

Abstract
The fate of most human endogenous retroviruses (HERVs) has been to undergo recombinational deletion. This process involves homologous recombination between the flanking long terminal repeats (LTRs) of a full-length element, leaving a relic structure in the genome termed a solo LTR. We examined loci in one family, HERV-K(HML2), and found that the deletion rate decreased markedly with age: the rate among recently integrated loci was almost 200-fold higher than that among loci whose insertion predated the divergence of humans and chimpanzees (8 × 10−5 and 4 × 10−7 recombinational deletion events per locus per generation, respectively). One hypothesis for this finding is that increasing mutational divergence between the flanking LTRs reduces the probability of homologous recombination and thus the rate of solo LTR formation. Consistent with this idea, we were able to replicate the observed rates by a simulation in which the probability of recombinational deletion was reduced 10-fold by a single mutation and 100-fold by any additional mutations. We also discuss the evidence for other factors that may influence the relationship between locus age and the rate of deletion, for example, host recombination rates and selection, and highlight the consequences of recombinational deletion for dating recent HERV integrations.
Endogenous retroviruses (ERVs) are the proviral form of exogenous viruses that have integrated into germ line cells and are passed vertically from parent to offspring (4). Each provirus is composed of several genes bounded by two noncoding regions termed long terminal repeats (LTRs), which are 500 to 1,000 bp in length and are identical upon insertion (integration). Approximately 98,000 human ERVs (HERVs), or fragments of HERVs, in the published human genome sequence have been located (reference 27; also see http://herv.img.cas.cz), and estimates of the percentage of the human genome that they represent range from 3 to 8% (19, 27). Many of these HERVs have existed for tens of millions of years, during which time approximately 85% of them have undergone recombinational deletion involving the two LTRs. This process results in the replacement of the full-length provirus by a single LTR sequence termed a solo LTR (20, 27, 34) and is a key determinant in the long-term outcome of HERV infection (17).
HERVs are divided into a relatively small number of lineages (or families), each of which is considered to represent the proliferation of an initial independent infection of the ancestor of the human genome up to 70 million years ago (mya) (36). With one possible exception, HERV-K(HML2), all these families have ceased proliferating and have effectively become extinct. The HERV-K(HML2) family, whose method of proliferation appears to be representative of that of other HERVs (2), is therefore the most suitable one for investigating the process of recombinational deletion. The family has homologues in Old World but not New World monkeys (24, 25, 29). The dates of the divergence of humans from Old World monkeys and of humans from New World monkeys are estimated to be approximately 21 to 25 mya and 32 to 36 mya, respectively (11). The family, therefore, must have initially invaded the ancestor of the human genome between these two periods, with 30 mya being a generally accepted estimate. The family appears to have been integrating continuously up to the present day (1, 3, 37) at an approximately steady rate (1).
The rate of recombinational deletion among HERVs may be expected to be related to the increasing mutational divergence between the LTRs as a provirus ages (22). With increasing age, the sequence similarity between the two LTRs of the provirus will decrease due to mutations acquired during host DNA replication. Recombinational deletion is likely to occur via intrachromosomal fold-back loops within the germ line, which are known to be dependent on the similarity between the two recombining sequences (33). Thus, increasing mutational divergence between the LTRs should reduce the probability of homologous pairing and hence of recombinational deletion.
We tested this prediction by determining the rates of recombinational deletion among loci of three different age classes in the family HERV-K(HML2), and we then attempted to recreate these rates in a simulation in which the probability of deletion was dependent upon the mutational divergence between the LTRs.
MATERIALS AND METHODS
Calculating the proportion of full-length proviruses.
