

Abstract
In prior studies, we have observed that HO activity protects astrocytes from heme-mediated injury, but paradoxically increases neuronal injury. In this study, we tested the hypothesis that an adenovirus encoding the human HO-1 gene driven by an enhanced glial fibrillary acidic protein promoter (Ad-GFAP-HO-1) would increase HO-1 expression selectively in astrocytes, and provide cytoprotection. Treatment with 100 MOI Ad-GFAP-HO-1 for 24h resulted in HO-1 expression that was 6.4-fold higher in cultured primary astrocytes than in neurons. Astrocyte HO activity was increased by approximately fourfold over baseline, which was sufficient to reduce cell death after 24 h hemin exposure by 60%, as assessed by both MTT and LDH release assays. A similar reduction in cell protein oxidation, quantified by carbonyl assay, was also observed. These results suggest that HO-1 transgene expression regulated by an enhanced GFAP promoter selectively increases HO-1 expression in astrocytes, and is cytoprotective. Further investigation of this strategy in vivo is warranted.
Keywords: cell culture, free radical, gene therapy, iron, hemoglobin toxicity, intracerebral hemorrhage, oxidative stress, stroke
Introduction
A growing body of experimental evidence supports the hypothesis that extracellular hemoglobin (Hb) contributes to iron-dependent oxidative injury after intracerebral hemorrhage. Since the heme groups of Hb are sequestered in hydrophobic pockets that limit their ability to participate in free radical reactions (Hebbel and Eaton, 1989), the deleterious effect of Hb is likely mediated by heme transfer to membrane lipids and proteins (Bunn and Jandl, 1967). This process is most efficient when Hb autoxidizes to methemoglobin (Balla et al., 1993), which occurs in intracranial hematomas (Bradley, 1993). Hemin, the oxidized form of heme, is a highly reactive compound that produces an iron-dependent injury to both neurons and astrocytes in cell culture and in vivo (Huang et al., 2002; Regan et al., 2002; Goldstein et al., 2003).
Under physiologic conditions, the cellular concentration of free heme is tightly regulated, and is generally maintained at <10−9M (Taketani, 2005). However, it increases significantly in CNS cells exposed to micromolar concentrations of extracellular Hb or hemin (Chen-Roetling et al., 2005; Chen-Roetling and Regan, 2006), suggesting that endogenous catabolic mechanisms may be insufficient to maintain homeostasis in cells adjacent to an intracerebral hematoma. Heme breakdown is mediated by the heme oxygenases (HO), which catalyze the rate-limiting step in its conversion to bilirubin, carbon monoxide, and iron. Two HO isoforms have been characterized in the CNS to date, which are the products of separate genes, but share similar mechanisms for substrate recognition and heme degradation (Chang et al., 2005). HO-1 is normally expressed at a very low level, but is rapidly induced in astrocytes, microglia, and some neurons by extravascular Hb, hemin, and a variety of oxidants (Matz et al., 1997; Turner et al., 1998). HO-2 is constitutively expressed, predominantly by neurons (Matz et al., 1997).
Prior cell culture studies suggest that HO has considerable but opposite effects on heme-mediated injury to astrocytes and neurons. Consistent with the benefit provided by HO in ischemia and trauma models (Doré et al., 1999; Chang et al., 2003), HO-1 knockout astrocytes were more vulnerable to Hb or hemin (Chen-Roetling et al., 2005; Chen-Roetling and Regan, 2006), while increasing astrocyte expression by genetic or pharmacologic means was protective (Teng et al., 2004; Chen and Regan, 2005). Putative protective mechanisms include the antioxidant effect of bilirubin production (Doré et al., 1999), and also the conversion of a lipid soluble oxidant, hemin, to iron, which is then sequestered in ferritin (Balla et al., 1992). Paradoxically, HO appears to accelerate Hb or hemin cytotoxicity in neurons in some cell culture and in vivo models (Rogers et al., 2003; Koeppen et al., 2004; Gong et al., 2006), presumably due to iron-mediated oxidative stress (Nakamura et al., 2004). Although the molecular basis for the discrepancy between neurons and astrocytes has not been precisely defined, it may reflect the limited ability of neurons to detoxify excess iron by increasing ferritin synthesis (Wu et al., 2003).
The disparate effect of HO on heme-mediated injury to neurons and astrocytes suggests that it may be a challenging therapeutic target. Although increasing HO expression in astrocytes has a potent and highly reproducible protective effect, nonselective approaches may also increase expression in neurons and thereby increase their vulnerability. A cell-specific approach may therefore be optimal. The glial fibrillary acidic protein (GFAP) promoter has previously been shown to direct gene expression with considerable specificity for astrocytes (Mucke et al., 1991; Brenner, 1994; Holland and Varmus, 1998; McKie et al., 1998; Morimoto et al., 2002). It is noteworthy, however, that this original GFAP promoter is relatively weak when compared with viral promoters such as the human CMV promoter (Morelli et al., 1999). A stronger driver of transgene expression may be preferable for mechanistic cell culture studies, and for in vivo investigation of the feasibility of gene therapy. Toward this end, novel second generation GFAP promoters have recently been described (de Leeuw et al., 2006). Due to redundancy of their enhancer regions, transgene expression driven by these promoters has been reported to be up to 75-fold greater than that provided by the parent GFAP promoter in the U251 glial cell line, without any loss of specificity (de Leeuw et al., 2006). However, their efficacy in cell injury models has not yet been reported. In the current study, we tested the hypothesis that transfer of the human HO-1 gene driven by an enhanced GFAP promoter would increase HO-1 specifically in primary cultured astrocytes, and lead to HO activity levels that would be sufficient to protect against heme-mediated oxidative injury.
Materials and Methods
Cell Cultures
Cortical primary astrocyte cultures were prepared from 1–2 day old BALB/c x 129/Sv mice as recently described in detail (Chen-Roetling et al., 2005). Plating medium contained Minimal Essential Medium (MEM, Invitrogen, Carlsbad, CA), 10% equine serum (Hyclone, Logan, UT), 10% fetal bovine serum (Hyclone), 2 mM glutamine, and 10 μg/ml epidermal growth factor (EGF, Sigma-Aldrich, St. Louis, MO). Two-thirds of the medium was replaced at 5 days in vitro and twice weekly thereafter. Feeding medium was similar to plating medium, except that it lacked fetal bovine serum and EGF, and contained 10% equine serum. Mixed neuron-astrocyte cultures were prepared from fetal mice at 15–17 days gestational age, also as previously described (Regan et al., 2004). They were fed twice weekly with the same maintenance medium used for astrocytes until 10 days in vitro, and subsequently were fed daily. Pure neuronal cultures were plated on 24-well plates coated with 50 μg/ml poly-D-lysine, in Neurobasal medium (Invitrogen) containing B27 supplement (Invitrogen). All cultures were incubated in a 5% CO2 atmosphere at 37°C.
