

Abstract
We have investigated a role for Escherichia coli DNA polymerase II (Pol II) in copying chromosomal and episomal DNA in dividing cells in vivo. Forward mutation frequencies and rates were measured at two chromosomal loci, rpoB and gyrA, and base substitution and frameshift mutation frequencies were measured on an F′(lacZ) episome. To amplify any differences in polymerase error rates, methyl-directed mismatch repair was inactivated. When wild-type Pol II (polB+) was replaced on the chromosome by a proofreading-defective Pol II exo− (polBex1), there was a significant increase in mutation frequencies to rifampicin resistance (RifR) (rpoB) and nalidixic acid resistance (NalR) (gyrA). This increased mutagenesis occurred in the presence of an antimutator allele of E. coli DNA polymerase III (Pol III) (dnaE915), but not in the presence of wild-type Pol III (dnaE+), suggesting that Pol II can compete effectively with DnaE915 but not with DnaE+. Sequencing the RifR mutants revealed a G → A hot spot highly specific to Pol II exo−. Pol II exo− caused a significant increase in the frequency of base substitution and frameshift mutations on F′ episomes, even in dnaE+ cells, suggesting that Pol II is able to compete with Pol III for DNA synthesis on F episomes.
DNA polymerase II (Pol II) from Escherichia coli was discovered 26 years ago (1), yet its primary function in the cell remains an enigma. Pol II is induced as part of the SOS regulon under control of the LexA repressor (2), and it has been identified as the product of the DNA damage-inducible dinA gene (3, 4). Pol II may be involved in repair of DNA damaged by UV radiation (5) or by oxidation (6), and it has been suggested that Pol II is needed to catalyze bypass of abasic lesions if heat shock genes cannot be induced (7).
Pol II synthesizes DNA with high processivity in vitro in the presence of the E. coli DNA polymerase III (Pol III) accessory proteins, the β sliding clamp and the clamp loading γ complex (8–10). These accessory proteins are known to play a central role during DNA synthesis as part of the Pol III holoenzyme complex (11). The potential for sharing the βγ complex with Pol III implies that Pol II has an important, although not essential, role to play during DNA synthesis in vivo. We have recently shown that Pol II is directly involved in DNA synthesis on an F′ episome in nondividing E. coli (12). In this paper, we provide genetic data demonstrating that Pol II is involved in DNA synthesis on the chromosome and on the F′ episome in actively dividing cells.
MATERIALS AND METHODS
Bacterial Strains, Plasmids, and Media.
Only relevant markers are listed. NR11555 (ara, leu::Tn10, Δprolac), NR9918 (zae502::Tn10, zae::Tn10dCam, mutL::Tn5), NR9915 (as NR9918, but dnaE915), HC203 (polBex1), and SH2101 (polBΔ1) have been described (6, 12). KA796 (ara, Δprolac) and NR9901 (dnaE911) were kindly provided by R. M. Schaaper (National Institute on Environmental Health Sciences).
The strains carrying episomes with lacZ frameshift and base substitution mutations, CC101–CC111 (13, 14) and their F′ parent, P90C [ara, Δ(lacproB)XIII], were obtained from J. Miller (University of California, Los Angeles). The scavenger strain, FC755, is P90C carrying F′ [ΔlacIZ, pro+]. Mutant alleles were placed into the desired strains by transduction with P1 bacteriophage. The dnaE915, dnaE911, and dnaE+ markers, flanked by the zae502::Tn10 and zea::Tn10dCam markers, were transduced by selecting for tetracycline resistance and screening for chloramphenicol resistance. mutL::Tn5 and mutH::Tn5 were transduced by selecting for kanamycin resistance. The episomes bearing the lacZ frameshift and base substitution mutations were mated into the relevant strains last. M9 minimal medium has been described (15, 16). In addition, M9 lactose plates were supplemented with 5 ml/liter of Luria–Bertani (LB) to allow for approximately three cell divisions before selection of lactose utilization. We also used M9 lactose plates that were not supplemented with LB. In the latter case, tiny colonies corresponding to Lac+ revertants were barely detectable after 24 h. For the latter case, colonies were counted after 48-h incubations. Antibiotic concentrations were as follows: kanamycin, 30 μg/ml; tetracycline, 10 μg/ml; chloramphenicol, 17 μg/ml; rifampicin, 100 μg/ml; nalidixic acid, 40 μg/ml, and streptomycin, 10 μg/ml. Genetic and molecular procedures were standard (16).