Loci in the oldest age category were detected and analyzed by mining the published human and chimpanzee genome sequences (3). We identified all HERV-K(HML2) loci in the human genome sequence whose homologues in the chimpanzee genome sequence are full-length proviruses (the loci we selected do not represent an exhaustive list because the chimpanzee genome project is incomplete). For the two younger age categories, we determined the mean proportion of full-length proviruses among 19 human DNA samples for 63 loci that are represented by solo LTRs in the published human genome sequence. Previously, we have screened these 19 samples for insertional polymorphisms (unfixed loci) (1). In the present study, we rescreened all samples that had an insertion, using a primer (5′ ATTTTACTTTTAGTTAGCCCC 3′) designed against a conserved region within the leader sequence, in order to determine whether the insertion was a full-length provirus or a solo LTR. Flanking primer sequences are available on request. PCR conditions were 40 cycles of 94°C for 1 min, 45 to 55°C for 1 min, and 72°C for 100 s and a final extension step for 10 min (25 pmol of each primer was used with 250 to 500 ng of template DNA). We then combined our findings with previously published data for an additional 11 loci that are represented by full-length proviruses in the published human genome sequence (16). This gave us the proportions of full-length proviruses for a total of 74 loci.
Simulation.
To simulate the mutational divergence between the LTRs, we used a background rate of mutation within humans of 2 × 10−8 per bp per generation (7). The integration rate was assumed to be constant (1), and mutations in the two LTRs (each 968 bp, the mean length for the family) were randomly acquired in accordance with a Poisson distribution. The probability of deletion per generation was set as 10−4, 10−5, and 10−6 for zero, one, and two or more mutations in the LTRs, respectively. For the category corresponding to ages of 6 to 0.8 million years, we excluded those loci that represented the youngest age category. For the oldest category, we simulated the integration and aging of loci over a period of 24 million years prior to the chimpanzee-human divergence, and then for those loci that had survived as full-length proviruses to 6 mya, we simulated the recombinational deletion that occurred over the subsequent 6 million years. The perl script to implement this simulation is available upon request.
Our simulation ignored the positions of mutations in the LTRs. If the critical determinant of homologous recombination is the length of a region of identical nucleotides, mutations occurring close together in an LTR (or near the corresponding position in the other LTR) may reduce the probability of homologous recombination less than mutations that occur farther apart. We investigated this idea within our simulation by ignoring mutations occurring less than 500 bp from an existing mutation. This practice led to a slightly increased rate of deletion in older loci but did not affect the overall pattern, giving proportions of full-length proviruses in the three age categories very similar to those produced without this modification (0.30, 0.04, and 0.69 compared to 0.33, 0.05, and 0.74, respectively).
Recombinational deletion and host recombination rate.
The effect of variation in local host recombination rates was investigated by assigning every HERV locus a recombination rate. For this assignment, we used a high-resolution recombination map (18) based on 5,136 microsatellite markers and calculated average recombination rates across 3-Mb windows centered on the markers. Each HERV locus was assigned a recombination rate based on the nearest marker (only loci within 1.5 Mb of a marker were included in the analysis), and the means of the recombination rates for the loci were compared using a t test.
RESULTS
Division of HERV-K(HML2) loci into three age categories.
We analyzed HERV-K(HML2) loci belonging to three age categories. (i) The first category comprised loci present as full-length proviruses in the published chimpanzee genome sequence that correspond to either a solo LTR or a full-length provirus at the orthologous position in the human sequence. These elements therefore integrated between 30 mya and the human-chimpanzee divergence, which is dated at 6 mya (11, 12). The other two categories were loci, either full-length proviruses or solo LTRs, whose insertion postdated the human-chimpanzee divergence (chimpanzees have the preintegration site) and which were either fixed (ii) or unfixed (iii) in samples from diverse human individuals. We estimated the cutoff point between the latter two age categories to be 0.8 mya, which is the average time of fixation for a neutral allele (14) given a long-term effective human population size of 10,000 and a generation time of 20 years (6, 13, 39).
Calculating rates of recombinational deletion.
