

Abstract
Background & Aims
Early growth response-1 (Egr-1), an immediate early gene/zinc-finger transcription factor, is required for maximal stimulation of tumor necrosis factor α (TNF-α) transcription in response to lipopolysaccharide (LPS). Because chronic ethanol exposure sensitizes macrophages to LPS-stimulated TNF-α expression, we have investigated the role of Egr-1 in mediating increased LPS-stimulated TNF-α expression after chronic ethanol feeding. Furthermore, because TNF-α contributes to alcoholic liver injury, we tested the hypothesis that Egr-1 is required for the development of ethanol-induced fatty liver injury in wild type and egr-1 −/− mice.
Methods
Wild-type and egr-1 −/− mice were fed ethanol-containing diets or pair-fed control diets for 6 weeks.
Results
Wild-type mice fed the ethanol diet developed hepatic steatosis characterized by micro- and macrovesicular lipid accumulation. However, egr-1 −/− mice did not develop steatosis after ethanol feeding. Alanine transferase and TNF-α concentrations in serum were increased after ethanol feeding in wild-type but not egr-1 −/− mice. In wild-type mice, challenge with LPS increased Egr-1 messenger RNA (mRNA) and DNA binding activity in liver; this response to LPS was enhanced after chronic ethanol feeding. LPS challenge also increased hepatic TNF-α mRNA and serum TNF-α to a greater extent after ethanol feeding compared with pair-fed wild-type mice. However, chronic ethanol feeding did not enhance LPS-stimulated TNF-α mRNA or serum TNF-α in egr-1 −/− mice.
Conclusions
These data show that Egr-1 contributes to increased LPS-mediated TNF-α expression after chronic ethanol and that the absence of Egr-1 prevents chronic ethanol-induced fatty liver, as well as increased sensitivity to LPS.
Production of tumor necrosis factor α (TNF-α) is one of the earliest responses of the liver to injury.1 TNF-α acts as a principal mediator of the inflammatory response in mammals, transducing differential signals that regulate cellular activation and proliferation, cytotoxicity, and apoptosis.2,3 In addition to its role in acute septic shock, TNF-α has been implicated in the pathogenesis of a wide variety of chronic inflammatory diseases4,5 and in the progression of ethanol-induced liver injury.1,6 Studies making use of transgenic mice lacking the TNF receptor I, as well as treatment of mice and rats with antibodies to TNF-α during chronic ethanol exposure, have shown an essential role for TNF-α in the progression of chronic ethanol-induced liver injury.1
Kupffer cells, the resident macrophage in the liver, are an important source of TNF-α and other inflammatory cytokines in the liver. Production of TNF-α is a highly regulated process; regulation occurs at the level of transcription, translation, and secretion.7 Lipopolysaccharide (LPS), present in the cell wall of gram-negative bacteria, is an important activator of TNF-α production by Kupffer cells.6 Transcriptional activation of TNF-α by LPS requires the activation of a distinct set of transcription factors binding to at least 2 regions of the TNF-α promoter, which include binding sites for nuclear factor κB (NF-κB), early growth response-1 (Egr-1), and activator protein-1 (AP-1).8,9 Although the exact array of transcription factors interacting with the TNF-α promoter is to some extent cell and species specific,10 recruitment of NF-κB and Egr-1, as well as increased c-jun binding to a cyclic adenosine monophosphate response element/AP-1 site, appear to be required for full activation of TNF-α expression in response to LPS in most types of macrophages.8,9
Circulating TNF-α is increased in the blood of alcoholics and in animals chronically exposed to ethanol.6,11 Although enhanced expression of TNF-α after ethanol exposure is thought to be caused in part by an increased exposure to LPS,6 chronic ethanol also increases the susceptibility of animals to LPS-induced liver injury.12–14 The mechanisms for this potentiation of LPS-mediated responses by chronic ethanol are not completely understood. After chronic ethanol feeding, LPS-stimulated TNF-α expression in isolated Kupffer cells is increased compared with expression in Kupffer cells from pair-fed control rats.15,16 Enhanced sensitivity to LPS after chronic ethanol exposure is caused, at least in part, by enhanced expression and DNA binding activity of the transcription factor Egr-1 in both Kupffer cells17 and RAW 264.7 cells, a macrophage-like cell line.18 Over-expression of a dominant negative form of Egr-1 in RAW 264.7 macrophages prevented increased accumulation of TNF-α messenger RNA (mRNA) in response to LPS after chronic ethanol exposure.18 These data suggest that Egr-1 expression and DNA binding activity are involved in mediating chronic ethanol-induced sensitization of macrophages to LPS.
Egr-1 (also known as nerve growth factor induced-A, Krox-24, ZIF268, ETR103, and TIS8) is a zinc-finger transcription factor discovered for its role in regulation of cell growth and proliferation.19 Expression of Egr-1, an immediate early gene, is rapidly and transiently induced in response to a variety of stimuli, including cytokines and growth factors, as well as environmental stress, including ischemic injury and tissue damage.19,20 Egr-1 binding to the TNF-α promoter is required for full activation of TNF-α transcription in response to LPS.9 Because chronic ethanol increases LPS-stimulated TNF-α expression via an Egr-1– dependent mechanism in isolated Kupffer cells, here we have investigated whether chronic ethanol feeding enhances LPS-stimulated Egr-1 expression and DNA binding activity in liver. Furthermore, because TNF-α is required for the development of chronic ethanol-induced liver injury, we made use of an egr-1 −/− mouse model to test the hypotheses that Egr-1 (1) contributes to increased LPS sensitivity after chronic ethanol exposure and (2) is required for the development of ethanol-induced liver injury.
Materials and Methods
Materials
Female C57BL/6 mice (8 –10 weeks old) were purchased from Jackson Labs (Bar Harbor, ME). Egr-1 −/− mice, developed on a C57BL/6 background, were from Dr J. Milbrandt (Washington University, St. Louis, MO) and have been previously described.21 Lieber-DeCarli ethanol and control diets were purchased from Dyets (Bethlehem, PA). Antibodies were from the following sources: CYP2E1 (Research Diagnostics, Inc, Flanders, NJ), IκB-α (Cell Signaling, Beverly, MA), ERK1/2 (Upstate Biotechnology, Lake Placid, NY), p65 subunit of NF-κB, and Egr-1 (Santa Cruz Biotechnology, Santa Cruz, CA). Anti-rabbit and mouse immunoglobulin G–peroxidase were purchased from Boehringer Mannheim (Indianapolis, IN). Oligonucleotides corresponding to the Egr-1 and AP-1 DNA binding sites in the murine TNF-α promoter were synthesized by IDT Technologies (Coralville, IA). The oligonucleotide corresponding to the NF-κB consensus binding site was purchased from Santa Cruz Biotechnology. LPS (Escherichia coli serotype 026:B6) was from Sigma-Aldrich (#L2654) (St. Louis, MO).
Ethanol Feeding
All procedures using animals were approved by the Case Western Reserve University Institutional Animal Care and Use Committee. Eight- to 10-week-old female and male wild-type (C57BL/6) and egr-1 −/− were housed in shoe-box cages (2 animals/cage) with microisolator lids. Standard microisolator handling procedures were used throughout the study. Mice were randomized into ethanol-fed and pair-fed groups and then adapted to control liquid diet for 2 days. The ethanol-fed group was allowed free access to ethanol containing diet with increasing concentrations of ethanol: 1% (vol/vol) and 2% for 2 days, then 4% ethanol for 7 days, and finally 5% ethanol for a further 4 weeks. Control mice were pair-fed diets that isocalorically substituted maltose dextrins for ethanol over the entire feeding period. Blood was taken from the tail-vein 3–3.5 hours into the feeding cycle for measurements of serum ethanol concentrations. At the conclusion of the feeding period, mice were injected intraperitoneally with 0.7 μg LPS/g body weight or an equivalent volume of sterile, endotoxin-free saline (0.09%). Mice were then anesthetized, blood taken from the retro-orbital sinus and the inferior vena cava, livers blanched with saline via the portal vein, and livers excised. Portions of each liver were then either fixed in formalin for histology, frozen in optimal cutting temperature compound (Sakura Finetek U.S.A., Inc, Torrance CA), flash frozen in liquid nitrogen or stored in RNAlater (Qiagen, Valencia, CA), and stored at −80°C until further analysis.
