered pp71 fails to reach the nucleus, where it binds to Daxx and induces Daxx degradation. The same virions that deliver pp71 to the nuclei of fibroblasts, T2RA cells, and macrophages fail to do so in NT2 and THP-1 cells, indicating that, in broad terms, the entry pathway is different in these undifferentiated cells. We speculate that this different entry pathway may reflect differences in tegument disassembly at the start of lytic and latent infections because the delivery of viral genomes to the nucleus once the virus has entered the cell is nearly as efficient in undifferentiated THP-1 cells as it is in their differentiated counterparts (Fig. 5). Also, pp71 proteins synthesized de novo in undifferentiated NT2 and THP-1 cells accumulate in the nucleus (Fig. 4A), indicating that pp71, in the absence of other viral proteins, is capable of nuclear entry in these cells. Lastly, prior infection with HCMV does not prevent pp71 synthesized de novo after rAD transduction from entering the nucleus (data not shown), indicating that HCMV does not alter the cellular environment so as to preclude individual pp71 molecules capable of entering the nucleus from localizing there. We are currently exploring the mechanisms that govern the subcellular localization of tegument-delivered pp71 in different cell types and are focusing on viral and cellular factors that may play a role in the disassembly of the tegument after viral entry.
The HSV-1 VP16 protein is a functional homolog of pp71 in that it is a tegument protein that activates viral IE gene expression. VP16 is transported to the nucleus when bound to the cellular HCF (host cell factor) protein (47), and a complex of VP16, HCF, and Oct-1 activates viral IE promoters (90). Interestingly, in neurons where HSV-1 establishes latency, HCF remains in the cytoplasm and IE genes are not expressed upon infection (44). Thus, maintaining important activators of viral gene expression in the cytoplasm may be a common approach used by alpha- and betaherpesviruses to establish latency. This mechanistic similarity also supports the suggestion that latency may be the default pathway upon herpesvirus infections. Having latency as a default pathway makes sense for viruses with long replication cycles that are under intense immune surveillance.
Daxx modulates the replication of divergent viral families.
The data presented here show that Daxx functions in the establishment of latent-like HCMV infections in two in vitro model systems. We have not yet examined if Daxx is required to maintain HCMV latency or if forced or natural pp71 expression can reactivate latent infections. However, the identification of an antisense transcript to the pp71 gene from cells latently infected in vivo (4) that could modulate pp71 expression during latency argues for the continued involvement of Daxx and pp71 past the establishment phase of latency. Because pp71 is expressed with early late kinetics during a lytic infection (28), for it to play a role during reactivation from latency, it would have to be expressed in a different temporal manner (i.e., before the IE genes). The timing of pp71 expression upon reactivation from latency (or that of any HCMV gene) has not been examined. However, early genes are expressed before IE genes when latent herpes simplex virus type 1 is induced to reactivate (84), making it possible that pp71 expression could precede IE gene expression when latent HCMV reactivates.
Interestingly, Daxx is absolutely required for proper early development in mice (61), and some Daxx expression may be required in most cells because depletion of Daxx sensitizes cells to apoptosis (10, 62). By using an essential cellular protein as a means to silence its genome and by encoding a viral assassin that can inactivate that protein, HCMV may ensure that it will always have a hospitable reservoir in which to establish a latent infection, as well as the means to reactivate itself from latency.
Our work focuses on the Daxx-mediated restriction of HCMV infections, but Daxx may also regulate the replication of other human herpesviruses (65, 71), the small DNA human tumor viruses (3, 94), certain retroviruses (23, 25), and even a bunyavirus (52). In addition, Daxx localizes to PML nuclear bodies and there is increasing evidence that the proteins which localize to these structures have inhibitory effects on viral replication (8, 9, 17, 23, 24, 50, 54, 72, 75, 76, 79, 85, 91). Our data indicate that a single protein found at these sites can have either positive or negative effects on HCMV replication, depending upon what phase of the viral life cycle is examined (latency or lytic replication, respectively). As pp71 and Daxx play critical roles in both phases of HCMV replication, they are attractive targets for antiviral therapies.