For a full-length provirus that inserted t generations earlier, the probability P that now it is still full-length (that is, it has not undergone recombinational deletion) is expressed as follows:
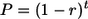 |
(1) |
where r is the probability of deletion in any one generation, assumed to be constant. For the oldest age class, there were 25 full-length proviruses (Table 1) in the common ancestor of chimpanzees and humans, estimated to have existed 6 mya, giving t of 3 × 105 generations. Twenty-two of them are still present as full-length proviruses in a single randomly chosen human genome, giving P of 0.88. Using equation 1, we therefore calculate r to be 4.3 × 10−7. The 2-unit support limits on P (equivalent to 95% confidence limits [8]) are 0.71 and 0.97, giving bounds on r of 1.0 × 10−7 and 1.1 × 10−6. These calculations ignore the possibility of independent recombinational deletion in both human and chimpanzee lineages, but assuming equal rates for all insertions, this probability is small. Similar results are obtained if we include full-length elements from the human lineage and their full-length or solo LTR orthologues in the chimpanzee, expanding the sample size by examining the proportion of shared, homologous loci that have been deleted in the chimpanzee lineage while remaining full-length in the human. We found that 2 of 24 such loci have undergone recombinational deletion in the chimpanzee lineage. Recent evidence suggests that a common generation time of 15 years can be used for much of the human and chimpanzee lineages (9), and applying this figure to the combined data set, we get a probability P of 0.90, with 2-unit support limits of 0.79 and 0.96. From these probabilities, we can derive a value of r of 2.6 × 10−7 (t = 4 × 105 generations) with bounds of 1.0 × 10−7 and 5.9 × 10−7.
TABLE 1.
Locations of all HERV-K(HML2) loci found to correspond to homologous full-length proviruses in the chimpanzee genomea
| Locus | Type of elementb | Chromosome | Start position | End position |
|---|---|---|---|---|
| 4c4 | F | 4 | 4097118 | 4106707 |
| 91c3 | F | 3 | 75683155 | 75691840 |
| 194c5 | F | 5 | 153995706 | 154004408 |
| 44c8 | F | 8 | 47294815 | 47302826 |
| 114c9 | F | 9 | 128692069 | 128699290 |
| 83c19 | F | 19 | 42289389 | 42298906 |
| 10c1 | F | 1 | 13424447 | 13434372 |
| 48c6 | F | 6 | 42969387 | 42979345 |
| 182c1 | F | 1 | 157470489 | 157486262 |
| 258c4 | F | 4 | 166274445 | 166281670 |
| 103c19 | F | 19 | 57935823 | 57945520 |
| 258c4 | F | 4 | 166274445 | 166281670 |
| 119c9 | F | 9 | 136950603 | 136960065 |
| 15c4 | F | 4 | 9335849 | 9345443 |
| 12c22 | F | 22 | 22204481 | 22215169 |
| 68c5 | F | 5 | 46035919 | 46045759 |
| 203c5 | F | 6 | 28758347 | 28768711 |
| 206c2 | F | 3 | 102893427 | 102902546 |
| 207c1 | F | 1 | 147418357 | 147421431 |
| 209c5 | F | 6 | 57730693 | 57736681 |
| 211c1 | F | 1 | 163306261 | 163311916 |
| 213c8 | F | 10 | 6906150 | 6915609 |
| 201c1 | S | 1 | 73307005 | 73307969 |
| 05c20 | S | 19 | 57616024 | 57616983 |
| 208c15 | S | 14 | 23511177 | 23512588 |
Locations are from the May 2004 build (NCBI build 35) at the University of California, Santa Cruz, Genome Bioinformatics site (http://genome.ucsc.edu).
Type of element in published human genome sequence. F, full-length provirus; S, solo LTR.
The intermediate age class includes elements that are present (either as full-length proviruses or as solo LTRs) in the published human genome sequence and in all other humans surveyed but are absent in chimpanzees (that is, chimpanzees have a preintegration site). We found 66 such elements (Table 2): for 7 of them, all humans surveyed still had the full-length provirus; for 56 of them, all humans surveyed had a solo LTR; and for 3 of them, we (or the authors of previous reports) found both full-length proviruses and solo LTRs, with frequencies of full-length proviruses of 0.46, 0.84, and 0.95 (average, 0.75). The (unweighted) average frequency of full-length proviruses across these loci thus corresponds to a P of 0.14. We assume that all these elements integrated in the period between the splitting of humans from chimpanzees and the average date for the coalescence of nuclear genes, that is, in the period between 6 and 0.8 mya, or 300,000 and 40,000 generations ago. If we assume a constant rate of insertion over this period and a constant rate of recombinational deletion between insertion and the present, then the expected proportion of full-length proviruses in modern humans is expressed as follows:
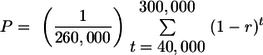 |
(2) |
Combining this equation with our observed value of P of 0.14, we get r of 1.5 × 10−5. The 2-unit support limits on P are approximately 0.067 and 0.25 (calculated assuming a binomial distribution with n of 66), giving bounds on r of 9.5 × 10−6 and 2.3 × 10−5.
