

Abstract
The nervous system-specific leucine-rich repeat Ig-containing protein LINGO-1 is associated with the Nogo-66 receptor complex and is endowed with a canonical EGF receptor (EGFR)-like tyrosine phosphorylation site. Our studies indicate that LINGO-1 expression is elevated in the substantia nigra of Parkinson's disease (PD) patients compared with age-matched controls and in animal models of PD after neurotoxic lesions. LINGO-1 expression is present in midbrain dopaminergic (DA) neurons in the human and rodent brain. Therefore, the role of LINGO-1 in cell damage responses of DA neurons was examined in vitro and in experimental models of PD induced by either oxidative (6-hydroxydopamine) or mitochondrial (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) toxicity. In LINGO-1 knockout mice, DA neuron survival was increased and behavioral abnormalities were reduced compared with WT. This neuroprotection was accompanied by increased Akt phosphorylation (p-Akt). Similar neuroprotective in vivo effects on midbrain DA neurons were obtained in WT mice by blocking LINGO-1 activity using LINGO-1-Fc protein. Neuroprotection and enhanced neurite growth were also demonstrated for midbrain DA neurons in vitro. LINGO-1 antagonists (LINGO-1-Fc, dominant negative LINGO-1, and anti-LINGO-1 antibody) improved DA neuron survival in response to MPP+ in part by mechanisms that involve activation of the EGFR/Akt signaling pathway through a direct inhibition of LINGO-1's binding to EGFR. These results show that inhibitory agents of LINGO-1 activity can protect DA neurons against degeneration and indicate a role for the leucine-rich repeat protein LINGO-1 and related classes of proteins in the pathophysiological responses of midbrain DA neurons in PD.
Keywords: dopamine neuron, substantia nigra, degeneration, neuroprotection, axon
New therapeutics are required that simultaneously preserve dopamine (DA) neurons and their functional connections to limit or eliminate the progression of the movement disorder of Parkinson's disease (PD) (1, 2). Several growth factors normally active in cell growth and survival during brain development have been shown to provide protection against cell death in animal models of PD (1–5). The phosphoinositide 3-kinases (PI3-Ks) and Akt (protein kinase B) signaling pathways have been shown to participate in such growth factor actions (1, 2, 5, 6). Recent studies also suggest that some leucine-rich repeat (LRR) Ig-containing proteins can influence growth factors by modulating EGF receptor (EGFR) signaling-related pathways (7, 8). LINGO-1 is a LRR-Ig protein first identified as a critical component of the NogoR1-p75NTR complex in RhoA activation, and it is responsible for some inhibition of axonal regeneration by myelin-associated factors (9, 10). Unlike NogoR1, LINGO-1 gene expression is increased when adult nerve cells are exposed to traumatic injuries (9), indicating that LINGO-1 may be involved in cell injury responses. LRRK2, another LRR protein, was recently genetically linked to PD and Lewy body disease (11, 12). As we describe here, LINGO-1 appears to regulate neurite growth and the structural integrity of neurons, in analogy with LRRK2 (9, 13).
In this study, elevated LINGO-1 levels were found after selective experimental damage to DA nerve terminals in the striatum of mice, and increased expressed levels of LINGO-1 were found in the substantia nigra (SN) of some PD patients. Using methods that reduced or eliminated the negative actions of LINGO-1, we demonstrate that midbrain DA neuron survival, growth, and function improve in primary in vitro cultures and in vivo experimental models of parkinsonism in mice. We also show that LINGO-1 normally binds to EGFR and negatively regulates the EGFR/Akt signaling pathway in cells and tissues relevant to these studies.
Results
Expression of LINGO-1 in Human and Rodent SN.