Serum Measurements
Serum samples were assayed for ethanol and alanine aminotransferase (ALT) using commercially available enzymatic assay kits (Sigma-Aldrich). Serum TNF-α peptide was measured by enzyme-linked immunosorbent assay (ELISA) (mouse Quantikine, R&D Systems, Minneapolis, MN). Endotoxin concentrations were measured in platelet-rich plasma prepared from blood collected from the retro-orbital sinus. Endotoxin was measured kinetically using a chromogenic test based on the limulus amebocyte lysate assay (Kinetic-QCL, BioWhittaker, Walkersville, MD) as described by Rivera et al.22
Liver Histology and Triglycerides
Formalin-fixed tissues were paraffin-embedded, sectioned, and stained with H&E. Sections were coded before analysis and examined by 2 independent individuals. For Oil Red O (Sigma, St. Louis, MO) staining, 4-μm sections were cut from frozen optimal cutting temperature samples, affixed to a microscope slide, and allowed to air dry overnight at room temperature. Liver sections were stained in fresh Oil Red O for 10 minutes, rinsed in water, and then counterstained with hematoxylin. Total liver triglycerides were measured using the Triglyceride Reagent Kit from Pointe Scientific Inc (Lincoln Park, MI).
Liver Homogenization and Isolation of Nuclear Proteins
Frozen liver tissue, 0.3– 0.6 g, was homogenized in 10 mL/g tissue in homogenizing buffer (10 mmol/L HEPES, pH 7.6, 25 mmol/L KCl, 1 mmol/L EDTA, 10% glycerol, 0.25 mol/L sucrose) with added protease inhibitors (Complete; Roche Diagnostics, Mannheim, Germany, 17.5 μg/mL aprotinin, 5 μg/mL bestatin, 10 μg/mL leupeptin, 1 mg/mL bacitracin, and 20 μg/mL E64) and phosphatase inhibitors (1 mmol/L vanadate and 10 mmol/L Na pyrophosphate) using 15 strokes in a glass on glass homogenizer (loose pestle). Homogenates were then filtered through a coarse nylon mesh to remove debris. Aliquots of homogenates were extracted in 1% Triton-X-100 and then flash frozen in liquid nitrogen until further use. The remainder of the homogenate was layered onto a 0.35-mol/L sucrose pad in homogenizing buffer. Samples were centrifuged at 1000 ×g for 5 minutes at 4°C. Aliquots of the postnuclear supernatant were flash frozen and the nuclear pellets layered again over a 0.35-mol/L sucrose pad and centrifuged as described previously. Nuclear pellets were then resuspended in 20 mmol/L HEPES, pH 7.9, 0.4 mol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 1 mmol/L Na orthovanadate, 10 mmol/L Na pyrophosphate, protease inhibitors (Complete), and 50% glycerol to extract nuclear proteins.23 After extraction on ice for 15 minutes, nuclei were centrifuged at 14,000 × g for 5 minutes and the supernatants flash frozen in liquid nitrogen. Protein concentrations were measured using a dye-binding assay (BioRad, Richmond, CA).
Western Blotting
Protein concentrations were normalized and samples prepared in Laemmli buffer and boiled for 5 minutes. Samples were then separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to membranes for Western blotting. Membranes were probed with specific antibodies overnight at 4°C and then washed and incubated for 1 hour in secondary antibodies coupled to horseradish peroxidase. Bound antibody was detected by chemiluminescence. Immunoreactive protein quantity was assessed by scanning densitometry.
Electrophoretic Mobility Shift Assays
Nuclear extracts were used in electrophoretic mobility shift assays (EMSAs) as previously described,18 except that 0.08% NP-40, 0.25% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), and 2.5 mg/mL bovine serum albumin were added to Egr-1 binding mixtures. Oligonucleotides corresponding to the Egr-1 (5′-AACCCTCTGCCCCCGCGATGGAG-3′) and AP-1 (5′-TCCACATGAGATCATGGTTT-3′) binding sites in the promoter region of the murine TNF-α gene and the consensus NF-κB binding sites (Santa Cruz) were used to measure the DNA binding activity of Egr-1, NF-κB, and AP-1.
Isolation of RNA and Real-Time Polymerase Chain Reaction
RNA was isolated from mouse liver using Nucleospin columns (BD Biosciences, Palo Alto, CA). Four hundred nanograms of total RNA was reverse transcribed using the RET-ROscript kit (Ambion, Austin, TX) with random decamers as primer. Briefly, the RNA and random decamer were incubated at 82°C for 3 minutes to melt RNA secondary structure then allowed to cool to 42° before adding the remaining reaction mixture components. First-strand synthesis was then performed during a 60-minute incubation at 42°C, followed by a 10-minute incubation at 92°C to inactivate the Moloney Murine Leukemia virus (MMLV)-reverse transcriptase enzyme. Real-time polymerase chain reaction (PCR) amplification was performed in an iCycler (BioRad, Hercules, CA), using the SYBR Green PCR Core Reagents (Applied Biosystems, Warrington, United Kingdom). Reactions were performed in duplicate in a 96-well optical reaction plate (BioRad). Each reaction mixture contained 3.5 mmol/L MgCl2, 200 μmol/L deoxynucleotides (dNTPs), 0.625 units of AmpliTaq Gold Taq polymerase, and 0.1 μmol/L of each primer (in separate reactions; see below for sequences) in a SYBR Green PCR buffer to which 1 μL of complementary DNA was added for a total volume of 25 μL per well. The thermal cycle was as follows: 94°C for 10 minutes, repeated once, and 94°C for 15 seconds, 60°C for 20 seconds, and 72°C for 20 seconds, repeated 40 times. The thermal cycle was followed by melt curve analysis to ensure homogeneity of the PCR products. The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to that of β-actin. Ct values were determined using the iCycler software package (BioRad).
The primer sequences are as follows: TNF-α forward: 5′-CAA GGA GGA GAA GTT CCC AA-3′, reverse: 5′-CTC TGC TTG GTG GTT TGC TA-3′; Egr-1 forward: 5′-GAA CAA CCC TAC GAG CAC CT-3′, reverse: 5′-GGG TAG TTT GGC TGG GAT AA-3′; Toll-like receptor 4 (TLR4) forward: 5′-ATG GCA TGG CTT ACA CCA CC-3′, reverse: 5′-GAG GCC AAT TTT GTC TCC ACA-3′; CD14 forward: 5′-CTG AAT CCC ACT CGG AGA AG-3′, reverse: 5′-CTC AGA AAC CAG GAG GAT GC-3′; β-actin forward: 5′-CTT TGC AGC TCC TTC GTT GC-3′, reverse: 5′-ACG ATG GAG GGG AAT ACA GC-3′. All primers used for real-time PCR analysis were synthesized by Integrated DNA Technologies, Inc (Coralville, NJ).
Statistical Analysis
Values reported are means ± standard error of the mean. Data were analyzed by a Student t test or general linear models procedure (SAS, Carey, IN), blocking for trial effects if data from more than one feeding trial was used in a data set. Data were log transformed if needed to obtain a normal distribution.
Results
Chronic ethanol exposure increases the sensitivity of hepatic macrophages (Kupffer cells) to LPS-stimulated TNF-α production.24 In Kupffer cells isolated from rats fed ethanol for 4 weeks, this sensitization to LPS is mediated in part by an increase in LPS-stimulated Egr-1 expression. Egr-1, an immediate early gene, is rapidly and transiently induced in Kupffer cells in response to LPS and contributes to TNF-α expression.18 Here we have asked whether chronic ethanol feeding also increases hepatic Egr-1 expression in response to LPS exposure in vivo. Female wild-type mice were fed ethanol as part of a complete liquid diet for 6 weeks or pair-fed control diets. Ethanol- and pair-fed mice gained body mass at the same rate (Table 1). When mice were injected with 0.7 μg/g body weight LPS, hepatic Egr-1 mRNA content increased rapidly and transiently in both pair- and ethanol-fed mice (Figure 1A). However, after chronic ethanol feeding, LPS-stimulated Egr-1 mRNA accumulation was increased by 3-fold in liver at 60 minutes after LPS treatment compared to pair-fed controls. This increased expression of Egr-1 mRNA was paralleled by increased accumulation of Egr-1 protein in nuclear extracts (Figure 1B). In contrast, translocation of the p65 subunit of NF-κB to the nucleus, although increased with LPS, was not affected by chronic ethanol feeding. Egr-1 DNA binding activity was increased at 60 minutes after LPS injection; LPS-stimulated Egr-1 DNA binding activity was 1.8 ± 0.3-fold higher (n = 6, P < .05) in ethanol-fed compared with pair-fed mice. Activation of NF-κB and AP-1 DNA binding activities were also increased in response to LPS; however, chronic ethanol feeding had no effect on LPS-stimulated DNA binding activity (Figure 1C). LPS-stimulated NF-κB and AP-1 DNA binding activity after chronic ethanol feeding were 83% ± 8% (n = 7) and 100% ± 5% (n = 4) of activity in pair-fed mice, respectively (Figure 1C). These data show that LPS rapidly induces the expression of Egr-1 in mouse liver in vivo and that chronic ethanol feeding increases the LPS-stimulated expression and DNA binding activity of Egr-1.