TABLE 2.
Details of HERV loci examined
| Locus(i) | Type of element in published human genome sequence | No. of alleles examined | No. of full-length alleles | No. of solo LTRs | Proportion of full-length alleles |
|---|---|---|---|---|---|
| Fixed loci | |||||
| 12q14a | Full-length provirus | 37 | 17 | 20 | 0.46 |
| 109a | Full-length provirus | 37 | 31 | 6 | 0.84 |
| 7 Unnamed locib | Full-length provirus | 147 | 147 | 0 | 1 |
| s480c10c | Solo LTR | 39 | 37 | 2 | 0.95 |
| 56 Other locic | Solo LTR | 2,184 | 0 | 2,184 | 0 |
| Total no. of alleles | 232 | 2,212 | |||
| Overall proportion of full-length allelesd | 0.10 | ||||
| Mean proportion of full-length alleles | 0.14 | ||||
| Unfixed loci | |||||
| 154c11 (11q22)a | Full-length provirus | 37 | 21 | 14 | 0.6 |
| 8c8 (K115)a | Full-length provirus | 46 | 2 | 0 | 1 |
| 165c5c | Solo LTR | 39 | 0 | 28 | 0 |
| 859c12c | Solo LTR | 39 | 19 | 4 | 0.83 |
| 399c8c | Solo LTR | 39 | 0 | 19 | 0 |
| 240c3c | Solo LTR | 39 | 0 | 36 | 0 |
| 1601c9c,e | Solo LTR | 39 | 0 | 36 | 0 |
| 2563c7c | Solo LTR | 39 | 0 | 10 | 0 |
| Total no. of alleles | 42 | 147 | |||
| Overall proportion of full-length allelesd | 0.22 | ||||
| Mean proportion of full-length alleles | 0.30 |
Data from Fig. 2 in reference 16. We excluded 2c7 (108), where a tandem duplication has occurred and the solo LTR may have arisen from this recombination event, and 1p13, which could not be categorized as fixed or unfixed. Diploid samples from a total of 18 individuals plus the published haploid human genome sequence were screened.
Represented by bands in the Southern blot in Fig. 1 of reference 16. We assumed that humans are homozygous for full-length proviruses at these loci. Diploid samples from a total of 10 individuals plus the published haploid human genome sequence were screened.
From our study; diploid samples from a total of 19 individuals plus the published haploid human genome sequence were screened. See reference 1 for the genomic location.
This figure is calculated from the total number of alleles and the number of these that are full-length.
Finally, the youngest age class includes elements that are present (either as full-length proviruses or as solo LTRs) in the published human genome sequence but were absent from at least one human in our survey (that is, some humans had a preintegration site). We found eight such elements (Table 2), and the average proportion of full-length proviruses among all insertions (ignoring alleles with a preintegration site) was 0.30. We assume that all these elements integrated in the time since the average coalescence of nuclear genes, that is, in the last 0.8 million years, or 40,000 generations. If we assume constant rates of insertion and recombinational deletion over this period, then the expected proportion of full-length proviruses in modern humans is expressed as follows:
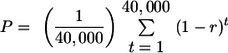 |
(3) |
Combining this equation with our observed value of P of 0.30, we get r of 8.0 × 10−5. It is not clear how to calculate support limits on this estimate, but if we assume a binomial distribution with a sample size of eight, we get conservative bounds on P of 0.044 and 0.72, giving bounds on r of 1.8 × 10−5 and 5.6 × 10−4.
The rate of recombinational deletion inferred from the observed mean proportions thus decreases markedly with the increasing age of the locus (Table 3). There is an almost 200-fold decrease between the rates of the youngest and the oldest age categories of 8.0 × 10−5 and 4.3 × 10−7 per locus per generation, respectively.
TABLE 3.
