

Abstract
MicroRNAs (miRNAs) are an abundant class of tiny RNAs thought to regulate the expression of protein-coding genes in plants and animals. In the present study, we describe a computational procedure to identify miRNA genes conserved in more than one genome. Applying this program, known as MiRscan, together with molecular identification and validation methods, we have identified most of the miRNA genes in the nematode Caenorhabditis elegans. The total number of validated miRNA genes stands at 88, with no more than 35 genes remaining to be detected or validated. These 88 miRNA genes represent 48 gene families; 46 of these families (comprising 86 of the 88 genes) are conserved in Caenorhabditis briggsae, and 22 families are conserved in humans. More than a third of the worm miRNAs, including newly identified members of the lin-4 and let-7 gene families, are differentially expressed during larval development, suggesting a role for these miRNAs in mediating larval developmental transitions. Most are present at very high steady-state levels—more than 1000 molecules per cell, with some exceeding 50,000 molecules per cell. Our census of the worm miRNAs and their expression patterns helps define this class of noncoding RNAs, lays the groundwork for functional studies, and provides the tools for more comprehensive analyses of miRNA genes in other species.
Keywords: miRNA, noncoding RNA, computational gene identification, Dicer
Supplemental material is available at http://www.genesdev.org.
Noncoding RNAs (ncRNAs) of ∼22 nucleotides (nt) in length are increasingly recognized as playing important roles in regulating gene expression in animals, plants, and fungi. The first such tiny regulatory RNA to be identified was the lin-4 RNA, which controls the timing of Caenorhabditis elegans larval development (Lee et al. 1993; Wightman et al. 1993). This 21-nt RNA pairs to sites within the 3′ untranslated region (UTR) of target mRNAs, specifying the translational repression of these mRNAs and triggering the transition to the next developmental stage (Lee et al. 1993; Wightman et al. 1993; Ha et al. 1996; Moss et al. 1997; Olsen and Ambros 1999). A second tiny riboregulator, let-7 RNA, is expressed later in development and appears to act in a similar manner to trigger the transition to late-larval and adult stages (Reinhart et al. 2000; Slack et al. 2000). The lin-4 and let-7 RNAs are sometimes called small temporal RNAs (stRNAs) because of their important roles in regulating the timing of larval development (Pasquinelli et al. 2000). The lin-4 and let-7 stRNAs are now recognized as the founding members of a large class of ∼22-nt ncRNAs termed microRNAs (miRNAs), which resemble stRNAs but do not necessarily control developmental timing (Lagos-Quintana et al. 2001; Lau et al. 2001; Lee and Ambros 2001).
Understanding the biogenesis and function of miRNAs has been greatly facilitated by analogy and contrast to another class of tiny ncRNAs known as small interfering RNAs (siRNAs), first identified because of their roles in mediating RNA interference (RNAi) in animals and posttranscriptional gene silencing in plants (Hamilton and Baulcombe 1999; Hammond et al. 2000; Parrish et al. 2000; Zamore et al. 2000; Elbashir et al. 2001a; Klahre et al. 2002). During RNAi, long double-stranded RNA (either a bimolecular duplex or an extended hairpin) is processed by Dicer, an RNAse III enzyme, into many siRNAs that serve as guide RNAs to specify the destruction of the corresponding mRNA (Hammond et al. 2000; Zamore et al. 2000; Bernstein et al. 2001; Elbashir et al. 2001a). Although these siRNAs are initially short double-stranded species with 5′ phosphates and 2-nt 3′ overhangs characteristic of RNAse III cleavage products, they eventually become incorporated as single-stranded RNAs into a ribonucleoprotein complex, known as the RNA-induced silencing complex (RISC; Hammond et al. 2000; Elbashir et al. 2001a,b; Nykäken et al. 2001; Martinez et al. 2002; Schwarz et al. 2002). The RISC identifies target messages based on perfect (or nearly perfect) antisense complementarity between the siRNA and the mRNA, and then the endonuclease of the RISC cleaves the mRNA at a site near the middle of the siRNA complementarity (Elbashir et al. 2001a,b). Similar pathways have been proposed for gene silencing in plants and fungi, with siRNAs targeting mRNA for cleavage during posttranscriptional gene silencing and heterochromatic siRNAs targeting chromatin for histone methylation, triggering heterochromatin formation and consequent transcriptional gene silencing (Hamilton and Baulcombe 1999; Vance and Vaucheret 2001; Hall et al. 2002; Hamilton et al. 2002; Pickford et al. 2002; Reinhart and Bartel 2002; Volpe et al. 2002; Zilberman et al. 2003).
miRNAs have many chemical and functional similarities to the siRNAs. Like siRNAs they are processed by Dicer, and so they are the same length and possess the same 5′-phosphate and 3′-hydroxyl termini as siRNAs (Grishok et al. 2001; Hutvágner et al. 2001; Ketting et al. 2001; Lau et al. 2001; Park et al. 2002; Reinhart et al. 2002). They are also incorporated within a ribonucleoprotein complex, known as the miRNP, which is similar if not identical to the RISC (Caudy et al. 2002; Hutvágner and Zamore 2002; Ishizuka et al. 2002; Martinez et al. 2002; Mourelatos et al. 2002). In fact, many plant miRNAs match their predicted mRNA targets with near-perfect antisense complementarity, as if they were functioning as siRNAs within a RISC complex (Rhoades et al. 2002), and the plant miR171 and miR165/166 have been shown to specify cleavage of their mRNA targets (Llave et al. 2002b; Tang et al. 2003). The C. elegans and Drosophila miRNAs do not have as pronounced a tendency to pair with their targets with near-perfect complementarity (Rhoades et al. 2002). Nonetheless, some might still direct cleavage of their targets, as suggested by the observation that miRNAs and siRNAs with 3–4 mismatches with their targets can still direct cleavage in plant and animal lysates (Tang et al. 2003). Furthermore, the let-7 miRNA is present within a complex that can cleave an artificial RNA target when such a target is perfectly complementary to the miRNA (Hutvágner and Zamore 2002). The known biological targets of lin-4 and let-7 RNAs have several mismatches within the central region of the miRNA complementary sites, perhaps explaining why in these particular cases, the miRNAs specify translational repression rather than mRNA cleavage during C. elegans larval development (Lee et al. 1993; Wightman et al. 1993; Ha et al. 1996; Moss et al. 1997; Olsen and Ambros 1999; Reinhart et al. 2000; Slack et al. 2000; Hutvágner and Zamore 2002).
Regulatory targets for most animal miRNAs have not yet been identified. Prediction of plant miRNA targets has led to the proposal that many plant miRNAs function to clear from differentiating cells mRNAs encoding key transcription factors, thereby facilitating plant development and organogenesis (Rhoades et al. 2002). Confident computational prediction of animal miRNA targets has relied on experimental evidence to first narrow the number of candidate mRNAs (Lai 2002). Nonetheless, as seen for the plant miRNAs, the sequences of the animal miRNAs are generally highly conserved in evolution. For example, 91 of the 107 miRNAs cloned from mammals are detected in the pufferfish (Fugu rubripes) genome, implying that they have important functions preserved during vertebrate evolution (Lim et al. 2003).
The first step in a systematic approach to identifying the biological roles of miRNAs is to find the miRNA genes themselves. Because gene-prediction programs had not been developed to identify miRNAs in genomic sequence, miRNA gene identification has been primarily achieved by cloning the small RNAs from size-fractionated RNA samples, sometimes specifically enriching in miRNAs by first immunoprecipitating the miRNP complex or by using a cloning protocol specific for the 5′ phosphate and 3′ hydroxyl found on Dicer products (Lagos-Quintana et al. 2001, 2002, 2003; Lau et al. 2001; Lee and Ambros 2001; Llave et al. 2002a; Mourelatos et al. 2002; Park et al. 2002; Reinhart et al. 2002). Once small RNAs have been cloned, the challenge is to differentiate the authentic miRNAs from other RNAs present in the cell, particularly from endogenous siRNAs. Because both miRNAs and siRNAs are Dicer products and both can act to specify mRNA cleavage, miRNAs cannot be differentiated based on their chemical composition or their functional properties. However, miRNAs can be distinguished from siRNAs based on their biogenesis and evolutionary conservation: (1) They are 20- to 24-nt RNAs that derive from endogenous transcripts that can form local RNA hairpin structures; (2) these hairpins are processed such that a single miRNA molecule ultimately accumulates from one arm of each hairpin precursor molecule; (3) the sequences of the mature miRNAs and their hairpin precursors are usually evolutionarily conserved; and (4) the miRNA genomic loci are distinct from and usually distant from those of other types of recognized genes, although a few are found within predicted introns but not necessarily in the same orientation as the introns. Endogenous siRNAs differ in that (1) they derive from extended dsRNA, (2) each dsRNA precursor gives rise to numerous different siRNAs, (3) they generally display less sequence conservation, and (4) they often perfectly correspond to the sequences of known or predicted mRNAs, transposons, or regions of heterochromatic DNA (Aravin et al. 2001; Djikeng et al. 2001; Elbashir et al. 2001a; Lau et al. 2001; Llave et al. 2002a; Mochizuki et al. 2002; Reinhart and Bartel 2002; Reinhart et al. 2002). Regarding this fourth criterion, miRNAs can also perfectly correspond to sequences of their mRNA targets, but when they do, they still derive from loci distinct from those of their mRNA targets (Llave et al. 2002a,b; Reinhart et al. 2002). Because miRNAs are primarily distinguished based on their biogenesis and evolutionary conservation, the current norms for identification and validation of miRNA genes include experimental evidence for endogenous expression of the miRNA, coupled with evidence of a hairpin precursor, preferably one that is evolutionarily conserved (Ambros et al. 2003).
Some miRNAs might be difficult to isolate by cloning, due to their low abundance or to biases in cloning procedures. Thus, computational identification of miRNAs from genomic sequences would provide a valuable complement to cloning. Recent advances have been made in the computational identification of ncRNA genes through comparative genomics, and complex algorithms have been developed to identify ncRNAs in general (Argaman et al. 2001; Rivas et al. 2001; Wassarman et al. 2001), as well as specific ncRNA families such as tRNAs and snoRNAs (Lowe and Eddy 1997, 1999).