Finally, the data presented here demonstrate how HCMV commandeers a cellular intrinsic immune defense intended to protect the host from viral infection and uses it to aid in the silencing of its genome in a process that is required for the establishment of a life-long latent infection. It will be interesting to see if there are also additional cases in which other pathogens eschew inactivating an immune defense in favor of using it to enhance specific stages of their replication cycles.
Acknowledgments
We thank Phil Balandyk for expert technical assistance; Bill Sugden for critical comments, reagents, and the use of his microscope; and Yoshi Kawaoka, Teresa Compton, Caroline Alexander, and Bert Vogelstein for reagents.
Research in the Kalejta laboratory is supported by start-up funds from the University of Wisconsin—Madison, a Scientist Development Grant from the American Heart Association, and the New Investigator Program of the Wisconsin Partnership Fund for a Healthy Future. R.F.K. is a Burroughs Wellcome Fund Investigator in Pathogenesis.
Footnotes
Published ahead of print on 27 June 2007.
REFERENCES
- 1.Albrecht, T., M. Nachtigal, S. C. St. Jeor, and F. Rapp. 1976. Induction of cellular DNA synthesis and increased mitotic activity in Syrian hamster embryo cells abortively infected with human cytomegalovirus. J. Gen. Virol. 30:167-177. [DOI] [PubMed] [Google Scholar]
- 2.Andrews, P. W., I. Damjanov, D. Simon, G. S. Banting, C. Carlin, N. C. Dracopoli, and J. Fogh. 1984. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab. Investig. 50:147-162. [PubMed] [Google Scholar]
- 3.Becker, K. A., L. Florin, C. Sapp, and M. Sapp. 2003. Dissection of human papillomavirus type 33 L2 domains involved in nuclear domains (ND) 10 homing and reorganization. Virology 314:161-167. [DOI] [PubMed] [Google Scholar]
- 4.Bego, M., J. Maciejewski, S. Khaiboullina, G. Pari, and S. St. Jeor. 2005. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J. Virol. 79:11022-11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bego, M. G., and S. St. Jeor. 2006. Human cytomegalovirus infection of cells of hematopoietic origin: HCMV-induced immunosuppression, immune evasion, and latency. Exp. Hematol. 34:555-570. [DOI] [PubMed] [Google Scholar]
- 6.Beisser, P. S., L. Laurent, J. L. Virelizier, and S. Michelson. 2001. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 75:5949-5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bresnahan, W. A., and T. E. Shenk. 2000. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 97:14506-14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantrell, S. R., and W. A. Bresnahan. 2006. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 80:6188-6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chelbi-Alix, M. K., F. Quignon, L. Pelicano, M. H. Koken, and H. de The. 1998. Resistance to virus infection conferred by the interferon-induced promyelocytic leukemia protein. J. Virol. 72:1043-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, L. Y., and J. D. Chen. 2003. Daxx silencing sensitizes cells to multiple apoptotic pathways. Mol. Cell. Biol. 23:7108-7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung, A. K., A. Abendroth, A. L. Cunningham, and B. Slobedman. 2006. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 108:3691-3699. [DOI] [PubMed] [Google Scholar]
- 12.Cinatl, J., J. U. Vogel, R. Kotchetkov, and H. W. Doerr. 2004. Oncomodulatory signals by regulatory proteins encoded by human cytomegalovirus: a novel role for viral infection in tumor progression. FEMS Microbiol. Rev. 28:59-77. [DOI] [PubMed] [Google Scholar]
- 13.Cotter, M. A., II, and E. S. Robertson. 2002. Molecular biology of Kaposi's sarcoma-associated herpesvirus. Front. Biosci. 7:d358-d375. [DOI] [PubMed] [Google Scholar]
- 14.Craig, K. L., and M. Tyers. 1999. The F-box: a new motif for ubiquitin dependent proteolysis in cell cycle regulation and signal transduction. Prog. Biophys. Mol. Biol. 72:299-328. [DOI] [PubMed] [Google Scholar]
- 15.Deb-Rinker, P., D. Ly, A. Jezierski, M. Sikorska, and P. R. Walker. 2005. Sequential DNA methylation of the Nanog and Oct-4 upstream regions in human NT2 cells during neuronal differentiation. J. Biol. Chem. 280:6257-6260. [DOI] [PubMed] [Google Scholar]
- 16.Dósa, R., K. Burian, and E. Gönczöl. 2005. Human cytomegalovirus latency is associated with the state of differentiation of the host cells: an in vitro model in teratocarcinoma cells. Acta Microbiol. Immunol. Hung. 52:397-406. [DOI] [PubMed] [Google Scholar]
