

Abstract
Here we model the Alzheimer β-peptide ion channel with the goal of obtaining insight into the mechanism of amyloid toxicity. The models are built based on NMR data of the oligomers, with the universal U-shaped (strand-turn-strand) motif. After 30-ns simulations in the bilayer, the channel dimensions, shapes and subunit organization are in good agreement with atomic force microscopy (AFM). The models use the Aβ17–42 pentamer NMR-based coordinates. Extension and bending of the straight oligomers lead to two channel topologies, depending on the direction of the curvature: 1), the polar/charged N-terminal β-strand of Aβ17–42 faces the water-filled pore, and the hydrophobic C-terminal β-strand faces the bilayer (CNpNC; p for pore); and 2), the C-terminal β-strand faces the solvated pore (NCpCN). In the atomistic simulations in a fully solvated DOPC lipid bilayer, the first (CNpNC) channel preserves the pore and conducts solvent; by contrast, hydrophobic collapse blocks the NCpCN channel. AFM demonstrated open pores and collapsed complexes. The final averaged CNpNC pore dimensions (outer diameter 8 nm; inner diameter ∼2.5 nm) are in the AFM range (8–12 nm; ∼2 nm, respectively). Further, in agreement with high-resolution AFM images, during the simulations, the channels spontaneously break into ordered subunits in the bilayer; however, we also observe that the subunits are loosely connected by partially disordered inner β-sheet, suggesting subunit mobility in the bilayer. The cationic channel has strong selective affinity for Ca2+, supporting experimental calcium-selective β-amyloid channels. Membrane permeability and consequent disruption of calcium homeostasis were implicated in cellular degeneration. Consequently, the CNpNC channel topology can sign cell death, offering insight into amyloid toxicity via an ion “trap-release” transport mechanism. The observed loosely connected subunit organization suggests that amyloid channel formation in the bilayer is a dynamic, fluid process involving subunit association, dissociation, and channel rearrangements.
INTRODUCTION
It is still a mystery how deposition of a normally soluble protein into an ordered aggregate is linked with neurotoxicity. The best known among the neurotoxic diseases is Alzheimer's (1), which is characterized by the deposition of insoluble fibril plaques in the extracellular space of the brain tissue. The major component of these plaques is a 39- to 42-residue β-peptide, called the β-amyloid (Aβ) peptide, whose predominant secondary structure in the fibril is a β-sheet. Although amyloid fibril formation is generally linked to fatal neurotoxicity, evidence suggests that oligomers, rather than the fully formed fibrils, are the toxic species (2–4). The pathogenic nature of amyloid oligomers is still unclear, and the fundamental mechanism by which the assembly process causes the neurotoxicity leading to cell death is a debated subject. Recent reviews suggest two potential scenarios for the neurotoxicity: 1), the oligomers form heterogeneous ion channels in the cell membrane leading to cellular degeneration (5–17); and 2), the cellular deposition of amyloid oligomers induces the dysfunction of an inherent channel or pump in the membrane (18–22). The mechanisms underlying Aβ-induced cell damage are closely related to dysregulation and/or destabilization of cellular ionic homeostasis.
Obtaining atomic-level structures of the amyloids is still an extremely challenging task because of their insolubility and noncrystalline nature (23,24). In 2002, our group proposed a detailed atomistic model of solvated Aβ oligomers of the Aβ16–35 peptides (25). In the model, the peptides in the protofibril were organized in parallel and bent at residues 24–27 with an intramolecular salt bridge between residues D23 and K28. This model was subsequently confirmed by Tycko and his colleagues in 2006 (26): using solid state NMR for the Aβ9-40 peptides, they obtained similar side-chain orientation as predicted in the simulations. In 2005, detailed amide hydrogen/deuterium exchange NMR data, combined with side-chain packing constraints from pairwise mutagenesis, solid-state NMR, and high-resolution cryoelectron microscopy led to an atomic level Aβ17–42 oligomer model (27), with the coordinates deposited in the Protein Data Bank (PDB) (id: 2BEG). Our current set of simulations were initiated in 2005, after the publication of that article. In the Lührs et al. (27) PDB structure, the Aβ17–42 peptide has two ordered β-strands (residues 18–26 and 31–42) and a connecting loop (residues 27–30). The tertiary structure of the protofibril also reveals two parallel in-register β-sheets; however, the D23/K28 salt bridge and side chain contacts are intermolecular. Thus, the N-terminal β-strand of the nth molecule interacts with the C-terminal β-strand at the (n − 1)th molecule at the dry interface (28). Residues 1–16 are disordered.
To relate the Aβ structure with cellular toxicity, the atomic-level structure of Aβ oligomers when associated with the membrane is essential. However, defining the atomistic structure in the membrane is even more difficult because of the complex nature of the lipid bilayer. Channel-like annular structures of synthetic Aβ oligomers were observed by electron microscopy (EM) (9,29,30). These images indicated an outer diameter of 8–12 nm and an inner diameter of the cavity pore of 2.0–2.5 nm. Recent experiments using atomic force microscopy (AFM) reported the three-dimensional structures of several amyloid oligomers, including Aβ1–40, α-synuclein, ABri, ADan, serum amyloid A (SAA), and amylin, in a 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) bilayer (11). Although AFM images cannot provide the detailed peptide conformation in the oligomers, a channel-like structure with a central pore is clearly observed for all imaged cases (11) with similar dimensions as obtained by EM, 8–12 nm for the outer diameter and ∼2.0 nm for the inner diameter. At higher resolution, the AFM images reveal that the amyloid channels are assemblies of several subunits, with the channel shape varying from rectangular with four subunits to octahedral with eight subunits (11). For the Aβ1–40 where a series of images were provided, it most frequently contains 5–6 subunits (6). The detailed series of AFM images obtained by Lal and his colleagues (6,11) present a consistent picture of subunit organization, establishing that amyloid channels in the membrane do not possess a perfect annular morphology. The results from the measurements of electrophysiological recordings also suggest that amyloid channels exhibit heterogeneous single-channel conductance, inducing ion-channel activities in the lipid bilayer. Similar Aβ channels with calcium selectivity have been extensively studied for the molecular basis of neurotoxicity in connection with the disruption of calcium homeostasis in the cell (13–17).