In the present study, we describe a computational procedure to identify miRNA genes. By using this procedure, together with extensive sequencing of clones (3423 miRNA clones were sequenced), we have detected 30 additional miRNA genes, including previously unrecognized lin-4 and let-7 homologs. Extrapolation of the computational analysis indicates that miRNA gene identification in C. elegans is now approaching saturation, and that no more than 120 miRNA genes are present in this species. We also identify those genes with intriguing expression patterns during larval development and conditions of nutrient stress, and we show that most miRNAs are expressed at very high levels, with some present in as many copies per cell as the highly abundant U6 snRNA. This extensive census of worm miRNAs and their expression patterns establishes the general properties of this gene class and provides resources and tools for studies of miRNA function in nematodes and other organisms.
Results
Computational prediction of C. elegans miRNA genes
We developed a computational tool to specifically identify miRNAs that are conserved in two genomes and have the features characteristic of known miRNAs. To identify miRNAs in nematodes, the C. elegans genome was first scanned for hairpin structures with sequences that were conserved in Caenorhabditis briggsae. About 36,000 hairpins were found that satisfied minimum requirements for hairpin structure and sequence conservation. This procedure cast a sufficiently wide net to capture 50 of the 53 miRNAs previously reported to be conserved in the two species (Lau et al. 2001; Lee and Ambros 2001). These 50 published miRNA genes served as a training set for the development of a program called MiRscan, which was then used to assign scores to each of the 36,000 hairpins, evaluating them based on their similarity to the training set with respect to the following features: base pairing of the miRNA portion of the fold-back, base pairing of the rest of the fold-back, stringent sequence conservation in the 5′ half of the miRNA, slightly less stringent sequence conservation in the 3′ half of the miRNA, sequence biases in the first five bases of the miRNA (especially a U at the first position), a tendency toward having symmetric rather than asymmetric internal loops and bulges in the miRNA region, and the presence of two to nine consensus base pairs between the miRNA and the terminal loop region, with a preference for 4–6 bp (Fig. 1A).
Figure 1.

Criteria used by MiRscan to identify miRNA genes among aligned segments of two genomes. (A) The seven components of the MiRscan score for mir-232 of C. elegans/C. briggsae. These components are annotated in the context of the MiRscan prediction for mir-232, with the residues of the predicted miRNA circled in purple and the residues of the validated miRNA (Table 2), circled in green. In parenthesis are the scores for each component, which were added together to give the total score of 13.9. MiRscan predictions are visualized within the consensus C. elegans/C. briggsae secondary structure, as generated by using ClustalW (Thompson et al. 1994) and Alidot (Hofacker and Stadler 1999). Shown is the C. elegans sequence with residues colored to indicate conserved sequence and pairing potential. Residues conserved in C. briggsae are red, residues that vary while maintaining their predicted paired or unpaired state are blue (with variant residues that maintain pairing also circled in black), and residues that maintain neither sequence nor pairing are in gray. (B) Estimated relative importance of each MiRscan criterion. Estimates were based on the relative entropy between the training set of 50 previously identified nematode miRNAs and the background set of ∼36,000 potential stem loops. Because pairing and conservation were used to identify the potential stem loops, the total contributions of these types of criteria for distinguishing miRNA genes from non-protein-coding genomic sequence were underestimated. Likewise, the total contribution of the distance from the loop was underestimated because only those candidates 2–9 bp from the loop were evaluated.
The distribution of MiRscan scores for the ∼36,000 hairpins illustrated the ability of MiRscan to discern the 50 miRNA genes of the training set, which fell mostly in the high-scoring tail of the distribution (Fig. 2). Of the features evaluated by MiRscan, base-pairing potential and sequence conservation played primary roles in distinguishing known miRNAs (Fig. 1B). Some of the other conserved hairpins also scored highly; 35 had scores exceeding 13.9, the median score of the 58 known miRNAs (Fig. 2B). These 35 hairpins were carried forward as the top miRNA candidates predicted by MiRscan.
Figure 2.

Computational identification of miRNA genes. (A) The distribution of MiRscan scores for 35,697 C. elegans sequences that potentially form stem loops and have loose conservation in C. briggsae. Note that the Y-axis is discontinuous so that the scores of the 50 previously reported miRNA genes that served as the training set for MiRscan can be more readily seen (red). Scores for these 50 genes were jackknifed to prevent inflation of their values because of their presence in the training set. (B) An expanded view of the high-scoring tail of the distribution. This view captures 49 of the 50 genes of the training set (red). The median score of the 58 previously reported miRNA loci that satisfy the current criteria for designation as miRNA genes (Ambros et al. 2003) is 13.9 (green arrow). Note that this median score was the midpoint between the scores of the 29th and 30th highest-scoring loci of the 50-member training set; namely, it was designated the median score after including the 8 previously reported miRNA genes that were not in the training set because they were lost during the identification of conserved hairpins, usually because they lacked sufficient C. briggsae homology. Scores of genes validated by cloning are indicated (yellow), as are scores of six genes that have not yet been cloned but were verified by Northern analysis (purple). (C) Examples of miRNA genes identified by MiRscan with the Northern blots that served to validate them. Stem-loops were annotated as in Figure 1A, except the DNA rather than RNA sequence is depicted. The Northern blots show analysis of RNA from either wild-type (N2) or dcr-1 worms, isolated using either our standard protocol (Std.) or an additional polyethylene glycol precipitation step to enrich for small RNAs (Enr.). Homozygous worms of the dcr-1 population have reduced Dicer activity, increasing the level of miRNA precursors (e.g., miR-250-L and miR-255-L), which facilitated the validation of miRNA loci, especially those for which the mature miRNA was not detected (e.g., miR-255). RNA markers (left lane) are 18, 21, 24, 60, 78, and 119 nt. The miR-250 stem loop shown received a MiRscan score of 14.7. The mir-250 reverse complement received an even greater score of 18.4, but was not detected by Northern analysis. Thus, the predicted mir-250 gene was assigned the score of the higher-scoring, although incorrect, alternative stem loop (Table 1; Fig. 2B).
Molecular identification of miRNA genes
Our initial cloning and sequencing of small RNAs from mixed-stage C. elegans had identified 300 clones that represented 54 unique miRNA sequences (Lau et al. 2001). For the present study, this approach for identifying miRNAs was scaled-up ∼10-fold. In an effort to identify miRNAs not normally expressed in mixed-stage logarithmically growing hermaphrodite worms, RNA was also cloned from populations of him-8 worms, starved L1, and dauer worms. The him-8 population was ∼40% males, whereas the normal (N2) population was nearly all hermaphrodites (Broverman and Meneely 1994). Starved L1 and dauer worms are arrested in development at larval stages L1 and L3, respectively, with dauer worms having undergone morphological changes that enhance survival after desiccation or other harsh conditions.
As before, some clones matched Escherichia coli, the food source of the worms, others corresponded to fragments of annotated C. elegans RNAs. Nevertheless, 3423 clones were classified as miRNA clones (Table 1). Most of these represented the 58 miRNA genes previously identified in C. elegans (Lau et al. 2001; Lee and Ambros 2001). For example, lin-4 was represented by 125 clones, let-7 by 17 clones, and mir-52 by 404 clones (Table 1). The remaining miRNA clones represented 23 newly identified miRNA loci.
Table 1.
Cloning frequency and MiRscan scores of Caenorhabditis elegans miRNAs
| miRNA
|
MiRscan score
|
Number of sequenced clones
|
||||
|---|---|---|---|---|---|---|
| mixed stage
|
dauer
|
starved L1
|
him-8
|
total
|
||
| let-7 RNA | 13.8 | 15 | 0 | 0 | 2 | 17 |
| lin-4 RNA | 15.8 | 48 | 46 | 4 | 27 | 125 |
| miR-1 | 14.7 | 43 | 17 | 7 | 9 | 76 |
| miR-2 | 6.2 | 138 | 46 | 20 | 9 | 213 |
| miR-34 | 14.1 | 13 | 25 | 5 | 9 | 52 |
| miR-35 | 14.4 | 23 | 0 | 1 | 2 | 26 |
| miR-36 | 14.6 | 21 | 0 | 1 | 5 | 27 |
| miR-37 | 9.6 | 8 | 0 | 1 | 2 | 11 |
| miR-38 | 8.9 | 10 | 0 | 1 | 0 | 11 |
| miR-39 | 9.5 | 11 | 0 | 0 | 1 | 12 |
| miR-40 | 15.4 | 12 | 0 | 4 | 2 | 18 |
| miR-41 | 12.0 | 2 | 0 | 0 | 0 | 2 |
| miR-42 | 9.5 | 10 | 4 | 3 | 1 | 18 |
| miR-43 | 17.5 | 8 | 1 | 9 | 0 | 18 |
| miR-44/45 | 16.6/17.4 | 22 | 3 | 3 | 4 | 32 |
| miR-46 | 11.3 | 14 | 11 | 9 | 3 | 37 |
