

Abstract
Foxa1, 2 and 3 (formerly HNF-3α, -β and -γ) constitute a sub-family of winged helix transcription factors with multiple roles in mammalian organ development. While all three Foxa mRNAs are present in endoderm derivatives including liver and pancreas, only Foxa3 is expressed in the testis. Here we demonstrate by genetic lineage tracing that Foxa3 is expressed in postmeiotic germ and interstitial Leydig cells. The germinal epithelium of Foxa3-deficient testes is characterized by a loss of germ cells secondary to an increase in germ cell apoptosis that ultimately leads to a Sertoli cell-only syndrome. Remarkably, not only the Foxa3−/− mice but also Foxa3+/− mice exhibited loss of germ cells. This cellular phenotype caused significantly reduced fertility and testis weight of both Foxa3−/− and Foxa3+/− mice. Using microarray analysis, we found a dramatic downregulation of the zinc finger protein 93 and the testicular tumor-associated paraneoplastic Ma antigen (PNMA) and increased expression of a number of genes including zinc finger protein 94 and several kallikrein 1-related peptidases which could account for at least part of the observed phenotype. In summary, we have identified Foxa3 as a transcriptional regulator with a dominant phenotype in germ cell maintenance and suggest FOXA3 as a potential candidate gene for subfertility in man.
Keywords: testis, Leydig cell, Sertoli cell, spermatogenesis, Foxa3, winged helix transcription factor, kallikreins, fertility, haploinsufficiency, paracrine signalling
Introduction
Subfertility and infertility in men are widespread yet poorly understood health problems. Approximately 15% of all couples of reproductive age are infertile, and the causes of infertility are roughly equally distributed between genders (Nieschlag, 2001). Germ cell production in the mammalian testis depends on intimate biochemical and physical interactions between somatic Sertoli cells and germ cells (Mruk and Cheng, 2004; Sharpe et al., 2003). In addition, normal Leydig cell function is also critical for proper sperm production in rodents and men (Andersson et al., 2004; Qian et al., 2001; Rich et al., 1979). Hence, it is well-established that there is a functional network between the somatic cell types, i.e. the Sertoli and the Leydig cells, and the germ cells (Gnessi et al., 2000; Gnessi et al., 1997; Huleihel and Lunenfeld, 2004; Jegou et al., 1984; O’Donnell et al., 2001; Weinbauer and Wessels, 1999).
Spermatogenesis can be divided into three phases: phase 1 consists of up to eleven mitotic divisions of the germinal stem cells (i.e. the spermatogonia), phase 2, which is meiosis of the spermatogonia, and phase 3, which comprises the postmeiotic differentiation of the haploid spermatids. Thus, theoretically each germinal stem cell division can result in the production of several thousand sperm cells (de Rooij, 2001). Due to the exponential nature of the process even a small decrease in the efficiency of spermatogonial proliferation or survival, caused for instance by abnormal Sertoli or Leydig cell function, can result in a dramatic reduction in sperm count and fertility.
While gene targeting studies in mice have identified many factors as being important in spermatogenesis (Cooke and Saunders, 2002; de Rooij and de Boer, 2003; Escalier, 2001; Matzuk and Lamb, 2002; Venables and Cooke, 2000), none of the Fox (Forkhead box) or winged helix DNA binding proteins have been shown to play a role in this process thus far. Here we demonstrate using genetic lineage tracing analysis that the transcription factor Foxa3, previously known as hepatocyte nuclear factor 3γ or HNF-3γ, is expressed in Leydig cells and in spermatids. Mice either homozygous or heterozygous for a Foxa3 null allele exhibit reduced male fertility secondary to increased germ cell apoptosis. We identify several candidate genes possibly linking the lack of Foxa3 with the observed phenotype and conclude that Foxa3 plays an important role in the maintenance of a healthy germinal epithelium. We suggest that mutations in the human FOXA3 gene might contribute to subfertility in man.
Results and Discussion
Foxa3 is expressed in Leydig cells and in spermatids
Foxa3 is the only member of the Foxa subfamily of transcription factors expressed in testes, and, as such, a candidate gene for a regulator of spermatogenesis (Kaestner et al., 1994; Lai et al., 1991). Foxa3 mRNA was expressed from day 6 to day 70 during postnatal testicular development in the mouse (Fig 1a), suggesting a role for Foxa3 in testicular development as well as the maintenance of the adult status. Because none of the available antibodies produced consistent staining patterns that were absent from Foxa3 null tissue (data not shown), we decided to localize Foxa3 expression further by genetic lineage tracing. In genetic lineage tracing (see scheme in Fig. 1b), the control elements of the gene of interest, here Foxa3, are used to drive the site-specific DNA recombinase Cre. In all cells where Cre is expressed, a loxP flanked stop sequence is removed, allowing for the expression of a marker gene, in our case β-galactosidase. Because the Cre activity produces a permanent mark in the genome, all descendants of the first cell that expressed Cre will be labeled as well. An essential requirement for this technique is a Cre transgene that accurately mimics the expression of the endogenous gene. Therefore, we employed a 170 kb yeast artificial chromosome (YAC) encompassing the entire Foxa3 locus to direct expression of Cre, as this transgene has been shown to faithfully recapitulate the expression of the endogenous Foxa3 gene (Hiemisch et al., 1997; Lee et al., 2005). As shown in Figure 1d,e, both Leydig cells and spermatids were β-galactosidase positive in Foxa3Cre/Rosa26 reporter mice. No staining was observed in Rosa26 reporter mice alone (Fig 1c). Not all Leydig cells were positive for lacZ in Foxa3Cre/Rosa26 reporter mice, which most likely reflects the heterogeneous physiological state of Leydig cells. For instance, it is well-established that Leydig cells also differ in the expression of the androgen receptor (AR), with a subpopulation of Leydig cells expressing no AR (Vornberger et al., 1994). Foxa3Cre expression in spermatids as evidenced by β-galactosidase staining starts shortly before nuclear condensation of spermatids. However, as judged by the phenotype of Foxa3 deficient mice described below, Foxa3 expression in spermatids is of minor physiological importance compared to its expression in Leydig cells.
Fig. 1.