Episomal Lac− → Lac+ Reversions.
Several independent isolates were tested for all episomal lacZ reversions. Cells were grown to saturation in M9 glucose medium, centrifuged, and resuspended in the same volume of 0.85% NaCl/0.001% gelatin. To titer the cell number, dilutions were plated in duplicate on LB and M9 glucose media. The cell suspensions were mixed with 109 scavenger cells (FC755) and plated on M9 lactose plates. Revertants were counted at 24 h for LB-supplemented plates and at 48 h for nonsupplemented plates. The data presented in Table 1 are for plates containing LB; however, the reversion frequencies were similar in the presence or absence of LB (data not shown). Reversion frequencies are the mean number of Lac+ revertants divided by the number of cells plated.
Table 1.
Spontaneous mutant frequencies in cells with various mutant polymerases
| Genotype* | Spontaneous mutants/ 106 cells†
|
|||
|---|---|---|---|---|
| RifR‡ | NalR§ | |||
| polB+dnaE+ |
|
|
||
| polBΔ1 dnaE+ |
|
|
||
| polB+dnaE915 |
|
|
||
| polBΔ1 dnaE915 |
|
|
||
| polB+dnaE+ |
|
ND | ||
| polBex1 dnaE+ |
|
ND | ||
| polB+dnaE911 |
|
ND | ||
| polBex1 dnaE911 |
|
ND | ||
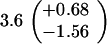
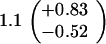
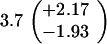
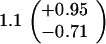
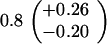
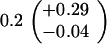
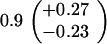
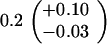
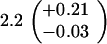
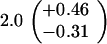
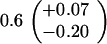
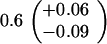
ND, not determined.
Genotype corresponds to isogenic parental strains for top and bottom half of table, respectively.
Medians of 15 independent cultures from 2–3 isolates each, grown in rich medium (top half of table) and minimal medium (bottom half of table). Errors represent 95% confidence intervals for the medians.
Mutations from rifampicin sensitivity to rifampicin resistance.
Mutations from nalidixic acid sensitivity to nalidixic acid resistance.
Chromosomal Mutation Frequencies.
Single colonies of the relevant strains were inoculated into 5 ml of LB containing tetracycline and grown overnight. Typically, for each strain 12–15 single colonies from 2–3 isolates were tested.
An isolate refers to an independent transductant carrying dnaE915 or dnaE+ and either polB+ or polBex1. Aliquots (0.1 ml) of each culture were plated on LB agar containing rifampicin or nalidixic acid to determine the number of mutants, and dilutions were plated on LB agar containing tetracycline and chloramphenicol to determine the total cell number. Plates were incubated at 37°C overnight and colonies counted at 24 h. Mutation frequencies are the median number of mutations divided by the average cell titer (17).
Chromosomal Mutation Rates to Rifampicin Resistance.
Overnight cultures were diluted 107-fold in LB and 0.2 ml of the dilutions inoculated into 96-well microtiter dishes and grown at 37°C overnight with shaking (18, 19). The low initial cell number increased the probability of starting without mutants. Dilutions from five to six randomly picked wells were plated on LB agar to determine the cell titer. The entire contents of 24–48 wells were each plated on LB agar containing rifampicin to determine the number of mutants. Colonies were counted 24 h after plating. Mutation rates were calculated using the maximum likelihood method (20, 21).
Sequencing of Rifampicin Resistant (RifR) Mutants.
Independent cultures were grown and plated as described above. To ensure that the same original clone was not sequenced repeatedly, only one RifR colony was picked at random from each plate for sequencing. Each colony was inoculated into 1 ml of LB and grown to saturation at 37°C. Genomic DNA was prepared using standard phenol/chloroform extractions followed by ethanol precipitation. An ≈200-bp section of the rpoB gene, including the two Rif clusters (22, 23) of each mutant, was amplified using PCR. The PCR primer sequences were GM175 (5′-GCCAAGCCGATTTCCGCAGCAGTGAAAGAG-3′), and GM176 (5′-GTATTCGTTAGTCTGTGCGTACACGGACAG-3′). The PCR-amplified DNA was purified using the Wizard PCR-Preps kit, and sequenced using the F-Mol Cycle Sequencing kit (Promega).