Proportions of HERV-K(HML2) loci that are full-length proviruses, and inferred recombinational deletion rates, among the three different age categoriesa
| Age (millions of yr) | No. of HERV loci examined | No. of host individuals examined | Mean proportion of full-length proviruses | Rate of recombinational deletion (no. of recombinational deletion events/locus/generation) | Confidence interval for rate of recombinational deletion | Mean proportion of full-length proviruses in simulation | |||
|---|---|---|---|---|---|---|---|---|---|
| 6 to 30 | 25 | 1d | 0.88 | 4.3 × 10−7 | 1.0 × 10−7-1.1 × 10−6 | 0.74 | |||
| 0.8 to 6 | 66b | 10-19e | 0.14 | 1.5 × 10−5 | 9.5 × 10−6-2.3 × 10−5 | 0.05 | |||
| Less than 0.8 | 8c | 10-19e | 0.30 | 8.0 × 10−5 | 1.8 × 10−5-5.6 × 10−4 | 0.33 |
Nine loci from reference 16 plus 57 examined in the present study.
Two loci from reference 16 plus six examined in the present study.
Data from the published human genome sequence.
Samples from 10 or 18 individuals were screened in the study reported in reference 16, and we screened or rescreened a total of 19 samples in the present study.
Reproduction of observed recombinational deletion rates by computer simulation.
We found that the decreasing rate of recombinational deletion with increasing locus age could be reproduced approximately in a simple simulation in which the probability of recombinational deletion of 10−4 per generation was reduced 10-fold by the acquisition of one mutation in the LTRs and reduced 100-fold by the acquisition of two or more mutations (Table 3 and Fig. 1). These parameter values were based on experimental data on the rates of homologous recombination within mammals. For example, it appears that 150 to 500 bp of uninterrupted sequence identity is required for full efficiency, with the rate of homologous recombination declining from threefold to more than 100-fold with a variety of 1- or 2-bp mismatches (21, 28, 38). The observed numbers of mutations in the LTRs of human-specific proviruses lend additional support for the accuracy of our simulation: we observed a mean of nine mutations in these LTRs (n = 17), and our simulation predicted a mean of six. We therefore suggest that mutational divergence determines the rate of recombinational deletion among HERVs. The marked decline in the deletion rate explains why most unfixed (insertionally polymorphic) HERV loci are represented only by a solo LTR (even though they are probably only a few hundred thousand years old) yet some proviruses can persist in their full-length state for tens of millions of years. In our simulation, 50% of integrations became solo LTRs within 150,000 years.
FIG. 1.

Comparison of the observed and simulated proportions of full-length proviruses in different age categories. Large dots (shown with confidence limits) representing the observed data have been placed at the midpoints of the provirus age groups. Small dots represent the means for 1,000 generations in the simulation. Note that the proportions for loci older than 6 million years are abruptly and massively elevated because we excluded deletions that occurred before 6 mya (we examined only loci that are represented by full-length proviruses in the chimpanzee genome). Simulated deletion probabilities per generation were 10−4, 10−5, and 10−6 for 0, 1, and >1 mutations, respectively.
DISCUSSION
There is an earlier and higher estimate (2 × 10−3 deletion events per locus per generation) of the rate of recombinational deletion among members of the HERV-K(HML2) family (16). This rate was calculated by counting the number of deletion events that have taken place in 13 human-specific loci and then using standard population genetics theory to extrapolate the number lost by genetic drift. Our figures, which are based on a larger sample size and a different method, are more in agreement with observed recombinational deletion rates among relatively recent integrations in the mouse. For example, the rate is 4.5 × 10−6 deletion events in the single ecotropic Emv-3 locus per generation (31) and averages around 4 × 10−6 deletion events per generation (1 in 250,000 meiotic generations) among 103 nonecotropic murine leukemia virus loci (10); both rates were calculated from direct observations of deletions among progeny. Our lower confidence limit for the two younger HERV-K(HML2) categories (ca. 10−5) is close to the upper confidence limits for the mouse loci, which are 8 ×10−6 for Emv-3 (31) and 10−5 for the other murine leukemia virus loci (our calculation from data presented in reference 10).