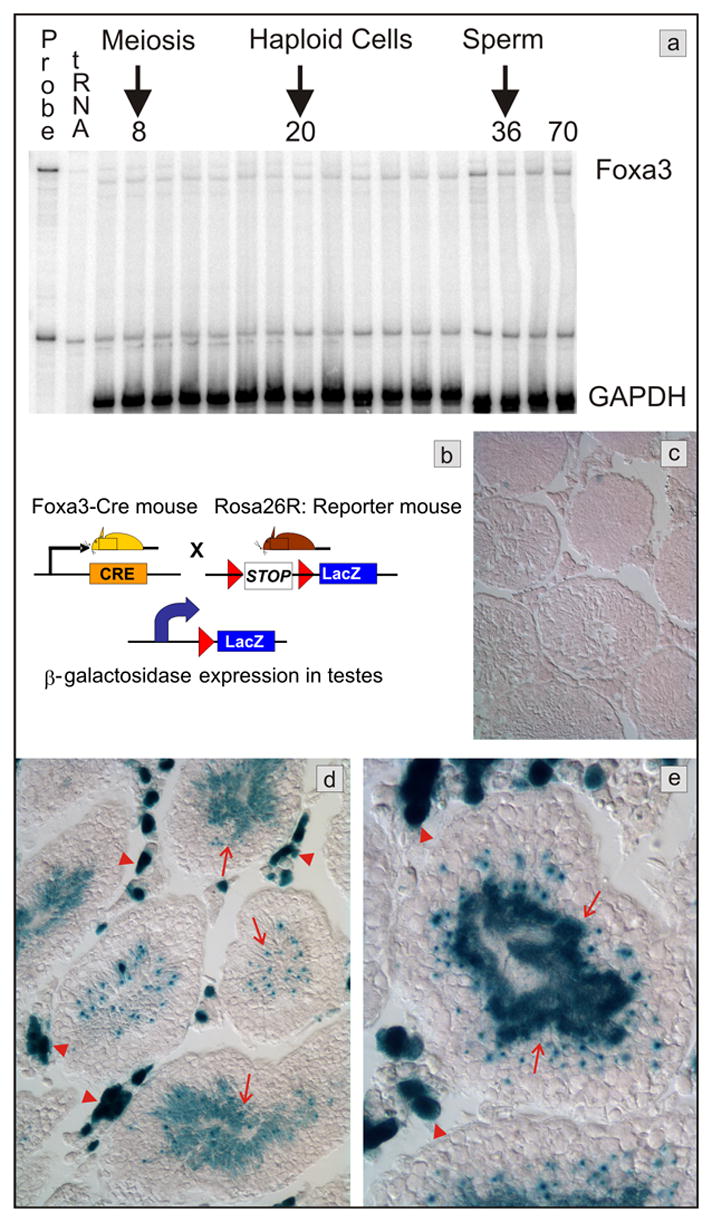
Testicular expression of Foxa3. a) Messenger RNA expression analysis by RNase protection assay. Foxa3 mRNA is detectable in the mouse testis throughout postnatal development. The mRNA abundance is increased approximately 2-fold in the adult testis when compared to the 6 days old testis (normalized to GAPDH mRNA). RNA was prepared from testis of postnatal day 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 36, 40, and 70, respectively. b) Scheme of the genetic lineage tracing employed to track Foxa3 expression in the testis. Beta-galactosidase expression occurs only in those cells that express Foxa3 or are the descendants of Foxa3-expressing cells. c) The control mouse (Rosa26R, no Cre), shows no β-galactosidase expression in the testes. d, e) Mice carrying both the β-galactosidase reporter and the Foxa3-Cre transgene (Rosa26R; Foxa3Cre mice) display robust staining in Leydig cells (red arrowheads) and in spermatids (arrows).
Foxa3 mutant mice show abnormal testicular development
Next we investigated testicular development in Foxa3 mutant mice. In contrast to normal mice, aging Foxa3-deficient testes could not support spermatogenesis throughout the entire germinal epithelium (Fig 2). At the postnatal age of 40 days, when the testis produces the first sperm cells, no histological defects were seen in Foxa3 mutant mice (data not shown). In Foxa3-deficient testes of three-months old mice a few abnormal, but no completely degenerated tubules were visible (Fig 2a,b). During the next months, the phenotype became more pronounced and in eight-months old mice a significant portion of tubules was degenerated (Fig 2c,d). In mice older than one year about a third of all tubular cross sections exhibited severe impairment of spermatogenesis. While the mutant testes at any age also contained tubules with apparently normal spermatogenesis, directly adjacent tubules were often degenerating (Fig 2d,e) or completely devoid of germ cells (Fig 2f). Degenerating tubules were randomly distributed over the testes. Remarkably, the last remaining germ cells in degenerating tubules were elongated spermatids and mature sperm cells (Fig 2e). This indicates that the observed phenotype is likely caused by impairment of early germ cell proliferation or survival, and not by a specific block during sperm maturation, even though Foxa3 is expressed during spermiogenesis. After passing an early critical checkpoint, the developing germ cells can complete meiosis and spermiogenesis, even if the following germ cell generations are completely missing and the germinal epithelium is almost atrophied (Fig 2e). Hence, histologically, the lack of Foxa3 in spermatids does not affect spermiogenesis. At the late stages of tubular degeneration as seen in Fig 2f, some of the Sertoli cell nuclei have lost the pyramidal shape normally found in Sertoli cells supporting ongoing spermatogenesis.
Fig. 2.
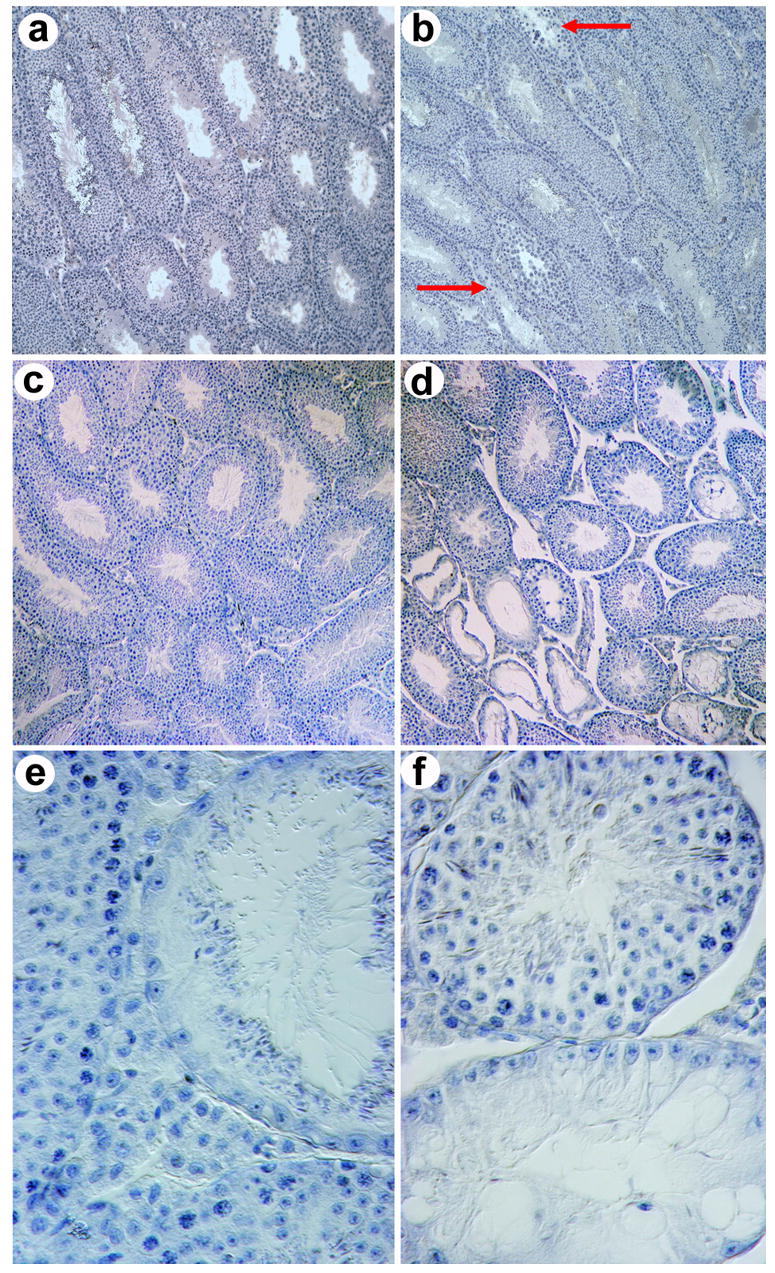
Testicular degeneration in Foxa3-deficient mouse testes. a) Histological section of a wild type mouse testis at the age of three months. All seminiferous tubules contain somatic Sertoli cells as well as germ cells and exhibit ongoing germ cell production. b) Mutant testis at the age of three months. Sporadic tubulus with early signs of degeneration, i.e. thinning of the germinal epithelium, can be seen (arrow), c) Histological section of a wild type mouse testis at the age of eight months showing normal spermatogenesis in all tubules. d,e,f) Histological sections of Foxa3-deficient mouse testes at eight months of age. The testis contains both tubules with normal appearance and ongoing spermatogenesis as well as severely degenerated tubules. The characteristic feature of the defective tubules is a selective loss of germ cells. The Sertoli cells remained within the tubule resulting in a focal Sertoli-cell-only syndrome. e) An almost completely atrophied seminiferous tubule. The only germ cells present are elongated spermatids. All spermatogonia, spermatocytes, and round spermatids are missing. The elongated spermatids are arrayed on the adluminal surface of the Sertoli cells as they usually are shortly before sperm release from the germinal epithelium (spermiation). f) A Sertoli cell only tubule (bottom) adjacent to a tubule with complete spermatogenesis (top). The Sertoli cell nuclei characterized by their triangular shape and their prominent nucleoli have aligned with the basal lamina indicating the absence of any germ cells. Heterozygous testes show histological pictures similar to the homozygous mutant testes. Magnification of a), b), c), and d) 100x; e) and f) 600x.