Table 1.
Body Weight, Liver to Body Weight Ratios, and Serum Ethanol Concentrations in Wild-type and egr-1−/−Mice After Chronic Ethanol Feeding
| Females
|
Males
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Wild type
|
egr-1 −/−
|
Wild type
|
egr-1 −/−
|
|||||
| Pair-fed | EtOH-fed | Pair-fed | EtOH-fed | Pair-fed | EtOH-fed | Pair-fed | EtOH-fed | |
| Body weight Initial | 17.9 ± 0.2 | 18.8 ± 0.2 | 20.1 ± 0.4 | 19.6 ± 0.2 | 25.4 ± 0.5 | 27.4 ± 0.5 | 25.3 ± 1.2 | 23.9 ± 0.3 |
| Final | 22.8 ± 0.3a | 23.0 ± 0.3a | 26.3 ± 0.1b | 26.4 ± 0.5b | 29.8 ± 0.8a | 29.4 ± 0.4a | 32.4 ± 2.2b | 33.6 ± 1.4b |
| Liver (mg)/body weight (g) | 51 ± 2 | 58 ± 1* | 46 ± 4 | 47 ± 4 | 47 ± 2 | 58 ± 3* | 44 ± 2 | 41 ± 3 |
| Serum EtOH (mmol/L) | ND | 2.4 ± 0.2 | ND | 3.5 ± 0.6 | ND | 2.4 ± 0.2 | ND | 4.0 ± 1.3 |
NOTE. Values represent means ± SEM, n = 3 per group. Values with different superscripts are significantly different, P < .05.
ND, not determined.
P < .05 compared with pair-fed within genotype.
Figure 1.
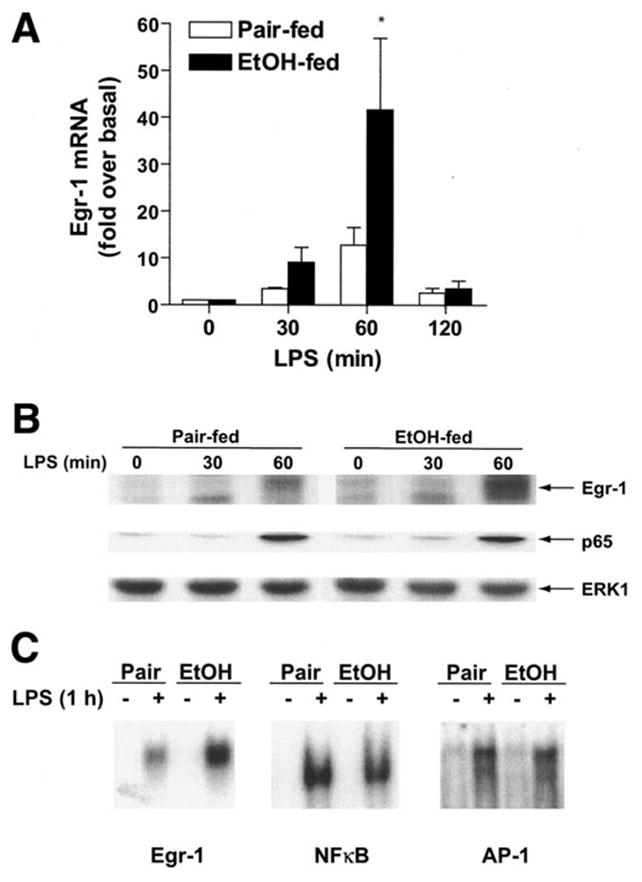
Chronic ethanol feeding enhances LPS-stimulated Egr-1 expression. (A) Hepatic Egr-1 mRNA content is increased in response to LPS. Female C57BL/6 mice were fed an ethanol-containing diet over 6 weeks or pair-fed control diets. Mice were then injected intraperitoneally with 0.7 μg/g body weight LPS and livers harvested after 30, 60, or 120 minutes. Mice not treated with LPS were injected with vehicle (0.09% saline) and livers harvested after 120 minutes. Total RNA was isolated and quantity of Egr-1 mRNA determined by real-time reverse-transcriptase PCR. Values represent means ± SEM, n = 4–6 per time point, *P < .05 compared with pair-fed. (B) Nuclear Egr-1 protein quantity is increased in response to LPS. Nuclear extracts were prepared from ethanol- and pair-fed mice and relative quantity of Egr-1 and p65 protein measured by Western blot. Total ERK1/2 immunoreactivity is shown as a loading control. Figure is representative of at least 3 sets of animals. (C) Treatment with LPS stimulates Egr-1, NF-κB, and AP-1 DNA binding activity in mouse liver. Nuclear extracts were prepared from ethanol- and pair-fed mice after treatment with or without LPS for 1 hour. The binding of nuclear proteins to oligonucleotides corresponding to the Egr-1 and AP-1 binding sites in the murine TNF-α promoter, as well as consensus sequence for NF-κB binding sites, were assessed by EMSA. Figures are representative of at least 3 sets of animals.
Egr-1 is an important contributor to maximal LPS-stimulated TNF-α expression in monocytes and macrophages.9,18 Because TNF-α is considered a critical component in the progression of ethanol-induced liver injury, we hypothesized that mice lacking Egr-1 (egr-1 −/−) should be protected from chronic ethanol-induced liver injury. Wild-type and egr-1 −/− mice were allowed free access to ethanol-containing diets for 6 weeks or pair-fed control diets. Body weights of wild-type and egr-1 −/− mice increased over the course of the ethanol feeding period. There were no differences in final body weight between pair-fed and ethanol-fed of either genotype, but egr-1 −/− mice were heavier than wild-type mice at the end of the feeding period (Table 1). Serum ethanol concentrations, measured 3–3.5 hours into the dark/feeding cycle, were approximately 2.5–5 mmol/L and did not differ between wild-type and egr-1 −/− mice (Table 1). Liver weight to body weight ratios were increased after ethanol feeding in wild-type mice but not in egr-1 −/− mice (Table 1).
Serum ALT concentrations were increased by 2-fold in response to ethanol feeding compared with pair-fed in wild-type mice (Figure 2A). In contrast, ALT concentrations did not increase with ethanol feeding in egr-1 −/− mice compared with pair-fed controls (Figure 2A). Chronic ethanol feeding also increased serum TNF-α concentrations in wild-type mice by 20-fold (Figure 2B). In egr-1 −/− mice, serum TNF-α concentrations remained at control concentrations even after ethanol feeding (Figure 2B). Responses to ethanol feeding did not differ between females and males in either wild-type or egr-1 −/− mice (Table 1, Figure 2). Figure 3A shows representative photomicrographs of liver histological sections stained with H&E from female wild-type and egr-1 −/− mice fed ethanol-containing diets or pair-fed control diets. In wild-type mice, chronic ethanol feeding increased the accumulation of lipid in the liver, characterized by the appearance of micro- and macrovesicular lipid droplets (Figure 3A). Oil Red O staining of frozen sections (Figure 3B) and biochemical analysis of total liver triglycerides (Figure 3C) show significantly increased lipid accumulation in the livers of wild-type mice after chronic ethanol feeding. Typical of ad libitum ethanol feeding to mice, little inflammation and no necrosis were observed in these ethanol-fed mice (Figure 3A). In contrast to the ethanol-induced steatosis in wild-type mice, ethanol-fed egr-1 −/− did not develop steatosis (Figure 3A). Although Oil Red O staining (Figure 3B) and liver triglycerides (Figure 3C) were increased slightly in egr-1 −/− after ethanol feeding, egr-1 −/− mice were protected from chronic ethanol-induced steatosis. Histology from male wild-type mice showed a similar pattern of micro- and macrovesicular steatosis after chronic ethanol feeding, whereas male egr-1 −/− mice were protected from ethanol-induced steatosis (data not shown).