The earlier study on HERV-K(HML2) (16) used sequence data to show that multiple deletion events have occurred at a single solo LTR locus (11q22, or 154c11). Furthermore, the three deletion events observed were estimated to have occurred up to 1.5 million years after the integration of the original full-length element (the LTRs had diverged by several mutations prior to each deletion event). We suggest that this particular locus is either an exception to the general trend that we have proposed (our oldest age category includes a few loci that underwent recombinational deletion at an even greater age) or, as suggested by the authors of the other study, that we may be observing the effect of recombination and/or gene conversion after a deletion event.
We have shown that the rate of recombinational deletion declines with locus age in a fashion that can be explained by the increasing mutational divergence between the LTRs. However, there are two other factors that may complicate this relationship.
First, the background local rate of recombination varies substantially across the human genome (18), and therefore, old, full-length proviruses may have persisted simply because they are situated in genomic regions experiencing low levels of recombination. A pooled analysis of all HERV families (17a) has shown that local variation in the host recombination rate does have an effect on the rate of recombinational deletion, but this effect is small: we estimate that it is sufficient to produce only an approximately threefold difference in the proportion of full-length HERV-K(HML2) proviruses, not the 200-fold difference observed. The effect of the background recombination rate would require a larger sample size and a longer time period to manifest itself, and we thus observe no tendency for the old HERV-K(HML2) proviruses to be in regions of lower host recombination rates than human-specific insertions represented today by solo LTRs (t test; P = 0.44).
Second, there may be greater selection against full-length proviruses than solo LTRs. There is considerable evidence that ERVs are generally harmful to their hosts (15, 23, 26, 32), and solo LTRs are likely to be less harmful than full-length replication-competent proviruses. Although solo LTRs are unlikely to be neutral in all cases (because the regulatory sequences capable of disrupting host gene expression are present in the LTR), they cannot themselves give rise to further insertions. Additionally, the somatic effects of some ERVs are known to be caused only by the full-length provirus: for example, the mutations d (dilute coat color) and hr (hairless) caused by ERV integration into the mouse are reversed following recombinational deletion (31, 35). Also, although a medical effect of HERV-K(HML2) is unproven, the injection of the accessory gene rec (cORF), which is found only in full-length HERV-K(HML2) proviruses, induces tumor formation in immunocompromised nude mice (5). The splice sites in the internal region may also interfere with host transcription. Therefore, among recent insertions, more full-length proviruses than solo LTRs may be lost from the host population as a result of selection factors acting on the host, and thus, fewer full-length viruses would drift towards fixation. If this phenomenon has occurred, it would lead us to overestimate the recombinational deletion rate by reducing the observed proportion of full-length proviruses. However, at the present we have no data on selection that would allow us to correct for this possibility. A point that arises from our analysis is that recently inserted full-length proviruses are perhaps as likely to be inactivated (in the sense of being unable to replicate further) by recombinational deletion as by point mutation. Our inferred recombinational deletion rate for the youngest age category approaches 10−4 per generation, and the probability of acquiring a point mutation approximates that figure (given a background mutation rate in humans of 10−8/bp and a typical provirus length of 104 bp), with perhaps 40% of these mutations being lethal (30).
One consequence of the differences in recombinational deletion rates demonstrated above is that the estimated dates of integration of full-length HERVs may, in general, be too old when there are only a few mutational differences between their paired LTRs. This is because such dates are typically estimated from the pairwise differences between LTRs by assuming a neutral average rate of human evolution since their integration. However, as we have shown, proviruses that acquire (by chance) mutations in their LTRs at, or soon after, integration are less likely to undergo recombinational deletion and therefore persist in their full-length state, whereas most other elements rapidly decay into solo LTRs.
Acknowledgments
This work was funded by the Wellcome Trust. J.W. and A.K. were supported by Natural Environment Research Council studentships, and A.K. was also supported by a Medical Research Council fellowship.
We thank Vini Pereira and Anna Dawson for help with the genome mining and experimental work, respectively.
Footnotes
Published ahead of print on 20 June 2007.