| miR-47 | 16.5 | 19 | 7 | 4 | 5 | 35 |
| miR-48 | 12.0 | 52 | 1 | 0 | 8 | 61 |
| miR-49 | 13.1 | 1 | 0 | 1 | 1 | 3 |
| miR-50 | 14.6 | 10 | 16 | 5 | 1 | 32 |
| miR-51 | 12.0 | 16 | 5 | 2 | 2 | 25 |
| miR-52 | 11.6 | 287 | 70 | 18 | 29 | 404 |
| miR-53 | 12.4 | 20 | 6 | 1 | 4 | 31 |
| miR-54 | 9.4 | 49 | 40 | 9 | 13 | 111 |
| miR-55 | 13.8 | 47 | 32 | 16 | 15 | 110 |
| miR-56 | NS | 40 | 16 | 9 | 6 | 71 |
| miR-57 | 12.1 | 31 | 11 | 8 | 3 | 53 |
| miR-58 | 17.5 | 181 | 51 | 27 | 31 | 290 |
| miR-59 | 18.5 | 1 | 0 | 0 | 0 | 1 |
| miR-60 | 14.1 | 20 | 6 | 3 | 7 | 36 |
| miR-61 | 13.7 | 8 | 5 | 1 | 3 | 17 |
| miR-62 | 15.1 | 4 | 4 | 6 | 0 | 14 |
| miR-63 | NS | 7 | 1 | 0 | 1 | 9 |
| miR-64 | NS | 11 | 4 | 8 | 3 | 26 |
| miR-65 | 7.4 | 22 | 7 | 3 | 2 | 34 |
| miR-66 | NS | 68 | 25 | 6 | 7 | 106 |
| miR-67 | 16.8 | 3 | 0 | 0 | 0 | 3 |
| miR-70 | 11.6 | 11 | 8 | 3 | 6 | 28 |
| miR-71 | 17.9 | 53 | 72 | 23 | 22 | 170 |
| miR-72 | NS | 49 | 22 | 10 | 9 | 90 |
| miR-73 | 11.3 | 13 | 7 | 1 | 1 | 22 |
| miR-74 | 17.9 | 35 | 12 | 6 | 7 | 60 |
| miR-75 | 12.6 | 14 | 3 | 2 | 2 | 21 |
| miR-76 | 14.9 | 1 | 2 | 6 | 3 | 12 |
| miR-77 | 14.2 | 17 | 3 | 0 | 2 | 22 |
| miR-78 | NS | 5 | 1 | 1 | 0 | 7 |
| miR-79 | 14.2 | 14 | 3 | 3 | 3 | 23 |
| miR-80 | 17.1 | 121 | 27 | 20 | 17 | 185 |
| miR-81 | 18.8 | 32 | 24 | 6 | 12 | 74 |
| miR-82 | 16.3 | 36 | 12 | 6 | 11 | 65 |
| miR-83 | 15.2 | 12 | 12 | 2 | 8 | 34 |
| miR-84 | −3.3 | 12 | 2 | 1 | 4 | 19 |
| miR-85 | 17.5 | 10 | 0 | 0 | 12 | 22 |
| miR-86 | 16.3 | 46 | 57 | 30 | 17 | 150 |
| miR-87 | 16.7 | 1 | 0 | 0 | 0 | 1 |
| miR-88 | −7.9 | 0 | ||||
| miR-90 | 14.0 | 5 | 37 | 14 | 9 | 65 |
| miR-124 | 15.7 | 7 | 16 | 7 | 5 | 35 |
| miR-228 | 17.5 | 1 | 13 | 8 | 3 | 25 |
| miR-229 | NS | 2 | 1 | 0 | 0 | 3 |
| miR-230 | 16.8 | 0 | 0 | 0 | 1 | 1 |
| miR-231 | 14.1 | 1 | 2 | 0 | 0 | 3 |
| miR-232 | 13.8 | 4 | 7 | 2 | 1 | 14 |
| miR-233 | 16.4 | 1 | 8 | 4 | 0 | 13 |
| miR-234 | 14.3 | 0 | 0 | 1 | 0 | 1 |
| miR-235 | 1.9 | 5 | 21 | 1 | 8 | 35 |
| miR-236 | 16.8 | 3 | 6 | 2 | 1 | 12 |
| miR-237 | 11.9 | 3 | 0 | 0 | 0 | 3 |
| miR-238 | 14.0 | 0 | 4 | 1 | 0 | 5 |
| miR-239a | 12.7 | 4 | 0 | 0 | 1 | 5 |
| miR-239b | 13.6 | 0 | ||||
| miR-240 | 12.5 | 0 | 0 | 0 | 1 | 1 |
| miR-241 | 14.9 | 7 | 0 | 0 | 3 | 10 |
| miR-242 | 9.9 | 0 | 0 | 1 | 1 | 2 |
| miR-243 | NS | 1 | 0 | 1 | 0 | 2 |
| miR-244 | 13.4 | 0 | 2 | 5 | 0 | 7 |
| miR-245 | 13.8 | 0 | 1 | 0 | 0 | 1 |
| miR-246 | 12.8 | 0 | 0 | 0 | 1 | 1 |
| miR-247 | NS | 0 | 2 | 0 | 0 | 2 |
| miR-248 | 14.6 | 0 | 2 | 0 | 0 | 2 |
| miR-249 | 13.7 | 0 | 2 | 1 | 0 | 3 |
| miR-250 | 18.4 | 0 | ||||
| miR-251 | 15.5 | 0 | ||||
| miR-252 | 17.7 | 0 | ||||
| miR-253 | 16.9 | 0 | ||||
| miR-254 | 15.7 | 0 | ||||
| miR-255 | 16.4 | 0 | ||||
| Total clones | 1821 | 851 | 363 | 388 | 3423 | |
A total of 3423 clones from logarithmically growing mixed-stage worms and worms from the indicated stages or mutant (dauer, starved L1, and him-8) represented 79 different miRNAs (and 80 different miRNA genes, because the miR-44/45 miRNA appears to be encoded at two loci). Genes not represented in the set of ∼36,000 stem loops did not receive scores (NS). Note that the previously reported miR-68 clone is not included. This RNA was not detected on Northern blots, and neither it nor its predicted precursor appears to be conserved in another species. Accordingly, it is now classified as an endogenous siRNA. Two other C. elegans loci previously thought to encode miRNAs (mir-69 and mir-89) also do not satisfy the current criteria for classification as miRNA genes (Ambros et al. 2003) and were not considered during the course of this study. One previously reported gene, mir-88, was not represented in our set of sequenced clones but is detected on Northern blots as a ∼22-nt RNA (V. Ambros, pers. comm.) and thus satisfies the current criteria for classification as an miRNA gene.
In total, 80 loci were represented by cloned miRNAs (Table 1). Of these, 77 had the classical features of C. elegans miRNA genes, in that they had the potential to encode stereotypic hairpin precursor molecules with the 20- to 25-nt cloned RNAs properly positioned within an arm of the hairpin so as to be excised during Dicer processing, and their expression was manifested as a detectable Northern signal in the 20- to 25-nt range. Three other loci, mir-41, mir-249, and mir-229, were also included. The mir-41 and mir-249 RNAs were not detected on Northern blots but were still classified as miRNAs because these RNAs and their predicted hairpin precursors appear to be conserved in C. briggsae.
The mir-229 locus was also classified as a miRNA gene, even though it appears to derive from an unusual fold-back precursor. Its precursor appears to be larger than normal, possibly because of an extra 35-nt stem loop protruding from the 3′ arm of the precursor stem loop (Supplementary Fig. 1). Nonetheless, miR-229 was detectable as a ∼25- to 26-nt species on Northern blots, and accumulation of its presumed precursor increased in the dcr-1 mutant, suggesting that Dicer processes this precursor despite the unusual predicted secondary structure (Supplementary Fig. 1). Furthermore, mir-229 is only 400 bp upstream of a previously recognized miRNA gene cluster, including mir-64, mir-65, and mir-66. miR-229 also has significant sequence identity with the miRNAs of this cluster. We provisionally classified mir-229 as a miRNA and a member of this C. elegans cluster. Greater confidence would be warranted if its unusual precursor structure were conserved in another species. A weakly homologous cluster of two potential miRNAs was found in C. briggsae, but neither of the predicted C. briggsae homologs appeared to have an unusual precursor resembling that of miR-229.
Validation of computationally predicted miRNAs
Of the 23 newly cloned miRNAs, 20 received MiRscan scores, and these scores are indicated in yellow in Figure 2B. The other three were not scored because orthologous sequences in C. briggsae were not identified. A Mann-Whitney test showed that the distribution of scores for these recently cloned miRNAs was not significantly different from that of the previously cloned miRNAs. Because the recently cloned miRNAs were not known during the development of MiRscan, their high scores gave added assurance that MiRscan was not over-fitting its training set. Ten of the 23 newly cloned miRNAs were among the set of 35 high-scoring miRNA gene candidates and served to validate these 10 candidates.
The remaining 25 candidate miRNAs that had not been cloned were tested by Northern blots. RNA from dcr-1 worms was included on the blots to enhance detection of precursor hairpins. Dicer-dependent processing of ∼70-nt precursors was detected for six candidates (as shown for miR-250 and miR-255; Fig. 2C), and ∼22-nt miRNAs were detected for miR-250, miR-251, and miR-252 . Despite prolonged exposure times and enrichment for small RNA by size fractionation, the Northern signals were generally weak, perhaps explaining why these miRNAs were missed in the current set of 3423 sequenced miRNA clones.
To investigate whether these miRNAs eventually would have been identified after further cloning and sequencing of our cDNA library of small RNA sequences, a PCR assay was used to detect the presence of these miRNAs in the library. By using a primer specific to the 3′ segment of the predicted miRNA, together with a second primer corresponding to the adapter sequence attached to the 5′ terminus of all the small RNAs, the 5′ segment of the miRNA was amplified, cloned, and sequenced. This procedure validated five of the six predicted miRNAs for which at least a precursor could be detected on Northerns, including two of the candidates (miR-253 and miR-254) for which a mature ∼22-nt RNA was not detected on Northern blots. In addition, it identified the 5′ terminus of these five miRNAs, which is difficult to achieve with confidence when using only bioinformatics and hybridization.
Combining the cloning and expression data, 16 of the 35 computationally identified candidates were validated (10 from cloning, five from Northerns plus the PCR assay, and one from Northerns only, which validated the precursor but did not identify the mature miRNA). Of the remaining 19 candidates, four could be readily classified as false positives. They appear to be nonannotated larger ncRNA genes, in that probes designed to hybridize to these candidates hybridized instead to high-molecular-weight species that remained constant in the samples from dcr-1 worms. The remaining 15 new candidates with high MiRscan scores but without any Northern signal might also be false positives, or they might be authentic miRNAs that are expressed at low levels or in only very specific cell types or circumstances. Considering the extreme case in which all the nonvalidated candidates are false positives, the minimum specificity of MiRscan for the C. elegans/C. briggsae analysis can be calculated as (29 + 16)/(29 + 35), or 0.70, at a sensitivity level that detects half of the 58 previously known miRNAs. A summary of the miRNA genes newly identified by validating computational candidates (16 genes) or by cloning alone (13 genes) is shown in Table 2, and predicted stem-loop precursors are shown in Supplemental Material. Table 2 also includes one additional gene, mir-239b, which was identified based on its homology with mir-239a and its MiRscan score of 13.6.
Table 2.