Figure 2.
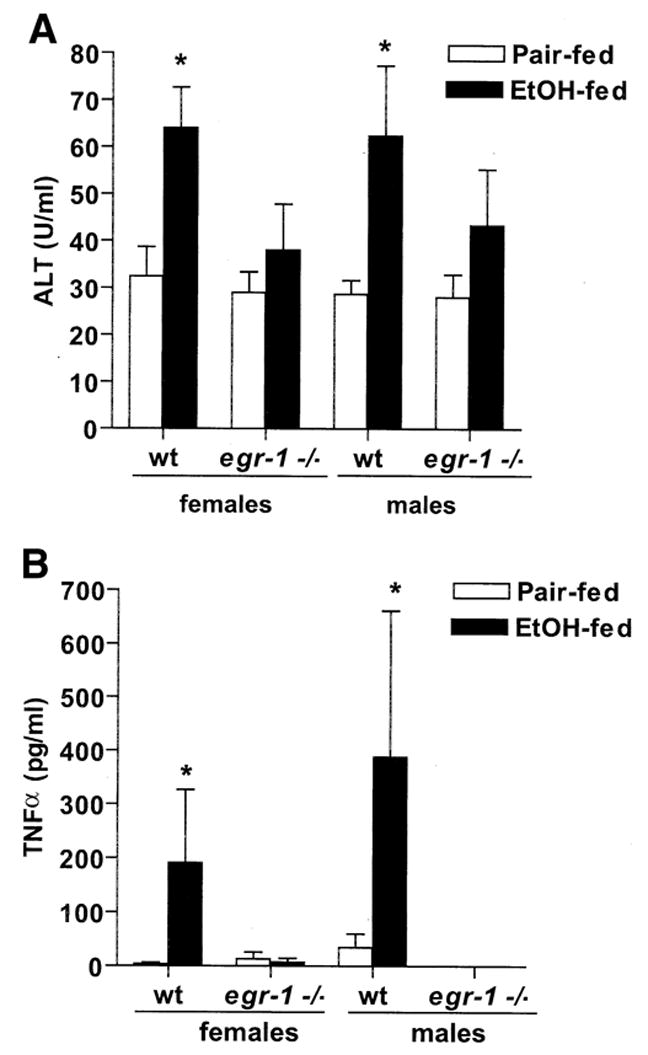
Chronic ethanol feeding increases serum ALT and serum TNF-α concentrations in wild-type but not egr-1 −/− mice. Mice were allowed free access to an ethanol-containing diet or pair fed a control diet for 6 weeks. ALT activity in serum was measured enzymatically. TNF-α was measured by ELISA. Values represent mean ± standard error of the mean, n = 11 for wild-type females, 5 for egr-1 −/− females, and 3 for wild-type and egr-1 −/− males. *P < .05 compared with pair-fed controls.
Figure 3.
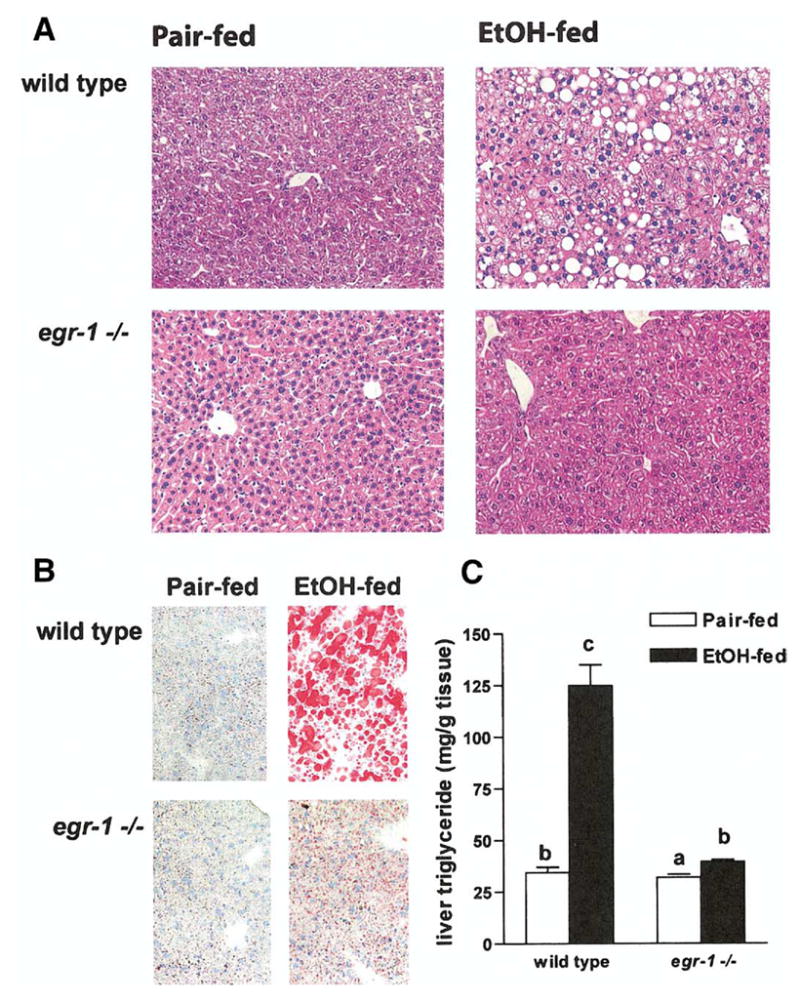
Liver histology and triglyceride content in wild-type and egr-1 −/− mice after chronic ethanol feeding. (A) Livers from mice allowed free access to ethanol-containing diets or pair-fed control diets were perfused with saline and then fixed in formalin. Liver sections were prepared and stained with H&E. Figures are representative of at least 5 mice in each treatment group. (B) Frozen liver sections were stained with Oil Red O and counterstained with hematoxylin. Figures are representative of 3 mice in each treatment group. (C) Liver triglycerides were measured. Values represent mean ± standard error of the mean, n = 4; values with different superscripts are significantly different, P < .05.
One current working model for the development of ethanol-induced liver injury6 hypothesizes that increased exposure to endotoxin/LPS during ethanol feeding increases the production of TNF-α by Kupffer cells.22,25,26 Increased TNF-α can then contribute to hepatic injury during chronic ethanol exposure.1 Thus, protection from chronic ethanol-induced liver injury in egr-1 −/− mice could result from a protective effect on the intestinal barrier function, resulting in a normalization of endotoxin exposure. Therefore, we compared the effects of ethanol feeding on endotoxin concentrations in platelet-rich plasma in male wild-type and egr-1 −/− mice. Chronic ethanol feeding increased endotoxin concentrations in both wild-type and egr-1 −/− male mice (Figure 4). These data suggest that protection from chronic ethanol-induced liver injury was not caused by differences in endotoxin exposure during ethanol feeding between wild-type and egr-1 −/− mice.
Figure 4.
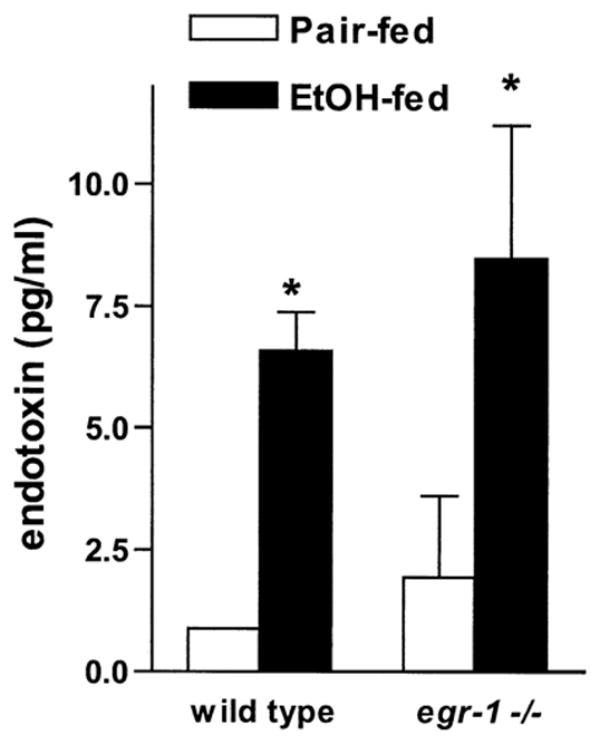
Endotoxin concentrations in platelet-rich plasma after chronic ethanol feeding. Platelet-rich plasma was prepared from blood taken from the retro-orbital sinus of male ethanol- and pair-fed wild-type and egr-1 −/− mice. Endotoxin concentrations were measured using a chromogenic assay. Values represent mean ± SEM, n = 3. *P < .05 compared with pair-fed controls.