Our attention focuses on the Aβ oligomer as a cationic channel because we believe that the Aβ channel can be directly or at least partially responsible for cell death in Alzheimer's (9–11). In the diseased state, the Aβ channel induces cellular degeneration by mediating specific ion conductance through the cellular membrane, destabilizing cellular ionic homeostasis, and ultimately leading to cell death. Here, we model the atomistic structures and investigate the dynamic properties of Aβ17–42 channels in annular topologies. The effects of the lipid environment on the annular conformation are studied by simulating the channels in explicit DOPC bilayer. With the Aβ17–42 monomers in the U-turn conformation (strand-turn-strand) based on NMR data (27) and the number of monomers extended from 5 (in the PDB NMR structure) to 24 (or more), two types of perfectly annular channel topologies are obtained depending on the direction of the curvature: CNpNC and NCpCN, where C and N are the C-terminal and N-terminal β-strands, respectively, and p denotes a central pore (Fig. 1 a). Such oligomeric channels are in agreement with the available Aβ solid-state NMR data of β-rich intermediates (31). For both annular topologies, the inner β-strands form a β-sheet encompassing the solvated central pore; on the other hand, because the outer diameter is larger and the monomers are equally spaced, no β-sheet can be formed on the outer lipid-contacting side. The Aβ17–42 channels are fully embedded in the DOPC bilayer because the height of the channels is comparable to the bilayer thickness. In this case, the first 16 residues (i.e., Aβ1–16), which are not included in the simulations, are extramembranous, located outside the bilayer membrane. The initial dimensions of both annular topologies consisting of 24 monomers are 6.4 nm for the outer diameter and 2.0 nm for the inner diameter. During the simulations monomers cluster to form the β-sheets at both the inner and the outer sides, spontaneously producing the subunit organization observed by AFM (11). At the end of the simulations, the averaged CNpNC channel obtains an 8-nm outer diameter and a ∼2.5-nm inner pore diameter; on the other hand, the NCpCN outer diameter remains at its starting value, and the inner diameter shrinks to 1.5 nm. For the CNpNC channel, even though the outer 8.0-nm diameter is at the lower margin of the 8-12 nm measured by AFM, the measured inner diameter is somewhat larger than that defined by AFM (∼2 nm). This could relate to the missing Aβ1–16 coordinates in the NMR structure (27) and consequently in the simulation. To investigate ion-channel activity, the ions are monitored in the pore for the different cations including Ca2+, K+, and Na+. If the construction and equilibration of the Aβ17–42 ion channel in the lipid bilayer are performed well, such simulations can offer considerable insight into the understanding of the mechanisms of amyloid toxicity, providing candidates for direct testing by experiment.
FIGURE 1.

(a) Topologies of the CNpNC (left) and NCpCN (right) Aβ17–42 channels; the coordinates of the monomers are taken from the NMR pentamer structure in the PDB (id: 2BEG). The monomer has a U-shaped conformation (strand-turn-strand). The initial annular channel topologies are shown as a cross section of a hollowed cylinder in gray with a cut along the pore axis. The Aβ17–42 peptide in a ribbon representation is projected into the cross-section area. The topology of each peptide is drawn by connected white arrows with C and N denoting the C- and N-termini, respectively. Different peptide orientations in the channels reflect the different channel topologies. The topology on the left is the toxic, ion-permeable, and Ca2+ selective amyloid channel. (b) The NMR pentamer (center) shown by the peptide backbones in ribbon representations. Two different directions of curvature yield the CNpNC (left) and NCpCN (right) channels. The up/down arrows from the pentamer indicate the direction of fibril growth. In the peptides, hydrophobic residues are shown in white, two polar (Ser26 and Asn27) and five Gly (25, 29, 33, 37, and 38) residues are shown in green, a positively charged residue (Lys28) is shown in blue, and two negatively charged residues (Glu22 and Asp23) are shown in red.
MATERIALS AND METHODS
We extracted the monomer conformation of the Aβ17–42 peptide from the NMR-based structure of the β-amyloid pentamer (PDB id: 2BEG) (27). The NMR-based monomer structure (residues 17–42) has two β-strands connected by a turn. In the PDB, the NMR structure contains five monomers (Fig. 1 b). We extended the number of monomers to 24 (also to 12 and 36, see below). Depending on how the monomers orient with respect to which strand is facing the bilayer and which strand is facing the solvated inner pore, two Aβ17–42 channels with different topologies may form (Fig. 1 a): CNpNC and NCpCN. The former has a central pore enclosed by the N-terminal β-strands, whereas the latter by the C-terminal β-strands. The C-terminal strand is largely hydrophobic; the N-terminal strand has polar/charged residues. The channel is minimized with a rigid body motion for the peptides to enhance the formation of backbone hydrogen bonds (H-bonds) within a β-sheet. The minimized channel is next embedded in the lipid bilayer containing 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC). A unit cell containing two layers of lipids with almost 150,000 atoms is constructed. In the bilayer construction, our method closely follows a previous method, which has been successfully applied to gramicidin (32), α-helical systems (33,34), rhodopsin (35), bacteriorhodopsin (36), and protegrin-1 (37,38). For the lipid bilayer, 300 DOPCs (150 DOPCs each side) constitute the lateral cell dimension of 116.0 Å × 116.0 Å. TIP3P waters were added and relaxed through a series of minimization dynamics. To neutralize the system, 24 counterions (Na+) were inserted. In addition to the counter ions, the system contains 15 mM calcium chloride, 15 mM potassium chloride, and 70 mM sodium chloride, which are added to satisfy a physiological salt concentration near 100 mM.