Adenovirus Preparation
A plasmid containing the enhanced GFAP promoter pGfa2(ABD)3 -nLacZ (de Leeuw et al., 2006) was kindly provided by Dr. Michael Brenner (University of Alabama, Birmingham). Shuttle vector pCMV-HO-1 was previously provided by Dr. Lee-Young Chau (Academia Sinica, Taiwan ROC). Both constructs were used to prepare the shuttle adenoviral vector pDUAL:GFAP-HO-1. The GFAP promoter of pGfa2(ABD)3 -nLacZ was excised by digestion with BglII on the 5′ end and BamHI on the 3′ end, and the HO-1 gene open reading frame was excised by digestion with BamHI on the 5′ end and HindIII on 3′ end from pCMV-HO-1. Both fragments were introduced to adenoviral shuttle vector pDUAL-CCM (Vector Biolabs, Philadelphia, PA) creating pDUAL-GFAP-HO-1. This construct was sequenced and transfected to primary cultured HO-1 knockout astrocytes for expression evaluation. Sequence analysis demonstrated promoter – insert orientation and insert identity to the human HO-1 sequence, accession number NM 002133, using the Chromas sequences analyzing software and NCBI- The Basic Local Alignment Search Tool (BLAST).
SwaI endonuclease was used to release the insert from shuttle pDUAL-GFAP-HO-1 and ligate it directly into the viral plasmid vector (pAd –VEC). Adenoviral plasmid clones pAd-VEC-GFAP-HO-1 were checked for expression before virus preparation by transfecting HO-1 knockout astrocytes (5 days in vitro, 1μg DNA per well) using Lipofectamine plus reagent in serum-free medium (OptiMEM, Invitrogen). HO-1 expression was assessed via immunoblotting as described below. The viral construct was also verified using sequencing primers and restrictional analysis. For viral packaging in HEK293 cells, pAd –VEC-GFAP-HO-1 was transfected in linear form (digested with Pac-I), to produce Ad-GFAP-HO-1. After propagation and harvesting, titer was quantified by cytopathic effect assay.
Adenovirus encoding human HO-1 controlled by the CMV promoter (Ad-CMV-HO-1, Juan et al., 2001) was provided as a gift by Dr. Lee-Young Chau. Adenoviral infection of astrocytes was accomplished in medium similar to feeding medium, except that it contained 3.3% equine serum. In a prior study using this culture system, treatment with 100 MOI of serotype 5 adenovirus in this medium resulted in transfection of approximately 80% of astrocytes (Teng et al., 2004); similar efficacy has been observed for cultured neurons (unpublished observations).
Immunoblotting
Cells were homogenized in cold cell lysis buffer (210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1mM EDTA, 0.1% sodium dodecyl sulfate) and sonicated. After protein assay (Pierce, Rockford, IL), 20–25 μg samples of total proteins were separated on a 15% polyacrylamide gel (Bio Rad, Laboratories, Hercules, CA) and transferred to a polyvinylidene difluoride Imobilon-P transfer membrane filter (Millipore, Billerica, MA) using a semi-dry transfer apparatus. HO-1 protein expression was detected with 1:5000 dilution of rabbit anti-HO-1 antibody and HO-2 protein expression was detected with 1:2000 dilution of rabbit anti-HO-2 antibody (incubation period of 16h at 4°C, Stressgen Biotechnologies Corp., San Diego, CA), followed by one hour incubation with goat anti-rabbit IgG (1:20,000, Pierce, Rockford, IL). Immunoreactive proteins were visualized using Super Signal West Femto Reagent (Pierce) and Kodak Gel Logic 2200.
Cytotoxicity Assays
Cultures prepared from wild-type mice were washed free of growth medium and virus, and were placed into medium consisting of MEM with 10 mM glucose (MEM10). Hemin (Sigma-Aldrich) was diluted in this medium to final concentrations that were based on previously determined concentration-toxicity relationships in these cultures (Regan et al., 2004; Chen-Roetling et al., 2005). Plates were then incubated for 24 hours, with minimal disturbance.
Cell death was quantified by measuring lactate dehydrogenase (LDH) activity in the serum-free culture medium, as previously described (Regan and Choi, 1994). This assay correlates well with cell counts of cultures stained with trypan blue, a marker of membrane disruption (Koh and Choi, 1988). LDH values in astrocyte cultures were scaled to the mean value in sister cultures subjected to treatment with 0.3% Triton X-100, which lyses all cells and therefore releases all LDH. Cell viability in astrocyte cultures was also assessed via the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, which measures the ability of cells to reduce MTT to formazan, also as recently described (Chen and Regan, 2004). After acid isopropanol extraction, formazan absorbance was quantified at 562 nm with a reference wavelength of 650 nm. The absorbance associated with 100% astrocyte survival was determined from sister cultures treated with MEM10 only; that associated with 0% survival was determined from sister cultures treated with 0.3% Triton X-100. Cell death in mixed neuron-astrocyte cultures treated with hemin concentrations that injure only neurons was normalized to that in sister cultures treated with 3μM hemin for 48h, which kills all neurons but does not injure astrocytes (Regan et al., 2004).
Protein Oxidation Assay
Wild-type cells were homogenized in cold cell lysis buffer and sonicated. After centrifugation, the protein concentration on an aliquot of supernatant was determined. Proteins in the remaining supernatant were protected from further oxidation by addition of β-mercaptoethanol (0.5% final concentration), and were then denatured in 12% sodium dodecyl sulfate. Carbonyl groups were derivatized to 2,4-dintrophenylhydrazone by reaction with 2,4-dinitrophenyhydrazine, using the Oxyblot™ kit (Chemicon, Inc., Temecula, CA), and following the manufacturer’s instructions. After separation on a 12% polyacrylamede gel, proteins were then transferred onto a polyvinylidene difluoride transfer membrane filter as described above. Carbonylated proteins were detected with rabbit anti-DNP (1:150, Chemicon) followed by goat anti-rabbit IgG (1:300). Immunoreactive proteins were visualized as described above.