Induction of CYP2E1 expression and associated production of reactive oxygen species is another important mechanism for chronic ethanol-induced liver injury.27 Chronic ethanol feeding increased the expression of hepatic CYP2E1, measured by Western blotting, in both wild-type and egr-1 −/− mice (Figure 5), suggesting that egr-1 is not required for CYP2E1 induction in response to chronic ethanol exposure.
Figure 5.
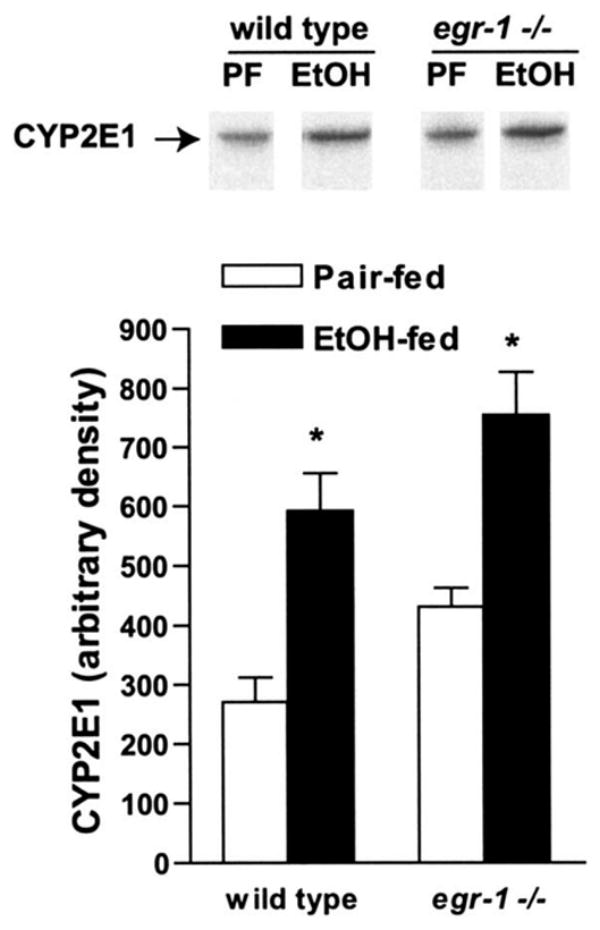
CYP2E1 expression is induced in livers of wild-type and egr-1 −/− mice after chronic ethanol feeding. Homogenates were prepared from livers of EtOH- and pair-fed mice and the relative expression of CYP2E1 assessed by Western blot analysis. Western blots were also probed with antibody against β-actin as a loading control (data not shown). Values represent mean ± standard error of the mean, n = 4. *P < .05 compared with pair-fed controls.
Taken together, these data suggested that egr-1 contributes to ethanol-induced liver injury at steps subsequent (ie, downstream) to increased endotoxin exposure and CYP2E1 induction. One essential downstream response of increased exposure to LPS/endotoxin is increased production of TNF-α. Because Egr-1 contributes to maximal stimulation of TNF-α expression in response to LPS, we hypothesized that egr-1 −/− mice, lacking functional Egr-1, would not develop a sensitization to LPS-stimulated TNF-α production after chronic ethanol feeding. The expression of TLR4 and CD14 mRNA was measured by real-time reverse-transcriptase PCR in wild-type and egr-1 −/− mice. The relative expression of TLR4 and CD14 mRNA to β-actin mRNA was 1.0 ± 0.04 for TLR4 and 1.02 ± 0.34 for CD14 in wild type and 1.04 ± 0.24 for TLR4 and 2.04 ± 0.58 in egr-1 −/− (n = 6). These data indicate that the lack of egr-1 did not affect the expression of these 2 major components of the LPS-receptor complex. Chronic ethanol feeding to wild-type mice increased LPS-stimulated TNF-α mRNA accumulation in liver compared with pair-fed controls (Figure 6A). LPS-stimulated serum TNF-α concentrations were also higher after chronic ethanol feeding in wild-type mice compared with pair-fed wild-type mice (Figure 6B). In egr-1 −/− mice, LPS stimulation of hepatic TNF-α mRNA accumulation was decreased by approximately 40% (Figure 6A). However, in contrast to wild-type mice, chronic ethanol feeding did not further sensitize the egr-1 −/− mice to LPS exposure (Figure 6A). Similarly, serum TNF-α concentrations were increased in both ethanol- and pair-fed egr-1 −/− mice in response to LPS, but with no additional sensitization to LPS after chronic ethanol feeding (Figure 6B).
Figure 6.
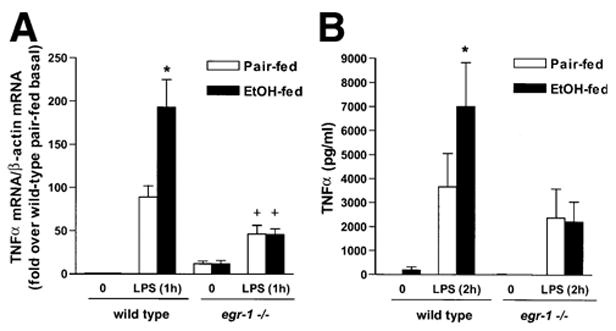
LPS-stimulated hepatic TNF-α mRNA and serum TNF-α concentrations. (A) Total RNA was isolated from livers from ethanol- and pair-fed wild-type and egr-1 −/− mice. Expression of TNF-α mRNA was measured by real-time PCR. Quantity of TNF-α mRNA is expressed relative to β-actin mRNA. LPS-stimulated TNF-α mRNA/β-actin mRNA ratios are expressed relative to basal ratios in wild-type mice and relative to pair-fed wild-type baseline values for egr-1 −/− mice. Values represent mean ± standard error of the mean, n = 7 for wild type and n = 3 for egr-1 −/−. *P < .05 compared with pair-fed, compared with wild type. (B) Serum TNF-α concentrations were measured by ELISA. Values represent mean ± SEM, n = 13 for wild-type and n = 6 for egr-1 −/− mice at the 2-hour time point. *P < .05 compared with pair-fed mice at the 2-hour time point.
Because NF-κB activity plays an important role in LPS-stimulated TNF-α expression,8 normalization of LPS-mediated TNF-α mRNA expression in egr-1 −/− mice fed chronic ethanol could result from impaired activation of NF-κB in response to LPS. To test this hypothesis, the binding of hepatic nuclear proteins to NF-κB DNA binding sites was compared between wild-type and egr-1 −/− mice. LPS increased the binding of nuclear proteins to the NF-κB binding site (Figure 7A). As in wild-type mice (Figure 1C), chronic ethanol feeding had no effect on LPS-stimulated activation of NF-κB DNA binding activity in egr-1 −/− mice (Figure 7A). Similarly, chronic ethanol feeding had no effect on LPS-stimulated degradation of IκB-α or translocation of p65 from the cytosol (Figure 7B). These data further support the hypothesis that the absence of Egr-1 transcriptional activation of TNF-α gene expression in egr-1 −/− mice is a critical factor in maintaining/normalizing LPS-stimulated TNF-α mRNA and protein expression after chronic ethanol feeding.
Figure 7.