Newly identified Caenorhabditis elegans miRNA genes
| miRNA gene
|
ID method
|
miRNA sequence
|
miRNA length (nt)
|
C. briggsae homology
|
Fold-back arm
|
Chr.
|
Distance to nearest gene
|
||
|---|---|---|---|---|---|---|---|---|---|
| mir-124 | MS, C, N | UAAGGCACGCGGUGAAUGCCA | 21 | +++ | 3′ | IV | within intron of | C29E6.2 | (s) |
| mir-228 | MS, C, N | AAUGGCACUGCAUGAAUUCACGG | 21–24 | +++ | 5′ | IV | 0.2 kb downstream of | T12E12.5 | (as) |
| mir-229 | C, N | AAUGACACUGGUUAUCUUUUCCAUCG | 25–27 | − | 5′ | III | 0.4 kb upstream of | mir-64 | (s) |
| mir-230 | MS, C, N | GUAUUAGUUGUGCGACCAGGAGA | 23 | ++ | 3′ | X | 0.4 kb downstream of | F13D11.3 | (as) |
| mir-231 | MS, C, N | UAAGCUCGUGAUCAACAGGCAGAA | 23–24 | ++ | 3′ | III | 10.4 kb upstream of | lin-39 | (s) |
| mir-232 | C, N | UAAAUGCAUCUUAACUGCGGUGA | 23–24 | +++ | 3′ | IV | 1.1 kb downstream of | F13H10.5 | (as) |
| mir-233 | MS, C, N | UUGAGCAAUGCGCAUGUGCGGGA | 19–23 | +++ | 3′ | X | within intron of | W03G11.4 | (s) |
| mir-234 | MS, C, N | UUAUUGCUCGAGAAUACCCUU | 21 | +++ | 3′ | II | 1.5 kb downstream of | Y54G11B.1 | (as) |
| mir-235 | C, N | UAUUGCACUCUCCCCGGCCUGA | 22 | + | 3′ | I | 0.6 kb upstream of | T09B4.7 | (s) |
| mir-236 | MS, C, N | UAAUACUGUCAGGUAAUGACGCU | 21–25 | +++ | 3′ | II | 0.3 kb downstream of | C52E12.1 | (as) |
| mir-237 | C, N | UCCCUGAGAAUUCUCGAACAGCUU | 23–24 | + | 5′ | X | 3.4 kb upstream of | F22F1.2 | (as) |
| mir-238 | MS, C, N | UUUGUACUCCGAUGCCAUUCAGA | 21–23 | ++ | 3′ | III | 2.0 kb upstream of | mir-80 | (s) |
| mir-239a | C, N | UUUGUACUACACAUAGGUACUGG | 22–23 | ++ | 5′ | X | 6.0 kb upstream of | C34E11.1 | (s) |
| mir-239b | H | UUUGUACUACACAAAAGUACUGG | n.d. | ++ | 5′ | X | 7.0 kb upstream of | C34E11.1 | (s) |
| mir-240 | C, N | UACUGGCCCCCAAAUCUUCGCU | 22 | ++ | 3′ | X | 1.7 kb upstream of | C39D10.3 | (s) |
| mir-241 | MS, C, N | UGAGGUAGGUGCGAGAAAUGA | 21 | ++ | 5′ | V | 1.8 kb upstream of | mir-48 | (s) |
| mir-242 | C, N | UUGCGUAGGCCUUUGCUUCGA | 21 | ++ | 5′ | IV | 0.9 kb downstream of | nhr-78 | (as) |
| mir-243 | C, N | CGGUACGAUCGCGGCGGGAUAUC | 22–23 | − | 3′ | IV | 1.0 kb upstream of | R08C7.1 | (s) |
| mir-244 | C, N | UCUUUGGUUGUACAAAGUGGUAUG | 23–25 | +++ | 5′ | I | 1.6 kb downstream of | T04D1.2 | (as) |
| mir-245 | C, N | AUUGGUCCCCUCCAAGUAGCUC | 22 | +++ | 3′ | I | 1.9 downstream of | F55D12.1 | (s) |
| mir-246 | C, N | UUACAUGUUUCGGGUAGGAGCU | 22 | ++ | 3′ | IV | 0.4 kb downstream of | ZK593.8 | (s) |
| mir-247 | C, N | UGACUAGAGCCUAUUCUCUUCUU | 22–23 | − | 3′ | X | 1.9 kb upstream of | C39E6.2 | (as) |
| mir-248 | MS, C, N | UACACGUGCACGGAUAACGCUCA | 23 | ++ | 3′ | X | within intron of | AH9.3 | (s) |
| mir-249 | C | UCACAGGACUUUUGAGCGUUGC | 22–23 | ++ | 3′ | X | 2.7 kb upstream of | Y41G9A.6 | (s) |
| mir-250 | MS, N, PCR | UCACAGUCAACUGUUGGCAUGG | ∼22 | ++ | 3′ | V | 0.1 kb downstream of | mir-61 | (s) |
| mir-251 | MS, N, PCR | UUAAGUAGUGGUGCCGCUCUUAUU | ∼24 | +++ | 5′ | X | 0.2 kb downstream of | F59F3.4 | (as) |
| mir-252 | MS, N, PCR | UAAGUAGUAGUGCCGCAGGUAAC | ∼23 | +++ | 5′ | II | 1.8 kb downstream of | VW02B12L.4 | (as) |
| mir-253 | MS, D, PCR | CACACCUCACUAACACUGACC | n.d. | ++ | 5′ | V | within intron of | F44E7.5 | (s) |
| mir-254 | MS, D, PCR | UGCAAAUCUUUCGCGACUGUAGG | n.d. | ++ | 3′ | X | within intron of | ZK455.2 | (s) |
| mir-255 | MS, D | — | n.d. | 1.5 kb upstream of | F08F3.9 | (as) | |||
For predicted stem-loop precursors, see Supplementary Fig. 2. Genes were identified and validated as indicated in the ID method column: MS, candidate gene had high MiRscan score (Table 1); C, miRNA was cloned and sequenced (Table 1); N, expression of the mature miRNA was detectable on Northern blots; D, the miRNA stem-loop precursor was detected on Northern blots and enriched in RNA from dcr-1 animals, but the mature miRNA was not detected; PCR, targeted PCR amplification and sequencing detected the miRNA in a library of C. elegans small RNAs; H, the locus was closely homologous to that of a validated miRNA. For the miRNAs cloned and sequenced, some miRNAs were represented by clones of different lengths, due to heterogeneity at the miRNA 3′ terminus. The observed range in length is indicated, and the sequence of the most abundant length is shown. For the RNAs that have not been cloned, the 5′ terminus was determined by the PCR assay, but the 3′ terminus was not determined. For mir-250, mir-251, and mir-252, the length of the miRNA sequence shown was inferred from the Northern blots; for other miRNAs not cloned, the length was not determined (n.d.). For mir-254, the PCR assay detected ∼22-nt RNAs from both sides of the fold-back, representing both the miRNA and the miRNA*. Their relative positions within the precursor suggest that the RNA from the 5′ arm is 22 nt and the RNA from the 3′ arm is 23 nt. The RNA from the 3′ arm was chosen as the miRNA because of its similarity to the human miR-19 gene family. The miR-255 gene is known only as the precursor, a conserved stem loop with Dicer-dependent processing (Fig. 2b). Comparison to C. briggsae shotgun traces from the C. briggsae Sequencing Consortium (obtained from www.ncbi.nlm.nih.gov) revealed miRNA orthologs with 100% sequence identity (+++) and potential orthologs with >90% (++) and >75% (+) sequence identity. To indicate the genomic loci of the genes, the chromosome (Chr.), distance to nearest annotated gene, and the orientation relative to that gene, sense (s) or antisense (as), are specified.
Evolutionary conservation of miRNAs
The 88 C. elegans miRNA genes identified to this point were grouped into 48 families, each comprising one to eight genes (data not shown). Within families, sequence identity either spanned the length of the miRNAs or was predominantly at their 5′ terminus. All but two of these families extended to the miRNAs of C. briggsae. The two families without recognizable C. briggsae orthologs each comprised a single miRNA (miR-78 and miR-243). Thus, nearly all (>97%) of the C. elegans miRNAs identified had apparent homologs in C. briggsae, and all but six of these C. elegans miRNAs (miR-72, miR-63, miR-64, miR-66, miR-229, and miR-247) had retained at least 75% sequence identity to a C. briggsae ortholog. Of the 48 C. elegans miRNA families, 22 also had representatives among the known human miRNA genes (Fig. 3). In that these 22 families included 33 C. elegans genes, it appears that at least a third (33/88) of the C. elegans miRNA genes have homologs in humans and other vertebrates.
Figure 3.

Alignments of C. elegans and human miRNA sequences that can be grouped together in families. Human miRNAs (Hs) are those identified in human cells (Lagos-Quintana et al. 2001; Mourelatos et al. 2002) or are orthologs of miRNAs identified in other vertebrates (Lagos-Quintana et al. 2002, 2003; Lim et al. 2003).
Developmental expression of miRNAs
The expression of 62 miRNAs during larval development was examined and compiled together with previously reported expression profiles (Lau et al. 2001) to yield a comprehensive data set for the 88 C. elegans miRNAs (Fig. 4). RNA from wild-type embryos, the four larval stages (L1 through L4), and young adults was probed, as was RNA from glp-4 (bn2) young adults, which are severely depleted in germ cells (Beanan and Strome 1992). Nearly two thirds of the miRNAs appeared to have constitutive expression during larval development (Fig. 4A). These miRNAs might still have differential expression during embryogenesis, or they might have tissue-specific expression, as has been observed for miRNAs of larger organisms in which tissues and organs can be more readily dissected and examined (Lee and Ambros 2001; Lagos-Quintana et al. 2002; Llave et al. 2002a; Park et al. 2002; Reinhart et al. 2002).
Figure 4.

Expression of C. elegans miRNAs during larval development. Total RNA was analyzed from mixed-stage N2 worms (M), embryos (E), larval stages (L1, L2, L3, L4), adults (A), glp-4(bn2) adults (G), N2 dauers (D), mixed-stage him-8(e1489) worms (H), and N2 starvation-arrested L1 larvae (sL1). Intense signals are represented as black rectangles and faint signals are represented as gray rectangles. Of the 87 C. elegans miRNAs identified, 6 could not be detected on developmental Northerns (miR-41, miR-78, miR-249, miR-253, miR-254, and miR-255). (A) miRNAs constitutively expressed throughout nematode development. (B) stRNAs, lin-4 and let-7, and similarly expressed miRNAs, which commence expression during larval development and remain expressed through adulthood. (C) miRNAs with discontinuous developmental expression patterns. (D) Northern analysis of miRNAs with enhanced expression in the dauer stage. To control for loading, the blot used for both miR-234 and miR-248 and the blot used for miR-247 were reprobed for the U6 snRNA (U6). Quantitation with a PhosphorImager showed that the lane-to-lane variation in U6 signal was as great as threefold. Normalizing to the U6 signal, the miR-248 signal was fourfold greater in dauer than in most other stages, except for glp-4 adults, in which it was twofold greater, whereas the miR-234 signal was highest in dauer and L1, with a signal in these stages about twofold greater than the average of the other stages. (E) Northern analysis of the lin-4 RNA and its paralog, miR-237.