HO Activity Assay
HO activity was measured by quantification of carbon monoxide (CO) production via gas chromatography (Vreman and Stevenson, 1988). Sixty microliters of 150 μM methemalbumin and 13–18 μg cell protein were placed into septum-sealed 2-mL vials (Alltech Associates, Deerfield, IL). Reactions were initiated by addition of 60 μl of 4.5 mM NADPH (total reaction volume 180 μl, reactant concentrations: 50 μM methemalbumin, 1.5 mM NADPH). After purging vials with CO-free air, reactions were run at 37°C for 15 minutes, and then were rapidly stopped by placing vials on dry ice. CO in the vial headspace was quantified using a gas chromatograph (Peak Laboratories, Mountain View, CA), and extrapolated from a CO standard curve. HO activity was expressed as nmol CO/mg protein/h.
Statistical Analysis
Data were analyzed with one-way analysis of variance when three or more groups were compared. Differences between groups were then assessed with the Bonferroni multiple comparisons test. The unpaired two-tailed t test was used for data sets containing only two groups. Significance was assigned to a P value less than 0.05.
Results
Ad-GFAP-HO-1 increases astrocyte HO expression and activity
In initial experiments, HO-1 expression produced by Ad-GFAP-HO-1 was directly compared in cultured neurons and astrocytes. In order to eliminate endogenous HO-1 expression from this analysis, HO-1 knockout astrocytes (Yet et al., 1999) were used exclusively for these initial studies. After treatment with 100 MOI (multiplicity of infection) Ad-GFAP-HO-1 for 24 hours, expression was consistently greater in astrocyte cultures (Fig. 1), with a mean density that was 6.4-fold higher than in identically-treated pure neuronal cultures. HO-1 expression in cultures treated with Ad-GFAP-HO-1 was similar to that in cultures treated with the same dose of Ad-CMV-HO-1 (Fig. 2), and was increased by treating cultures with dibutyryl cAMP, which increases GFAP promoter activity in cultured astrocytes (Shafit-Zagardo et al., 1988). In wild-type astrocyte cultures, 100 MOI Ad-GFAP-HO-1 increased HO-1 protein expression 11-fold when compared with that in sister cultures treated with an empty control adenovirus (Fig. 3). In wild-type neuron cultures, the same treatments resulted in a 1.3-fold increase. Consistent with prior observations, HO-2 was constitutively expressed in astrocyte cultures; its level was not altered by virus treatment (Fig. 3).
Figure 1.
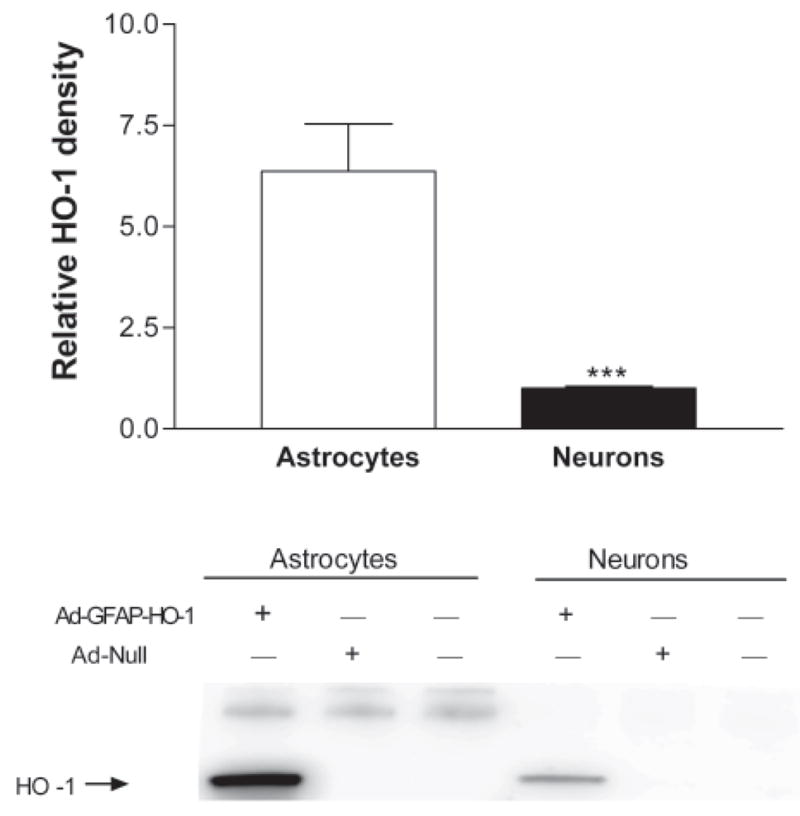
Effect of Ad-GFAP-HO-1 on HO-1 expression in neurons and astrocytes. Pure astrocyte and pure neuron cultures derived from HO-1 knockout mice were treated with 100 MOI Ad-GFAP-HO-1 for 24 hours. Control cultures were treated with empty adenovirus (Ad-Null) or subjected to media exchanges only. Bars represent mean density of HO-1 expression (± S.E.M., n = 10–11/condition). ***P = 0.0001, unpaired two-tailed t test.
Figure 2.
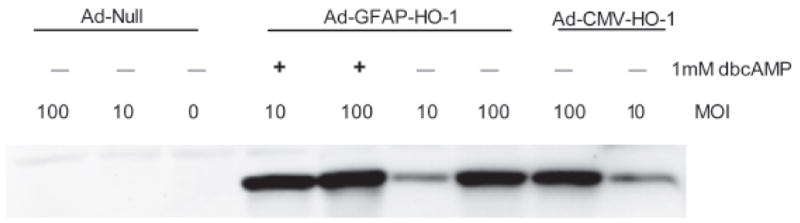
Comparison of HO-1 expression after treatment with Ad-GFAP-HO-1 or Ad-CMV-HO-1. HO-1 knockout astrocyte cultures were treated with 0, 10, or 100 MOI Ad-Null, Ad-CMV-HO-1, or Ad-GFAP-HO-1 for 24 hours. Additional cultures that were treated with Ad-GFAP-HO-1 were also treated with 1 mM dibutyryl cAMP (dbcAMP) to increase GFAP promoter activity.