Foxa3 mutant testes exhibit increased apoptosis in the germinal epithelium
The maintenance of a normally composed germinal epithelium depends on a finely tuned balance between proliferation, differentiation, and apoptosis of the germ cells (de Rooij, 2001). We analyzed germ cell proliferation using BrdU incorporation as a marker for DNA synthesis in replicating cells. Two hours after injection mitotically dividing spermatogonia and primary spermatocytes were labeled. There was no difference in the number of BrdU-positive cells between wild type and mutant testes (Fig 3a), indicating that the defect in the Foxa3−/− mice was not caused by decreased proliferation of germ cells. Even in strongly degenerated tubules BrdU-positive germ cells were seen. In order to quantify germ cell apoptosis we carried out TUNEL assays on testicular tissue sections (Fig 3b,c). We found a significantly increased number of TUNEL-positive cells per TUNEL-positive tubular cross-section in the Foxa3-mutant as well as in the heterozygous testes compared to controls (p<0.02 wt vs. heterozygous and homozygous mutants) (Fig 3d). Furthermore, the percentage of tubules containing TUNEL-positive cells was slightly increased (wt: 15.1 %, het: 17.1 %, mut: 18.9 % of TUNEL-positive tubules). Altogether, the number of apoptotic germ cells in the Foxa3-deficient testes was increased 1.77-fold. This difference becomes even more relevant taking in consideration that mostly spermatogonia are affected by apoptosis as judged from the position of the labeled cells in the germinal epithelium. The extinction of even one Asingle spermatogonium, the stem cell for germ cell production, will result in the loss of the entire germ cell population within a region of the germinal epithelium. Thus increased germ cell apoptosis is expected to result in seminiferous tubules devoid of germ cells as seen in the Foxa3−/− mice (Fig 2).
Fig. 3.
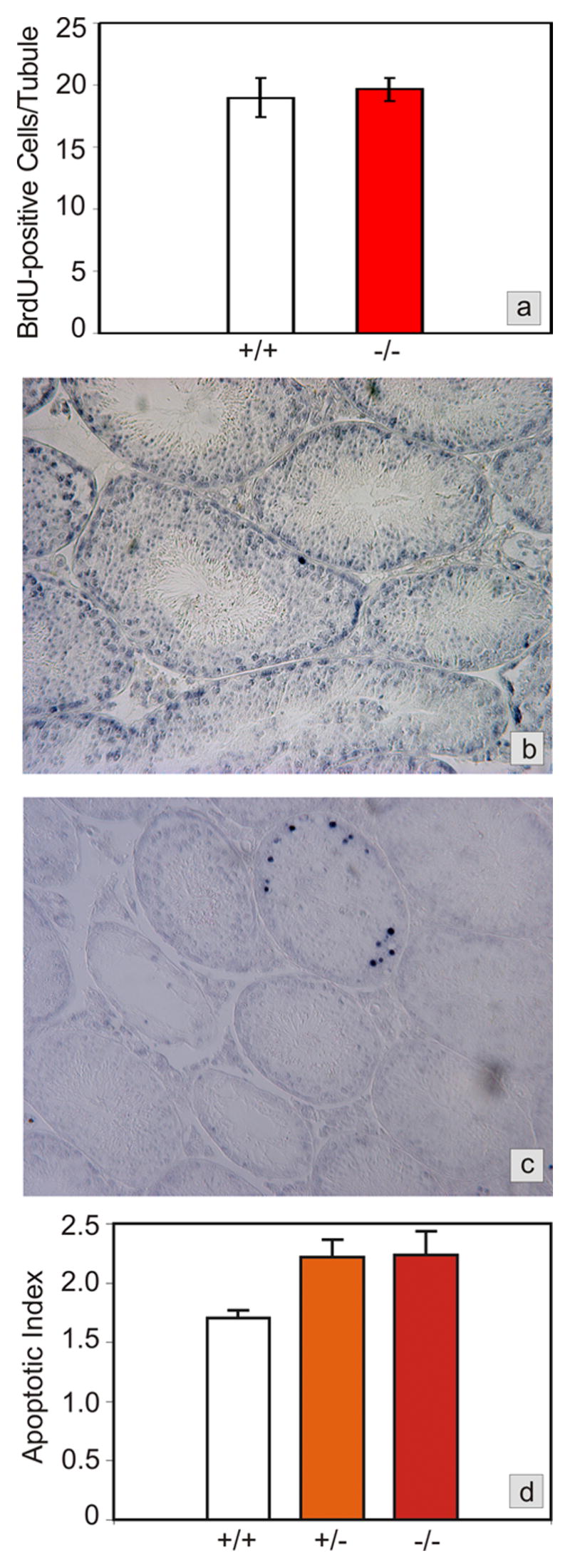
Testicular proliferation and apoptosis in Foxa3-deficient mice. a) Proliferation rates, as determined by BrdU labeling of cells in S-phase during spermatogenic stages VII and VIII, are not different between wild type and Foxa3-deficient mice (p>0.35). b) Apoptosis in a wild type mouse testis as revealed by the TUNEL-technique. Only a minor proportion of all tubules contains one or more TUNEL-positive cells. All other tubules are devoid of TUNEL-positive cells. As judged from the appearance and the location of the cells in the seminiferous tubule the apoptotic cells are almost exclusively spermatogonia and spermatocytes. c) Foxa3-deficient mice loose their germ cell population by apoptosis. The tubule undergoing degeneration exhibits a high number of apoptotic germ cells, again mainly spermatogonia and spermatocytes. Note the already degenerated tubules which are devoid of TUNEL-positive cells. d) Apoptotic index expressed as TUNEL-positive cells per tubule containing TUNEL-positive cells. The mutant as well as the heterozygous testes show a significant increase in the number of apoptotic cells (p<0.05).