- 17.Everett, R. D., S. Rechter, P. Papior, N. Tavalai, T. Stamminger, and A. Orr. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80:7995-8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gault, E., Y. Michel, A. Dehee, C. Belabani, J. C. Nicolas, and A. Garbarg-Chenon. 2001. Quantification of human cytomegalovirus DNA by real-time PCR. J. Clin. Microbiol. 39:772-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gawn, J. M., and R. F. Greaves. 2002. Absence of IE1 p72 protein function during low-multiplicity infection by human cytomegalovirus results in a broad block to viral delayed-early gene expression. J. Virol. 76:4441-4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gönczöl, E., P. W. Andrews, and S. A. Plotkin. 1984. Cytomegalovirus replicates in differentiated but not in undifferentiated human embryonal carcinoma cells. Science 224:159-161. [DOI] [PubMed] [Google Scholar]
- 21.Goodrum, F. D., C. T. Jordan, K. High, and T. Shenk. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. USA 99:16255-16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greaves, R. F., and E. S. Mocarski. 1998. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus ie1 mutant. J. Virol. 72:366-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greger, J. G., R. A. Katz, A. M. Ishov, G. G. Maul, and A. M. Skalka. 2005. The cellular protein Daxx interacts with avian sarcoma virus integrase and viral DNA to repress viral transcription. J. Virol. 79:4610-4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guldner, H. H., C. Szostecki, T. Grotzinger, and H. Will. 1992. IFN enhance expression of Sp100, an autoantigen in primary biliary cirrhosis. J. Immunol. 149:4067-4073. [PubMed] [Google Scholar]
- 25.Gurer, C., L. Berthoux, and J. Luban. 2005. Covalent modification of human immunodeficiency virus type 1 p6 by SUMO-1. J. Virol. 79:910-917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hahn, G., R. Jores, and E. S. Mocarski. 1998. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 95:3937-3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hänfler, J., K. A. Kreuzer, K. Laurisch, N. Rayes, P. Neuhaus, C. A. Schmidt, and H. Oettle. 2003. Quantitation of cytomegalovirus (hCMV) DNA and beta-actin DNA by duplex real-time fluorescence PCR in solid organ (liver) transplant recipients. Med. Microbiol. Immunol. 192:197-204. [DOI] [PubMed] [Google Scholar]
- 28.Hensel, G. M., H. H. Meyer, I. Buchmann, D. Pommerehne, S. Schmolke, B. Plachter, K. Radsak, and H. F. Kern. 1996. Intracellular localization and expression of the human cytomegalovirus matrix phosphoprotein pp71 (ppUL82): evidence for its translocation into the nucleus. J. Gen. Virol. 77(Pt. 12):3087-3097. [DOI] [PubMed] [Google Scholar]
- 29.Hofmann, H., H. Sindre, and T. Stamminger. 2002. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 76:5769-5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang, T. H., T. Oka, T. Asai, T. Okada, B. W. Merrills, P. N. Gertson, R. H. Whitson, and K. Itakura. 1996. Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res. 24:1695-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang, J., and R. F. Kalejta. 27 June 2007, posting date. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology doi: 10.1016/j.virol.2007.05.037. [DOI] [PubMed]
- 32.Ioudinkova, E., M. C. Arcangeletti, A. Rynditch, F. De Conto, F. Motta, S. Covan, F. Pinardi, S. V. Razin, and C. Chezzi. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384:120-128. [DOI] [PubMed] [Google Scholar]
- 33.Jenkins, C., A. Abendroth, and B. Slobedman. 2004. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 78:1440-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalejta, R. F., J. T. Bechtel, and T. Shenk. 2003. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol. Cell. Biol. 23:1885-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalejta, R. F., and T. Shenk. 2003. The human cytomegalovirus UL82 gene product (pp71) accelerates progression through the G1 phase of the cell cycle. J. Virol. 77:3451-3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalejta, R. F., and T. Shenk. 2003. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc. Natl. Acad. Sci. USA 100:3263-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamiya, S., J. Tanaka, T. Ogura, H. Sato, H. Ogura, T. Yoshie, and M. Hatano. 1985. Abortive infection with human cytomegalovirus induces an alteration of growth pattern: morphological changes with cytocidal effect in rabbit kidney epithelial cells. Brief report. Arch. Virol. 86:143-150. [DOI] [PubMed] [Google Scholar]
- 38.Keller, M. J., A. W. Wu, J. I. Andrews, P. W. McGonagill, E. E. Tibesar, and J. L. Meier. 2007. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic-AMP signaling pathway. J. Virol. 81:6669-6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khaiboullina, S. F., J. P. Maciejewski, K. Crapnell, P. A. Spallone, A. Dean Stock, G. S. Pari, E. D. Zanjani, and S. S. Jeor. 2004. Human cytomegalovirus persists in myeloid progenitors and is passed to the myeloid progeny in a latent form. Br. J. Haematol. 126:410-417. [DOI] [PubMed] [Google Scholar]
- 40.Kieff, E. D., and A. B. Rickinson. 2007. Epstein-Barr virus and its replication, p. 2603-2700. In D. M. Knipe and P. M. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA.