Figure 3.
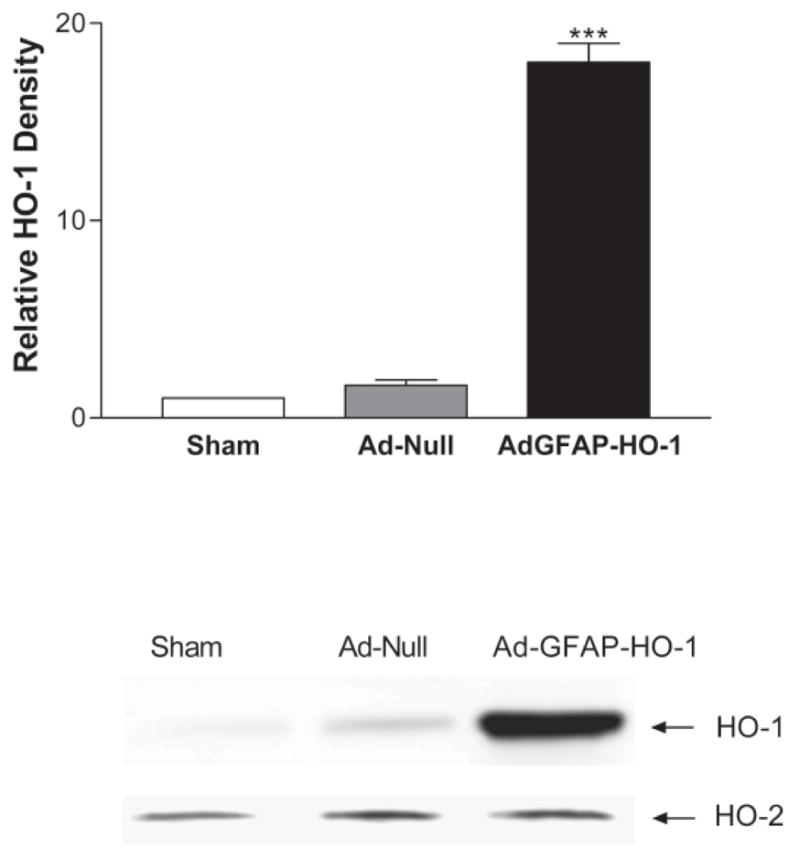
Effect of adenovirus treatment on HO-1 and HO-2 expression in wild-type astrocyte cultures. Cultures were treated with 100 MOI Ad-GFAP-HO-1 for 24 hours. Control cultures were treated with empty adenovirus (100 MOI Ad-Null) or were subjected to medium exchange only (Sham). Representative immunoblots were stained with anti-HO-1 or anti-HO-2. Bars represent mean HO-1 band density (± S.E.M., n = 4/condition). ***P <0.001 vs. culture treated with control Ad-Null, Bonferroni multiple comparisons test.
HO-1 gene transfer using a viral vector has been reported to produce HO activity that is considerably weaker than would be expected based on analysis of HO-1 protein expression (Quan et al., 2002). HO activity was therefore quantified by carbon monoxide production assay in wild-type astrocytes treated with 100 MOI Ad-GFAP-HO-1, Ad-CMV-HO-1, or a control empty adenovirus (Ad-Null). Compared with the latter, Ad-GFAP-HO-1 increased HO activity by approximately fourfold, compared with a 5.8-fold increase produced by exposure to the same dose of Ad-CMV-HO-1 (Fig. 4).
Figure 4.

HO activity is increased by adenoviral gene transfer in wild-type astrocytes. Mean rate of CO production (± SEM) in sonicates of cultures treated for 24 hours with 100 MOI of control virus (Ad-Null), or same dose of adenovirus encoding the human HO-1 gene under either the CMV (Ad-CMV-HO-1) or GFAP (Ad-GFAP-HO-1) promoters. The difference in activity between cultures treated with Ad-CMV-HO-1 and Ad-GFAP-HO-1 is not statistically significant. *P < 0.05, **P < 0.01 v. activity in cultures treated with Ad-Null, Bonferrroni multiple comparisons test.
Ad-GFAP-HO-1 protects astrocytes from heme-mediated oxidative injury
Consistent with prior observations, treatment of wild-type astrocyte cultures with hemin for 24 hours resulted in cell body retraction, nuclear pyknosis, and detachment of some cells from the culture plate (Fig. 5), resulting in disruption of the culture monolayer. The cell monolayer in sister cultures subjected to medium exchange only remained intact, and was unchanged from baseline. The morphologic appearance of cultures pretreated with 100 MOI of a control empty adenovirus followed by hemin was similar to that of cultures treated with hemin only, while pretreatment with 100 MOI Ad-GFAP-HO-1 largely preserved the normal culture appearance despite subsequent hemin treatment.
Figure 5.
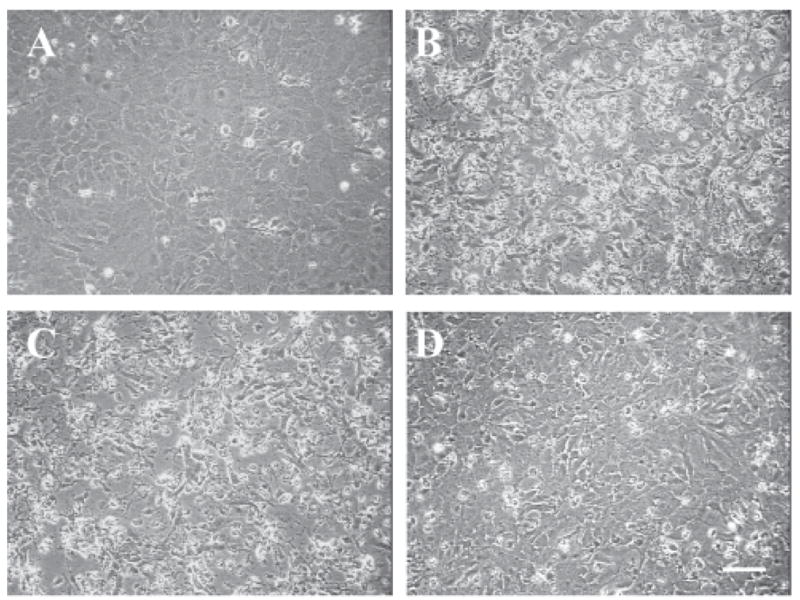
Ad-GFAP-HO-1 protects wild-type astrocytes from hemin. Phase-contrast photomicrographs of astrocyte cultures, 24 hours after: A) medium exchange only; B) treatment with 10 μM hemin; the culture monolayer is no longer confluent, and most remaining astrocytes have developed a granular appearance with pyknotic nuclei; C) treatment with 10 μM hemin after 24 hour incubation with 100 MOI Ad-Null; D) treatment with 10 μM hemin after 24 hour incubation with 100 MOI Ad-GFAP-HO-1; culture monolayer remains intact. Scale bar = 50 μm.