Our simulations for the initial construction and the pre-equilibration employed the NPAT (constant number of atoms, pressure, surface area, and temperature) ensemble, an effective (time-averaged) surface tension with a constant normal pressure applied in the direction perpendicular to the membrane. An alternative protocol would involve using variable surface area controlled by constant surface tension with the NPγT (constant number of atoms, pressure, surface tension, and temperature) ensemble, where γ is the applied surface tension. When γ = 0, NPγT is equivalent to NPT. Although the experimentally measured macroscopic property of γ should be close to or zero for the nonstressed lipid bilayers (39), a nonzero surface tension must be employed in the NPγT simulations because of the presence of long-wavelength undulation for the microscopic membrane patch (40). In the NPT ensemble, simulations with CHARMM27 (41) parameter sets reproduce incorrectly the reduced surface area (42,43). However, a simulation with a correctly parameterized constant surface area with the NPAT ensemble can be directly comparable to an applied constant surface tension (44,45). In the production simulations, the dynamics were performed on both the NPAT and NPγT with γ = 0. However, no significant differences in the critical results were found for the use of different ensembles.
The CHARMM program (41) and Charmm 27 force field were used to construct the set of starting points and to relax the systems to a production-ready stage. In the pre-equilibrium stages, the initial configurations were gradually relaxed, with the channels held rigid. A series of dynamic cycles were performed with the harmonically restrained peptides in the channels, and then the harmonic restraints were gradually diminished with the full Ewald electrostatics calculation and constant temperature (Nosé-Hoover) thermostat/barostat at 300 K. The entire pre-equilibration cycle took 5 ns to yield the starting point. For the production runs of 30 ns, any constraint applied to the peptides was removed, and the simulations were performed with the same parameter sets as used in the pre-equilibrium simulations. The system reached equilibration after the initial 5 ns. The NAMD code (46) on a Biowulf cluster (this study utilized the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov)) was used for the starting point with the same Charmm 27 force field in the production simulations. Averages were taken after 10 ns, discarding initial transient of the first 5 ns. Channels with different sizes (36- and 12-mers) were also simulated with the same method mentioned above. Here, the simulation results are presented only for the 24-mers, as the others are still ongoing.
RESULTS
The β-amyloid channel topologies in the lipid bilayer
To examine the conformational evolution of the Aβ channel, we have performed 30-ns molecular dynamics (MD) simulations on Aβ17–42 channels embedded in a fully solvated DOPC bilayer membrane. Initially the channels have perfect annular shapes (Fig. 1 a). The C-terminal β-strand of the Aβ17–42 peptide is largely hydrophobic, whereas the N-terminal β-strand includes one polar (Ser26) and two negatively charged residues (Glu22 and Asp23). In the CNpNC topology, the central pore is enclosed by a β-sheet formed by the N-terminal strands, whereas the NCpCN pore is lined by the C-terminal β-strands. The lipid-contacting outer β-strands do not form a β-sheet because of the larger curvature at the channel periphery. The initial frustration in the annular conformation is gradually removed via relaxations of the lipid bilayer, and environmentally relaxed peptides in the channels can be observed after 5 ns (Fig. 2). The CNpNC channel seems to extend its overall size, whereas the NCpCN channel retains its size, but with an apparently collapsed central pore. The perfect annular shape disappears in both relaxed channel structures (Fig. 2). The size of the channels in our simulations is comparable to amyloid channels as defined from the AFM images (see the section below) (11).
FIGURE 2.

Snapshots of Aβ17–42 channels in the DOPC bilayer taken at simulation times of t = 0, 10, 20, and 30 ns for the (a) CNpNC and (b) NCpCN Aβ17–42 channels (see Fig. 1 for the topology). The cartoons representing the peptide backbone are in stereo view, and each peptide backbone in the cartoons is in a different color.
The relaxed channel conformations imply that the lipid environment does not support perfect annular morphologies. To reduce the curvature of the channel periphery, the ordered peptides cluster into subunits, and the annular structure breaks at the disordered peptide regions. Fig. 3 shows the averaged channel structures in a ribbon representation with a transparent surface (left panels). All channels are viewed from the top leaflet of the lipid bilayer and colored according to the B-factor (or temperature factor), which can be calculated from the RMS fluctuations relative to the starting point during the simulations with a simple correlation of 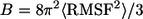 (47). The degree of residue fluctuations increases with the color codes in the order of blue → cyan → green. The color is scaled relative to each channel, i.e., blue color for the CNpNC channel represents a more ordered state than the same color for the NCpCN channel. For the CNpNC channel, we observed formation of a triangular shape with three distinct subunits. The ordered subunits (blue-colored regions in the surface representation, left panels) are defined by calculating the RMS fluctuations of the peptide residues and the β-strand order parameters (right panels). The β-strand order parameter measures the “straightness” of the β-strands, which can be defined by an equation,
(47). The degree of residue fluctuations increases with the color codes in the order of blue → cyan → green. The color is scaled relative to each channel, i.e., blue color for the CNpNC channel represents a more ordered state than the same color for the NCpCN channel. For the CNpNC channel, we observed formation of a triangular shape with three distinct subunits. The ordered subunits (blue-colored regions in the surface representation, left panels) are defined by calculating the RMS fluctuations of the peptide residues and the β-strand order parameters (right panels). The β-strand order parameter measures the “straightness” of the β-strands, which can be defined by an equation,
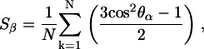 |
(1) |
where θα is the angle between the positional vectors connecting two Cα atoms, and N is the total number of vector pairs. In the calculation, the Cα atoms located in the N-terminal (residues 18–26) and C-terminal (residues 31–42) β-strands are considered separately. In the ordered subunits, peptides have higher values of the β-strand order parameter for both the interior N-terminal and the exterior C-terminal β-strands. The outer β-sheet, not formed in the starting conformation because of the large curvature at the channel periphery, begins to form in addition to the retention of the inner β-sheet. The ordered subunits are twisted, showing the common cross β-structure of amyloid fibrils (48). Between the subunits, the backbone H-bonds in the inner β-sheet are straddled, marginally preventing the dissociation of the subunits, and the H-bonds in the outer β-sheet are broken at the borders of the subunits. The surface representation of the CNpNC channel (cartoon image in Fig. 3 a, left panel) illustrates the similarity of the obtained channel to the AFM images (11), confirming that the amyloid channel is an assembly of subunits in the lipid bilayer. In contrast, for the NCpCN channel we failed to identify ordered subunits, though a rectangular shape (Fig. 3 b) was observed in the relaxed channels. The interior C-terminal β-strands are relatively ordered, although the N-terminal β-strands that interact with lipids are largely disordered in the NCpCN channel.