Foxa3 deficiency reduces male fertility
To test whether the morphological defects described above had an impact on the reproductive capability of the Foxa3 mutant mice we mated three-months old Foxa3+/+, Foxa3+/−, and Foxa3−/− males (n = 8 in each group) for five months to wild-type females. Interestingly, the average litter size of the homozygous mutant as well of the heterozygous group was significantly smaller than in the wild type group (p<0.05) (Fig 4a). The litter sizes of the Foxa3+/− and Foxa3−/− groups did not differ significantly. The biological relevance of the defect in spermatogenesis is emphasized by the fact that not all males were fertile in the mutant and in the heterozygous group.
Fig. 4.
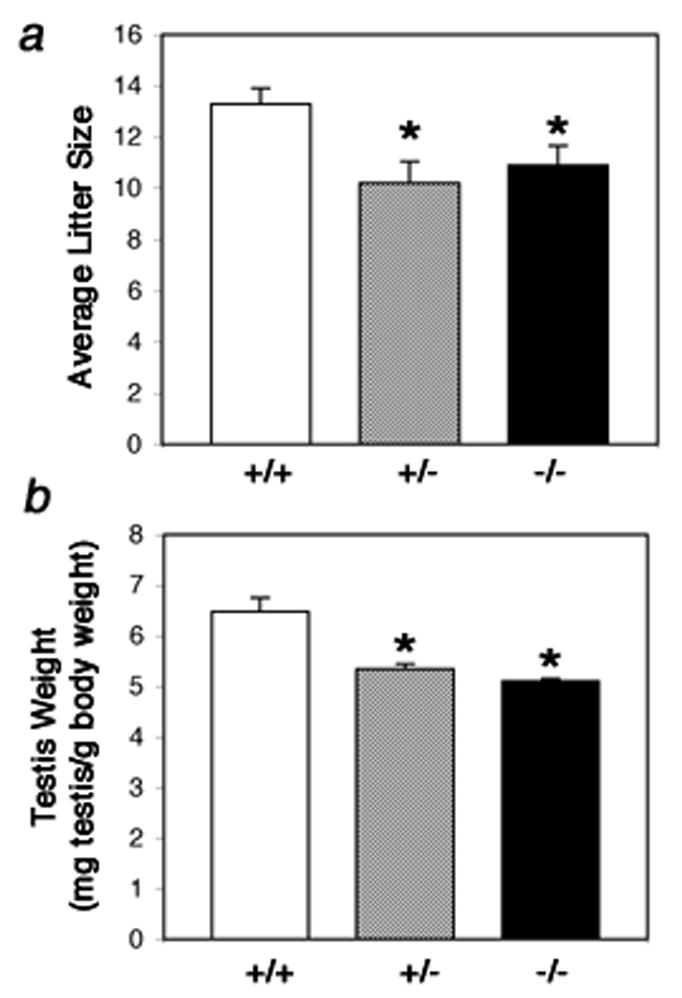
Reduced male fertility and testis weight in Foxa3-deficient mice. a) The testicular defect in Foxa3-deficient mice results in reduced fertility. Male age-matched mice of each genotype were housed together with a female CD1 wt mouse. The average litter size in the mutant (p<0.05) and in the heterozygous group (p<0.02) was significantly lower than in the wt group. b) The Foxa3-mutant (p=0.003) as well as the heterozygous (p=0.007) mice show a significantly reduced testis weight compared to the wild type controls. Data are presented as paired testis weight [mg] per body weight [g].
The mating data correlate well with the testicular weight in relation to body weight at 8 months of age. While this coefficient is 6.48 for the wt group (mg paired testis weight per g bodyweight), the coefficient for the Foxa3+/− and the Foxa3−/− group is 5.43 and 5.10, respectively (P≤0.0001) (Fig 4b). The bodyweight did not differ significantly among the three experimental groups (P≥0.2).
Hormonal analysis
Qualitatively and quantitatively normal spermatogenesis is dependent on several hormones. We analyzed circulating FSH, estradiol, and testosterone but found no significant changes between the Foxa3 mutant mice and their littermate controls (data not shown). Moreover, androgen receptor (AR) expression was also unchanged in Leydig and Sertoli cells (see below and Fig 6e, f). The unchanged serum levels of these hormones suggest that the abnormal spermatogenesis in Foxa3-deficient mice is not an endocrine defect, but does not preclude a local paracrine defect in growth factor or cytokine signaling within the testis (see below).
Fig. 6.
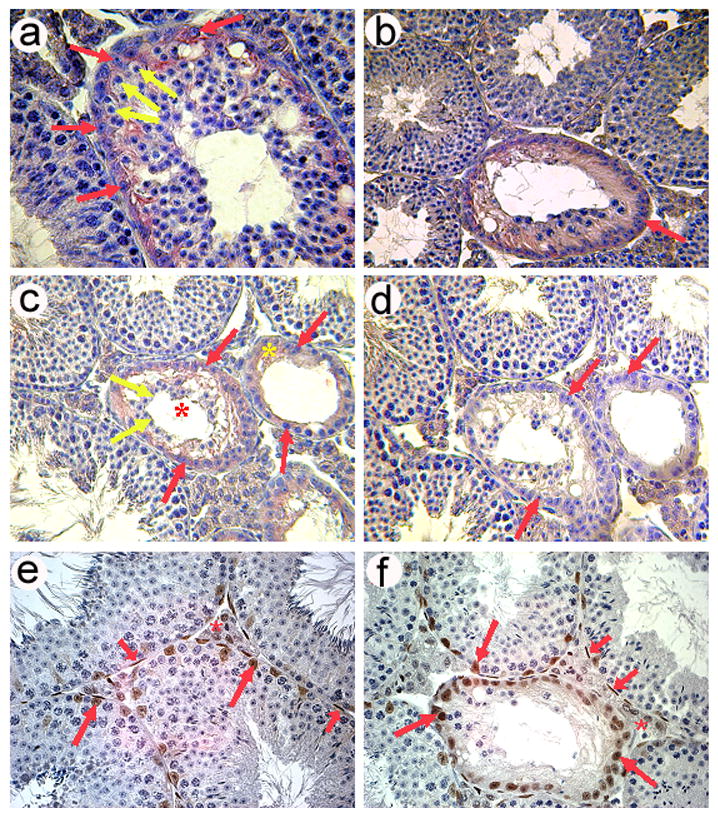
Degenerating seminiferous tubules in Foxa3-deficient testes re-express Anti-Muellerian hormone (AMH) as revealed by immunohistochemistry (a–d). a) A seminiferous tubule in the early phase of degeneration. The thickness of the germinal epithelium appears to be normal, although vacuolization of the epithelium has already occurred. Germ cells present in this tubule are mostly round spermatids as judged from nuclear morphology. Earlier germ cell stages (spermatogonia and spermatocytes) are rather rare. Germ cells are devoid of any pink stain (yellow arrows) but the surrounding cytoplasm of the Sertoli cells is clearly stained (red arrows). Tubules with normal appearance do not exhibit any staining (lower left part). b) A degenerating tubule in a more advanced stage than that shown in a). One generation of spermatocytes (presumably maturing to spermiation) is still present in this tubule. Sertoli cell nuclei have aligned along the tubular wall and the cytoplasm is clearly stained by the AMH antibody (red arrow) c) A tubule during degeneration (red asterisk) still containing some germ cells (yellow arrows) and a tubule at the final stage of degeneration (yellow asterisk), which exhibits a Sertoli cell only-phenotype characterized by the complete loss of germ cells. At this stage the Sertoli cells have flattened and extend little beyond their nuclei. Again, AMH immunoreactivity is widespread. Red arrows point out AMH positive Sertoli cells. d) Negative control omitting the AMH antibody from the diluent. Also, the adult wild type control mice exhibited no staining while Sertoli cells in young testes up to p10 showed strong cytoplasmic Sertoli cell staining (data not shown). e) Expression of the androgen receptor (AR; brown stain) in a wt testis as revealed by immunohistochemistry. Sertoli cells (long red arrows), peritubular myoid cells (short red arrows) and Leydig cells (red asterisk) are AR positive. f) AR expression in an 8 months old mutant testis showing staining in the same cell types as the age-matched wildtype control. Remarkably, also Sertoli cells in almost completely degenerated tubules still express the AR.