- 41.Kondo, K., H. Kaneshima, and E. S. Mocarski. 1994. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 91:11879-11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kondo, K., and E. S. Mocarski. 1995. Cytomegalovirus latency and latency-specific transcription in hematopoietic progenitors. Scand. J. Infect. Dis. Suppl. 99:63-67. [PubMed] [Google Scholar]
- 43.Kondo, K., J. Xu, and E. S. Mocarski. 1996. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. USA 93:11137-11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kristie, T. M., J. L. Vogel, and A. E. Sears. 1999. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc. Natl. Acad. Sci. USA 96:1229-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulesza, C. A., and T. Shenk. 2004. Human cytomegalovirus 5-kilobase immediate-early RNA is a stable intron. J. Virol. 78:13182-13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuntz-Simon, G., and G. Obert. 1995. Sodium valproate, an anticonvulsant drug, stimulates human cytomegalovirus replication. J. Gen. Virol. 76(Pt. 6):1409-1415. [DOI] [PubMed] [Google Scholar]
- 47.La Boissière, S., T. Hughes, and P. O'Hare. 1999. HCF-dependent nuclear import of VP16. EMBO J. 18:480-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.LaFemina, R., and G. S. Hayward. 1986. Constitutive and retinoic acid-inducible expression of cytomegalovirus immediate-early genes in human teratocarcinoma cells. J. Virol. 58:434-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LaFemina, R. L., and G. S. Hayward. 1988. Differences in cell-type-specific blocks to immediate early gene expression and DNA replication of human, simian and murine cytomegalovirus. J. Gen. Virol. 69(Pt. 2):355-374. [DOI] [PubMed] [Google Scholar]
- 50.Lavau, C., A. Marchio, M. Fagioli, J. Jansen, B. Falini, P. Lebon, F. Grosveld, P. P. Pandolfi, P. G. Pelicci, and A. Dejean. 1995. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene 11:871-876. [PubMed] [Google Scholar]
- 51.Lee, C. H., G. C. Lee, Y. J. Chan, C. J. Chiou, J. H. Ahn, and G. S. Hayward. 1999. Factors affecting human cytomegalovirus gene expression in human monocyte cell lines. Mol. Cells 9:37-44. [PubMed] [Google Scholar]
- 52.Li, X. D., T. P. Makela, D. Guo, R. Soliymani, V. Koistinen, O. Vapalahti, A. Vaheri, and H. Lankinen. 2002. Hantavirus nucleocapsid protein interacts with the Fas-mediated apoptosis enhancer Daxx. J. Gen. Virol. 83:759-766. [DOI] [PubMed] [Google Scholar]
- 53.Liu, R., J. Baillie, J. G. Sissons, and J. H. Sinclair. 1994. The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic Acids Res. 22:2453-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maul, G. G. 1998. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 20:660-667. [DOI] [PubMed] [Google Scholar]
- 55.Meier, J. L. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 75:1581-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meier, J. L., and M. F. Stinski. 1997. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J. Virol. 71:1246-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meier, J. L., and M. F. Stinski. 1996. Regulation of human cytomegalovirus immediate-early gene expression. Intervirology 39:331-342. [DOI] [PubMed] [Google Scholar]
- 58.Mendelson, M., S. Monard, P. Sissons, and J. Sinclair. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 77(Pt. 12):3099-3102. [DOI] [PubMed] [Google Scholar]
- 59.Michaelis, M., N. Kohler, A. Reinisch, D. Eikel, U. Gravemann, H. W. Doerr, H. Nau, and J. Cinatl, Jr. 2004. Increased human cytomegalovirus replication in fibroblasts after treatment with therapeutical plasma concentrations of valproic acid. Biochem. Pharmacol. 68:531-538. [DOI] [PubMed] [Google Scholar]