REFERENCES
- 1.Belshaw, R., A. L. A. Dawson, J. Woolven-Allen, J. Redding, A. Burt, and M. Tristem. 2005. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity. J. Virol. 79:12507-12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belshaw, R., A. Katzourakis, J. Pačes, A. Burt, and M. Tristem. 2005. High copy number in human endogenous retrovirus families is associated with copying mechanisms in addition to reinfection. Mol. Biol. Evol. 22:814-817. [DOI] [PubMed] [Google Scholar]
- 3.Belshaw, R., V. Pereira, A. Katzourakis, G. Talbot, J. Pačes, A. Burt, and M. Tristem. 2004. Long-term reinfection of the human genome by endogenous retroviruses. Proc. Natl. Acad. Sci. USA 101:4894-4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boeke, J. D., and J. P. Stoye. 1997. Retrotransposons, endogenous retroviruses, and the evolution of retroelements, p. 343-435. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. CSHL Press, New York, NY. [PubMed]
- 5.Boese, A., M. Sauter, U. Galli, B. Best, H. Herbst, J. Mayer, E. Kremmer, K. Roemer, and N. Mueller-Lantzsch. 2000. Human endogenous retrovirus protein cORF supports cell transformation and associates with the promyelocytic leukemia zinc finger protein. Oncogene 19:4328-4336. [DOI] [PubMed] [Google Scholar]
- 6.Chen, F. C., E. J. Vallender, H. Wang, C. S. Tzeng, and W. H. Li. 2001. Genomic divergence between human and chimpanzee estimated from large-scale alignments of genomic sequences. J. Hered. 92:481-489. [DOI] [PubMed] [Google Scholar]
- 7.Drake, J. W., B. Charlesworth, D. Charlesworth, and J. F. Crow. 1998. Rates of spontaneous mutation. Genetics 148:1667-1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edwards, A. W. F. 1992. Likelihood, expanded ed. John Hopkins University Press, Baltimore, MD.
- 9.Elango, N., J. W. Thomas, and S. V. Yi. 2006. Variable molecular clocks in hominoids. Proc. Natl. Acad. Sci. USA 103:1370-1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frankel, W. N., J. P. Stoye, B. A. Taylor, and J. M. Coffin. 1990. A linkage map of endogenous murine leukemia proviruses. Genetics 124:221-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glazko, G. V., and M. Nei. 2003. Estimation of divergence times for major lineages of primate species. Mol. Biol. Evol. 20:424-434. [DOI] [PubMed] [Google Scholar]
- 12.Goodman, M., C. A. Porter, J. Czelusniak, S. L. Page, H. Schneider, J. Shoshani, G. Gunnell, and C. P. Groves. 1998. Toward a phylogenetic classification of primates based on DNA evidence complemented by fossil evidence. Mol. Phylogenet. Evol. 9:585-598. [DOI] [PubMed] [Google Scholar]
- 13.Harpending, H. C., M. A. Batzer, M. Gurven, L. B. Jorde, A. R. Rogers, and S. T. Sherry. 1998. Genetic traces of ancient demography. Proc. Natl. Acad. Sci. USA 95:1961-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartl, D. L., and A. G. Clark. 1997. Principles of population genetics. Sinauer, Sunderland, MA.
- 15.Hirsch, H. H., A. P. K. Nair, and C. Moroni. 1993. Suppressible and nonsuppressible autocrine mast-cell tumors are distinguished by insertion of an endogenous retroviral element (IAP) into the interleukin-3 gene. J. Exp. Med. 178:403-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hughes, J. F., and J. M. Coffin. 2004. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc. Natl. Acad. Sci. USA 101:1668-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katzourakis, A., A. Rambaut, and O. G. Pybus. 2005. The evolutionary dynamics of endogenous retroviruses. Trends Microbiol. 13:463-468. [DOI] [PubMed] [Google Scholar]
- 17a.Katzourakis, A., V. Pereira, and M. Tristem. Effects of recombination rate on human endogenous retrovirus fixation and persistence. J. Virol., in press. [DOI] [PMC free article] [PubMed]
- 18.Kong, A., D. F. Gudbjartsson, J. Sainz, G. M. Jonsdottir, S. A. Gudjonsson, B. Richardsson, S. Sigurdardottir, J. Barnard, B. Hallbeck, G. Masson, A. Shlien, S. T. Palsson, M. L. Frigge, T. E. Thorgeirsson, J. R. Gulcher, and K. Stefansson. 2002. A high-resolution recombination map of the human genome. Nat. Genet. 31:241-247. [DOI] [PubMed] [Google Scholar]
- 19.Lander, E. S., L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860-921. [DOI] [PubMed] [Google Scholar]