Altered gene expression in Foxa3 null testes
In order to investigate the molecular link between Foxa3 deficiency and atrophy of the seminiferous tubules, we performed expression profiling with testis RNA from wild type and Foxa3−/− mice at the age of eight months. Because of our experience that isolation and culture of primary cells often causes artefacts in microarray analyses, we decided to use total testes RNA samples for expression profiling. While this strategy is less sensitive to modulations of certain mRNAs that are selectively changed in only one cell type and remain unchanged in others, using this approach ensures that the results obtained are based on biological effects and not on cell purification or culture artefacts.
Overall, the gene expression profiles were similar between wild type and homozygous mutant mice, indicating that there is no global mis-regulation of transcription in Foxa3-deficient testes. However, the mRNA levels of several genes specifically expressed in germ and Leydig cells were significantly affected by the absence of Foxa3 (Table I). The changes in steady state mRNA levels of the most relevant genes detected by microarray analysis were confirmed independently by real time quantitative RT-PCR (Table I). The down-regulation of the genes identified in this study was selective and not due to the loss of the cell types expressing these genes since the steady state mRNA levels of the vast majority of genes expressed in Leydig cells or spermatids were not altered (e.g. protamines in spermatids and enzymes of the testosterone synthesis pathway in Leydig cells).
Table 1.
Foxa3-dependent gene expression in testis. The genes listed were found to be differentially expressed by microarray analysis and real time quantitative RT-PCR as described in Methods and Materials.
| Microarray | QPCR | ||||
|---|---|---|---|---|---|
| Affy ID | Gene Symbol | Fold Change | q-value(%) | Fold Change | P-value |
| 1439289_s_at | 0710005I19Rik | −4.0 | 0.0 | −3.8 | 0.02 |
| 1428781_at | 1110014F24Rik | −4.8 | 0.0 | −1.6 | 0.14 |
| 1457458_at | BC057627 | −3.3 | 3.4 | −1.2 | 0.27 |
| 1428173_at | Eml2 | −2.0 | 3.4 | −2.7 | 0.066 |
| 1451607_at | Klk1b21 | 2.1 | 4.4 | 3.0 | 0.001 |
| 1420770_at | Klk1b24 | 2.9 | 6.4 | 1.8 | 0.04 |
| 1421587_at | Klk1b27 | 2.6 | 4.4 | 1.7 | 0.056 |
| 1441876_x_at | Zfp93 | −2.8 | 3.4 | −1.9 | 0.09 |
| 1419521_at | Zfp94 | 3.8 | 0.0 | 2.2 | 0.002 |
Since the Foxa proteins are known as transcriptional activators, we searched the promoter sequences of the genes exhibiting reduced mRNA levels in the absence of Foxa3 for consensus Foxa binding sites. We found such binding sites in the putative promoter regions of all genes exhibiting reduced steady state mRNA levels, suggesting a direct effect of the lack of Foxa3 on the transcription of these genes (Fig 5). The precise mechanism of these regulatory relationships will need to be established experimentally in the future.
Fig. 5.
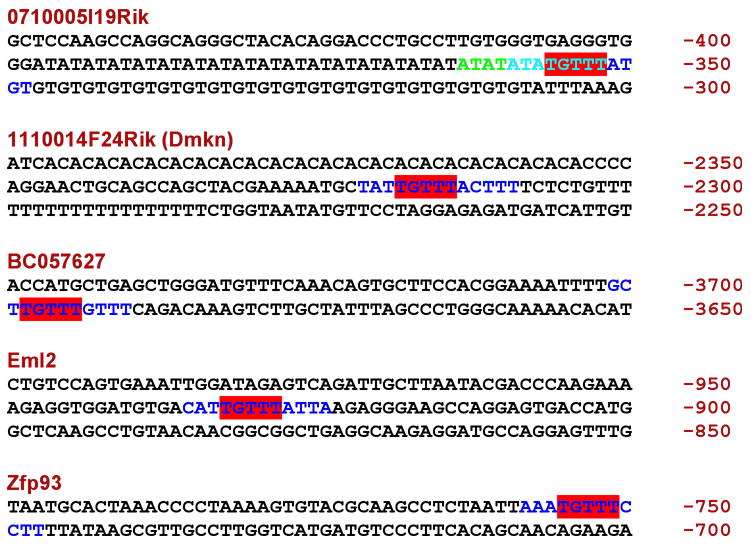
Foxa3 binding sites in the putative promotors of genes showing reduced steady state mRNA levels in Foxa3-mutant testes. Binding sites are shown in color. Numbers on the right give the position relative to the transcriptional start site. For 0710005I19Rik, there are 2 overlapping sites. ATATATATGTTT (starts in green, ends in light blue) and ATATGTTTATGT (starts in light blue, ends in dark blue). The part in light blue is the overlap (ATATGTTT). For all others the full predicted sites (12 bp) are in color. TGTTT is the core motif for Foxa binding.
The changes in gene expression, especially of the kallikrein genes, provide a possible explanation of the phenotypic changes described above. The kallikreins are a subgroup of the serine protease enzyme family (Clements et al., 2004; Diamandis and Yousef, 2002; Lundwall et al., 2006). Kallikreins are involved in propeptide processing (Blaber et al., 1987; Lundwall et al., 2006; Thomas et al., 1981) and have clinical relevance as prognostic factors for several types of endocrine-related tumors such as breast, ovarian and prostate cancer (Clements et al., 2004; Diamandis and Yousef, 2002). Moreover, kallikreins have been associated with such diverse biological processes as skin desquamation, tooth development, neural plasticity, and Alzheimer’s disease (Clements et al., 2004). Matsui and colleagues (Matsui et al., 2000; Matsui and Takahashi, 2001; Matsui et al., 2005) have isolated three cDNAs from the mouse testis, Klk1b21, Klk1b24, and Klk1b27 (formerly mGK-21 or mKlk21, mGK-24 or mKlk24, and mGK27 or mKlk27, respectively; Lundwall et al., 2006), which are related to human kallikrein-1 and are expressed in the testis exclusively in Leydig cells. We found all three kallikreins expressed in mouse testes significantly upregulated in the absence of Foxa3. Therefore, it is conceivable that massively deregulated kallikrein expression disturbs testicular function in Foxa3-deficient mice by paracrine mechanisms. In fact, the kallikrein-kinin system has been suggested previously to play an important role in paracrine regulation of the seminiferous epithelium (Monsees et al., 1997). The relatively minor changes in the testicular transcriptome of the mutant testes accompanied by the significant histological impairments may be explained by the fact that the effects of increased kallikrein activity in the testis affect primarily the proteome.