- 60.Michaelis, M., T. Suhan, A. Reinisch, A. Reisenauer, C. Fleckenstein, D. Eikel, H. Gumbel, H. W. Doerr, H. Nau, and J. Cinatl, Jr. 2005. Increased replication of human cytomegalovirus in retinal pigment epithelial cells by valproic acid depends on histone deacetylase inhibition. Investig. Ophthalmol. Vis. Sci. 46:3451-3457. [DOI] [PubMed] [Google Scholar]
- 61.Michaelson, J. S., D. Bader, F. Kuo, C. Kozak, and P. Leder. 1999. Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev. 13:1918-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Michaelson, J. S., and P. Leder. 2003. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J. Cell Sci. 116:345-352. [DOI] [PubMed] [Google Scholar]
- 63.Mittereder, N., K. L. March, and B. C. Trapnell. 1996. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J. Virol. 70:7498-7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mocarski, E. S., T. Shenk, and R. F. Pass. 2007. Cytomegaloviruses, p. 2701-2772. In D. M. Knipe and P. M. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA.
- 65.Murakami, Y., S. Yamagoe, K. Noguchi, Y. Takebe, N. Takahashi, Y. Uehara, and H. Fukazawa. 2006. Ets-1-dependent expression of vascular endothelial growth factor receptors is activated by latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus through interaction with Daxx. J. Biol. Chem. 281:28113-28121. [DOI] [PubMed] [Google Scholar]
- 66.Murphy, J. C., W. Fischle, E. Verdin, and J. H. Sinclair. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 21:1112-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nelson, J. A., and M. Groudine. 1986. Transcriptional regulation of the human cytomegalovirus major immediate-early gene is associated with induction of DNase I-hypersensitive sites. Mol. Cell. Biol. 6:452-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nowak, B., C. Sullivan, P. Sarnow, R. Thomas, F. Bricout, J. C. Nicolas, B. Fleckenstein, and A. J. Levine. 1984. Characterization of monoclonal antibodies and polyclonal immune sera directed against human cytomegalovirus virion proteins. Virology 132:325-338. [DOI] [PubMed] [Google Scholar]
- 69.Ory, D. S., B. A. Neugeboren, and R. C. Mulligan. 1996. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. USA 93:11400-11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pawelec, G., S. Koch, C. Franceschi, and A. Wikby. 2006. Human immunosenescence: does it have an infectious component? Ann. N. Y. Acad. Sci. 1067:56-65. [DOI] [PubMed] [Google Scholar]
- 71.Preston, C. M., and M. J. Nicholl. 2005. Human cytomegalovirus tegument protein pp71 directs long-term gene expression from quiescent herpes simplex virus genomes. J. Virol. 79:525-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Preston, C. M., and M. J. Nicholl. 2006. Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 87:1113-1121. [DOI] [PubMed] [Google Scholar]
- 73.Reeves, M. B., P. J. Lehner, J. G. Sissons, and J. H. Sinclair. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 86:2949-2954. [DOI] [PubMed] [Google Scholar]
- 74.Reeves, M. B., P. A. MacAry, P. J. Lehner, J. G. Sissons, and J. H. Sinclair. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 102:4140-4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Regad, T., and M. K. Chelbi-Alix. 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20:7274-7286. [DOI] [PubMed] [Google Scholar]
- 76.Regad, T., A. Saib, V. Lallemand-Breitenbach, P. P. Pandolfi, H. de The, and M. K. Chelbi-Alix. 2001. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J. 20:3495-3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reinhardt, B., R. Minisini, and T. Mertens. 2002. Opinion article: cytomegalovirus is a risk factor in atherogenesis. Herpes 9:21-23. [PubMed] [Google Scholar]