Over one third of the miRNAs had expression patterns that changed during larval development (Fig. 4B,C), and there were examples of miRNA expression initiating at each of the four larval stages (Fig. 4B). Expression profiles for miR-48 and miR-241 (which are within 2 kb of each other in the C. elegans genome) were similar to those previously reported for let-7 RNA and miR-84 (Fig. 4B; Reinhart et al. 2000; Lau et al. 2001). In fact, these four miRNAs appear to be paralogs, with all four miRNAs sharing the same first eight residues (Fig. 3). Another newly identified miRNA, miR-237, is a paralog of the other canonical stRNA, lin-4 RNA (Fig. 3), although miR-237 exhibited an expression pattern distinct from lin-4 RNA (Fig. 4E). The existence of these paralogs, as well as other families of miRNAs with expression initiating at the different stages of larval development, supports the idea that lin-4 and let-7 miRNAs are not the only stRNAs with important roles in the C. elegans heterochronic pathway.
Expression usually remained constant once it initiated, as has been seen for lin-4 and let-7 miRNA expression (Fig. 4A,B). Exceptions to this trend included the miRNAs of the mir-35–mir-41 cluster, which were expressed transiently during embryogenesis (Lau et al. 2001); miR-247, which was expressed transiently in larval stage 3 (and dauer); and miR-248, which was most highly expressed in dauer (Fig. 4C,D). miR-234 was expressed in all stages, but expression was highest in both L1 worms (which had been starved shortly before harvest to synchronize the worm developmental staging) and dauer worms, suggesting that this miRNA might be induced as a consequence of nutrient stress.
Molecular abundance of miRNAs
The very high cloning frequency of certain miRNAs (e.g., miR-52, represented by >400 clones) raised the question as to the molecular abundance of these and other miRNA species. In addition, there was the question of whether the actual molecular abundance of miRNAs in nematodes was proportionally reflected in the numbers of clones sequenced. To address these questions, quantitative Northerns were used to examine the molecular abundance of 12 representative miRNAs, picked so as to span the range of frequently and rarely cloned sequences and differing 3′ and 5′ terminal residues (Fig. 5).
Figure 5.

Quantitative analysis of miRNA expression. (A) Northern blot used to quantify the abundance of miR-66. RNA prepared from the wild-type (N2) mixed-stage worms used in cloning and from glp-4(bn2) young adult worms were run in duplicate with a concentration course of synthetic miRNA standard. The signal from the standard did not change when total RNA from HeLa cells replaced E. coli tRNA as the RNA carrier, showing that the presence of other miRNAs did not influence membrane immobilization of the miRNA or hybridization of the probe. (B) Standard curve from quantitation of miR-66 concentration course. The best fit to the data is a line represented by the equation y = 3.3x0.96 (R2 = 0.99). Interpolation of the average signal in the glp-4 lanes indicates that the glp-4 samples contain 240 pg of miR-66 (broken lines). (C) Molecular abundance of miRNAs and U6 snRNA. Amounts of the indicated RNA species in the glp-4 samples were determined as shown in A and B. The average number of molecules per cell was then calculated considering the number of animals used to prepare the sample, and the yield of a radiolabeled miRNA spiked into the preparation at an early stage of RNA preparation. Analogous experiments were performed to determine the amounts of the indicated human miRNAs in HeLa RNA samples. (D) Correlation between miRNA molecular abundance and cloning frequency. The number of molecules in the mixed-stage RNA samples was determined as described for the glp-4 samples and then plotted as a function of the number of times the miRNAs was cloned from this mixed-stage population (Table 1). The line is best fit to the data and is represented by the equation y = 0.32x (R2 = 0.78).
To determine the molecular abundance of these 12 miRNAs in the adult worm soma, the hybridization signals for RNA from a known number of glp-4 young adult worms were compared with standard curves from chemically synthesized miRNAs (Fig. 5; Hutvágner and Zamore 2002). Accounting for RNA extraction yields and dividing the number of miRNA molecules per worm by the total number of cells in the worms, yielded averages of up to 50,000 molecules per cell, with the most abundant miRNAs as plentiful as the U6 snRNA of the spliceosome (Fig. 5C). These are much higher numbers than those for the typical worm mRNAs, estimated to average ∼100 molecules per cell for the 5000 most highly expressed genes in the cell. [This estimate was calculated based on our yield of 20 pg total RNA per worm cell, assuming that the 5000 most highly expressed genes have mRNAs averaging 2 kb in length and represent 3% of the total RNA in an adult worm; it was consistent with estimates based on hybridization kinetics of mRNAs from mouse tissues (Hastie and Bishop 1976).] Perhaps high concentrations of miRNAs are needed to saturate the relevant complementary sites within the target mRNAs, which might be recognized with low affinity because of the noncanonical pairs or bulges that appear to be characteristic of the animal miRNA–target interactions.
Because these numbers represent molecular abundance averaged over all the cells of the worm, including cells that might not be expressing the miRNA, there are likely to be some cells that express even more molecules of the miRNA. To examine the abundance in a single cell type, HeLa RNA was probed for representative human miRNAs, yielding a similar range of molecular abundance (Fig. 5C). The high number of miRNA molecules in human cells increases the mystery as to why miRNAs had gone undetected for so long, which raises the question of whether other classes of highly expressed ncRNAs might yet remain to be discovered. A recent large-scale analysis of full-length cDNAs from mouse indicates the possible existence of hundreds or thousands of expressed ncRNAs in vertebrates (Okazaki et al. 2002).
To address the extent to which the actual molecular abundance of miRNAs in nematodes is proportionally reflected in the numbers of clones sequenced, the abundance of the miRNA within the mixed-stage RNA preparation was compared with the number of clones generated from that preparation (Fig. 5D). The strong positive correlation observed between the molecular abundance and the number of times the miRNAs were cloned indicated that systematic biases in the cloning procedure were not major. At most, these miRNAs were over- or underrepresented fivefold in the sequenced set relative to their actual abundance as measured by quantitative Northerns. We cannot rule out the possibility that certain miRNAs not yet cloned might be refractory to our cloning procedure, for example, because of a propensity to form secondary structures that preclude adaptor ligation reactions. Nonetheless, on the whole, the cloning frequencies can be used to approximate the molecular abundance of the miRNAs, and we have no reason to suspect that the set of miRNAs identified by cloning differs in any substantive way, other than an overall higher steady-state expression level, from the complete set of C. elegans miRNAs.
Other endogenous ∼22-nt RNAs of C. elegans
Of the 4078 C. elegans clones, a large majority represented authentic miRNAs (3423 clones, Table 1). The next most abundant class represented degradation fragments of larger ncRNAs, such as tRNA and rRNA (447 clones) and introns (18 clones). The remaining clones represented potential Dicer products that were not classified as miRNAs. Some corresponded to sense (18 clones) or antisense (23 clones) fragments of known or predicted mRNAs and might represent endogenous siRNAs. Others (143 clones) corresponded to regions of the genome not thought to be transcribed; these might represent another type of endogenous siRNAs, known as heterochromatic siRNAs (Reinhart and Bartel 2002). The possible roles of the potential siRNAs and heterochromatic siRNAs in regulating gene expression are still under investigation. The remaining clones were difficult to classify because they matched more than one locus, and their loci were of different types (six clones).
A fourth class of potential Dicer products (38 clones, representing 14 loci) corresponded to miRNA precursors but derived from the opposite arm of the hairpin than the more abundantly expressed miRNA, as has been reported previously for miR-56 in C. elegans, miR156d and miR169 in plants, and several vertebrate miRNAs (Lau et al. 2001; Lagos-Quintana et al. 2002, 2003; Mourelatos et al. 2002; Reinhart et al. 2002). Our current data add another 13 examples of this phenomenon (Fig. 6). In all of our cases, the ∼22-nt RNA from one arm of the fold-back was cloned much more frequently than that from the other and was far more readily detected on Northern blots. We designated the less frequently cloned RNA as the miRNA-star (miRNA*) fragment (Lau et al. 2001).
Figure 6.

miRNA (red) and miRNA* (blue) sequences within the context of their predicted fold-back precursors. The number of sequenced clones is shown in parentheses. For each miRNA and miRNA*, colored residues are those for the most frequently cloned species. There was 3′ heterogeneity among the sequenced clones for some miRNA*s and most miRNAs. Heterogeneity at the 5′ terminus was not seen among the sequenced clones for the miRNA*s and was rare among those for the miRNAs; when it occurred, it was not observed for more than one of the many clones representing each miRNA.
Discussion
We have developed a computational procedure for identifying miRNA genes conserved in two genomes. By using this procedure, together with extensive sequencing of clones from libraries of small RNAs, we have now identified 87 miRNA genes in C. elegans (Tables 1, 2). Together with mir-88 (Lee and Ambros 2001), which we have not yet cloned or found computationally, the number of validated C. elegans genes stands at 88. More than a third of these genes have human homologs (Fig. 3), and a similar fraction, including previously unrecognized lin-4 and let-7 paralogs, is differentially expressed during larval development (Fig. 4). Most miRNAs accumulated to very high steady-state levels, with some at least as plentiful as the U6 snRNA (Fig. 5). Below, we discuss some implications of these results with regard to some of the defining features of miRNA genes in animals, the processing of miRNA precursors, and the number of miRNA genes remaining to be identified.