In cultures pretreated with Ad-Null followed by 10 μM hemin, 81.4±3.9% of cellular LDH was released into the culture medium by 24 hours (Fig. 6). Medium LDH activity was reduced to 28.2±2.9% of full-kill control cultures by pretreatment with 100 MOI Ad-GFAP-HO-1. Cell viability, as detected by the MTT assay, in cultures pretreated with Ad-Null followed by hemin for 24 hours was 23.8±1.4% of that in control cultures subjected to medium change only, and was increased to 59.8±2.1% by pretreatment with 100 MOI Ad-GFAP-HO-1.
Figure 6.
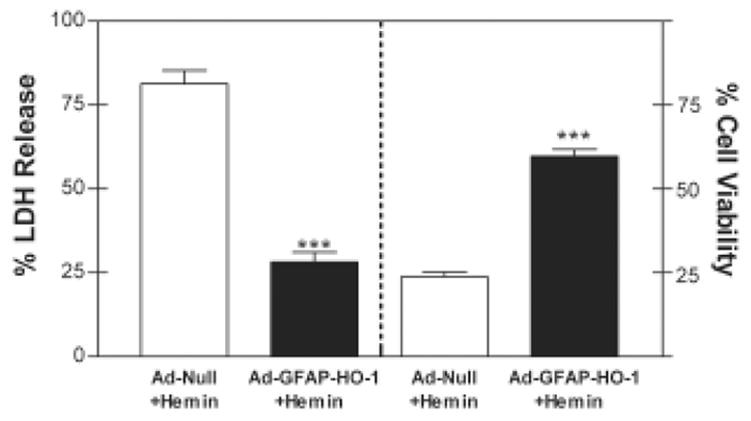
Quantification of the protective effect of Ad-GFAP-HO-1. Mean culture percentage LDH release (left axis) and percentage cell viability (right axis) as assessed by MTT assay (± SEM, n = 7–10/condition), 24 hours after treatment with 10 μM hemin, preceded by 24 hour treatment with 100 MOI Ad-GFAP-HO-1 or 100 MOI control Ad-Null. ***P < 0.001, unpaired two-tailed t test.
Quantification of protein oxidation by carbonyl assay is an accurate marker of heme-mediated injury in this culture system (Chen-Roetling et al., 2005). In cultures pretreated with Ad-Null followed by hemin, the mean carbonyl signal intensity was approximately 14-fold higher than that in controls subjected to medium exchange only (Fig. 7). Approximately three-fourths of this increase was prevented by pretreatment with Ad-GFAP-HO-1, while Ad-Null had no significant effect.
Figure 7.

Astrocyte protein oxidation after hemin treatment is reduced by Ad-GFAP-HO-1. Upper figure is a representative immunoblot of lysates from astrocyte cultures treated with: Sham, medium exchange only; Hemin/AdNull, 10 μM hemin for 24 hours, after pretreatment for 24 hours with 100 MOI Ad-Null; Hemin, 10 μM hemin only for 24 hours, without virus pretreatment; Hemin/AdGFAP, 10 μM hemin for 24 hours, after pretreatment for 24 hours with 100 MOI Ad-GFAP-HO-1. Bars represent mean carbonyl signal intensities (± SEM, n = 5/condition). ***P < 0.001 v. hemin alone, # # #P < 0.001 v. sham, Bonferroni multiple comparisons test.
Effect of Ad-GFAP-HO-1 on heme-mediated neuronal injury
Mixed neuron-astrocyte cultures were exposed to 3 μM hemin for 24 hours or to 10 μM hemin for 4 hours. Prior experiments had determined that these exposures produce cell injury in mixed cultures that is limited to neurons (Regan et al., 2004), and thereby permit specific investigation of heme-mediated neuronal injury under conditions that may be somewhat more physiologic than those produced by pure neuronal cultures. Treatment with 10 μM hemin for 4 hours released 20.6±4.6% of neuronal LDH when preceded by 24 h treatment with 100 MOI Ad-Null, and 26.8±5.4% after pretreatment with 100 MOI Ad-GFAP-HO-1 (P = 0.39). The difference was likewise insignificant when the 24 h exposure paradigm was used (59.4±7.9% and 75.4±8.2%, respectively, P = 0.18).
Discussion
The results of this study suggest three main conclusions. First, transfer of the HO-1 gene driven by the enhanced GFAP promoter increases HO-1 protein expression in astrocytes, with considerable but not absolute specificity when compared with neuronal expression. Second, this HO-1 gene transfer results in a marked increase in HO activity, as quantified with a sensitive carbon monoxide assay. Third, this activity level is sufficient to protect astrocytes from heme-mediated injury, while having no significant effect on neuronal injury.
Although the GFAP promoter was a potent driver of HO-1 expression in astrocytes, it was not completely specific, since a low level of expression was observed in pure neuron cultures. In agreement with this observation, some neuronal gene expression directed by the first generation GFAP promoter has previously been observed by Su et al.(Su et al., 2004), and varied considerably with transfer of different reporter genes. In that study, very high neuronal expression was observed for the protective protein/cathepsin A minigene, while moderate expression was noted for lacZ; green fluorescent protein expression was below the detection limit. The present results suggest that neuronal HO-1 expression driven by a second generation GFAP promoter is detectable but weak, and in this model did not directly alter neuronal vulnerability to hemin. However, neuronal injury was assessed in mixed neuron-astrocyte cultures after exposure to a low concentration of hemin that kills only neurons. Since this situation is unlikely to occur after intracranial hemorrhage in vivo (Letarte et al., 1993), reduction of astrocyte injury may actually provide secondary benefit to neurons, due to a variety of trophic effects (Swanson et al., 2004).