Zinc finger proteins (Zfp) 93, 94, 95, and 96 are closely related DNA binding proteins with predominant or exclusive expression in testis (Shannon et al., 1996; Shannon and Stubbs, 1998; Weissig et al., 2003). Zfp94 steady state mRNA levels were increased approximately fourfold in Foxa3-mutant mice compared to controls. In contrast, Zfp93 is strongly down-regulated in the mutant testis. The Riken cDNA 0710005i19, which was down-regulated four-fold in the Foxa3-deficient testes, encodes the mouse homolog of the human MA2 antigen. Expression of MA2 in healthy tissues in humans is restricted to the brain. However, in patients with testicular cancer exhibiting concomitant neurological disorders (paraneoplastic limbic and/or brain-stem encephalitis), MA2 antigen was also present in testicular tumors, suggesting a role for MA2 during testicular tumorigenesis (Voltz et al., 1999). Whether reduced MA antigen mRNA expression in Foxa3 mutant mice contributes to testicular germ cell loss remains to be resolved.
Sertoli cells in degenerating tubules re-express Anti-Müllerian Hormone
Although Anti-Müllerian Hormone (AMH) itself is not essential for Sertoli cell differentiation (Behringer et al., 1994), it is switched on very early in fetal Sertoli cells and is a very reliable marker for the maturation state of this cell type (Sharpe et al., 2003). In normal testis, AMH is specifically expressed in immature Sertoli cells until puberty. With the appearance of meiotic germ cells and androgen sensitivity of Sertoli cells, AMH expression is strongly down-regulated in the normal testis and mature Sertoli cells are devoid of AMH (Sharpe et al., 2003). Interestingly, the Sertoli cells within the Sertoli cell-only tubules in the Foxa3 mutant testes expressed AMH protein as revealed by immunohistochemistry (Fig 6a–d). Moreover, AMH protein expression appeared to be induced even where meiotic and postmeiotic germ cells were still present (Fig 6a, b), indicating that the loss of germ cells was not the primary cause of AMH protein accumulation. Likewise, transcriptional reactivation of the AMH locus is not a likely explanation for the reappearance of AMH protein, as mRNA levels were not altered in the mutants (data not shown). The molecular mechanisms underlying the reappearance of the AMH antigenicity in Sertoli cells remain to be investigated, but it is tempting to speculate that increased kallikrein activity, which alters the local crosstalk between Leydig and Sertoli cells, may contribute to this effect.
Usually, AMH is dowregulated when Sertoli cells start expressing the androgen receptor as a sign of maturity (Sharpe et al. 2003). Interestingly, in Foxa3-deficient mice the Sertoli cells in degenerated tubules still express the AR (Fig 6e,f) indicating an intermediate state of the Sertoli cells between maturity and dedifferentiation.
Summary and model of the progression of the testicular lesion
Foxa3 is expressed in spermatids and in Leydig cells. According to our current view the lack of Foxa3 in Leydig cells is causative for the phenotype described above. Since spermiogenesis proceeds normal even in the absence of Foxa3 in spermatids, this transcription factor appears to be dispensable in germ cells for completed spermatogenesis. However, lack of Foxa3 in Leydig cells causes significantly increased Kallikrein mRNA levels. We hypothesize that increased Kallikrein activity in the mutant testes severely disturbs paracrine communication between testicular cells. As a consequence, Sertoli cells may fail to maintain their fully differentiated state, as evidenced by the presence of AMH antigenicity, and lose their ability to support and regulate the early steps of spermatogenesis, which leads to an increase in germ cell apoptosis and eventually to degeneration of seminiferous tubules.
Conclusions
In conclusion, we have shown that Foxa3 is necessary for the maintenance of the testicular germ cell population and male fertility in mice and provide evidence that the germinal epithelium in testes lacking Foxa3 is not able to maintain its differentiated phenotype. Surprisingly, Foxa3 is haploinsufficient in the testes, an unusual finding for transcription factors in this organ. We have identified several differentially expressed genes which may contribute to the phenotypic findings. Strikingly, the mutant testes express increased levels of all testicular kallikrein proteases, which may disturb the local communication between the Leydig cells and the cells of the seminiferous tubules. Approximately 8 % of all men are infertile and in a large subset of these patients the underlying cause is unknown (Nieschlag, 2001). The phenotype of Foxa3-deficient mice resembles a human spermatogenic disorder called mixed atrophy, which is characterized by a variable degree of degeneration of the seminiferous tubules. The molecular mechanisms causing this disorder are unknown. However, it is tempting to speculate that mixed atrophy in a subset of patients may be caused by mutations in the FOXA3 locus.
Methods and Materials
Mice
The derivation of the Foxa3 null allele has been described (Kaestner et al., 1998). Mice were genotyped by PCR on tail DNA as described previously. The Foxa3 null allele was backcrossed to C57BL6 inbred mice for 11 generations to obtain an incipient congenic strain (>99% C57BL6 contribution) prior to the onset of this study. Use of an inbred background reduces the variability of the phenotype, as all potential modifier loci are homozygous in all animals studied. For genetic lineage tracing experiments, Foxa3-Cre mice (Lee et al., 2005) were bred to Rosa26 lacZ reporter mice (Soriano, 1999). Testis from 3 month old mice were prepared for β-galactosidase staining as described (Hiemisch et al., 1997).
Mating experiments
Age-matched males (n=8 in each group; +/+, +/−, −/−) from our Foxa3 breeding colony were housed together with age-matched CD1 females (Charles River). At the outset of the study male mice were 3 months of age, i.e. when the phenotype became histologically evident. The study was continued for 5 months. Three times a week all cages were inspected and pups counted. Two to three weeks after birth the offspring were removed from the cages. At the termination of the study the body weight and the paired testis weight of the male mice was determined.
Hormone analysis
At termination of the mating experiments serum was collected from age-matched males (n = 6 per group) and levels of FSH, testosterone, and estradiol were determined by AniLytics, Inc. (Gaithersburg, MD).
Histology and immunohistochemistry
After sacrifice of the mice the testes were removed and cut into halves. Two halves were fixed in Bouin’s solution for 4 hours. One half-testis was fixed overnight in 4% PFA, while the remaining half of was frozen at −80°C. The fixed tissue was paraffin-embedded and used for histological evaluation, detection of apoptotic cells, and BrdU-localization. Immunohistochemistry was performed on Bouin-fixed tissue essentially as described (Behr and Weinbauer, 1999) using the primary antibody (sc-6886; Santa Cruz Biotechnology Inc.; CA, USA) to detect Anti-Müllerian hormone. As positive control we used newborn mouse testis, where we obtained specific and exclusive staining of the Sertoli cells. In negative controls the primary antibody was omitted. Androgen receptor was detected similarly using the AR (N-20) antibody (sc-816; Santa Cruz Biotechnology Inc.; CA, USA).