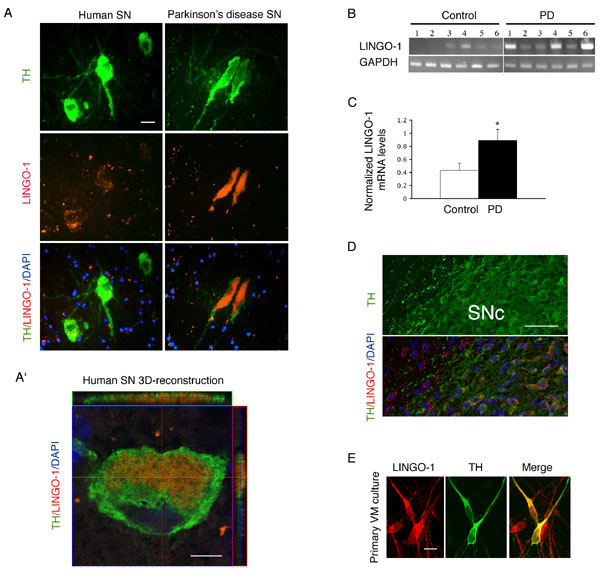
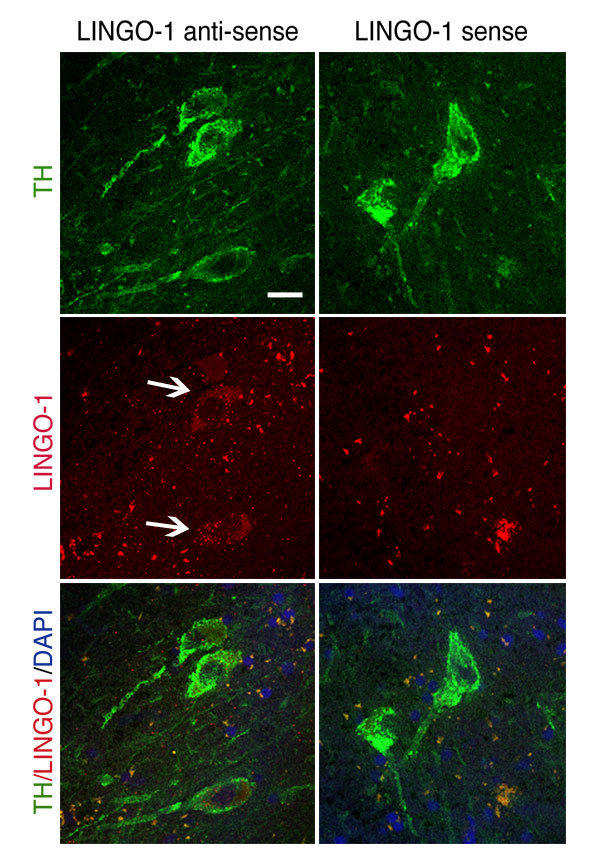
LINGO-1 expression was found in both tyrosine hydroxylase (TH) and non-TH neurons in human SN and rodent ventral midbrain (VM). LINGO-1 expression was examined by in situ hybridization in the SN of PD and age-matched controls [supporting information (SI) Figs. 4 A and A′ and 5] and semiquantitative RT-PCR (SI Fig. 4 B and C). LINGO-1 is expressed in remaining DA neurons in the SN of PD patients (SI Fig. 4 A and B). Preliminary data from PD patients indicate that LINGO-1 mRNA levels are higher in PD SN tissue than controls [unpaired Student's t test, t(10) = 2.280, P < 0.05] (SI Fig. 4 B and C). Data relevant to these PD patient and control cases are listed in SI Table 1. A detailed evaluation of LINGO-1 in the adult rat brain and rat primary embryonic cultures (embryonic day 15, 1 day in vitro) was obtained by in situ hybridization (SI Fig. 4D) and/or immunohistochemistry (SI Fig. 4E). In normal adult VM, LINGO-1 mRNA was expressed in many neuronal types and also colocalized with TH-positive DA neurons (SI Fig. 4D). In primary VM cultures, LINGO-1 protein was also shown to be present in TH neurons (SI Fig. 4E).
Enhanced Survival and Function of DA Neurons in Murine Models of PD by Elimination of LINGO-1.
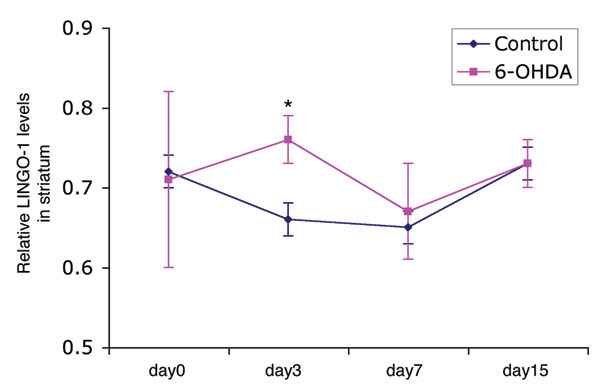
To examine the function of LINGO-1 in VM DA neuronal degeneration, we used the 6-hydroxydopamine (6-OHDA) (14, 15) and N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (16, 17) neurotoxin-induced experimental models of PD. LINGO-1 knockout (KO) mice display no gross anatomical abnormalities and physical alterations in behavior and locomotion (18). 6-OHDA was stereotaxically injected into the DA terminal striatum region of KO (n = 13) and WT (n = 13) littermates to produce a gradual and progressive neurodegenerative loss of DA terminals, axons, and cell bodies (14, 15). Apomorphine-induced motor asymmetry (rotational behavior) was measured 1, 2, 3, and 4 weeks after lesion. Motor asymmetry was significantly lower in the KO mice compared with WT littermate controls at all of the time points examined (Fig. 1A) (two-way ANOVA, F3,96 = 37.129, P < 0.001). In the postmortem analysis, the number of TH neurons was stereologically counted in the SN pars compacta (SNc, A9 area) regions using an unbiased optical fractionator method (19). There was no difference between KO and WT mice (F1,48 = 1.321, P > 0.05) in the total number of TH neurons present in nonlesioned VM (A9). The 6-OHDA lesion produced a significant loss of TH neurons in the SNc of WT and KO mice (F1,48 = 24.88, P < 0.0001) (Fig. 1 B and C). The number of TH neurons on the lesion side was normalized to the nonlesion side in each animal to prevent any influence of genotype. Statistical analysis showed a higher number of surviving TH neurons in the KO (mean ± SEM: 82 ± 3.8%) compared with WT mice (52 ± 4.0%) [t(24) = 2.40, P = 0.02], indicating a neuroprotective effect by eliminating the LINGO-1 response in the KO mice. In WT mice, using this selective 6-OHDA-induced experimental parkinsonism model, LINGO-1 protein levels were significantly increased in the striatum 3 days after injury (SI Fig. 6), a response previously shown in other types of neural injury models (9).