FIGURE 3.

Channel structures averaged over the simulation in a ribbon representation with a transparent surface are shown in the left panels. All channels are viewed from top leaflet of the lipid bilayer and colored according to the B-factor (or temperature factor, see text). The first and last peptides (peptides 1 and 24) of the annular channel are marked. The right panels illustrate the β-strand order parameters for the N-terminal and C-terminal β-strands for the (a) CNpNC and (b) NCpCN Aβ17–42 channel topologies (see Fig. 1). Red circles denote the order parameter for the N-terminal β-strand, whereas cyan circles denote the order parameter for the C-terminal β-strand of each peptide in the channels.
Dimensions of the β-amyloid channels
The CNpNC channel increases both the outer diameter and the inner pore sizes, whereas the NCpCN channel retains its size, but its pore size is greatly decreased. Fig. 4 shows the averaged outer channel diameter (upper panels) and the pore diameter (lower panels) for (a) the CNpNC and (b) NCpCN channels. The cross-sectional area of the channels is measured by the CHARMM program (41) as a function of the distance along the pore central axis. If the cross section of the channels is seen as a circle, the averaged outer channel diameter can be calculated. Because the shape of the channels is not homogeneous, the averaged outer channel diameter is slightly below the actual channel diameter by direct measurement. The averaged pore diameter is calculated by the HOLE program (49). In the starting conformations, both CNpNC and NCpCN channels have similar dimensions, with the outer diameter ∼6.4 nm, and the pore diameter is ∼2.0 nm. For the final structure of the CNpNC channel, both averaged outer and pore diameters increase to ∼8.0 nm and ∼2.7 nm, respectively. The outer dimension of the channel is in the 8–12 nm range of the experimentally defined amyloid channels (9,11,29,30), but the pore diameter is found to be slightly larger than the ∼2 nm measured by AFM. This discrepancy may result from the different length of the β-amyloid peptide used in the channel construction (Aβ17–42 versus Aβ1–40 in the AFM images). By contrast to the CNpNC channel, the NCpCN channel remains in its starting value of ∼6.4 nm, and the averaged pore diameter decreases to ∼1.5 nm in the final structure.
FIGURE 4.

Averaged outer channel diameter (open symbols at upper panels) and pore diameter (solid lines at lower panels) as a function of the distance along the pore center axis for the (a) CNpNC and (b) NCpCN Aβ17–42 channels. A black line with solid symbols in the outer diameter and a black solid line in the pore diameter represent the initial channel dimension at the starting points. The pore diameters are measured at selected simulation times of t = 5 (red lines), 10 (green lines), 15 (yellow lines), 20 (blue lines), 25 (pink lines), and 30 (cyan lines) ns.
The CNpNC pore resembles a fat tube (Fig. 5 a; the pore structure created by the HOLE program (49)). With such a large, well-defined pore, the CNpNC channel is ready for active ion conductance and thus can be labeled a pore-preserving channel. Compared to the CNpNC channel, the relaxed NCpCN channel contains more disordered Aβ peptides. This is because of the hydrophobic mismatch between the peptide and lipid. The NCpCN pore is significantly collapsed by the pore-facing hydrophobic C-terminal β-strands at the bottom leaflet of the lipid bilayer (Fig. 5 b). With such a collapsed pore, the NCpCN channel becomes a pore-collapsing channel, reduced to a β-amyloid oligomer complex blocking membrane transport. In agreement with these results, AFM experiments reveal that oligomeric complexes lacking central pores can also be observed in the membrane (11).
FIGURE 5.

Pore structures calculated by the HOLE program (49) at t = 0 and 30 ns for the (a) CNpNC and (b) NCpCN Aβ17–42 channels. For the pore structures in the surface representation, red denotes pore radius of r < 0.9 nm, green denotes pore radius in the range, 0.9 nm ≤ r ≤ 1.3 nm, and blue denotes pore radius of r > 1.3 nm. In the surface representation for the channels, the front part of the channels in the lateral view has been removed to allow a view of the pore. In the channel structures, hydrophobic residues are shown in white, two polar (Ser26 and Asn27) and five Gly (25, 29, 33, 37, and 38) residues are shown in green, a positively charged residue (Lys28) is shown in blue, and two negatively charged residues (Glu22 and Asp23) are shown in red.