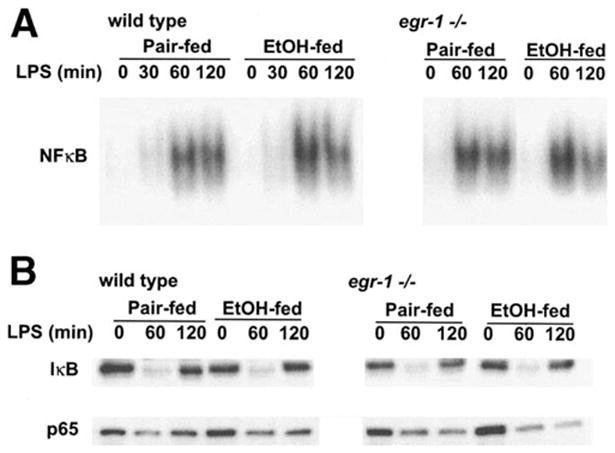
(A) LPS-stimulated NF-κB DNA binding activity in livers from wild-type and egr-1 −/− mice. Nuclear extracts were prepared from livers of ethanol- and pair-fed mice after treatment with or without LPS for 30–120 minutes. The binding of nuclear proteins to an oligonucleotide corresponding to the consensus sequence for the NF-κB binding site was assessed by EMSA. Figures are representative of at least 3 sets of animals. (B) Activation of NF-κB by LPS in wild-type and egr-1 −/− mice. Postnuclear supernatants were prepared from livers of ethanol- and pair-fed mice after treatment with or without LPS for 60–120 minutes. Immunoreactivity of IκB-α and p65 were measured by Western blot analysis. Figures are representative of at least 3 sets of animals.
Discussion
Although a number of mediators have been implicated in ethanol-induced liver injury, the role of TNF-α in the pathogenesis of alcohol-induced liver disease is thought to be of particular importance.1,6 Circulating TNF-α is increased in the blood of alcoholics and in animals chronically exposed to ethanol.6,11 Treatment of rats exposed to ethanol via intragastric feeding with antibiotics decreases TNF-α expression and ethanol-induced liver injury,6 suggesting that increased TNF-α after ethanol exposure is caused, at least in part, by increased exposure to LPS. Here we have shown that Egr-1, a transcription factor essential for maximal transcriptional activation of TNF-α expression in response to LPS,9,18 is required for the development of ethanol-induced fatty liver injury in mice. Chronic ethanol feeding to mice enhanced LPS-stimulated Egr-1 mRNA and DNA binding activity, contributing to increased LPS-induced TNF-α mRNA and protein expression observed in mice after chronic ethanol feeding. Elimination of Egr-1 activity in egr-1 −/− mice not only prevented the development of steatosis after ethanol feeding but also normalized LPS-stimulated TNF-α mRNA and protein expression. These data are the first to identify a specific transcription factor as a requirement in the development of chronic ethanol-induced fatty liver injury.
Synthesis of Egr-1, an immediate early gene, is rapidly and transiently induced in response to a variety of stimuli.28 Egr-1 has been characterized as a transcription factor that coordinates cellular responses to environmental stress, mediating increased expression of a number of target genes, including tissue factor, transforming growth factor β, platelet-derived growth factor, chemokines, and adhesion molecules.20 These gene products are required for progression of tissue repair, involving the acute inflammatory response and the release of cytokines and growth factors, neutrophil migration, collagen synthesis, and extracellular matrix remodeling.28 Recent data show that egr-1 −/− mice exhibit delayed liver regeneration in response to partial hepatectomy,29 consistent with a role for Egr-1 in wound healing. Egr-1 is also required for maximal stimulation of TNF-α expression in response to LPS in monocytes and macrophages.9,18
Here we have used LPS-stimulated TNF-α expression as a tool to identify the impact of Egr-1– dependent changes in gene expression during chronic ethanol feeding. Although TNF-α has been implicated in the progression of ethanol-induced liver injury,1,6 additional pathways clearly contribute to ethanol-induced liver injury. For example, a recent study by Ji et al30 suggests that TNF-α may only make a small contribution to ethanol-induced liver injury and that additional factors, including endoplasmic reticulum stress, are also major contributors to liver injury during chronic ethanol exposure. Although our data clearly show that Egr-1 is involved in ethanol-induced changes in TNF-α expression, Egr-1 may also contribute to additional pathways mediating liver injury. Of particular interest, Egr-1 directly regulates the expression of the intracellular adhesion molecule-1.31 Intracellular adhesion molecule-1 expression is increased during chronic ethanol feeding to rodents32 and has been implicated in the development of ethanol-induced liver injury.33 Future studies will be required to fully define the contributions of Egr-1–dependent gene expression in the multiple pathways contributing to ethanol-induced liver injury.
We have previously shown that chronic ethanol feeding increases LPS-stimulated Egr-1 DNA binding activity in Kupffer cells isolated from rats fed an ethanol containing diet for 4 weeks.17 Here we report that chronic ethanol feeding to mice also increases LPS-stimulated Egr-1 DNA binding activity in the livers of mice injected with LPS in vivo (Figure 1). Although hepatocytes are known to increase Egr-1 expression in response to epidermal growth factor treatment34 and Egr-1 expression is observed early in liver regeneration,35 hepatocytes are considered to be low-level responders to LPS.36 Although a contribution by hepatocytes to increased Egr-1 expression cannot be ruled out at the doses of LPS used in these studies,37 it is likely, because Kupffer cells are the primary cell type mediating the hepatic response to LPS,38 that most of the hepatic LPS-induced Egr-1 expression is produced by Kupffer cells.
Egr-1–null mice grow normally and do not exhibit any major phenotypic abnormalities under normal conditions, except that female egr-1 −/− mice are sterile because of impaired luteinizing hormone synthesis.39,40 Macrophage development and differentiation are normal in egr-1 −/− mice.20 Interestingly, although Egr-1 is involved in a number of inflammatory and immune responses,41,42 Egr-1 does not contribute to the early inflammatory responses of lung and kidney to endotoxemia.43 Although LPS potently increases Egr-1 expression in lung and kidney, LPS-treated wild-type and egr-1 −/− mice exhibit similar increases in serum TNF-α during endotoxemia.43 Furthermore, Egr-1 deficiency does not affect survival times in a model of acute endotoxemia.44 In liver, we found that egr-1 −/− mice pair-fed control diets exhibit a moderately suppressed, yet still robust, induction of hepatic TNF-α mRNA in response to LPS (Figure 6). Serum TNF-α concentrations after LPS exposure were not affected by the absence of Egr-1 (Figure 6). This maintenance of LPS-stimulated hepatic TNF-α mRNA and serum TNF-α in egr-1 −/− mice fed control diets compared with wild-type mice is likely caused by redundancy in the signaling pathways regulating the in vivo response to LPS. In contrast, after chronic ethanol feeding, there was a greater difference in LPS-stimulated TNF-α mRNA and circulating TNF-α between wild-type and egr-1 −/− mice (Figure 6). Taken together, these data suggest that chronic ethanol feeding enhances the role of Egr-1 in mediating LPS-stimulated TNF-α expression, with Egr-1 becoming a major contributing factor in the enhanced sensitivity of ethanol-fed mice to LPS.
In contrast to increased LPS-stimulated Egr-1 DNA binding activity, LPS-stimulation of NF-κB or AP-1 DNA binding activity after 60 minutes of LPS exposure was not increased after chronic ethanol feeding (Figure 2). Indeed, a suppression of LPS-stimulated NF-κB activity after chronic ethanol feeding to mice has been previously reported after 90 minutes of LPS exposure.12 Koteish et al13 speculated that, although LPS-stimulated activation of NF-κB DNA binding activity may be decreased after chronic ethanol feeding, the response was still robust and likely adequate for maintaining NF-κB–mediated responses. However, because LPS-stimulated NF-κB activation, measured by IκB-α degradation, movement of p65 from cytosol to nucleus, or DNA binding activity (Figures 1 and 7), is not increased after chronic ethanol feeding, it is unlikely that NF-κB activity can account for increased LPS-stimulated TNF-α production after chronic ethanol feeding. Our data suggest that, in addition to the key role played by NF-κB in mediating LPS-dependent responses, Egr-1 also plays an essential role in the enhanced sensitivity of mice to LPS after chronic ethanol feeding.