Fig. 1.

Effects of 6-OHDA or MPTP on SNc DA neurons in LINGO-1 KO mice. (A) Motor asymmetry induced by a DA agonist (apomorphine 0.4 mg/kg) was assessed at 1, 2, 3, and 4 weeks after the 6-OHDA DA lesion of the left striatum in WT and KO mice (n = 13 for each group). (B) Representative coronal midbrain sections of WT and KO mice after 6-OHDA lesion on the left side. (Scale bar: 700 μm.) (C) TH neuron number in the nonlesioned and lesioned sides of SNc of 6-OHDA-induced experimental parkinsonism in WT and KO genotype mice. There was a significant lesion effect in both genotypes (two-way ANOVA: ∗, P < 0.0001). Statistical analysis showed a relatively higher number of remaining TH neurons in the KO than WT mice (#, P = 0.046) after 6-OHDA-induced lesions. (D) TH neuron numbers in SNc of WT or KO mice treated with vehicle (WT, n = 7; KO, n = 8) or MPTP (n = 10 in each group). Statistical analyses showed that MPTP treatment reduced the loss of TH neurons in KO mice compared with WT mice (two-way ANOVA: #, P < 0.05). Both genotypes had reduced numbers of TH neurons after i.p. MPTP treatment compared with saline i.p. infusion (∗, P < 0.05). (E) Striatal DA levels (μg/mg) were significantly reduced by MPTP in WT but not LINGO-1 KO mice (∗, P < 0.05 vs. saline control). (F) In response to MPTP i.p. infusions, the number of TH neurons in the SNc of WT mice was significantly higher after LINGO-1 inhibition by LINGO-1-Fc injection into striatum on one side compared with the control side (∗, P < 0.05; n = 9). No difference was found in the Fc-injected mice. (G) Striatal DA levels (μg/mg) were greater on the LINGO-1-Fc-injected side compared with the control side (∗, P < 0.05; n = 8). DA levels were not different between the Fc-injected side and the control side. (H) As a control for toxin concentrations, striatal MPP+ levels were found to be the same between LINGO-1-Fc-injected and control sides. (I) Representative Western blots from dissected VM tissue of phosphorylated Akt (p-Akt), Akt, and β-actin in LINGO-1 WT (+/+) or LINGO-1 KO (−/−) mice after i.p. saline (−) or MPTP (+) infusion (n = 6 for each group). (J) Graph showing that p-Akt levels were significantly higher in the VM of MPTP-treated KO mice at 7 days compared with MPTP-treated WT mice (∗, P < 0.05) or saline-treated KO mice (#, P < 0.05; n = 6 for each group).
To extend the observations of neuroprotection seen in the 6-OHDA lesion in vivo model of PD, KO (n = 10) and WT (n = 10) littermates were also evaluated in the MPTP in vivo model of PD (16). WT (n = 7) and KO (n = 8) mice injected with i.p. saline (vehicle) served as controls. Postmortem stereological analysis at 7 days after i.p. MPTP infusions demonstrated that the number of TH neurons was significantly more reduced in the vulnerable SNc region of the WT relative to LINGO-1 KO mice (Fig. 1D) (two-way ANOVA, F1,31 = 4.60, P < 0.05). Saline treatment did not alter the total TH cell number in SNc of WT or KO mice (Fig. 1D). Consistent with such histological findings, striatal DA levels were lower in the MPTP-treated WT compared with vehicle-treated WT mice (Fig. 1E) (two-way ANOVA, P < 0.01) or vehicle-treated KO mice (P < 0.05). In KO mice, DA levels were not statistically different for saline and MPTP treatments, which is consistent with the DA neuroprotection observed in the in vivo 6-OHDA paradigm (see Fig. 1C). DA levels were not different between WT and KO mice treated with MPTP (Fig. 1E). In the MPTP paradigm, Nissl staining in the SNc was used to confirm the cell loss assessed by TH staining. Indeed, stereological analysis showed the cell number to be significantly less than in WT compared with KO mice [saline-treated WT, 9,808 ± 786; saline-treated KO, 9,166 ± 911; MPTP-treated WT, 4,497 ± 402; MPTP-treated KO, 6,869 ± 547 (P < 0.001 and P < 0.05 in WT and KO, MPTP vs. saline, respectively)]. As a control for MPTP toxicity effects by genotype, we measured the conversion of MPTP to the actively toxic form MPP+ (20). Striatal MPP+ levels in LINGO-1 KO or WT mice (n = 6 each group) measured at peak toxin levels 90 min after MPTP treatment were not significantly different [unpaired Student's t test, t(10) = 1.69, P > 0.05, mean ± SEM 39.68 ± 3.92 for WT and 62.06 ± 12.67 for KO]. Because