MiRscan accuracy and the defining features of miRNAs
As calculated in the Results section, the specificity of MiRscan was ≥0.70 at a sensitivity that detects half the previously known C. elegans miRNAs, when starting from an assembled C. elegans genome and C. briggsae shotgun reads. This accuracy was sufficient to identify new genes and obtain an upper bound on the total number of miRNA genes in the worm genome (described later). However, it was not sufficient to reliably identify all the conserved miRNA genes in C. elegans. The accuracy of MiRscan appears to be at least as high as that of general methods to identify ncRNA genes in bacteria (Argaman et al. 2001; Rivas et al. 2001; Wassarman et al. 2001), but is lower than that of algorithms designed to identify protein-coding genes or specialized programs that predict tRNAs and snoRNAs (Lowe and Eddy 1997, 1999; Burge and Karlin 1998). The relative difficulty in identifying miRNAs can be explained by the low information content inherent in their small size and lack of strong primary sequence motifs. The performance of MiRscan will improve with a more complete and assembled C. briggsae genome. We anticipate that using only those sequences conserved in a syntenic alignment of the two genomes would capture fewer of the background sequences, enabling the authentic miRNAs to be more readily distinguished from the false positives.
Improvement would also come from bringing in a third nematode genome, particularly a genome more divergent than those of C. elegans and C. briggsae. The advantage of such an additional genome is illustrated by our application of MiRscan to the identification of vertebrate miRNAs using three genomes. The version of MiRscan described here, which had been trained on the set of 50 miRNAs conserved in worms, was applied to the assembled human genome, shotgun reads of the mouse genome, and the assembled pufferfish (Fugu) genome (Lim et al. 2003). This analysis had a specificity of ≥0.71 at a sensitivity that detected three fourths of the previously known vertebrate miRNAs. The accuracy of the vertebrate analysis was therefore substantially improved over that of the C. elegans/C. briggsae analysis, even though the vertebrate genomes are 4–30 times larger than those of C. elegans and C. briggsae, and are expected to have a correspondingly higher number of background sequences. This improved performance can be attributed to using three genomes, as well as to the evolutionary distance between the mammalian and fish genomes, which are distant enough to reduce the number of fortuitously high scoring sequences, yet close enough to retain most of the known miRNAs.
Other improvements in the computational identification of miRNAs will come with the definition of additional sequence and structural features that specify which sequences are transcribed, processed into miRNAs, and loaded into the miRNP. With the exception of sequence conservation, the features that MiRscan currently uses to identify miRNAs (Fig. 1A) are among those that the cell also uses to specify the biogenesis of miRNAs and miRNPs. The utility of these parameters for MiRscan (Fig. 1B) is a function of both the degree to which these features are correctly modeled (or have already been used to restrict the number of miRNA candidates; see Fig. 1B legend) and their relative importance in vivo. Clearly, much of what defines a miRNA in vivo remains to be determined. Sequence elements currently unavailable for MiRscan include transcriptional promoter and termination signals. Additional sequence and structural features important for processing of the primary transcript and the hairpin precursors also remain to be identified (Lee et al. 2002).
miRNA biogenesis
The presence of miRNA* species, observed now for 14 of the C. elegans miRNAs (Fig. 6; Lau et al. 2001), provides evidence for the idea that Dicer processing of miRNA precursors resembles that of siRNA precursors (Hutvágner and Zamore 2002; Reinhart et al. 2002). We suspect that with more extensive sequencing of clones, miRNA* sequences will be found for a majority of the miRNA precursors, a notion supported by the identification of additional miRNA* sequences using our PCR assay (data not shown). As observed for both MIR156d and MIR169 in plants (Reinhart et al. 2002), the miRNA:miRNA* segments are typically presented within the predicted precursor, paired to each other with 2-nt 3′ overhangs (Fig. 6)—a structure analogous to that of a classical siRNA duplex. This is precisely the structure that would be expected if both the miRNA and the miRNA* were excised from the same precursor molecule, and the miRNA* fragments were transient side-products of productive Dicer processing. An alternative model for miRNA biogenesis and miRNA* formation, which we do not favor but cannot rule out, is that the Dicer complex normally excises a ∼22-nt RNA from only one side of a miRNA precursor but it sometimes binds the precursors in the wrong orientation and excises the wrong side. In an extreme version of the favored model, the production of the miRNA* would be required for miRNA processing and miRNP assembly; in a less extreme version, miRNA* production would be an optional off-pathway phenomenon. The idea that ∼22-nt RNAs might be generally excised from both sides of the same precursor stem loop brings up the question of why the miRNAs and miRNA*s are present at such differing levels. With the exception of miR-34* (sequenced 17 times), none of the miRNA*s is represented by more than three sequenced clones. Perhaps the miRNAs are stabilized relative to their miRNA* fragments because they preferentially enter the miRNP/RISC complex. Alternatively, both the miRNA and the miRNA* might enter the complex, but the miRNA might be stabilized by interactions with its targets.
Five of the newly identified miRNAs are within annotated introns, all five in the same orientation as the predicted mRNAs. When considered together with the previously identified miRNAs found within annotated introns (Lau et al. 2001), 10 of 12 known C. elegans miRNAs predicted to be in introns are in the same orientation as the predicted mRNAs. This bias in orientation, also reported recently for mammalian miRNAs (Lagos-Quintana et al. 2003), suggests that some of these miRNAs are not transcribed from their own promoters but instead derive from the excised pre-mRNA introns (as are many snoRNAs), and it is easy to imagine regulatory scenarios in which the coordinate expression of a miRNA with an mRNA would be desirable.
The number of miRNA genes in C. elegans and other animals
In addition to providing a set of candidate miRNAs, MiRscan scoring provides a means to estimate the total number of miRNA genes in C. elegans. A total of 64 loci have scores greater than the median score of the 58 initially reported C. elegans miRNAs (Fig. 2B). Note that this set of 58 miRNAs includes not only the 50 conserved miRNAs of the training set but also the eight previously reported miRNAs that were not in our set of 36,000 potential stem loops, usually because they lacked easily recognizable C. briggsae orthologs. Thus, the estimate calculated below takes into account the poorly conserved miRNAs without MiRscan scores. Four of the 64 high-scoring loci are known to be false positives. Thus, the upper bound on the number of miRNA genes in C. elegans would be 2 × (64 − 4), or 120. This upper bound of ∼120 genes remained stable when extrapolating from points other than the median, ranging from the top 25th–55th percentiles. For this estimate, we made the assumption that the set of all C. elegans miRNAs has a distribution of MiRscan scores similar to the distribution of initially reported miRNAs. Such an assumption might be called into question, particularly when considering that the initially reported miRNAs served as a training set for the development of MiRscan (even though the scores of the training-set loci have been jackknifed to prevent overfitting). However, this assumption is supported by two observations. First, the set of newly cloned miRNAs did indeed have a distribution of scores indistinguishable from that of the training set of previously reported miRNAs (Fig. 2B). Second, there is no correlation between the number of times that a miRNA has been cloned and its MiRscan score (Fig. 7). The absence of a correlation between cloning frequency and MiRscan score lessens our concern that miRNAs that are difficult to clone, including those still not present in our set of 3423 sequenced clones, might represent a population of miRNAs that are refractory to computational analysis as well.
Figure 7.

Plot illustrating the absence of a correlation between the MiRscan score of a cloned miRNA and the number of times that miRNA was cloned and sequenced. Nine of 80 cloned loci of Table 2 were not scored (left) because potential homologs of these genes were not identified among the available C. briggsae sequencing reads.
This estimate of 120 genes is an upper bound and would decrease if additional high-scoring candidates were shown to be false positives. The extreme scenario, in which all are false positives, places the lower bound of miRNA genes near the number of validated genes, adding perhaps another five genes to account for the low-scoring counterparts of the five computational candidates validated only by Northerns and PCR, yielding a lower bound on the number of C. elegans miRNAs of ∼93.
Our count of 105 ± 15 miRNA genes in C. elegans might underestimate the true count if there are miRNAs with unusual fold-back precursors that were cloned but dismissed as endogenous siRNAs or degradation fragments. To investigate this possibility, we examined the expression of each small RNA that was cloned more than once but did not appear to derive from a canonical miRNA precursor as predicted by RNAfold. Because most (72 of 88) of the authentic miRNAs identified to date were represented by multiple clones (Table 1), this analysis should uncover most of the miRNAs coming from nonconventional precursors. This broader analysis detected only a single additional miRNA, miR-229. All of the other sequences that we cloned more than once were minor degradation fragments or processing byproducts of larger ncRNAs (e.g., the 5′ leader sequence of a tRNA). Thus, the number of miRNAs that derive from nonconventional precursors is not sufficient to significantly influence the miRNA gene count.
The estimated number of miRNA genes represents between 0.5% and 1% of the genes identified in the C. elegans genome, a fraction similar to that seen for other very large gene families with presumed regulatory roles, such as those encoding nuclear hormone receptors (270 predicted genes), C2H2 Zinc-finger proteins (157 predicted genes), and homeodomain proteins (93 predicted genes; Chervitz et al. 1998; C. elegans Sequencing Consortium 1998). Extending our analysis to vertebrate genomes revealed that 230 ± 30 of the human genes are miRNAs, also nearly 1% of the genes in the genome (Lim et al. 2003). The miRNA genes are also among the most abundant of the ncRNA gene families in humans, comparable in number to the genes encoding rRNAs (∼650–900 genes), tRNAs (∼500 genes), snRNAs (∼100 genes), and snoRNAs (∼100–200 genes; Lander et al. 2001). For rRNAs, tRNAs, and snRNAs, the hundreds of gene copies in the human genome represent only relatively few distinct genes, probably <100 distinct genes for all three classes combined. For the miRNAs and snoRNAs, there are many more distinct genes, and each is present in only one or a few copies.
Unlike the other large ncRNA gene families and many of the transcription-factor gene families, there is no indication that miRNAs are present in single-celled organisms such as yeast. A pilot attempt to clone miRNAs from Schizosaccharomyces pombe did not detect any miRNAs (Reinhart and Bartel 2002), and there is no evidence that the proteins (such as Dicer) needed for miRNA accumulation in plants and animals are present in Saccharomyces cerevisiae. Given the known roles of miRNAs in C. elegans development (Lee et al. 1993; Wightman et al. 1993; Reinhart et al. 2000) and the very probable roles of miRNAs in plant development (Rhoades et al. 2002), it is tempting to speculate that the substantial expansion of miRNA genes in animals (and the apparent loss of miRNA genes in yeast) is related to their importance in specifying cell differentiation and developmental patterning, and that the extra layer of gene regulation afforded by miRNAs was crucial for the emergence of multicellular body plans. The identification of most of the worm miRNAs and the quantitation of the number of genes remaining to be found are important steps toward understanding the evolution of this intriguing class of genes and placing them within the gene regulatory circuitry of these and other animals.