- 20.Lower, R., J. Lower, and R. Kurth. 1996. The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc. Natl. Acad. Sci. USA 93:5177-5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lukacsovich, T., and A. S. Waldman. 1999. Suppression of intrachromosomal gene conversion in mammalian cells by small degrees of sequence divergence. Genetics 151:1559-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mager, D. L., and P. Medstrand. 2003. Retroviral repeat sequences, p. 57-63. In D. Cooper (ed.), Encyclopedia of the human genome, vol. 5. Nature Publishing Group, London, United Kingdom. [Google Scholar]
- 23.Marchetti, A., J. Robbins, G. Campbell, F. Buttitta, F. Squartini, M. Bistocchi, and R. Callahan. 1991. Host genetic background effect on the frequency of mouse mammary tumor virus-induced rearrangements of the int-1 and int-2 loci in mouse mammary tumors. J. Virol. 65:4550-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mariani-Costantini, R., T. M. Horn, and R. Callahan. 1989. Ancestry of a human endogenous retrovirus family. J. Virol. 63:4982-4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Medstrand, P., and D. L. Mager. 1998. Human-specific integrations of the HERV-K endogenous retrovirus family. J. Virol. 72:9782-9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Medstrand, P., L. N. van de Lagemaat, and D. L. Mager. 2002. Retroelement distributions in the human genome: variations associated with age and proximity to genes. Genome Res. 12:1483-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pačes, J., A. Pavlíček, and V. Pačes. 2002. HERVd: database of human endogenous retroviruses. Nucleic Acids Res. 30:205-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reiter, L. T., P. J. Hastings, E. Nelis, P. De Jonghe, C. Van Broeckhoven, and J. R. Lupski. 1998. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am. J. Hum. Genet. 62:1023-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reus, K., J. Mayer, M. Sauter, H. Zischler, N. Müller-Lantzsch, and E. Meese. 2001. HERV-K(OLD): ancestor sequences of the human endogenous retrovirus family HERV-K(HML-2). J. Virol. 75:8917-8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanjuán, R., A. Moya, and S. F. Elena. 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. USA 101:8396-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seperack, P. K., M. C. Strobel, D. J. Corrow, N. A. Jenkins, and N. G. Copeland. 1988. Somatic and germ-line reverse mutation rates of the retrovirus-induced dilute coat-color mutation of DBA mice. Proc. Natl. Acad. Sci. USA 85:189-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spence, S. E., D. J. Gilbert, D. A. Swing, N. G. Copeland, and N. A. Jenkins. 1989. Spontaneous germ line virus infection and retroviral insertional mutagenesis in eighteen transgenic Srev lines of mice. Mol. Cell. Biol. 9:177-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stankiewicz, P., and J. R. Lupski. 2002. Genome architecture, rearrangements and genomic disorders. Trends Genet. 18:74-82. [DOI] [PubMed] [Google Scholar]
- 34.Stoye, J. P. 2001. Endogenous retroviruses: still active after all these years? Curr. Biol. 11:R914-R916. [DOI] [PubMed] [Google Scholar]
- 35.Stoye, J. P., S. Fenner, G. E. Greenoak, C. Moran, and J. M. Coffin. 1988. Role of endogenous retroviruses as mutagens: the hairless mutation of mice. Cell 54:383-391. [DOI] [PubMed] [Google Scholar]
- 36.Tristem, M. 2000. Identification and characterization of novel human endogenous retrovirus families by phylogenetic screening of the Human Genome Mapping Project database. J. Virol. 74:3715-3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner, G., M. Barbulescu, M. Su, M. I. Jensen-Seaman, K. K. Kidd, and J. Lenz. 2001. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol. 11:1531-1535. [DOI] [PubMed] [Google Scholar]
- 38.Waldman, A. S., and R. M. Liskay. 1988. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol. Cell. Biol. 8:5350-5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wall, J. D. 2003. Estimating ancestral population sizes and divergence times. Genetics 163:395-404. [DOI] [PMC free article] [PubMed] [Google Scholar]