MPP+ enters DA terminals via the DA transporter, levels of this protein were examined by genotype. Western blot analysis showed that DA transporter levels were equivalent in WT and LINGO-1 KO mice (SI Fig. 7). As a physiological confirmation in WT mice of the neuroprotection observed in LINGO-1 KO animals, LINGO-1-Fc protein (6.5 μg/μl; total 2 μl), which is the truncated form of LINGO-1 functioning as a dominant negative (DN) molecule (9, 18), was unilaterally injected into the striatum region of C57BL/6 mice (n = 9). Injection of the Fc fragment served as controls (n = 11). Seven days after this surgery, the mice received i.p. MPTP. At postmortem analysis 1 week after systemic MPTP, a higher number of TH neurons remained in the SNc of the side injected with the LINGO-1-blocking agent (LINGO-1-Fc) (Fig. 1F) [paired Student's t test, t(8) = 3.99, P = 0.004]. Congruent with this finding, DA levels were determined to be higher in the LINGO-1-Fc-injected striatum (Fig. 1G) [paired Student's t test, t(7) = 2.9, P = 0.02]. In the control experiment, injection of the Fc fusion protein did not alter the number of TH neurons or striatal DA levels (Fig. 1 F and G). Control experiments showed that striatal MPP+ levels examined 90 min after systemic MPTP injections were similar in the LINGO-1-Fc-injected and contralateral sides of the brain (Fig. 1H) [t(8) = 0.14, P = 0.89], as was previously shown for MPTP to MPP+ conversion in WT and LINGO-1 KO mice. The neuroprotective phosphorylated form of Akt (p-Akt) was increased in vivo in the VM of MPTP-treated KO mice compared with those of MPTP-treated WT mice (Fig. 1 I and J) (F3,20 = 6.84, P < 0.01) or vehicle-treated KO mice (Fig. 1 I and J) (P < 0.05), whereas there were no differences between levels of p-Akt in vehicle-treated KO and WT mice (n = 6 each group) (Fig. 1 I and J).
Trophic Effects on Neurite Outgrowth and Survival of DA Neurons in Vitro.
To analyze the mechanisms and processes underlying the effects observed in vivo, we first studied LINGO-1 function in DA neurons in vitro in the presence of LINGO-1 antagonists. To provide LINGO-1 inhibition, cultured rodent VM neurons were transduced with lentivirus producing the full-length (FL) LINGO-1, or DN-LINGO-1 to block the endogenous LINGO-1 function, or with a vector control. Notably, in DN-LINGO-1-transduced VM cultures, TH neurons had longer neurites than similar control cultures (Fig. 2 A and B) (F2,6 = 7.872, P < 0.05). TH neurons showed no differences in neurite lengths when transduced with FL-LINGO-1 (Fig. 2 A and B). These observations show that DN-LINGO-1 induces neurite outgrowth by inhibition of endogenous LINGO-1 function. Indeed, LINGO-1-Fc protein, like DN-LINGO-1, also promoted neurite outgrowth of DA neurons (Fig. 2 A and B) [t(4) = 6.23, P = 0.003]. To confirm and extend the observations of increased p-Akt responses seen in vivo associated with LINGO-1 inhibition, we determined p-Akt levels in cell cultures exposed to DN-LINGO-1 (21–24). A significant increase in p-Akt was observed in primary VM cultures after DN-LINGO-1 transduction compared with FL-LINGO-1 (or control transductions using a lenti-GFP vector) accompanied by elevated levels of EGFR (Fig. 2 C and D) (ANOVA, P < 0.01), which is consistent with p-Akt elevation observed in vivo in LINGO-1 KO mice after the MPTP lesion (see Fig. 1J). This indicates that DN-LINGO-1 influences growth of TH neurons, at least in part by involvement of the Akt signaling pathway.
Fig. 2.

Blocking LINGO-1 function activities promotes DA neurite outgrowth. (A) Effects of LINGO-1-Fc and DN-LINGO-1 on neurite outgrowth of TH neurons. (B) TH neurite length was significantly higher in cultures treated with DN-LINGO-1 and LINGO-1-Fc compared with those treated with FL-LINGO-1, control lentivirus (∗, P < 0.05), and control Fc (∗, P < 0.003), respectively. (C) EGFR, p-Akt, total Akt, and HA-LINGO-1 expression in VM primary cultures infected with FL-LINGO-1, DN-LINGO-1, control lentivirus, and vehicle control, as detected by Western blotting. Antibody to the HA tag confirmed infection by FL-LINGO-1 and DN-LINGO-1 in the cultures. (D) p-Akt was significantly higher in the cultures incubated with DN-LINGO-1 compared with controls (∗, P < 0.05). (Scale bar: 100 μm.)