The mechanisms by which chronic ethanol feeding specifically increases LPS-stimulated Egr-1 DNA binding activity relative to NF-κB and AP-1 are not clear. It is widely accepted that reactive oxygen species (ROS) play a critical role in the development of alcoholic liver injury45,46; however, the targets of ROS during ethanol exposure have not been completely elucidated. ROS have been implicated in the activation of NF-κB during ethanol exposure47 (independent of the ability of LPS to activate NF-κB).48 However, additional signaling pathways are also targeted by ROS, including activation of ERK1/2 and Egr-1.49 LPS-stimulated production of ROS is required for activation of ERK1/2 in macrophages and is involved in LPS-stimulated inflammatory cytokine expression.44 Because increased LPS-stimulated Egr-1 activity after chronic ethanol exposure is dependent on ERK1/2 in Kupffer cells,17 it is possible that increased ROS production, either as a result of ethanol metabolism and/or in response to LPS stimulation, may contribute to increased Egr-1 activity after chronic ethanol exposure.
Because of the critical role of TNF-α in the development of alcohol-induced liver disease, clinical investigations of the therapeutic efficacy of using antibodies to TNF-α to treat patients with acute alcoholic steatohepatitis are currently under way.50 However, TNF-α also mediates important immuno- and hepatoprotective functions. Because TNF-α is a critical component of normal immune function, infectious disease is a primary concern during anti–TNF-α therapy.51 By understanding the specific mechanisms by which chronic ethanol enhances hepatic sensitivity to LPS-stimulated TNF-α production, very specific therapeutic strategies can be developed that target the molecular site(s) of ethanol action leading to “overproduction” of TNF-α. Strategies to normalize, rather than eliminate, TNF-α activity would allow for maintenance of normal immune function during treatment to resolve liver injury, as well as retain the hepatoprotective function of TNF-α. Here we have identified Egr-1 as a potential therapeutic target for normalizing TNF-α expression in response to ethanol exposure. After chronic ethanol feeding, the role of Egr-1 in mediating LPS-stimulated TNF-α production was greatly enhanced relative to its contribution in control mice. Importantly, the absence of functional Egr-1 transcriptional activity suppressed, but did not eliminate, LPS-stimulated TNF-α production. Furthermore, mice lacking functional Egr-1 did not develop chronic ethanol-induced fatty liver injury. These data suggest that strategies to regulate Egr-1 expression and/or functional activity would be expected to normalize the enhanced expression of inflammatory cytokines involved in the development of ethanol-induced liver injury.
Acknowledgments
Supported by NIH grants AA013868 and AA11975 to LEN.
The authors thank Dr J. Milbrandt (Washington University, St. Louis, MO) for the egr-1 +/− breeding pair, Dr Chantal Rivera (Baylor College of Medicine) for advice on the assay of endotoxin, and Dr Gretta Jacobs (Case Western Reserve University) for assistance in interpreting liver histology.
Abbreviations used in this paper
- ALT
alanine aminotransferase
- AP-1
activator protein-1
- Ct
comparative threshold
- Egr-1
early growth response-1 protein
- EMSA
electrophorectic mobility shift assay
- LPS
lipopolysaccharide
- NF-κB
nuclear factor kappa B
- PCR
polymerase chain reaction
- ROS
reactive oxygen species
- SEM
standard error of the mean
- TLR4
toll-like receptor 4
- TNF-α
tumor necrosis factor α
References
- 1.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B. TNF, immunity and inflammatory disease: lessons of the past decade. J Invest Med. 1995;43:227–235. [PubMed] [Google Scholar]
- 3.Jacob CO. Tumor necrosis factor alpha in autoimmunity: pretty girl or old witch? Immunol Today. 1992;13:122–125. doi: 10.1016/0167-5699(92)90107-i. [DOI] [PubMed] [Google Scholar]
- 4.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reimund JM, Wittersheim C, Dumont S, Muller CD, Baumann R, Poindron P, Duclos B. Mucosal inflammatory cytokine production by intestinal biopsies in patients with ulcerative colitis and Crohn’s disease. J Clin Immunol. 1996;16:144–150. doi: 10.1007/BF01540912. [DOI] [PubMed] [Google Scholar]
- 6.Thurman RG. Mechanisms of hepatic toxicity II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 7.Papadakis KA, Targan SR. Tumor necrosis factor: biology and therapeutic inhibitors. Gastroenterology. 2000;119:1148–1157. doi: 10.1053/gast.2000.18160. [DOI] [PubMed] [Google Scholar]
- 8.Tsai EY, Falvo JV, Tsytsykova AV, Barczak AK, Reimold AM, Glimcher LH, Fenton MJ, Gordon DC, Dunn IF, Goldfeld AE. A lipopolysaccharide-specific enhancer complex involving Ets, Elk-1, Sp1 and CREB binding protein and p300 is recruited to the tumor necrosis factor alpha promoter in vivo. Mol Cell Biol. 2000;20:6084–6094. doi: 10.1128/mcb.20.16.6084-6094.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yao J, Mackman N, Edgington TS, Fan ST. Lipopolysaccharide induction of the tumor necrosis factor α promoter in human monocytic cells. Regulation by egr-1, c-jun and NFκB transcription factors. J Biol Chem. 1997;272:17795–17801. doi: 10.1074/jbc.272.28.17795. [DOI] [PubMed] [Google Scholar]
- 10.Means TK, Pavlovich RP, Roca D, Vermeulen MW, Fenton MJ. Activation of TNFα transcription utilizes distinct MAP kinase pathways in different macrophage populations. J Leukocyte Biol. 2000;67:885–893. doi: 10.1002/jlb.67.6.885. [DOI] [PubMed] [Google Scholar]
- 11.Khoruts A, Stahnke L, McClain CJ, Logan G, Allen JI. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology. 1991;13:267–276. [PubMed] [Google Scholar]
- 12.Honchel R, Ray M, Marsano L, Cohen D, Lee E, Shedlofsky S, McClain CJ. Tumor necrosis factor in alcohol enhanced endotoxin liver injury. Alcohol Clin Exp Res. 1992;16:665–669. doi: 10.1111/j.1530-0277.1992.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 13.Koteish A, Yang S, Lin H, Huang X, Diehl AM. Chronic ethanol exposure potentiates lipopolysaccharide liver injury despite inhibiting Jun N-terminal kinase and caspase 3 activation. J Biol Chem. 2002;277:13037–13044. doi: 10.1074/jbc.M101632200. [DOI] [PubMed] [Google Scholar]
- 14.Mathurin P, Deng Q-G, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology. 2000;32:1008–1017. doi: 10.1053/jhep.2000.19621. [DOI] [PubMed] [Google Scholar]
- 15.Cao Q, Mak KM, Lieber CS. Dilinoleoylphosphatidylcholine decreases LPS-induced TNFα generation in Kupffer cells of ethanol-fed rats: respective roles of MAPKs and NFκB. Biochem Biophys Res Commun. 2002;294:849–853. doi: 10.1016/S0006-291X(02)00586-7. [DOI] [PubMed] [Google Scholar]
- 16.Kishore R, McMullen MR, Nagy LE. Stabilization of TNFα mRNA by chronic ethanol: role of A+U rich elements and p38 mitogen activated protein kinase signaling pathway. J Biol Chem. 2001;276:41930–41937. doi: 10.1074/jbc.M107181200. [DOI] [PubMed] [Google Scholar]
- 17.Kishore R, Hill JR, McMullen MR, Frenkel J, Nagy LE. ERK1/2 and Egr-1 contribute to increased TNFα production in rat Kupffer cells after chronic ethanol feeding. Am J Phys. 2002;282:G6–G15. doi: 10.1152/ajpgi.00328.2001. [DOI] [PubMed] [Google Scholar]
- 18.Shi L, Kishore R, McMullen M, Nagy LE. Chronic ethanol increases LPS-stimulated Egr-1 expression in RAW 264.7 macrophages: contribution to enhanced TNFα production. J Biol Chem. 2002;277:14777–14785. doi: 10.1074/jbc.M108967200. [DOI] [PubMed] [Google Scholar]