Materials and methods
Computational identification of stem loops
Potential miRNA stem loops were located by sliding a 110-nt window along both strands of the C. elegans genome (WormBase release 45, http://www.wormbase.org) and folding the window with the secondary structure-prediction program RNAfold (Hofacker et al. 1994) to identify predicted stem-loop structures with a minimum of 25 bp and a folding free energy of at least 25 kcal/mole (ΔG°folding ≤ −25 kcal/mole). Sequences that matched repetitive elements were discarded, as were those with skewed base compositions not observed in known miRNA stem loops and those that overlapped with annotated coding regions. Stem loops that had fewer base pairs than overlapping stem loops were also culled. C. briggsae sequences with at least loose sequence similarity to the remaining C. elegans sequences were identified among C. briggsae shotgun sequencing reads (November 2001 download from http://www.ncbi.nlm.nih.gov/Traces) using WU-BLAST with default parameters and a nonstringent cutoff of E < 1.8 (W. Gish, http://blast.wustl.edu). These C. briggsae sequences were folded with RNAfold to ensure that they met the minimal requirements for a hairpin structure as described above. This procedure yielded ∼40,000 pairs of potential miRNA hairpins. For each pair of potential miRNA hairpins, a consensus C. elegans/C. briggsae structure was generated using the alidot and pfrali utilities from the Vienna RNA package (Hofacker et al. 1998; Hofacker and Stadler 1999; http://www.tbi.univie.ac.at/∼ivo/RNA). To create RNA consensus structures, alidot and pfrali combine a Clustal alignment (Thompson et al. 1994) of a pair of sequences with either the minimum free energy structures of these sequences (alidot) derived using the Zuker algorithm (Zuker 1994) or the base pairing probability matrices of these sequences (pfrali) derived using the McCaskill algorithm (McCaskill 1990).
MiRscan
Of the ∼40,000 pairs of hairpins, 35,697 had the minimal conservation and base pairing needed to receive a MiRscan score. Among this set were 50 of the 53 previously published miRNAs that were reported to be conserved between C. elegans and C. briggsae (Lau et al. 2001; Lee and Ambros 2001). [miR-53 is included as a previously reported conserved miRNA because it is nearly identical to miR-52, which has a highly conserved C. briggsae ortholog (Lau et al. 2001; Lee and Ambros 2001). The three conserved genes missing from the ∼36,000 pairs of hairpins were mir-56, mir-75, and mir-88. The reverse complements of mir-75 and mir-88 were later observed among the ∼36,000 hairpins and given scores (Table 1).] The MiRscan program was developed to discriminate these 50 known miRNA hairpins from background sequences in the set of ∼36,000 hairpins. For a given 21-nt miRNA candidate, MiRscan makes use of the seven features derived from the consensus hairpin structure illustrated in Figure 1A: x1, “miRNA base pairing,” the sum of the base-pairing probabilities for pairs involving the 21-nt candidate miRNA; x2, “extension of base pairing,” the sum of the base-pairing probabilities of the pairs predicted to lie outside the 21-nt candidate miRNA but within the same helix; x3, “5′ conservation,” the number of bases conserved between C. elegans and C. briggsae within the first 10 bases of the miRNA candidate; x4, “3′ conservation,” the number of conserved bases within the last 11 bases of the miRNA candidate; x5, “bulge symmetry,” the number of bulged or mismatched bases in the candidate miRNA minus the number of bulged or mismatched bases in the corresponding segment on the other arm of the stem loop; x6, “distance from loop,” the number of base pairs between the loop of the stem loop and the closest end of the candidate; and x7, “initial pentamer,” the specific bases at the first five positions at the candidate 5′ terminus.
For a given feature i with a value xi, MiRscan assigns a log-odds score
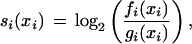 |
where fi(xi) is an estimate of the frequency of feature value xi in miRNAs derived from the training set of 50 known miRNAs, and gi(xi) is an estimate of the frequency of feature value xi among the background set of ∼36,000 hairpin pairs. The overall score assigned to a candidate miRNA is simply the sum of the log-odds scores for the seven features:
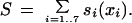 |
To score a given hairpin, MiRscan slides a 21-nt window representing the candidate miRNA along each arm of the hairpin, assigns a score to each window, and then assigns the hairpin the score of its highest-scoring window. In order to be evaluated, a window was required to be two to nine consensus base pairs away from the terminal loop.
For features x1, x3, x4, x5, and x6, fi and gi were obtained by smoothing the empirical frequency distributions from the training and background sets, respectively, using the R statistical package (http://lib.stat.cmu.edu/R/CRAN) with a triangular kernel. Because x1 and x2 are not independent of each other, the relative contribution of x2 was decreased by computing f2 and g2 separately subject to the conditions x1 ≥ 9 and x1 < 9, in order to account for this dependence. For x7, a weight matrix model (WMM) was generated for the five positions at the miRNA 5′ terminus. The background WMM, g7, was set equal to the base composition of the background sequence set. The miRNA WMM, f7, was derived from the position-specific base frequencies of the 50 training set sequences, using standard unit pseudo-counts and normalizing for the contributions of related miRNAs.
Because both strands of the C. elegans genome were analyzed, both a hairpin sequence and its reverse complement were sometimes included in the set of ∼36,000 stem loops. For representation in Figure 2, in such cases both sequences were considered as a single locus that received the score of the higher scoring hairpin. Also, to prevent overscoring of the 50 known miRNA loci within the training set, each known miRNA locus was assigned a jackknife score calculated by using a training set consisting of the other 49 miRNAs. MiRscan is available for use (http://genes.mit.edu/mirscan).
RNA cloning and bioinformatic analyses
Small RNAs were cloned as described previously (Lau et al. 2001), using the protocol available on the Web (http://web.wi.mit.edu/bartel/pub). Sequencing was performed by Agencourt Bioscience. Sequences of known C. elegans tRNA and rRNA were removed, and the remaining clones were clustered based on the location of their match to the C. elegans genome (C. elegans Sequencing Consortium 1998), downloaded from WormBase (http://www.wormbase.org). Genomic loci not previously reported to encode miRNAs were examined by using the RNA-folding program RNAfold (Hofacker et al. 1994). Two sequences were folded for each locus: one included 15 nt upstream and 60 nt downstream of the most frequently cloned sequence from that locus; the other included 60 nt upstream and 15 nt downstream. Sequences for which the most stable predicted folding resembled the stem-loop precursors of previously validated miRNAs were carried forward as candidate miRNA loci. Sequences without classical stem-loop precursors were also analyzed further (see Discussion), but only one, miR-229, was classified as a miRNA. The clones classified as representing potential fragments of mRNAs (18 clones) and potential antisense fragments of mRNAs (23 clones) corresponded to predicted ORFs (as annotated in GenBank) or probable UTR segments (100 bp upstream or 200 bp downstream of the predicted ORF).
Northerns
Expression of candidate miRNA loci was examined by using Northern blots and radiolabeled DNA probes (Lau et al. 2001). To maintain hybridization specificity without varying hybridization or washing conditions, the length of probes for different sequences was adjusted so that the predicted melting temperatures of the miRNA-probe duplexes did not exceed 60°C (Sugimoto et al. 1995). Probes not corresponding to the entire miRNA sequence were designed to hybridize to the 3′ region of the miRNA, which is most divergent among related miRNA sequences.
PCR validation
A PCR assay was performed to detect the sequences of predicted miRNAs within a cDNA library constructed from 18- to 26-nt RNAs expressed in mixed-stage worms. This library, the same as that used for cloning (Lau et al. 2001), consisted of PCR-amplified DNA that comprised the 18- to 26-nt sequences flanked by 3′- and 5′-adaptor sequences. For each miRNA candidate, a primer specific to the predicted 3′ terminus of the candidate and a primer corresponding to the 5′-adaptor sequence common to all members of the library (ATCGTAG GCACCTGAAA) were used at concentrations of 1.0 μM and 0.1 μM, respectively (100 μL PCR reaction containing 5 μL of a 400-fold dilution of the PCR reaction previously used to amplify all members of the cDNA library). The specific primer was added after the initial denaturation incubation had reached 80°C. After 20 PCR cycles, the reaction was diluted 20-fold into a fresh PCR reaction for another 20 cycles. PCR products were cloned and sequenced to both identify the 5′ terminus of the miRNA and ensure that the amplified product was not a primer-dimer or other amplification artifact. Specific primers for the reactions that successfully detected candidate miRNAs were ACCATGCCAACAGTTG (miR-250), TAAGAGCGGCACCA CTAC (miR-251), TACCTGCGGCACTACTAC (miR-252), GTCAGTGTTAGTGAGG (miR-253), TACAGTCGGAAAGA TTTG (miR-254), and GTGGAAATCTATGCTTC (miR-254*).
Quantitative Northerns
miRNA standards (purchased from Dharmacon) were diluted to appropriate concentrations in the presence of 1.0 μg/μL carrier RNA in the form of either E. coli tRNA or HeLa cell total RNA. Northern analysis was performed (Lau et al. 2001), loading 30 μg of RNA per lane, in the format shown for miR-66 (Fig. 5A). Signals were quantitated using phosphor imaging, standard curves (linear through at least three orders of magnitude, including the region of interpolation) were constructed, and absolute amounts of miRNAs per sample were determined, as illustrated for miR-66 (Fig. 5B). The average number of miRNA molecules per glp-4 adult nematode was calculated using 19 ng as the average amount of total RNA extracted per worm. This number was determined as the average of three independent extraction trials, from known numbers of synchronized, 2-day-old adult glp-4(bn2) hermaphrodites, the same frozen worm population used for the quantitative Northern blots. All extractions were performed as described previously (Lau et al. 2001), except during two of the trials a radiolabeled miRNA was spiked into the preparation during worm lysis. At least 90% of this RNA was recovered, indicating near quantitative yield. Having calculated the number of each miRNA per worm, the average number of miRNAs per cell was calculated using 989 as number of cells per worm. The 989 cells per worm is based on the 959 somatic nuclei of the adult hermaphrodites plus the 30 germ nuclei of 2-day-old adult glp-4(bn2) animals (Sulston et al. 1983; Beanan and Strome 1992). Total RNA from known numbers of HeLa cells was determined in an analogous fashion.