Consistent with the in vivo data above, DN-LINGO-1 significantly promoted DA neuron survival, compared with control or FL-LINGO-1-treated cultures exposed to 10 μM MPP+ (Fig. 3 A and C) (F5,18 = 25.155, P < 0.01). Additional evidence for the specificity of the LINGO-1 effects was obtained by inhibiting LINGO-1 using LINGO-1-Fc protein or a LINGO-1-blocking antibody named 1A7. After MPTP treatment, both LINGO-1-Fc protein and 1A7-treated VM cultures contained a relatively larger number of TH neurons than control Fc protein or IgG-treated cultures (Fig. 3 B and D) (P < 0.05 for LINGO-1 Fc and P < 0.01 for 1A7). To see whether non-TH cells, which also express LINGO-1, were affected in this paradigm, the DAPI-stained nuclei were counted (see Methods). The estimated cell number per coverslip was obtained by counting eight fields using randomized rotation (vehicle control, 180,133 ± 20,606; MPP+, 191,022 ± 28,594; control Fc, 191,511 ± 11,980; LINGO-1-Fc, 190,933; IA7, 170,578 ± 16,162; IgG, 206,667 ± 36,084; n = 4, P > 0.05, ANOVA). These in vitro results confirmed that inhibition of endogenous LINGO-1 protects DA neurons against MPP+-induced cell damage.
Fig. 3.

Inhibition of LINGO-1 function promotes DA neuronal survival. (A) Cultured VM cells incubated with FL-LINGO-1, DN-LINGO-1, and control lentivirus were treated with 10 μM MPP+ at the fourth day in vitro and fixed at the sixth day in vitro. (B) Cultured VM cells treated with LINGO-1-Fc protein or 1A7 (a LINGO-1-blocking antibody) and control Fc or antibody. (Scale bar: 100 μm.) (C) The number of TH neurons was significantly higher in cultures incubated with DN-LINGO-1 than in cultures incubated with FL-LINGO-1 or control vectors after MPP+ treatment (∗, P < 0.01). (D) After exposure to MPP+, the number of TH neurons was higher in LINGO-1-Fc- or 1A7-treated VM cultures compared with cultures treated with vehicle, control Fc, or IgG. ∗, P < 0.05 for LINGO-1-Fc and P < 0.01 for 1A7. (E) COS7 cells were infected with FL-LINGO-1 lentivirus at 0, 1, and 5 multiplicities of infection per cell for 2 days, and EGFR levels were examined. LINGO-1 decreased EGFR expression levels in a dose-dependent manner. (F) Coimmunoprecipitation of EGFR and LINGO-1 in cultured cells transfected with LINGO-1 and/or EGFR. (G) Coimmunoprecipitation of EGFR and LINGO-1 in the KO and WT VM tissues. (H) 1A7 blocks binding of LINGO-1 to EGFR in a cotransfected cell line. Transfection of oligodendrocyte–myelin glycoprotein was used as a negative control. (I) Western blots of EGFR, p-Akt, total Akt, and β-actin of VM cultures treated with LINGO-1-Fc or LINGO-1 antibody (1A7). (J) Statistical analysis showed a significant elevation of p-Akt levels in 1A7- and LINGO-1-Fc-treated cultures compared with control antibody and control Fc protein, respectively. Western blots were done at least in duplicate. IB, immunoblotting; IP, immunoprecipitation.
Further Mechanisms Involved in the LINGO-1 Effects.
In further analyzing the molecular mechanisms involved in LINGO-1 actions, we reasoned that LINGO-1 and EGFR could potentially interact because the cytoplasmic domain of LINGO-1 contains a canonical EGFR-like tyrosine phosphorylation site (9). We tested this hypothesis and found that LINGO-1 decreases EGFR protein levels in a dose-dependent manner (Fig. 3E). Direct interaction between LINGO-1 and EGFR was demonstrated by immunoprecipitation using anti-LINGO-1 or anti-EGFR antibodies in (i) cultured cells or (ii) WT or KO VM brain tissues (Fig. 3 F and G). The LINGO-1-blocking antibody 1A7 also abolished binding of LINGO-1 to EGFR, whereas the nonblocking anti-LINGO-1 antibody 2F3 did not have such effects. These data are evidence that LINGO-1 can reduce EGFR levels by a direct physical interaction (Fig. 3H). Consistent with this interpretation, both EGFR and p-Akt levels increased when MPP+-treated VM cultures were incubated with LINGO-1 inhibitors, such as 1A7 or LINGO-1-Fc protein, compared with control IgG or control Fc protein treatments of the cultures (Fig. 3 I and J) (F3,12 = 23.645, P < 0.001 for 1A7; F3,12 = 18.89, P < 0.001 for LINGO-1-Fc). Furthermore, EGFR activation was suppressed by LINGO-1, and 1A7 attenuated such inhibitory effects (SI Figs. 8 and 9).
Discussion
These results demonstrate that midbrain DA neurons can be protected in vivo and in vitro against parkinsonism-inducing agents by independent methods of inhibiting endogenous brain LINGO-1, including (i) an antibody against LINGO-1, (ii) LINGO-1 KO mice, (iii) DN-LINGO-1 transduction, or (iv) the addition of LINGO-1-Fc protein. The experiments also demonstrate that the endogenous LINGO-1 effects involve the activation of the EGFR/Akt intracellular signaling pathways. Finally, we believe that our findings may be particularly relevant, because another protein in the LRR family, LRRK2, has recently been genetically linked to PD (11, 12). Endogenous LRRK2, like LINGO-1, appears to function as a growth inhibitor of neurites and to be involved in the structural plasticity and integrity of the DA neurons (13).