Proliferation assay
Proliferation of cells was detected by the incorporation of BrdU into genomic DNA. A dose of 10mg BrdU (Zymed)/kg bodyweight was intraperitonally injected 2 hours before sacrifice. The tissue was fixed in 4% PFA and paraffin-embedded. After re-hydration and antigen retrieval by cooking in a microwave oven in 10mM citric acid buffer, the BrdU antigen was detected using an alkaline-phosphatase conjugated BrdU-specific antibody (Roche Molecular Biochemicals). Histologically normal seminiferous tubules in spermatogenic stages VII and VIII were evaluated. During these stages all tubules exhibit BrdU-positive cells. Labeled cells were counted in at least 20 round tubular cross sections per animal (n = 6 per group).
Apoptosis assay
Apoptotic cells were detected on Bouin-fixed tissue sections using the TUNEL technique (Wijsman et al., 1993). Paraffin-embedded sections were dewaxed and microwaved in 10 mM citric acid buffer for 8 minutes. For visualization of DNA double strand breaks the in situ cell death detection kit (Roche Molecular Biochemicals) was used according to the manufacturer’s protocol.
Microarray and RNA analysis
Four wild type and three Foxa3−/− testes samples, each containing 1 μg of total RNA, were hybridized after a single round of amplification to Affymetrix MOE430v2 oligo chips. Data was provided as absent/present calls and intensity values. We normalized the data using the GeneChip Robust Multi-Array Analysis (GCRMA) algorithm (Wu et al., 2003), implemented in a R package (http://www.bioconductor.org/repository/devel/vignette/gcrma.pdf). Fold-changes were calculated as the ratio of the geometric mean between wild type and Foxa3−/− intensities. Statistical analysis of the microarray data was completed using the significance of microarrays (SAM) (Tusher et al., 2001) package with a false discovery rate (FDR) of 10% and fold change cutoff of absolute value of 2.0. Annotation for each ‘spot’ was downloaded from the Affymetrix web site. The data has been deposited into ArrayExpress. Individual steady state transcript levels were determined by real time PCR (QPCR) using the SYBR Green Master Mix from Stratagene on a Stratagene MX3000 QPCR instrument. RNase protection analysis was basically carried out as described previously (Kaestner et al., 1994) using the RPAII kit from Ambion (Austin, TX, USA). The protected RNA fragments were separated on 8 M urea denaturing 6% acrylamide gels and the radioactive bands visualized using a PhosphoImager (Molecular Dynamics). Primer sequences used can be obtained from Kaestner Lab website (http://www.med.upenn.edu/kaestnerlab/reagents.shtml).
Acknowledgments
We thank Drs. J.A. Blendy, J. Friedman and N. B. Hecht for critically reviewing the manuscript. Our studies were facilitated by the University of Pennsylvania Diabetes Center (P30-DK19525) and the Penn Center for Molecular Studies in Digestive and Liver Disease (P30-DK50306). This work was supported by the NIDDK (R01 DK055342 and P01 DK049210) and the Deutsche Forschungsgemeinschaft (Be2296/2-1).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson AM, Jorgensen N, Frydelund-Larsen L, Rajpert-De Meyts E, Skakkebaek NE. Impaired Leydig cell function in infertile men: a study of 357 idiopathic infertile men and 318 proven fertile controls. J Clin Endocrinol Metab. 2004;89:3161–7. doi: 10.1210/jc.2003-031786. [DOI] [PubMed] [Google Scholar]
- Behr R, Weinbauer GF. Germ cell-specific cyclic adenosine 3′,5′-monophosphate response element modulator expression in rodent and primate testis is maintained despite gonadotropin deficiency. Endocrinology. 1999;140:2746–54. doi: 10.1210/endo.140.6.6764. [DOI] [PubMed] [Google Scholar]
- Behringer RR, Finegold MJ, Cate RL. Mullerian-inhibiting substance function during mammalian sexual development. Cell. 1994;79:415–25. doi: 10.1016/0092-8674(94)90251-8. [DOI] [PubMed] [Google Scholar]
- Blaber M, Isackson PJ, Bradshaw RA. A complete cDNA sequence for the major epidermal growth factor binding protein in the male mouse submandibular gland. Biochemistry. 1987;26:6742–9. doi: 10.1021/bi00395a025. [DOI] [PubMed] [Google Scholar]
- Clements JA, Willemsen NM, Myers SA, Dong Y. The tissue kallikrein family of serine proteases: functional roles in human disease and potential as clinical biomarkers. Crit Rev Clin Lab Sci. 2004;41:265–312. doi: 10.1080/10408360490471931. [DOI] [PubMed] [Google Scholar]
- Cooke HJ, Saunders PT. Mouse models of male infertility. Nat Rev Genet. 2002;3:790–801. doi: 10.1038/nrg911. [DOI] [PubMed] [Google Scholar]
- de Rooij DG. Proliferation and differentiation of spermatogonial stem cells. Reproduction. 2001;121:347–54. doi: 10.1530/rep.0.1210347. [DOI] [PubMed] [Google Scholar]
- de Rooij DG, de Boer P. Specific arrests of spermatogenesis in genetically modified and mutant mice. Cytogenet Genome Res. 2003;103:267–76. doi: 10.1159/000076812. [DOI] [PubMed] [Google Scholar]
- Diamandis EP, Yousef GM. Human tissue kallikreins: a family of new cancer biomarkers. Clin Chem. 2002;48:1198–205. [PubMed] [Google Scholar]
- Escalier D. Impact of genetic engineering on the understanding of spermatogenesis. Hum Reprod Update. 2001;7:191–210. doi: 10.1093/humupd/7.2.191. [DOI] [PubMed] [Google Scholar]
- Gnessi L, Basciani S, Mariani S, Arizzi M, Spera G, Wang C, Bondjers C, Karlsson L, Betsholtz C. Leydig cell loss and spermatogenic arrest in platelet-derived growth factor (PDGF)-A-deficient mice. J Cell Biol. 2000;149:1019–26. doi: 10.1083/jcb.149.5.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnessi L, Fabbri A, Spera G. Gonadal peptides as mediators of development and functional control of the testis: an integrated system with hormones and local environment. Endocr Rev. 1997;18:541–609. doi: 10.1210/edrv.18.4.0310. [DOI] [PubMed] [Google Scholar]
- Hiemisch H, Schütz G, Kaestner KH. Transcriptional regulation in endoderm development: Characterization of an enhancer controlling Hnf3g expression by transgenesis and targeted mutagenesis. EMBO J. 1997;16:3995–4006. doi: 10.1093/emboj/16.13.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huleihel M, Lunenfeld E. Regulation of spermatogenesis by paracrine/autocrine testicular factors. Asian J Androl. 2004;6:259–68. [PubMed] [Google Scholar]