Acknowledgments
We thank the C. briggsae Sequencing Consortium for the availability of sequencing reads, WormBase (http://www.wormbase.org) for annotation of the C. elegans genome, Compaq for computer resources, V. Ambros for communicating unpublished data, C. Mello for the dcr-1 strain, S. Griffiths-Jones and the miRNA Gene Registry for assistance with gene names, P. Zamore for helpful comments on this manuscript, and R.F. Yeh, H. Houbaviy, and G. Ruvkun for advice and helpful discussions. Supported by grants from the NIH and the David H. Koch Cancer Research Fund (D.P.B) and a grant from the NIH (C.B.B.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
Corresponding authors.
Article published online ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.1074403.
References
- Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy S, Griffiths-Jones S, Matzke M, et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Naumova NM, Tulin AA, Rozovsky YM, Gvozdev VA. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in D. melanogaster germline. Curr Biol. 2001;11:1017–1027. doi: 10.1016/s0960-9822(01)00299-8. [DOI] [PubMed] [Google Scholar]
- Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol. 2001;11:941–950. doi: 10.1016/s0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- Beanan MJ, Strome S. Characterization of a germ-line proliferation mutation in C. elegans. Development. 1992;116:755–766. doi: 10.1242/dev.116.3.755. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:295–296. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- Broverman SA, Meneely PM. Meiotic mutants that cause a polar decrease in recombination on the X chromosome in Caenorhabditis elegans. Genetics. 1994;136:119–127. doi: 10.1093/genetics/136.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge CB, Karlin S. Finding the genes in genomic DNA. Curr Opin Struct Biol. 1998;8:346–354. doi: 10.1016/s0959-440x(98)80069-9. [DOI] [PubMed] [Google Scholar]
- Caudy AA, Myers M, Hannon GJ, Hammond SM. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes & Dev. 2002;16:2491–2496. doi: 10.1101/gad.1025202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Chervitz SA, Aravind L, Sherlock G, Ball CA, Koonin EV, Dwight SS, Harris MA, Dolinski K, Mohr S, Smith T, et al. Comparison of the complete protein sets of worm and yeast: Orthology and divergence. Science. 1998;282:2022–2028. doi: 10.1126/science.282.5396.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng A, Shi H, Tschudi C, Ullu E. RNA interference in Trypanosoma brucei: Cloning of small interfering RNAs provides evidence for retroposon-derived 24–26-nucleotide RNAs. RNA. 2001;7:1522–1530. [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes & Dev. 2001a;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001b;20:6877–6888. doi: 10.1093/emboj/20.23.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, Mello CC. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell. 2001;106:23–34. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- Ha I, Wightman B, Ruvkun G. A bulged lin-4/lin-14 RNA duplex is sufficient for Caenorhabditis elegans lin-14 temporal gradient formation. Genes & Dev. 1996;10:3041–3050. doi: 10.1101/gad.10.23.3041. [DOI] [PubMed] [Google Scholar]
- Hall IM, Shankaranarayana GD, Noma K, Ayoub N, Cohen A, Grewal SI. Establishment and maintenance of a heterochromatin domain. Science. 2002;297:2232–2237. doi: 10.1126/science.1076466. [DOI] [PubMed] [Google Scholar]
- Hamilton AJ, Baulcombe DC. A novel species of small antisense RNA in posttranscriptional gene silencing. Science. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Voinnet O, Chappell L, Baulcombe D. Two classes of short interfering RNA in RNA silencing. EMBO J. 2002;21:4671–4679. doi: 10.1093/emboj/cdf464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SC, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates posttranscriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- Hastie ND, Bishop JO. The expression of three abundance classes of messenger RNA in mouse tissues. Cell. 1976;9:761–774. doi: 10.1016/0092-8674(76)90139-2. [DOI] [PubMed] [Google Scholar]
- Hofacker IL, Stadler PF. Automatic detection of conserved base pairing patterns in RNA virus genomes. Comput Chem. 1999;15:401–414. doi: 10.1016/s0097-8485(99)00013-3. [DOI] [PubMed] [Google Scholar]
- Hofacker IL, Fontana W, Stadler PF, Bonhoeffer S, Tacker M, Schuster P. Fast folding and comparison of RNA secondary structures. Monatshefte f Chemie. 1994;125:167–188. [Google Scholar]
- Hofacker IL, Fekete M, Flamm C, Huynen MA, Rauscher S, Stolorz PE, Stadler PF. Automatic detection of conserved RNA structure elements in complete RNA virus genomes. Nucleic Acids Res. 1998;26:3825–3836. doi: 10.1093/nar/26.16.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvágner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002;297:2056–2060. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- Hutvágner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- Ishizuka A, Siomi MC, Siomi H. A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes & Dev. 2002;16:2497–2508. doi: 10.1101/gad.1022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketting RF, Fischer SEJ, Bernstein E, Sijen T, Hannon GJ, Plasterk RHA. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes & Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klahre U, Crete P, Leuenberger SA, Iglesias VA, Meins F. High molecular weight RNAs and small interfering RNAs induce systemic posttranscriptional gene silencing in plants. Proc Natl Acad Sci. 2002;99:11981–11986. doi: 10.1073/pnas.182204199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T. New microRNAs from mouse and human. RNA. 2003;9:175–179. doi: 10.1261/rna.2146903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC. MicroRNAs are complementary to 3′UTR motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30:363–364. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, Fitz Hugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP. Vertebrate microRNA genes. Science. 2003;299:1540. doi: 10.1126/science.1080372. [DOI] [PubMed] [Google Scholar]
- Llave C, Kasschau KD, Rector MA, Carrington JC. Endogenous and silencing-associated small RNAs in plants. Plant Cell. 2002a;14:1605–1619. doi: 10.1105/tpc.003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llave C, Xie Z, Kasschau KD, Carrington JC. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002b;297:2053–2056. doi: 10.1126/science.1076311. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— A computational screen for methylation guide snoRNAs in yeast. Science. 1999;283:1168–1171. doi: 10.1126/science.283.5405.1168. [DOI] [PubMed] [Google Scholar]
- Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T. Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell. 2002;110:563–574. doi: 10.1016/s0092-8674(02)00908-x. [DOI] [PubMed] [Google Scholar]
- McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- Mochizuki K, Fine NA, Fujisawa T, Gorovsky MA. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in Tetrahymena. Cell. 2002;110:689–699. doi: 10.1016/s0092-8674(02)00909-1. [DOI] [PubMed] [Google Scholar]
- Moss EG, Lee RC, Ambros V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell. 1997;88:637–646. doi: 10.1016/s0092-8674(00)81906-6. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G. miRNPs: A novel class of ribonucleoproteins containing numerous microRNAs. Genes & Dev. 2002;16:720–728. doi: 10.1101/gad.974702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykäken A, Haley B, Zamore PD. ATP requirements and small interfering RNA structure in the RNA interference pathway. Cell. 2001;107:309–321. doi: 10.1016/s0092-8674(01)00547-5. [DOI] [PubMed] [Google Scholar]
- Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- Olsen PH, Ambros V. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol. 1999;216:671–680. doi: 10.1006/dbio.1999.9523. [DOI] [PubMed] [Google Scholar]
- Park W, Li J, Song R, Messing J, Chen X. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol. 2002;12:1484–1495. doi: 10.1016/s0960-9822(02)01017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S, Fleenor J, Xu S, Mello C, Fire A. Functional anatomy of a dsRNA trigger: Differential requirement for the two trigger strands in RNA interference. Mol Cell. 2000;6:1077–1087. doi: 10.1016/s1097-2765(00)00106-4. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda M, Maller B, Srinivasan A, Fishman M, Hayward D, Ball E, et al. Conservation across animal phylogeny of the sequence and temporal regulation of the 21 nucleotide let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- Pickford AS, Catalanotto C, Cogoni C, Macino G. Quelling in Neurospora crassa. Adv Genet. 2002;46:277–303. doi: 10.1016/s0065-2660(02)46010-5. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Bartel DP. Small RNAs correspond to centromere heterochromatic repeats. Science. 2002;297:1831. doi: 10.1126/science.1077183. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Bettinger JC, Pasquinelli AE, Rougvie AE, Horvitz HR, Ruvkun G. The 21 nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP. MicroRNAs in plants. Genes & Dev. 2002;16:1616–1626. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110:513–520. doi: 10.1016/s0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- Rivas E, Klein RJ, Jones TA, Eddy SR. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr Biol. 2001;11:1369–1373. doi: 10.1016/s0960-9822(01)00401-8. [DOI] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Haley B, Zamore PD. Evidence that siRNAs function as guides, not primers, in the Drosophila and human RNAi pathways. Mol Cell. 2002;10:537–548. doi: 10.1016/s1097-2765(02)00651-2. [DOI] [PubMed] [Google Scholar]
- Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell. 2000;5:659–669. doi: 10.1016/s1097-2765(00)80245-2. [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Nakano S, Katoh M, Matsumura A, Nakamuta H, Ohmichi T, Yoneyama M, Sasaki M. Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry. 1995;34:11211–11216. doi: 10.1021/bi00035a029. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Tang G, Reinhart BJ, Bartel DP, Zamore PD. A biochemical framework for RNA silencing in plants. Genes & Dev. 2003;17:49–63. doi: 10.1101/gad.1048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance V, Vaucheret H. RNA silencing in plants: Defense and counterdefense. Science. 2001;292:2277–2280. doi: 10.1126/science.1061334. [DOI] [PubMed] [Google Scholar]
- Volpe T, Kidner C, Hall I, Teng G, Grewal S, Martienssen R. Heterochromatic silencing and histone H3 lysine 9 methylation are regulated by RNA interference. Science. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. Identification of novel small RNAs using comparative genomics and microarrays. Genes & Dev. 2001;15:1637–1651. doi: 10.1101/gad.901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: Double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- Zilberman D, Cao X, Jacobsen SE. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science. 2003;299:716–719. doi: 10.1126/science.1079695. [DOI] [PubMed] [Google Scholar]
- Zuker M. Prediction of RNA secondary structure by energy minimization. Methods Mol Biol. 1994;25:267–294. doi: 10.1385/0-89603-276-0:267. [DOI] [PubMed] [Google Scholar]