How does LINGO-1 regulate DA neurite outgrowth? LINGO-1 is a coreceptor of NogoR1 and p75NTR or TROY and can inhibit neurite growth in dorsal root ganglion and cerebellar granular neurons in the presence of myelin-associated inhibitor(s) (9, 10). In such neurons, LINGO-1 normally inhibits process elongation and axonal growth. LINGO-1-Fc reverses the inhibitory properties of endogenous LINGO-1, and LINGO-1-Fc promotes axonal outgrowth by antagonizing the Nogo-R signaling pathway (9, 10). Interestingly, our studies of LINGO-1-mediated intracellular signaling demonstrated that inhibiting LINGO-1 activities increased EGFR and p-Akt levels in the absence of myelin-associated inhibitor(s). Such data show that LINGO-1 can reduce EGFR expression, indicating that endogenous LINGO-1 may be a negative regulator of the EGFR/Akt signaling pathways. However, it is not clear how LINGO-1 inhibits EGFR expression and function. We propose that LINGO-1 may regulate EGFR signaling pathways by accelerating EGFR internalization and degradation, thus decreasing the availability of EGFR, or that LINGO-1 directly inhibits EGFR phosphorylation and thereby reduces PI3-K/Akt signaling activity (see SI Fig. 10). Previous (9) and present data demonstrate that LINGO-1 is up-regulated after neuronal damage or cell death in vivo, suggesting that LINGO-1 participates in pathophysiological responses. EGFR has multiple ligands including EGF, TGF-α, heparin-binding EGF-like growth factor, and amphiregulin (25). Significantly, TGF-α mutant mice exhibit significant reductions in SNc DA neurons (19). Furthermore, EGF and EGFR protein levels are significantly decreased in the striatum of PD patients (2), and EGF treatment has been shown to protect DA neurons in primary VM cultures and animal models of PD (2, 4, 26–28). Familial PD can be caused by mutant dysfunctional parkin (PARK2 in PD gene classification), and normal parkin (29, 30) can delay EGFR internalization and degradation and promote PI3-K/Akt signaling (31). Intracellular signaling of EGFR is mediated by the PI3-K pathway (21), which increases phosphorylation and activation of Akt (p-Akt) (24). Growth factor-activated PI3-K/Akt signaling pathways have been previously documented to enhance neuronal survival and axonal regeneration. The mechanisms are complex and involve regulation of cytoplasmic cell death machinery, genes in cell death and survival, and metabolic pathways associated with cell survival (1, 22, 23, 32). Furthermore, overexpression of p-Akt in the SNc DA neurons provides potent protection against 6-OHDA-induced cellular damages and function of DA neurons (23), consistent with our neuroprotection data. In addition to EGFR, inhibition of LINGO-1 may reduce p75NTR activity that has been linked to cell death (33). Because LINGO-1 is also expressed by non-DA cells, those cells may also have positive influences on the DA neuronal function when LINGO-1 activity is inhibited. It has also been shown that LINGO-1 activates RhoA-GTP. Such RhoA activation can by itself cause cell death (34), thus providing another alternative mechanism for LINGO-1-associated cellular demise and dysfunction, because LINGO-1 antagonists can reduce RhoA (9, 18). The current experiments revealed that inhibition of endogenous LINGO-1 (by DN-LINGO-1 or LINGO-Fc protein) stimulated neurite extension of DA neurons cultured in vitro in the absence of myelin inhibitors. Given that FL-LINGO-1 overexpression did not by itself inhibit neurite growth, this indicates that the LINGO-1 inhibitory system is saturated at baseline or that other molecular partners are required to further block neurite growth.
It is of utmost importance to find treatments that reduce neuronal degeneration and maintain neuronal pathways and physiological circuits in PD. Using in vivo and in vitro PD models, this work demonstrates that LINGO-1 can be part of the injury response of DA neurons. Inhibition of endogenous LINGO-1 exerted neuroprotective and neurite growth-stimulating effects on the DA neurons that typically degenerate in PD. In a PD lesion animal model, LINGO-1 was also regionally elevated after 6-OHDA induced DA neuronal degeneration. Preliminary data also indicate that LINGO-1 is up-regulated in the brains of human PD cases. These LINGO-1 findings, combined with recent data showing the related LRRK2 protein's physiological role (13) and genetic link to PD (11, 12), demonstrate that LRR protein family members can play critical roles in the structural and functional integrity of neurons involved in neurodegenerative diseases. LINGO-1 and related functional proteins may therefore become useful targets for developing new treatments.
Methods
Generation of Recombinant Proteins, Viruses, and Antibodies.
Control Fc and LINGO-1-Fc were prepared as described (9). Briefly, LINGO-1-Fc (residues 1–532 of human LINGO-1 fused to the hinge and Fc region of human IgG1) was expressed in CHO cells and purified on protein A Sepharose (Amersham Pharmacia, Rockville, MD). The purified protein (>95% pure) ran on SDS/PAGE with Mr = 90 kDa under reducing conditions and Mr = 180 kDa under nonreducing conditions. FL mouse LINGO-1 (FL-LINGO-1, amino acid residues 34–614; and DN-LINGO-1, amino acid residues 34–548) DNA sequence was inserted into a lentiviral vector as described (9). FL-LINGO-1 and DN-LINGO-1 plasmids were transfected into 293T cells to produce lentivirus (9). The 1A7 and 2F3 monoclonal anti-LINGO-1 antibodies were generated by using soluble LINGO-1-Fc as an antigen.