- Jegou B, Laws AO, de Kretser DM. Changes in testicular function induced by short-term exposure of the rat testis to heat: further evidence for interaction of germ cells, Sertoli cells and Leydig cells. Int J Androl. 1984;7:244–57. doi: 10.1111/j.1365-2605.1984.tb00781.x. [DOI] [PubMed] [Google Scholar]
- Kaestner KH, Hiemisch H, Luckow B, Schutz G. The HNF-3 gene family of transcription factors in mice: gene structure, cDNA sequence, and mRNA distribution. Genomics. 1994;20:377–85. doi: 10.1006/geno.1994.1191. [DOI] [PubMed] [Google Scholar]
- Kaestner KH, Hiemisch H, Schutz G. Targeted disruption of the gene encoding hepatocyte nuclear factor 3gamma results in reduced transcription of hepatocyte-specific genes. Mol Cell Biol. 1998;18:4245–51. doi: 10.1128/mcb.18.7.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai E, Prezioso VR, Tao WF, Chen WS, Darnell JE., Jr Hepatocyte nuclear factor 3 alpha belongs to a gene family in mammals that is homologous to the Drosophila homeotic gene fork head. Genes-Dev. 1991;5:416–27. doi: 10.1101/gad.5.3.416. [DOI] [PubMed] [Google Scholar]
- Lee CS, Sund NJ, Behr R, Herrera PL, Kaestner KH. Foxa2 is required for the differentiation of pancreatic alpha-cells. Dev Biol. 2005;278:484–95. doi: 10.1016/j.ydbio.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Lundwall A, Band V, Blaber M, Clements JA, Courty Y, Diamandis EP, Fritz H, Lilja H, Malm J, Maltais LJ, Olsson AY, Petraki C, Scorilas A, Sotiropoulou G, Stenman UH, Stephan C, Talieri M, Yousef GM. A comprehensive nomenclature for serine proteases with homology to tissue kallikreins. Biol Chem. 2006;387:637–41. doi: 10.1515/BC.2006.082. [DOI] [PubMed] [Google Scholar]
- Matsui H, Moriyama A, Takahashi T. Cloning and characterization of mouse klk27, a novel tissue kallikrein expressed in testicular Leydig cells and exhibiting chymotrypsin-like specificity. Eur J Biochem. 2000;267:6858–65. doi: 10.1046/j.1432-1033.2000.01786.x. [DOI] [PubMed] [Google Scholar]
- Matsui H, Takahashi T. Mouse testicular Leydig cells express Klk21, a tissue kallikrein that cleaves fibronectin and IGF-binding protein-3. Endocrinology. 2001;142:4918–29. doi: 10.1210/endo.142.11.8505. [DOI] [PubMed] [Google Scholar]
- Matsui H, Takano N, Takahashi T. Characterization of mouse glandular kallikrein 24 expressed in testicular Leydig cells. Int J Biochem Cell Biol. 2005;37:2333–43. doi: 10.1016/j.biocel.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Lamb DJ. Genetic dissection of mammalian fertility pathways. Nat Cell Biol. 2002;4(Suppl):s41–9. doi: 10.1038/ncb-nm-fertilityS41. [DOI] [PubMed] [Google Scholar]
- Monsees TK, Schill WB, Miska W. Protease-protease inhibitor interactions in Sertoli cell-germ cell crosstalk. Adv Exp Med Biol. 1997;424:111–23. doi: 10.1007/978-1-4615-5913-9_20. [DOI] [PubMed] [Google Scholar]
- Mruk DD, Cheng CY. Sertoli-Sertoli and Sertoli-germ cell interactions and their significance in germ cell movement in the seminiferous epithelium during spermatogenesis. Endocr Rev. 2004;25:747–806. doi: 10.1210/er.2003-0022. [DOI] [PubMed] [Google Scholar]
- Nieschlag E. Scope and Goals of Andrology. In: Nieschlag H, Behre E, editors. Andrology: Male Reproductive Health and Dysfunction. Springer; Berlin, Heidelberg, New York: 2001. [Google Scholar]
- O’Donnell L, Robertson KM, Jones ME, Simpson ER. Estrogen and spermatogenesis. Endocr Rev. 2001;22:289–318. doi: 10.1210/edrv.22.3.0431. [DOI] [PubMed] [Google Scholar]
- Qian YM, Sun XJ, Tong MH, Li XP, Richa J, Song WC. Targeted disruption of the mouse estrogen sulfotransferase gene reveals a role of estrogen metabolism in intracrine and paracrine estrogen regulation. Endocrinology. 2001;142:5342–50. doi: 10.1210/endo.142.12.8540. [DOI] [PubMed] [Google Scholar]
- Rich KA, Kerr JB, de Kretser DM. Evidence for Leydig cell dysfunction in rats with seminiferous tubule damage. Mol Cell Endocrinol. 1979;13:123–35. doi: 10.1016/0303-7207(79)90013-3. [DOI] [PubMed] [Google Scholar]
- Shannon M, Ashworth LK, Mucenski ML, Lamerdin JE, Branscomb E, Stubbs L. Comparative analysis of a conserved zinc finger gene cluster on human chromosome 19q and mouse chromosome 7. Genomics. 1996;33:112–20. doi: 10.1006/geno.1996.0166. [DOI] [PubMed] [Google Scholar]
- Shannon M, Stubbs L. Analysis of homologous XRCC1-linked zinc-finger gene families in human and mouse: evidence for orthologous genes. Genomics. 1998;49:112–21. doi: 10.1006/geno.1998.5230. [DOI] [PubMed] [Google Scholar]
- Sharpe RM, McKinnell C, Kivlin C, Fisher JS. Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood. Reproduction. 2003;125:769–84. doi: 10.1530/rep.0.1250769. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. letter. [DOI] [PubMed] [Google Scholar]
- Thomas KA, Baglan NC, Bradshaw RA. The amino acid sequence of the gamma-subunit of mouse submaxillary gland 7 S nerve growth factor. J Biol Chem. 1981;256:9156–66. [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP, Cooke HJ. Lessons from knockout and transgenic mice for infertility in men. J Endocrinol Invest. 2000;23:584–91. doi: 10.1007/BF03343780. [DOI] [PubMed] [Google Scholar]
- Voltz R, Gultekin SH, Rosenfeld MR, Gerstner E, Eichen J, Posner JB, Dalmau J. A serologic marker of paraneoplastic limbic and brain-stem encephalitis in patients with testicular cancer. N Engl J Med. 1999;340:1788–95. doi: 10.1056/NEJM199906103402303. [DOI] [PubMed] [Google Scholar]
- Vornberger W, Prins G, Musto NA, Suarez-Quian CA. Androgen receptor distribution in rat testis: new implications for androgen regulation of spermatogenesis. Endocrinology. 1994;134:2307–16. doi: 10.1210/endo.134.5.8156934. [DOI] [PubMed] [Google Scholar]
- Weinbauer GF, Wessels J. ‘Paracrine’ control of spermatogenesis. Andrologia. 1999;31:249–62. doi: 10.1046/j.1439-0272.1999.00295.x. [DOI] [PubMed] [Google Scholar]
- Weissig H, Narisawa S, Sikstrom C, Olsson PG, McCarrey JR, Tsonis PA, Del Rio-Tsonis K, Millan JL. Three novel spermatogenesis-specific zinc finger genes. FEBS Lett. 2003;547:61–8. doi: 10.1016/s0014-5793(03)00669-0. [DOI] [PubMed] [Google Scholar]
- Wijsman JH, Jonker RR, Keijzer R, van de Velde CJ, Cornelisse CJ, van Dierendonck JH. A new method to detect apoptosis in paraffin sections: in situ end-labeling of fragmented DNA. J Histochem Cytochem. 1993;41:7–12. doi: 10.1177/41.1.7678025. [DOI] [PubMed] [Google Scholar]
- Wu Z, Irizarry R, Gentleman R, Murillo F, Spencer F. A Model Based Background Adjustment for Oligonucleotide Expression Arrays. Technical Report. Johns Hopkins University; Baltimore: 2003. [Google Scholar]