Lipids support the channel conformation
Cytotoxic peptides including the Aβ and antimicrobial peptides can form pores in the membrane. Depending on the mechanism of the peptide activity in the membrane, three different membrane pore models have been postulated as possible: 1), barrel-stave, 2), toroidal, and 3), carpet models (50). Pore-forming peptides also form ion channels in the membrane, inducing cell death. To analyze the membrane pore mediated by the Aβ channels, the three-dimensional densities for the positions of the lipid headgroups are calculated during the 30-ns simulation. Fig. 6 shows the three-dimensional density maps of the lipid headgroups (left panels). The density map is a lateral view of the bilayer. The headgroup density map reflects the average shape of the bilayer surface during the simulation. For the CNpNC channel in the lateral view of the bilayer, the density map clearly demonstrates that the CNpNC channel induces the barrel-stave pore (50). In this pore model, the hydrophobic peptide regions align with the lipid core region of the bilayer, and the hydrophilic peptide regions form the interior region of the pore. For the NCpCN channel, although the channel seems to satisfy the barrel-stave pore, wide distributions of the lipid headgroups near the channel edges indicate a slight thinning of the bilayer. This is caused by the disordered lipids in contact with the channel because of the hydrophobic mismatch that occurs between the peptides and the lipid. Nevertheless, there is no significant thinning of the bilayers containing both the Aβ17–42 channels. Lipids play important roles in supporting the conformation and dynamics of channels in the bilayer. The average positions of lipid groups provide useful information regarding the environmental response to the dynamics of channels during the simulation. The position probability distribution functions (P) for five different component groups of DOPC as a function of the distance along the pore axis are presented (Fig. 6, right panels). The DOPC headgroup is divided into four subunits, choline (PChol, black lines), phosphate (PPO4, red lines), glycerol (PGlyc, green lines), and carbonyl (PCarb, yellow lines). The tail group involves two fatty acids with terminal methyls (PCH3, blue lines). For the bilayer containing the CNpNC channel, the symmetric distributions of the lipid headgroups at both sides of the bilayer indicate the typical pure DOPC bilayer. There are no disturbances in the lipid arrangement induced by the channel. Thus, the lipid bilayer strongly supports the CNpNC channel topology. However, for the NCpCN channel, there are some disturbances in the lipid arrangement, especially at the bottom leaflet of the lipid bilayer, because of the hydrophobic mismatch between the peptide and lipid.
FIGURE 6.

Three-dimensional density maps (left panels) of the lipid headgroup of the DOPC bilayer (white surface) and probability distribution functions (P) (right panels) for different component groups of lipid (PChol (choline, black lines), PPO4 (phosphate, red lines), PGlyc (glycerol, green lines), PCarb (carbonyls, yellow lines), and PCH3 (methyl, blue lines)) as a function of the distance along the pore center axis for the (a) CNpNC and (b) NCpCN Aβ17–42 channels. The density maps represent the lateral view of the time-averaged bilayer structure. The Aβ17–42 channels in a surface representation with the B-factor coloring (see Fig. 3) are embedded in the density maps.
To investigate the average structure in the interior of the bilayer, the deuterium order parameter, SCD, was calculated using
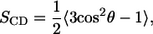 |
(2) |
where θ is the angle between the C–H-bond vector and the membrane normal, and the angular brackets indicate averaging over time and over lipids. Fig. 7 shows the order parameters for the DOPC chains for (a) CNpNC and (b) NCpCN channels. Lipids at the top leaflet (open symbols) and the bottom leaflet (solid symbols) of the lipid bilayer are considered separately. The bars represent the standard deviation errors. For the CNpNC channel, the order parameters for the lipids on both sides are similar. However, for the NCpCN channel, the order parameters for the lipids at the top leaflet are higher than those for the lipids at the bottom leaflet. Lower order parameters for the lipids at the bottom leaflet result from the environmental incompatibility between the N-termini of the channel and the lipids.
FIGURE 7.

Deuterium order parameters, SCD, for the lipid chains at the top leaflet (open symbols) and at the bottom leaflet (solid symbols) of the lipid bilayer composed of DOPC for the (a) CNpNC and (b) NCpCN Aβ17–42 channels.
Cationic ring and ion-channel activity
We analyze the cation activity in the pore cavity. Because the NCpCN channel has a collapsed pore that prevents cations from entering through the gates, we focus on the CNpNC channel. In the simulations, the system contains three different cations, Ca2+, K+, and Na+ in different concentrations. These cations are easily attracted to the negatively charged Glu22 side chain, located at the upper half of the pore in the top lipid bilayer leaflet. The cations are trapped by the side chain, creating a cationic ring in the pore as seen in the ion density maps (Fig. 8). The density map indicates popular interaction sites for Ca2+ (blue) with probabilities of 0.01/0.003 (surface/mesh), K+ (red) with probabilities of 0.01/0.003 (surface/mesh), Na+ (yellow) with probabilities of 0.02/0.005 (surface/mesh), and Cl− (gray) with probability of 0.03 (surface). In the pore, Ca2+ is immobile, as indicated by the large peak in the position probability distribution (Fig. 8 d) of the cations. In the simulation, K+ has the same concentration as Ca2+, but the behavior of K+ in the pore is quite different. Compared to Ca2+, K+ is very mobile, spending limited time in the pore, as shown in Fig. 8. The behavior of Na+ is similar to that of K+, but the concentration of Na+ is higher than the others. In addition to the cations, the system also contains Cl− anions, which are observed only near both channel gates with no deep pore penetration. However, we observed few cations transporting from one side of the bilayer to the other during the simulation.
FIGURE 8.

Three-dimensional density map of Ca2+ (blue surface and mesh), K+ (red surface and mesh), Na+ (yellow surface and mesh), and Cl− (gray surface) for the CNpNC Aβ17–42 channel in the (a) top, (b) angle, and (c) lateral views. The averaged channel structure is shown as cartoons in gray. (d) Probability distribution functions for Ca2+ (blue line), K+ (red line), Na+ (yellow line), and Cl− (gray line) as a function of the distance along the pore center axis. The Ca2+ trapped at the negatively charged Glu22 side chain enter from the top channel gate in the top lipid bilayer leaflet, whereas the Ca2+ near the bottom channel gate are trapped by the carboxyl termini.
The time series of the charged state induced by the ions in the pore is shown in Fig. 9. In the figure, the total charges increase as the pore size increases and then fluctuate near a specific value after 10 ns. Once Ca2+ binds to the interaction site, it stays in the strong electrostatic attraction site during the simulations, producing flat fluctuation in total charges by Ca2+. However, other cations, K+ and Na+, move into and out of the interaction site as indicated by the large fluctuations in the charged states.
FIGURE 9.