6-OHDA Lesion Model and Rotation Testing.
LINGO-1 KO mice were generated with a GFP/neo (neomycin-resistant gene) replacement vector that targeted the entire single-exon coding sequence of LINGO-1 (18). Thirteen male LINGO-1 KO mice and 13 littermate controls were used. Mice were anesthetized with ketamine/xylazine (100 mg/kg; Fort Dodge Animal Health, Fort Dodge, IA) and received unilateral striatal injection of 6-OHDA (14). A concentration of 10 μg/μl free base 6-OHDA dissolved in 0.02% ascorbate/saline (Sigma, St. Louis, MO) was injected in the left striatum (total dose, 10 μg) at the following coordinates (calculated from bregma): AP, +0.4; L, +1.5; DV, −2.5. The injection was performed over 2 min at a rate of 0.5 μl/min using a 26-gauge 10-μl Hamilton syringe. After injection the needle was left in place for an additional 2 min before withdrawal. The wound was sealed with one auto clip, and the mice were placed in a cage on a warming pad and observed until they awoke from anesthesia. Rotational testing was conducted at 1, 2, 3, and 4 weeks after lesion. Apomorphine (Sigma) in 0.02% of ascorbate was injected s.c. (0.4 mg/kg), and rotations contralateral to the lesion side were counted over 30 min.
MPTP Treatment of LINGO-1 KO and WT Mice.
Groups of male LINGO-1 KO mice and WT littermate controls were injected i.p. with 25 mg/kg MPTP hydrochloride (Sigma) four times 2 h apart (16, 17). Saline-treated LINGO-1 KO and WT mice with the same i.p. infusion schedule served as controls. All animals were killed 7 days after injection. MPTP-treated KO and WT (n = 10 each group) and saline-treated KO (n = 8) and WT (n = 7) mice were processed for histology and stereological analysis (20, 35) to determine specific neuroprotective effects on TH neurons in the VM. To examine protective intracellular mechanisms, VM and striatum tissues were collected from saline-treated mice (n = 6 each genotype) and MPTP-treated KO and WT mice (n = 6 each group) for Western blot analysis. For measurement of the conversion of MPTP to MPP+, LINGO-1 KO and WT mice (n = 6 each group) were killed 90 min after MPTP injection followed by dissection of striatum (20) (see SI Methods).
LINGO-1-Fc Injection and MPTP Treatment.
LINGO-1-Fc (6.5 μg/μl, 2 μl) (n = 9) and control Fc protein (n = 11) were unilaterally injected in the striatum of male C57BL/6 mice (2.5–3 months old) as follows: AP, +0.62; L, 1.5; DV, −3.3 and −2.5 (calculated from bregma). These mice received MPTP 7 days after surgery and were allowed to survive for another 7 days. Both striata were dissected out for the HPLC assay of DA at the time the mice were killed, and the brains were fixed in 4% paraformaldehyde in 0.1 M PB and were processed for stereological analysis of the number of VM DA neurons. Another group of mice (n = 9) was treated with MPTP (30 mg/kg), and striatum was dissected 90 min after MPTP injection for MPP+ measurements (see SI Methods).
Statistical Analysis.
Unpaired or paired Student's t test, one-way ANOVA with post hoc test, and two-way ANOVA were used to analyze data when appropriate (InStat; GraphPad, San Diego, CA; JMP, Cary, NC). P < 0.05 was considered significantly different.
Supplementary Material
Acknowledgments
We thank Jane Relton and Dorothy Kester for their technical assistance in the 6-OHDA and MPTP animal studies and Yalda Sadeghi and Shelley Yang for Western blot analysis. This work was supported by National Institute of Neurological Disorders and Stroke Grant NS39793 and the Orchard Foundation, the Consolidated Anti-Aging Foundation, and Michael K. Stern Parkinson's Research Foundation (O.I.). H.I. is the recipient of Yamada Science Foundation support for a long-term visit and a Uehara Memorial Foundation research fellowship.
Abbreviations
- LRR
leucine-rich repeat
- PD
Parkinson's disease
- SN
substantia nigra
- SNc
SN pars compacta
- DA
dopamine/dopaminergic
- MPTP
N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- TH
tyrosine hydroxylase
- VM
ventral midbrain
- 6-OHDA
6-hydroxydopamine
- PI3-K
phosphoinositide 3-kinase
- KO
knockout
- FL
full-length
- DN
dominant negative
- EGFR
EGF receptor.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0700901104/DC1.