- 78.Revello, M. G., and G. Gerna. 2002. Diagnosis and management of human cytomegalovirus infection in the mother, fetus, and newborn infant. Clin. Microbiol. Rev. 15:680-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saffert, R. T., and R. F. Kalejta. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863-3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shelbourn, S. L., S. K. Kothari, J. G. Sissons, and J. H. Sinclair. 1989. Repression of human cytomegalovirus gene expression associated with a novel immediate early regulatory region binding factor. Nucleic Acids Res. 17:9165-9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sinclair, J., and P. Sissons. 2006. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 87:1763-1779. [DOI] [PubMed] [Google Scholar]
- 82.Söderberg-Nauclér, C., K. N. Fish, and J. A. Nelson. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119-126. [DOI] [PubMed] [Google Scholar]
- 83.Söderberg-Nauclér, C., D. N. Streblow, K. N. Fish, J. Allan-Yorke, P. P. Smith, and J. A. Nelson. 2001. Reactivation of latent human cytomegalovirus in CD14+ monocytes is differentiation dependent. J. Virol. 75:7543-7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tal-Singer, R., T. M. Lasner, W. Podrzucki, A. Skokotas, J. J. Leary, S. L. Berger, and N. W. Fraser. 1997. Gene expression during reactivation of herpes simplex virus type 1 from latency in the peripheral nervous system is different from that during lytic infection of tissue cultures. J. Virol. 71:5268-5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tavalai, N., P. Papior, S. Rechter, M. Leis, and T. Stamminger. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 80:8006-8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taylor-Wiedeman, J., J. G. Sissons, L. K. Borysiewicz, and J. H. Sinclair. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 72(Pt. 9):2059-2064. [DOI] [PubMed] [Google Scholar]
- 87.Taylor-Wiedeman, J., P. Sissons, and J. Sinclair. 1994. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 68:1597-1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weinshenker, B. G., S. Wilton, and G. P. Rice. 1988. Phorbol ester-induced differentiation permits productive human cytomegalovirus infection in a monocytic cell line. J. Immunol. 140:1625-1631. [PubMed] [Google Scholar]
- 89.White, K. L., B. Slobedman, and E. S. Mocarski. 2000. Human cytomegalovirus latency-associated protein pORF94 is dispensable for productive and latent infection. J. Virol. 74:9333-9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wilson, A. C., M. A. Cleary, J. S. Lai, K. LaMarco, M. G. Peterson, and W. Herr. 1993. Combinatorial control of transcription: the herpes simplex virus VP16-induced complex. Cold Spring Harbor Symp. Quant. Biol. 58:167-178. [DOI] [PubMed] [Google Scholar]
- 91.Woodhall, D. L., I. J. Groves, M. B. Reeves, G. Wilkinson, and J. H. Sinclair. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 281:37652-37660. [DOI] [PubMed] [Google Scholar]
- 92.Wright, E., M. Bain, L. Teague, J. Murphy, and J. Sinclair. 2005. Ets-2 repressor factor recruits histone deacetylase to silence human cytomegalovirus immediate-early gene expression in non-permissive cells. J. Gen. Virol. 86:535-544. [DOI] [PubMed] [Google Scholar]
- 93.Yee, L. F., P. L. Lin, and M. F. Stinski. 2007. Ectopic expression of HCMV IE72 and IE86 proteins is sufficient to induce early gene expression but not production of infectious virus in undifferentiated promonocytic THP-1 cells. Virology 363:174-188. [DOI] [PubMed] [Google Scholar]
- 94.Zhao, L. Y., A. L. Colosimo, Y. Liu, Y. Wan, and D. Liao. 2003. Adenovirus E1B 55-kilodalton oncoprotein binds to Daxx and eliminates enhancement of p53-dependent transcription by Daxx. J. Virol. 77:11809-11821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu, H., Y. Shen, and T. Shenk. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69:7960-7970. [DOI] [PMC free article] [PubMed] [Google Scholar]