Time series of the charged state in the pore of the CNpNC Aβ17–42 channel for Ca2+ (blue line), K+ (red line), Na+ (yellow line), Cl− (gray line), and total charges (black). The number of charges are counted in the pore with a cutoff, −1.5 nm < z < 1.5 nm, from the bilayer center along the pore axis.
DISCUSSION AND CONCLUSIONS
We extracted the coordinates of Aβ17–42 monomers from the NMR-based pentamer structure in the PDB. Extending the 5 monomers to 24, two annular β-amyloid channels, CNpNC and NCpCN, may form depending on the direction of the curvature and consequently which strand (N-terminal or C-terminal) faces the lipid bilayer. Both are simulated in an explicit membrane environment. In the lipid bilayer simulations, the CNpNC channel abandons the perfect annular morphology. Instead, the annular structure divides into several subunits that contain ordered β-amyloid peptides. In agreement with the AFM images (11), this spontaneously obtained morphology points to the amyloid channel as an assembly of subunits and provides a detailed atomistic model to the AFM images. The channel dimensions are also in good agreement with high-resolution AFM images (11) and other experimentally defined amyloid channels (9,29,30). The ordered subunits are geometrically similar to the β-amyloid oligomer based on hydrogen/deuterium exchange NMR data (27), except the no twist is present in the NMR-based pentamer. By contrast, these subunits have a cross β-structure commonly observed in amyloid fibrils (48), suggesting that the β-sheet twist is responsible for the channel shape. The peptide-lipid interactions in the CNpNC channel produce a large well-defined pore, enclosed by the inner β-sheet consisting of the N-terminal β-strands. The outer β-sheet of the annular channel consisting of the C-terminal β-strands, which was not formed in the starting conformation because of the large curvature at the outer channel periphery, is recovered when the annular channel breaks into the ordered subunits. Neighboring ordered subunits interact with each other through the inner β-sheet backbone H-bonds, preventing the channel dissociation. The outer β-sheet H-bond network discontinuity defines the subunits of the channel. Rapid pore collapse is prevented by electrostatic repulsion between the negatively charged side chains of the Glu22 residues, which are arranged circularly in the pore inducing the cationic ring. On the other hand, the collapsed (NCpCN topology) channel fails to preserve its initial pore, reducing to an oligomer complex. This is caused by the hydrophobic mismatch between the peptide and lipid. The hydrophobic C-terminal β-strands in the inner β-sheet of the channel collapse, avoiding contact with the bulk water and ions in the pore, and block the pore transport. Oligomeric complexes without pores are also populated in the membrane bilayer (11).
The CNpNC channel exhibits ion-channel activity. The negatively charged side chains of Glu22 in the pore are a key factor in conducting the ions through the membrane. The side chains located at the upper half portion of the pore in the top leaflet of the lipid bilayer stretch toward the central pore axis and strongly attract cations from the bulk region. Among the cations inserted into the system, Ca2+ ions are dominantly trapped by the side chains and contribute to generating a cationic ring together with other cations in the pore. One can imagine that the trapped Ca2+ ions are released to the other side of the lipid bilayer (bottom leaflet) through the bombardment of incoming Ca2+ from the top leaflet bulk. This ion permeation of the “trap-release” mechanism is similar to the “knock-on” mechanism that is observed in biological channels in the cell membrane, in which ions can jump over the quantized binding sites in the pore with low free energy profiles (51). The Glu22 side chains clearly provide the binding sites for Ca2+ with low free energy. If the top leaflet of the lipid bilayer is regarded as the extracellular side of the plasma membrane, the CNpNC channel presents a model for the calcium uptake mechanism by the β-amyloid channel. Experimental results suggest that β-amyloid channels are calcium permeable and selective, taking up Ca2+ from the extracellular side and releasing Ca2+ to the cytoplasmic side (13–17). We note, however, that observing single-channel ion conductance as experimentally recorded for the amyloid channels requires simulations over micro- or milliseconds, reflecting long-term stability of β-amyloid channels as in AFM/EM images (6,9,11). Increasing the cellular calcium to a toxic level is responsible for the cellular pathophysiology and degeneration leading to cell death.
Here we propose the atomistic structure of the cationic, nonhomogeneous channel in the lipid bilayer, which consists of several β-amyloid subunits. The presence of subunits in the channel structure of the β-amyloid oligomer is also demonstrated by simulating channels with different sizes (not shown here). A channel with 36 Aβ17–42 peptides in the CNpNC topology also generates (five) subunits and a channel with only 12 Aβ17–42 peptides reduces to a two-subunit association without opening a pore. Further, even a second simulation of 24 strands also led to five subunits. This suggests that the marginal size of a toxic channel formation in the lipid bilayer requires at least three subunits. The CNpNC channel exhibits ion-permeable characteristics with calcium selectivity, which can correlate with the cytotoxic mechanism of neurodegenerative diseases. The model of simulated β-amyloid channel in the bilayer offers insight into molecular mechanisms of amyloid toxicity and reveals structural information that can promote drug discovery to inhibit the formation of such toxic channels in the cell membrane. We further note that although all simulations presented here are based on the Aβ17–42 (27), we have recently obtained from R. Tycko (National Institutes of Health) the coordinates of the U-shaped strand-turn-strand Aβ9–40 oligomer (26). Preliminary results from the ongoing simulations on 24 monomers of Aβ9–42 suggest that although the details differ, given the different lengths of the Aβ17–42 versus Aβ9–42 and the starting conformation, overall a similar channel picture is obtained.
Amyloid channels are generally believed to be rigid structures, formed to stay. By contrast, the topology and atomistic structure we obtain suggest a mobile mechanism of channel formation via diffusion and assembly of subunits and/or single peptides. The loosely assembled oligomeric subunits in the channels suggest that channel formation and breakage are dynamic processes in the bilayer. The Aβ U-shaped strand-loop-strand (25–27) conformation appears to be a general motif for amyloids (52–54), and the channel morphology by AFM (11) similarly illustrates common shared features. This leads us to propose that similar nonhomogeneous annular structures consisting of loosely attached and mobile subunits with ion channel activities may be general phenomena in pore-forming amyloid ion topologies in the lipid bilayer. As with all models, proper modeling procedures and visual and quantitative consistency with available experimental data merely qualify a model as a valid candidate; however, to be proven correct, direct and thorough testing by experiment is essential.