Prior studies have demonstrated that hyperexpression of HO-1 by genetic or pharmacologic approaches may result in a relatively weak increase in cell HO activity, suggesting that much of the expressed protein is nonfunctional (Quan et al., 2002; Quan et al., 2004; Teng et al., 2004; Botros et al., 2005). Quantification of activity may therefore be a more accurate index of the therapeutic potential of a drug or construct in a particular cell population than measurement of protein expression per se. The fourfold increase in HO activity produced by 100 MOI Ad-GFAP-HO-1 in primary cultured astrocytes is similar to the maximal effect that has been reported in other gene transfer studies (Abraham et al., 2002; Liu et al., 2002). The molecular basis for the discrepancy in HO-1 protein expression and activity has not been defined. Although HO-1 is a phosphoprotein (Salim et al., 2001), the relationship between its phosphorylation state and its catalytic activity has not been reported. It is noteworthy that HO-1 activity may be diminished by interaction with HO-2 (Weng et al., 2003), which may be relevant to cell populations expressing high levels of the latter. Further investigation of the post-translational regulation of HO-1 catalytic activity seems warranted.
The antioxidant effect of HO is usually attributed to an increased concentration of cell bilirubin, which is generated as a consequence of heme breakdown. While this protective mechanism may be highly relevant to neurons and perhaps other cell types (Doré et al., 1999), prior studies in astrocytes have demonstrated that exogenous bilirubin has no effect on heme-mediated oxidative injury (Regan et al., 2000; Chen-Roetling and Regan, 2006). Although bilirubin is clearly a potent antioxidant (Stocker et al., 1987), its efficacy in astrocytes may be specifically limited by two mechanisms: rapid mitochondrial catabolism and rapid export from cells via multi-drug resistance protein-1 (Hansen and Allen, 1997; Gennuso et al., 2004). The protective effect of HO in astrocytes may rather be due, at least in part, to conversion of the potent pro-oxidant hemin to iron, which can then be detoxified by sequestration in ferritin (Balla et al., 1992). Consistent with this hypothesis, cultured astrocytes are significantly more sensitive to hemin than to low molecular weight iron, particularly when lacking either the HO-1 or HO-2 genes (Chen and Regan, 2004; Chen-Roetling et al., 2005). In addition, release of carbon monoxide may contribute to the beneficial effect of HO (Ryter and Otterbein, 2004).
The increase in HO-1 expression and activity provided by Ad-GFAP-HO-1 is consistent with our prior observations that over 95% of cells in this astrocyte culture system express GFAP that is detectable by immunostaining, and that approximately 80% are transfected when treated with 100 MOI of serotype 5 adenovirus for 24 hours (Teng et al., 2004). Although the present results suggest that gene transfer driven by the enhanced GFAP promoter may be a useful tool for in vitro experiments, its efficacy in vivo remains to be defined. Precedent for therapeutic gene transfer using a viral vector in CNS hemorrhage models has been provided by Ono et al., who observed that adenoviral transfer of the HO-1 gene attenuated basilar artery vasospasm in a rat model of subarachnoid hemorrhage (Ono et al., 2002). In addition, Masada et al. reported that intraventricular injection of an adenovirus encoding interleukin-1 receptor antagonist attenuated tissue edema and the local inflammatory response after experimental striatal hemorrhage (Masada et al., 2001). Hemorrhagic stroke may be more amenable to gene therapy than ischemic stroke, since the component of injury produced by hemoglobin is delayed for 2–3 days (Xi et al., 1998), which may reflect the time required for erythrocyte lysis and hemoglobin oxidation. This interval may be sufficient for introduction of a viral vector and transgene expression. It is noteworthy that the HO-1 hyperexpression produced by Ad-GFAP-HO-1 in this culture system is very similar to that produced by hemin alone (Benvenisti-Zarom et al., 2006), although the latter is quite toxic per se. The timing of HO-1 hyperexpression, i.e. prior to toxic hemin exposure, appears to be a critical determinant of its protective effect (Teng et al., 2003). The results of this study suggest that HO-1 hyperexpression can be rapidly accomplished in astrocytes by HO-1 gene transfer driven by an enhanced GFAP promoter. Further investigation of this approach in experimental intracerebral hemorrhage models therefore seems warranted.
Acknowledgments
This study was supported by a grant from the National Institutes of Health (NS50662) to RFR, and by a postdoctoral fellowship from the Pennsylvania/Delaware affiliate of the American Heart Association to LBZ (award number 0525457U). The authors thank Dr. Michael Brenner, University of Alabama, Birmingham, for providing a plasmid containing the enhanced GFAP promoter. They also thank Dr Lee-Young Chau, Academia Sinica, Taiwan ROC, for providing shuttle vector pCMV-HO-1 and Ad-CMV-HO-1. They also thank Ms. Wei Wang for mouse genotyping and other technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham NG, Quan S, Mieyal PA, Yang L, Burke-Wolin T, Mingone CJ, Goodman AI, Nasjletti A, Wolin MS. Modulation of cGMP by human HO-1 retrovirus gene transfer in pulmonary microvessel endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1117–1124. doi: 10.1152/ajplung.00365.2001. [DOI] [PubMed] [Google Scholar]
- Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, Vercellotti GM. Ferritin: A cytoprotective strategem of endothelium. J Biol Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci U S A. 1993;90:9285–9289. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenisti-Zarom L, Chen-Roetling J, Regan RF. Inhibition of the ERK/MAP kinase pathway attenuates heme oxygenase-1 expression and heme-mediated neuronal injury. Neurosci Lett. 2006;398:230–234. doi: 10.1016/j.neulet.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Botros FT, Schwartzman ML, Stier CT, Jr, Goodman AI, Abraham NG. Increase in heme oxygenase-1 levels ameliorates renovascular hypertension. Kidney Int. 2005;68:2745–2755. doi: 10.1111/j.1523-1755.2005.00745.x. [DOI] [PubMed] [Google Scholar]
- Bradley WG., Jr MR appearance of hemorrhage in the brain. Radiology. 1993;189:15–26. doi: 10.1148/radiology.189.1.8372185. [DOI] [PubMed] [Google Scholar]
- Brenner M. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn HF, Jandl JH. Exchange of heme along hemoglobins and between hemoglobin and albumin. J Biol Chem. 1967;243:465–475. [PubMed] [Google Scholar]
- Chang EF, Claus CP, Vreman HJ, Wong RJ, Noble-Haeusslein LJ. Heme regulation in traumatic brain injury: relevance to the adult and developing brain. J Cereb Blood Flow Metab. 2005;25:1401–1417. doi: 10.1038/sj.jcbfm.9600147. [DOI] [PubMed] [Google Scholar]
- Chang EF, Wong RJ, Vreman HJ, Igarashi T, Galo E, Sharp FR, Stevenson DK, Noble-Haeusslein LJ. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J Neurosci. 2003;23:3689–3696. doi: 10.1523/JNEUROSCI.23-09-03689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Regan RF. Heme oxygenase-2 gene deletion increases astrocyte vulnerability to hemin. Biochem Biophys Res Commun. 2004;318:88–94. doi: 10.1016/j.bbrc.2004.03.187. [DOI] [PubMed] [Google Scholar]
- Chen J, Regan RF. Increasing expression of heme oxygenase-1 by proteasome inhibition protects astrocytes from heme-mediated oxidative injury. Curr Neurovasc Res. 2005;2:189–196. doi: 10.2174/1567202054368344. [DOI] [PubMed] [Google Scholar]
- Chen-Roetling J, Benvenisti-Zarom L, Regan RF. Cultured astrocytes from heme oxygenase-1 knockout mice are more vulnerable to heme-mediated oxidative injury. J Neurosci Res. 2005;82:802–810. doi: 10.1002/jnr.20681. [DOI] [PubMed] [Google Scholar]
- Chen-Roetling J, Regan RF. Effect of heme oxygenase-1 on the vulnerability of astrocytes and neurons to hemoglobin. Biochem Biophys Res Commun. 2006;350:233–237. doi: 10.1016/j.bbrc.2006.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw B, Su M, ter Horst M, Iwata S, Rodijk M, Hoeben RC, Messing A, Smitt PS, Brenner M. Increased glia-specific transgene expression with glial fibrillary acidic protein promoters containing multiple enhancer elements. J Neurosci Res. 2006;83:744–753. doi: 10.1002/jnr.20776. [DOI] [PubMed] [Google Scholar]
- Doré S, Takahashi M, Ferris CD, Hester LD, Guastella D, Snyder SH. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci USA. 1999;96:2445–2450. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennuso F, Fernetti C, Tirolo C, Testa N, L’Episcopo F, Caniglia S, Morale MC, Ostrow JD, Pascolo L, Tiribelli C, Marchetti B. Bilirubin protects astrocytes from its own toxicity by inducing up-regulation and translocation of multidrug resistance-associated protein 1 (Mrp1) Proc Natl Acad Sci U S A. 2004;101:2470–2475. doi: 10.1073/pnas.0308452100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein L, Teng ZP, Zeserson E, Patel M, Regan RF. Hemin induces an iron-dependent, oxidative injury on human neuron-like cells. J Neurosci Res. 2003;73:113–121. doi: 10.1002/jnr.10633. [DOI] [PubMed] [Google Scholar]
- Gong Y, Tian H, Xi G, Keep RF, Hoff JT, Hua Y. Systemic zinc protoporphyrin administration reduces intracerebral hemorrhage-induced brain injury. Acta Neurochir Suppl. 2006;96:232–236. doi: 10.1007/3-211-30714-1_50. [DOI] [PubMed] [Google Scholar]
- Hansen TW, Allen JW. Oxidation of bilirubin by brain mitochondrial membranes--dependence on cell type and postnatal age. Biochem Mol Med. 1997;60:155–160. doi: 10.1006/bmme.1996.2565. [DOI] [PubMed] [Google Scholar]
- Hebbel RP, Eaton JW. Pathobiology of heme interaction with the erythrocyte membrane. Sem Hematol. 1989;26:136–149. [PubMed] [Google Scholar]
- Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci USA. 1998;95:1218–1223. doi: 10.1073/pnas.95.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- Juan SH, Lee TS, Tseng KW, Liou JY, Shyue SK, Wu KK, Chau LY. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;104:1519–1525. doi: 10.1161/hc3801.095663. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Dickson AC, Smith J. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J Neuropathol Exp Neurol. 2004;63:587–597. doi: 10.1093/jnen/63.6.587. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Vulnerability of cultured cortical neurons to damage by excitotoxins: Differential susceptibility of neurons containing NADPH-diaphorase. J Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letarte PB, Lieberman K, Nagatani K, Haworth RA, Odell GB, Duff TA. Hemin: levels in experimental subarachnoid hematoma and effects on dissociated vascular smooth muscle cells. J Neurosurg. 1993;79:252–255. doi: 10.3171/jns.1993.79.2.0252. [DOI] [PubMed] [Google Scholar]
- Liu XM, Chapman GB, Wang H, Durante W. Adenovirus-mediated heme oxygenase-1 gene expression stimulates apoptosis in vascular smooth muscle cells. Circulation. 2002;105:79–84. doi: 10.1161/hc0102.101369. [DOI] [PubMed] [Google Scholar]
- Masada T, Hua Y, Xi G, Yang GY, Hoff JT, Keep RF. Attenuation of intracerebral hemorrhage and thrombin-induced brain edema by overexpression of interleukin-1 receptor antagonist. J Neurosurg. 2001;95:680–686. doi: 10.3171/jns.2001.95.4.0680. [DOI] [PubMed] [Google Scholar]
- Matz PG, Weinstein PR, Sharp FR. Heme oxygenase-1 and heat shock protein-70 induction in glia and neurons throughout rat brain after experimental intracerebral hemorrhage. Neurosurgery. 1997;40:152–160. doi: 10.1097/00006123-199701000-00034. [DOI] [PubMed] [Google Scholar]