- 19.Gashler A, Sukhatme VP. Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog Nucleic Acid Res Mol Biol. 1995;50:191–224. doi: 10.1016/s0079-6603(08)60815-6. [DOI] [PubMed] [Google Scholar]
- 20.Yan SF, Fujita T, Lu J, Okada K, Zou YS, Mackman N, Pinsky DJ, Stern DM. Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat Med. 2000;6:1355–1361. doi: 10.1038/82168. [DOI] [PubMed] [Google Scholar]
- 21.Lee SL, Wang Y, Milbrandt J. Unimpaired macrophage differentiation and activation in mice lacking the zinc finger transplantation factor NGFI-A (EGR1) Mol Cell Biol. 1996;16:4566–4572. doi: 10.1128/mcb.16.8.4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivera CA, Bradford BU, Seabra V, Thurman RG. Role of endotoxin in the hypermetabolic state after acute ethanol exposure. Am J Physiol Gastrointest Liver Physiol. 1998;275:G1252–G1258. doi: 10.1152/ajpgi.1998.275.6.G1252. [DOI] [PubMed] [Google Scholar]
- 23.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ’mini-extracts prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagy LE. New insights into the role of the innate immune response in the development of alcoholic liver disease. Exp Biol Med. 2003;228:882–890. doi: 10.1177/153537020322800803. [DOI] [PubMed] [Google Scholar]
- 25.Merial C, Bouloumie A, Trocheris V, Lafontan M, Galitzky J. Nitric oxide-dependent downregulation of adipocyte UCP-2 expression by tumor necrosis factor-α. Am J Physiol Cell Physiol. 2000;279:C1100–C1106. doi: 10.1152/ajpcell.2000.279.4.C1100. [DOI] [PubMed] [Google Scholar]
- 26.Nanji AA, Khettry U, Sadrzadeh SMH, Yamanaka T. Severity of liver injury in experimental alcoholic liver disease: correlation with plasma endotoxin, prostaglandin E2, leukotriene B4 and throm-boxane B2. Am J Pathol. 1993;142:367–373. [PMC free article] [PubMed] [Google Scholar]
- 27.Guha M, O’Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, Stern D, Mackman N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor α expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood. 2001;98:1429–1439. doi: 10.1182/blood.v98.5.1429. [DOI] [PubMed] [Google Scholar]
- 28.Braddock M. The transcription factor Egr-1: a potential drug in wound healing and tissue repair. Ann Med. 2001;33:313–318. doi: 10.3109/07853890109002083. [DOI] [PubMed] [Google Scholar]
- 29.Liao Y, Shikapwashya ON, Shteyer E, Dieckgraefe BK, Hruz PW, Rudnick DA. Delayed hepatocellular mitotic progression and impaired liver regeneration in early growth response-1-deficient mice. J Biol Chem. 2004;279:43107–43116. doi: 10.1074/jbc.M407969200. [DOI] [PubMed] [Google Scholar]
- 30.Ji C, Deng Q, Kaplowitz N. Role of TNFα in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology. 2004;40:442–451. doi: 10.1002/hep.20309. [DOI] [PubMed] [Google Scholar]
- 31.Maltzman JS, Carmen JA, Monroe JG. Transcriptional regulation of the Icam-1 gene in antigen receptor- and phorbol ester-stimulated B lymphocytes: role for transcription factor EGR1. J Exp Med. 1996;183:1747–1759. doi: 10.1084/jem.183.4.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nanji AA, Greenberg SS, Tahan SR, Fogt F, Loscalzo J, Sadrzadeh SMH, Xie J, Stamler JS. Nitric oxide production in experimental alcoholic liver disease in the rat: role in protection from injury. Gastroenterology. 1995;109:899–907. doi: 10.1016/0016-5085(95)90400-x. [DOI] [PubMed] [Google Scholar]
- 33.Kono H, Uesugi T, Froh M, Rusyn I, Bradford BU, Thurman RG. ICAM-1 is involved in the mechanism of alcohol-induced liver injury: studies with knock-out mice. Am J Physiol. 2001;280:G1289–1295. doi: 10.1152/ajpgi.2001.280.6.G1289. [DOI] [PubMed] [Google Scholar]
- 34.Tsai JC, Liu L, Zhang J, Spokes KC, Topper JN, Aird WC. Epidermal growth factor induces Egr-1 promoter activity in hepatocytes in vitro and in vivo. Am J Physiol. 2001;281:G1271–G1278. doi: 10.1152/ajpgi.2001.281.5.G1271. [DOI] [PubMed] [Google Scholar]
- 35.Peng Y, Du K, Ramirez S, Diamond RH, Taub R. Mitogenic up-regulation of the PRL-1 protein-tyrosine phosphatase gene by Egr-1. Egr-1 activation is an early event in liver regeneration. J Biol Chem. 1999;274:4513–4520. doi: 10.1074/jbc.274.8.4513. [DOI] [PubMed] [Google Scholar]
- 36.Volpes R, van den Oord JJ, Desmet VJ. Can hepatocytes serve as ‘activated’ immunomodulating cells in the immune response? J Hepatol. 1992;16:228–240. doi: 10.1016/s0168-8278(05)80121-7. [DOI] [PubMed] [Google Scholar]
- 37.Liu S, Gallo DJ, Green AM, Williams DL, Gong X, Shapiro RA, Gambotto AA, Humphris EL, Vodovotz Y, Billiar TR. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect Immunol. 2002;70:3433–3442. doi: 10.1128/IAI.70.7.3433-3442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 2002;283:G256–G265. doi: 10.1152/ajpgi.00550.2001. [DOI] [PubMed] [Google Scholar]
- 39.Lee SL, Sadovsky Y, Swirnoff AH, Polish JA, Goda P, Gavrilina G, Milbrandt J. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1) Science. 1996;273:1219–1221. doi: 10.1126/science.273.5279.1219. [DOI] [PubMed] [Google Scholar]
- 40.Lee SL, Tourtellotte LC, Wesselschmidt RL, Milbrandt J. Growth and differentiation proceeds normally in cells deficient in the immediate early gene NGFI-A. J Biol Chem. 1995;270:9971–9977. doi: 10.1074/jbc.270.17.9971. [DOI] [PubMed] [Google Scholar]
- 41.Fitzgerald KA, O’Neill LA. Characterization of CD44 induction by IL-1: a critical role for Egr-1. J Immunol. 1999;162:4920–4927. [PubMed] [Google Scholar]
- 42.McMahon SB, Monroe JG. The role of early growth response gene 1 (egr-1) in regulation of the immune response. J Leukoc Biol. 1996;60:159–166. doi: 10.1002/jlb.60.2.159. [DOI] [PubMed] [Google Scholar]
- 43.Pawlinski R, Pedersen B, Kehrle B, Aird WC, Frank RD, Guha M, Mackman N. Regulation of tissue factor and inflammatory mediators by Egr-1 in a mouse endotoxemia model. Blood. 2003;101:3940–3947. doi: 10.1182/blood-2002-07-2303. [DOI] [PubMed] [Google Scholar]
- 44.Hsu HY, Wen MH. Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem. 2002;277:22131–22139. doi: 10.1074/jbc.M111883200. [DOI] [PubMed] [Google Scholar]
- 45.Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi: 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 47.Nanji AA, Jokelainen K, Rahemtulla A, Miao L, Fogt F, Matsumoto H, Tahan SR, Su GL. Activation of nuclear factor kappa B and cytokine imbalance in experimental alcoholic liver disease in the rat. Hepatology. 1999;30:934–943. doi: 10.1002/hep.510300402. [DOI] [PubMed] [Google Scholar]
- 48.Hill DB, Devalaraja R, Joshi-Barve S, Barve S, McClain CJ. Antioxidants attenuate nuclear factor-kappa B activation and tumor necrosis factor-alpha production in alcoholic hepatitis patient monocytes and rat Kupffer cells, in vitro. Clin Biochem. 1999;32:563–570. doi: 10.1016/s0009-9120(99)00056-9. [DOI] [PubMed] [Google Scholar]
- 49.Chan ED, Riches DW, White CW. Redox paradox: effect of N-acetylcysteine and serum on oxidation reduction-sensitive mitogen-activated protein kinase signaling pathways. Am J Respir Cell Mol Biol. 2001;24:627–632. doi: 10.1165/ajrcmb.24.5.4280. [DOI] [PubMed] [Google Scholar]
- 50.Jalan R, Williams R, Kaser A, Davies NA, Zoller H, Hodges SJ, Graziadei I, Shawcorss D, Vogel W, Alisa A, Ludwickzek O, Tilg H. Clinical and cytokine response to anti-TNF antibody therapy in severe alcoholic hepatitis. Hepatology. 2001;34:441A. [Google Scholar]
- 51.Keane J, Gerson S, Wise RP, Miabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. Tuberculosis associated with infliximab, a tumor necrosis factor α–neutralizing agent. N Engl J Med. 2001;345:1098–1104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]