Acknowledgments
We thank Dr. Robert Tycko for providing us with the coordinates of the Aβ9–40 oligomer. We thank Drs. Buyong Ma and Chung-Jung Tsai for discussions and useful suggestions.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract No. NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Dept. of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. This research was supported (in part) by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. This study utilized the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov).
Editor: John E. Straub.
References
- 1.Selkoe, D. J. 1991. Alzheimer's disease. In the beginning. Nature. 354:432–433. [DOI] [PubMed] [Google Scholar]
- 2.Kirkitadze, M. D., G. Bitan, and D. B. Teplow. 2002. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J. Neurosci. Res. 69:567–577. [DOI] [PubMed] [Google Scholar]
- 3.Bucciantini, M., E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson, and M. Stefani. 2002. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 416:507–511. [DOI] [PubMed] [Google Scholar]
- 4.Walsh, D. M., I. Klyubin, J. V. Fadeeva, W. K. Cullen, R. Anwyl, M. S. Wolfe, M. J. Rowan, and D. J. Selkoe. 2002. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 416:535–539. [DOI] [PubMed] [Google Scholar]
- 5.Lin, H., Y. J. Zhu, and R. Lal. 1999. Amyloid β protein (1–40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles. Biochemistry. 38:11189–11196. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia, R., H. Lin, and R. Lal. 2000. Fresh and globular amyloid β protein (1–42) induces rapid cellular degeneration: evidence for AβP channel-mediated cellular toxicity. FASEB J. 14:1233–1243. [DOI] [PubMed] [Google Scholar]
- 7.Zhu, Y. J., H. Lin, and R. Lal. 2000. Fresh and nonfibrillar amyloid β protein(1–40) induces rapid cellular degeneration in aged human fibroblasts: evidence for AβP-channel-mediated cellular toxicity. FASEB J. 14:1244–1254. [DOI] [PubMed] [Google Scholar]
- 8.Lin, H., R. Bhatia, and R. Lal. 2001. Amyloid β protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 15:2433–2444. [DOI] [PubMed] [Google Scholar]
- 9.Lashuel, H. A., D. Hartley, B. M. Petre, T. Walz, and P. T. Lansbury Jr. 2002. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 418:291. [DOI] [PubMed] [Google Scholar]
- 10.Lashuel, H. A., D. M. Hartley, B. M. Petre, J. S. Wall, M. N. Simon, T. Walz, and P. T. Lansbury Jr. 2003. Mixtures of wild-type and a pathogenic (E22G) form of Aβ40 in vitro accumulate protofibrils, including amyloid pores. J. Mol. Biol. 332:795–808. [DOI] [PubMed] [Google Scholar]
- 11.Quist, A., I. Doudevski, H. Lin, R. Azimova, D. Ng, B. Frangione, B. Kagan, J. Ghiso, and R. Lal. 2005. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 102:10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lau, T. L., E. E. Ambroggio, D. J. Tew, R. Cappai, C. L. Masters, G. D. Fidelio, K. J. Barnham, and F. Separovic. 2006. Amyloid-β peptide disruption of lipid membranes and the effect of metal ions. J. Mol. Biol. 356:759–770. [DOI] [PubMed] [Google Scholar]
- 13.Arispe, N., E. Rojas, and H. B. Pollard. 1993. Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA. 90:567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arispe, N., H. B. Pollard, and E. Rojas. 1994. β-Amyloid Ca2+-channel hypothesis for neuronal death in Alzheimer disease. Mol. Cell. Biochem. 140:119–125. [DOI] [PubMed] [Google Scholar]
- 15.Arispe, N., H. B. Pollard, and E. Rojas. 1996. Zn2+ interaction with Alzheimer amyloid β protein calcium channels. Proc. Natl. Acad. Sci. USA. 93:1710–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhee, S. K., A. P. Quist, and R. Lal. 1998. Amyloid β protein-(1–42) forms calcium-permeable, Zn2+-sensitive channel. J. Biol. Chem. 273:13379–13382. [DOI] [PubMed] [Google Scholar]
- 17.Kawahara, M., and Y. Kuroda. 2000. Molecular mechanism of neurodegeneration induced by Alzheimer's β-amyloid protein: channel formation and disruption of calcium homeostasis. Brain Res. Bull. 53:389–397. [DOI] [PubMed] [Google Scholar]
- 18.Etcheberrigaray, R., E. Ito, K. Oka, B. Tofel-Grehl, G. E. Gibson, and D. L. Alkon. 1993. Potassium channel dysfunction in fibroblasts identifies patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA. 90:8209–8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss, J. H., C. J. Pike, and C. W. Cotman. 1994. Ca2+ channel blockers attenuate β-amyloid peptide toxicity to cortical neurons in culture. J. Neurochem. 62:372–375. [DOI] [PubMed] [Google Scholar]
- 20.Lambert, M. P., A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft, and W. L. Klein. 1998. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacManus, A., M. Ramsden, M. Murray, Z. Henderson, H. A. Pearson, and V. A. Campbell. 2000. Enhancement of 45Ca2+ influx and voltage-dependent Ca2+ channel activity by β-amyloid-(1–40) in rat cortical synaptosomes and cultured cortical neurons. Modulation by the proinflammatory cytokine interleukin-1β. J. Biol. Chem. 275:4713–4718. [DOI] [PubMed] [Google Scholar]
- 22.Kayed, R., Y. Sokolov, B. Edmonds, T. M. McIntire, S. C. Milton, J. E. Hall, and C. G. Glabe. 2004. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 279:46363–46366. [DOI] [PubMed] [Google Scholar]
- 23.Serpell, L. C. 2000. Alzheimer's amyloid fibrils: structure and assembly. Biochim. Biophys. Acta. 1502:16–30. [DOI] [PubMed] [Google Scholar]
- 24.Tycko, R. 2004. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr. Opin. Struct. Biol. 14:96–103. [DOI] [PubMed] [Google Scholar]