- McKie EA, Graham DI, Brown SM. Selective astrocytic expression in vitro and in vivo from the GFAP promoter in a HSV RL1 null mutant vector-potential glioblastoma targeting. Gene Therapy. 1998;5:440–450. doi: 10.1038/sj.gt.3300621. [DOI] [PubMed] [Google Scholar]
- Morelli AE, Larregina AT, Smith-Arica J, Dewey RA, Southgate TD, Ambar B, Fontana A, Castro MG, Lowenstein PR. Neuronal and glial cell type-specific promoters within adenovirus recombinants restrict the expression of the apoptosis-inducing molecule Fas ligand to predetermined brain cell types, and abolish peripheral liver toxicity. J Gen Virol. 1999;80 (Pt 3):571–583. doi: 10.1099/0022-1317-80-3-571. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Cassell MD, Sigmund CD. Glia- and neuron-specific expression of the renin-angiotensin system in brain alters blood pressure, water intake, and salt preference. The Journal of Biological Chemistry. 2002;277:33235–33241. doi: 10.1074/jbc.M204309200. [DOI] [PubMed] [Google Scholar]
- Mucke L, Oldstone MB, Morris JC, Nerenberg MI. Rapid activation of astrocyte-specific expression of GFAP-lacZ transgene by focal injury. New Biol. 1991;3:465–474. [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004;100:672–678. doi: 10.3171/jns.2004.100.4.0672. [DOI] [PubMed] [Google Scholar]
- Ono S, Komuro T, Macdonald RL. Heme oxygenase-1 gene therapy for prevention of vasospasm in rats. J Neurosurg. 2002;96:1094–1102. doi: 10.3171/jns.2002.96.6.1094. [DOI] [PubMed] [Google Scholar]
- Quan S, Yang L, Shenouda S, Jiang H, Balazy S, Schwartzman ML, Shibahara I, Shinohara KNGA. Functional expression of human heme oxygenase-1 (HO-1) driven by HO-1 promoter in vitro and in vivo. J Cell Biochem. 2002;85:410–421. [PubMed] [Google Scholar]
- Quan S, Yang L, Shnouda S, Schwartzman ML, Nasjletti A, Goodman AI, Abraham NG. Expression of human heme oxygenase-1 in the thick ascending limb attenuates angiotensin II-mediated increase in oxidative injury. Kidney Int. 2004;65:1628–1639. doi: 10.1111/j.1523-1755.2004.00562.x. [DOI] [PubMed] [Google Scholar]
- Regan RF, Chen J, Benvenisti-Zarom L. Heme oxygenase-2 gene deletion attenuates oxidative stress in neurons exposed to extracellular hemin. BMC Neurosci. 2004;5:34. doi: 10.1186/1471-2202-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan RF, Choi DW. The effect of NMDA, AMPA/kainate, and calcium channel antagonists on traumatic cortical neuronal injury in culture. Brain Res. 1994;633:236–242. doi: 10.1016/0006-8993(94)91544-x. [DOI] [PubMed] [Google Scholar]
- Regan RF, Guo YP, Kumar N. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci Lett. 2000;282:1–4. doi: 10.1016/s0304-3940(00)00817-x. [DOI] [PubMed] [Google Scholar]
- Regan RF, Kumar N, Gao F, Guo YP. Ferritin induction protects cortical astrocytes from heme-mediated oxidative injury. Neuroscience. 2002;113:985–994. doi: 10.1016/s0306-4522(02)00243-9. [DOI] [PubMed] [Google Scholar]
- Rogers B, Yakopson V, Teng ZP, Guo Y, Regan RF. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Rad Biol Med. 2003;35:872–881. doi: 10.1016/s0891-5849(03)00431-3. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Otterbein LE. Carbon monoxide in biology and medicine. Bioessays. 2004;26:270–280. doi: 10.1002/bies.20005. [DOI] [PubMed] [Google Scholar]
- Salim M, Brown-Kipphut BA, Maines MD. Human biliverdin reductase is autophosphorylated, and phosphorylation is required for bilirubin formation. J Biol Chem. 2001;276:10929–10934. doi: 10.1074/jbc.M010753200. [DOI] [PubMed] [Google Scholar]
- Shafit-Zagardo B, Kume-Iwaki A, Goldman JE. Astrocytes regulate GFAP mRNA levels by cyclic AMP and protein kinase C-dependent mechanisms. Glia. 1988;1:346–354. doi: 10.1002/glia.440010507. [DOI] [PubMed] [Google Scholar]
- Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Su M, Hu H, Lee Y, d’Azzo A, Messing A, Brenner M. Expression specificity of GFAP transgenes. Neurochem Res. 2004;29:2075–2093. doi: 10.1007/s11064-004-6881-1. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004;4:193–205. doi: 10.2174/1566524043479185. [DOI] [PubMed] [Google Scholar]
- Taketani S. Aquisition, mobilization and utilization of cellular iron and heme: endless findings and growing evidence of tight regulation. Tohoku J Exp Med. 2005;205:297–318. doi: 10.1620/tjem.205.297. [DOI] [PubMed] [Google Scholar]
- Teng ZP, Chen J, Chau LY, Galunic N, Regan RF. Adenoviral transfer of the heme oxygenase-1 gene protects cortical astrocytes from heme-mediated oxidative injury. Neurobiol Dis. 2004;17:179–187. doi: 10.1016/j.nbd.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Teng ZP, Wang HM, Chau LY, Regan RF. Adenoviral transfer of the heme oxygenase-1 gene protects astrocytes from heme-mediated oxidative injury. Acad Em Med. 2003;10:480–481. doi: 10.1016/j.nbd.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Turner CP, Bergeron M, Matz P, Zegna A, Noble LJ, Panter SS, Sharp FR. Heme oxygenase-1 is induced in glia throughout brain by subarachnoid hemoglobin. J Cereb Blood Flow Metab. 1998;18:257–273. doi: 10.1097/00004647-199803000-00004. [DOI] [PubMed] [Google Scholar]
- Vreman HJ, Stevenson DK. Heme oxygenase activity as measured by carbon monoxide production. Anal Biochem. 1988;168:31–38. doi: 10.1016/0003-2697(88)90006-1. [DOI] [PubMed] [Google Scholar]
- Weng YH, Yang G, Weis S, Dennery PA. Interaction between heme oxygenase-1 and 2 proteins. J Biol Chem. 2003;278:50999–51005. doi: 10.1074/jbc.M307644200. [DOI] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Nakemura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]
- Xi GH, Keep RF, Hoff JT. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg. 1998;89:991–996. doi: 10.3171/jns.1998.89.6.0991. [DOI] [PubMed] [Google Scholar]
- Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee ME. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest. 1999;103:R23–29. doi: 10.1172/JCI6163. [DOI] [PMC free article] [PubMed] [Google Scholar]