References
- 1.Brunet A, Datta SR, Greenberg ME. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- 2.Iwakura Y, Piao YS, Mizuno M, Takei N, Kakita A, Takahashi H, Nawa H. J Neurochem. 2005;93:974–983. doi: 10.1111/j.1471-4159.2005.03073.x. [DOI] [PubMed] [Google Scholar]
- 3.Ericson C, Georgievska B, Lundberg C. Eur J Neurosci. 2005;22:2755–2764. doi: 10.1111/j.1460-9568.2005.04503.x. [DOI] [PubMed] [Google Scholar]
- 4.Hanke M, Farkas LM, Jakob M, Ries R, Pohl J, Sullivan AM. Neuroscience. 2004;124:757–766. doi: 10.1016/j.neuroscience.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 5.Onyango IG, Tuttle JB, Bennett JP., Jr Neurobiol Dis. 2005;20:141–154. doi: 10.1016/j.nbd.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Farkas LM, Krieglstein K. J Neural Transm. 2002;109:267–277. doi: 10.1007/s007020200022. [DOI] [PubMed] [Google Scholar]
- 7.Gur G, Rubin C, Katz M, Amit I, Citri A, Nilsson J, Amariglio N, Henriksson R, Rechavi G, Hedman H, et al. EMBO J. 2004;23:3270–3281. doi: 10.1038/sj.emboj.7600342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldoni S, Iozzo RA, Kay P, Campbell S, McQuillan A, Agnew C, Zhu JX, Keene DR, Reed CC, Iozzo RV. Oncogene. 2006;26:368–381. doi: 10.1038/sj.onc.1209803. [DOI] [PubMed] [Google Scholar]
- 9.Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, Levesque M, Allaire N, Perrin S, Sands B, et al. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- 10.Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, Thill G, Levesque M, Sah D, McCoy JM, et al. Neuron. 2005;45:353–359. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 11.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, et al. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 12.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Macleod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 14.Brundin P, Isacson O, Gage FH, Prochiantz A, Bjorklund A. Brain Res. 1986;366:346–349. doi: 10.1016/0006-8993(86)91316-8. [DOI] [PubMed] [Google Scholar]
- 15.Liang Q, Smith AD, Pan S, Tyurin VA, Kagan VE, Hastings TG, Schor NF. Biochem Pharmacol. 2005;70:1371–1381. doi: 10.1016/j.bcp.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 16.Battaglia G, Busceti CL, Pontarelli F, Biagioni F, Fornai F, Paparelli A, Bruno V, Ruggieri S, Nicoletti F. Neuropharmacology. 2003;45:155–166. doi: 10.1016/s0028-3908(03)00146-1. [DOI] [PubMed] [Google Scholar]
- 17.Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, et al. Proc Natl Acad Sci USA. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, Chang J, Thill G, Levesque M, Zhang M, et al. Nat Neurosci. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- 19.Blum M. Nat Neurosci. 1998;1:374–377. doi: 10.1038/1584. [DOI] [PubMed] [Google Scholar]
- 20.Cleren C, Starkov AA, Calingasan NY, Lorenzo BJ, Chen J, Beal MF. Neurobiol Dis. 2005;20:701–708. doi: 10.1016/j.nbd.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 21.Arcaro A, Zvelebil MJ, Wallasch C, Ullrich A, Waterfield MD, Domin J. Mol Cell Biol. 2000;20:3817–3830. doi: 10.1128/mcb.20.11.3817-3830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D'Astous M, Mendez P, Morissette M, Garcia-Segura LM, Di Paolo T. Mol Pharmacol. 2006;69:1492–1498. doi: 10.1124/mol.105.018671. [DOI] [PubMed] [Google Scholar]
- 23.Ries V, Henchcliffe C, Kareva T, Rzhetskaya M, Bland R, During MJ, Kholodilov N, Burke RE. Proc Natl Acad Sci USA. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, McCullough KD, Franke TF, Holbrook NJ. J Biol Chem. 2000;275:14624–14631. doi: 10.1074/jbc.275.19.14624. [DOI] [PubMed] [Google Scholar]
- 25.Iwamoto R, Mekada E. Cell Struct Funct. 2006;31:1–14. doi: 10.1247/csf.31.1. [DOI] [PubMed] [Google Scholar]
- 26.Hadjiconstantinou M, Fitkin JG, Dalia A, Neff NH. J Neurochem. 1991;57:479–482. doi: 10.1111/j.1471-4159.1991.tb03776.x. [DOI] [PubMed] [Google Scholar]
- 27.Ventrella LL. J Neurosurg Sci. 1993;37:1–8. [PubMed] [Google Scholar]
- 28.Seroogy KB, Numan S, Gall CM, Lee DC, Kornblum HI. NeuroReport. 1994;6:105–108. doi: 10.1097/00001756-199412300-00028. [DOI] [PubMed] [Google Scholar]
- 29.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 30.Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, et al. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 31.Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, et al. Nat Cell Biol. 2006;8:834–842. doi: 10.1038/ncb1441. [DOI] [PubMed] [Google Scholar]
- 32.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E, Vincenz C. J Biol Chem. 2001;276:33812–33820. doi: 10.1074/jbc.M010548200. [DOI] [PubMed] [Google Scholar]
- 34.Dubreuil CI, Winton MJ, McKerracher L. J Cell Biol. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez-Pernaute R, Ferree A, Cooper O, Yu M, Brownell AL, Isacson O. J Neuroinflammation. 2004;1:6–14. doi: 10.1186/1742-2094-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}