- 25.Ma, B., and R. Nussinov. 2002. Stabilities and conformations of Alzheimer's β-amyloid peptide oligomers (Aβ16–22, Aβ16–35, and Aβ10–35): Sequence effects. Proc. Natl. Acad. Sci. USA. 99:14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petkova, A. T., W. M. Yau, and R. Tycko. 2006. Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry. 45:498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luhrs, T., C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert, and R. Riek. 2005. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 102:17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson, R., M. R. Sawaya, M. Balbirnie, A. O. Madsen, C. Riekel, R. Grothe, and D. Eisenberg. 2005. Structure of the cross-β spine of amyloid-like fibrils. Nature. 435:773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haass, C., and D. J. Selkoe. 2007. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8:101–112. [DOI] [PubMed] [Google Scholar]
- 30.Bitan, G., M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek, and D. B. Teplow. 2003. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA. 100:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chimon, S., and Y. Ishii. 2005. Capturing intermediate structures of Alzheimer's β-amyloid, Aβ(1–40), by solid-state NMR spectroscopy. J. Am. Chem. Soc. 127:13472–13473. [DOI] [PubMed] [Google Scholar]
- 32.Woolf, T. B., and B. Roux. 1994. Molecular dynamics simulation of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. USA. 91:11631–11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woolf, T. B. 1997. Molecular dynamics of individual α-helices of bacteriorhodopsin in dimyristol phosphatidylocholine. I. Structure and dynamics. Biophys. J. 73:2376–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woolf, T. B. 1998. Molecular dynamics simulations of individual α-helices of bacteriorhodopsin in dimyristoylphosphatidylcholine. II. Interaction energy analysis. Biophys. J. 74:115–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crozier, P. S., M. J. Stevens, L. R. Forrest, and T. B. Woolf. 2003. Molecular dynamics simulation of dark-adapted rhodopsin in an explicit membrane bilayer: coupling between local retinal and larger scale conformational change. J. Mol. Biol. 333:493–514. [DOI] [PubMed] [Google Scholar]
- 36.Jang, H., P. S. Crozier, M. J. Stevens, and T. B. Woolf. 2004. How environment supports a state: molecular dynamics simulations of two states in bacteriorhodopsin suggest lipid and water compensation. Biophys. J. 87:129–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jang, H., B. Ma, T. B. Woolf, and R. Nussinov. 2006. Interaction of protegrin-1 with lipid bilayers: membrane thinning effect. Biophys. J. 91:2848–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang, H., B. Ma, and R. Nussinov. 2007. Conformational study of the Protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Structure. 7:21. [DOI] [PMC free article] [PubMed]
- 39.Jahnig, F. 1996. What is the surface tension of a lipid bilayer membrane? Biophys. J. 71:1348–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feller, S. E., and R. W. Pastor. 1996. On simulating lipid bilayers with an applied surface tension: periodic boundary conditions and undulations. Biophys. J. 71:1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. Charmm—a program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4:187–217. [Google Scholar]
- 42.Jensen, M. O., O. G. Mouritsen, and G. H. Peters. 2004. Simulations of a membrane-anchored peptide: structure, dynamics, and influence on bilayer properties. Biophys. J. 86:3556–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benz, R. W., F. Castro-Roman, D. J. Tobias, and S. H. White. 2005. Experimental validation of molecular dynamics simulations of lipid bilayers: a new approach. Biophys. J. 88:805–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feller, S. E., and R. W. Pastor. 1999. Constant surface tension simulations of lipid bilayers: The sensitivity of surface areas and compressibilities. J. Chem. Phys. 111:1281–1287. [Google Scholar]
- 45.Skibinsky, A., R. M. Venable, and R. W. Pastor. 2005. A molecular dynamics study of the response of lipid bilayers and monolayers to trehalose. Biophys. J. 89:4111–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comp. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wriggers, W., E. Mehler, F. Pitici, H. Weinstein, and K. Schulten. 1998. Structure and dynamics of calmodulin in solution. Biophys. J. 74:1622–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sipe, J. D., and A. S. Cohen. 2000. Review: history of the amyloid fibril. J. Struct. Biol. 130:88–98. [DOI] [PubMed] [Google Scholar]
- 49.Smart, O. S., J. M. Goodfellow, and B. A. Wallace. 1993. The pore dimensions of gramicidin A. Biophys. J. 65:2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brogden, K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238–250. [DOI] [PubMed] [Google Scholar]
- 51.Roux, B. 2005. Ion conduction and selectivity in K+ channels. Annu. Rev. Biophys. Biomol. Struct. 34:153–171. [DOI] [PubMed] [Google Scholar]
- 52.Ferguson, N., J. Becker, H. Tidow, S. Tremmel, T. D. Sharpe, G. Krause, J. Flinders, M. Petrovich, J. Berriman, H. Oschkinat, and A. R. Fersht. 2006. General structural motifs of amyloid protofilaments. Proc. Natl. Acad. Sci. USA. 103:16248–16253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwata, K., T. Fujiwara, Y. Matsuki, H. Akutsu, S. Takahashi, H. Naiki, and Y. Goto. 2006. 3D structure of amyloid protofilaments of β2-microglobulin fragment probed by solid-state NMR. Proc. Natl. Acad. Sci. USA. 103:18119–18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng, J., B. Ma, and R. Nussinov. 2006. Consensus features in amyloid fibrils: sheet-sheet recognition via a (polar or nonpolar) zipper structure. Phys Biol. 3:1–4. [DOI] [PubMed] [Google Scholar]