Oligonucleotide Probing of rpoB Mutations.
One hundred and fifty independent RifR mutants in each genetic background were further analyzed by oligonucleotide hybridization (12, 24) for the G → A mutation at position 1583 (R529H) and the A → G mutation at position 1544 (D516G). The oligonucleotides were complimentary to the mutant, MU19 (5′-GCGGAGATATGACGTTTGT-3′), and wild type, WT19 (5′-GCGGAGATACGACGTTTGT-3′), sequence at position 1583 and the mutant, SRWT (5′-GTTTATGGGCCAGAACAAC-3′), and wild type, SRMU (5′GTTTATGGACCAGAACAAC3′), sequence at position 1544. The 32P-labeled oligomers were hybridized in situ to genomic DNA immobilized in a grid pattern on filter paper (24). Three filters, each containing DNA obtained from 50 independently isolated RifR mutants were used for each strain. The probes hybridized with high selectivity to their complementary sequences. The location of the 32P-labeled spots corresponding to a positive hybridization signal was determined by phosphorimaging.
RESULTS
To investigate the involvement of Pol II in DNA synthesis in dividing E. coli, three genetic loci were examined, two on the E. coli chromosome and the other on an F′(lac) episome. The ability of Pol II to carry out chromosomal synthesis was assayed by measuring forward mutation rates to RifR and nalidixic acid resistance (NalR) in strains with different combinations of mutant and wild-type alleles of polB (Pol II) and dnaE (Pol III α subunit). To detect the ability of Pol II to copy F′(lac), we measured changes in reversion frequencies of lacZ base substitution and frameshift mutations in different Pol II and Pol III backgrounds.
Exonuclease-Deficient DNA Pol II Increases Spontaneous Mutation to RifR and NalR in a dnaE915 Antimutator Background.
Polymerase errors are efficiently corrected by postreplication methyl-directed mismatch repair (25). To observe differences in the intrinsic error rates of mutant polymerases, we inactivated mismatch repair by using a strain that was mutL::Tn5. In addition, an antimutator allele of dnaE, dnaE915 (17), was used to minimize the contribution that Pol III makes to mutation frequencies. In this background, a chromosomal replacement of wild-type Pol II (polB+) with a proofreading-defective Pol II derivative (polBex1) resulted in a significant increase in spontaneous mutation frequencies to RifR and NalR of 7.8-fold and 9.3-fold, respectively (Fig. 1). Similar results were obtained in cells grown in minimal medium, and if the cells were mutH::Tn5 instead of mutL::Tn5 (data not shown). We also observed that Pol II exo− stimulated the mutation frequency to RifR 3- to 4-fold in mismatch repair-proficient strains (data not shown).
Figure 1.

DNA Pol II spontaneous mutant frequencies in wild-type and antimutator Pol III genetic backgrounds. All strains were mutL::Tn5. We measured spontaneous mutant frequencies to rifampicin and nalidixic acid resistance at the rpoB and gyrA loci, respectively, in various polB and dnaE backgrounds. The data represent an average of the medians from three experiments, each with 10–20 independent cultures grown from two to three isolates. Error bars indicate ±95% confidence intervals for the medians.
These data suggest that Pol II is able to copy chromosomal DNA in dividing cells in the presence of the DnaE915 antimutator DNA Pol III. We performed a fluctuation analysis (see Materials and Methods) that provided additional evidence that exonuclease-deficient Pol II strongly enhanced RifS → RifR mutation rates in the dnaE915 background (data not shown).
In the presence of wild-type Pol III, mutation frequencies to RifR and NalR appeared to be slightly higher in polBex1 compared with polB+ cells (Fig. 1). However, in contrast to the clear effect in the dnaE915 background, these differences in mutation frequencies are too small to demonstrate that Pol II is carrying out chromosomal DNA synthesis in the presence of wild-type Pol III. The ability of Pol II exo− to increase the frequency of chromosomal mutations also depended on the specific Pol III antimutator allele used.
There was no detectable increase in RifR mutation frequency in the presence of the dnaE911 (17) Pol III antimutator allele (Table 1), in contrast to the 7.8-fold increase in the frequency of RifR mutants in the dnaE915 background (Fig. 1). A possible explanation for this result is that Pol II can gain access to the chromosome much more easily in the presence of the DnaE915 protein because this mutant polymerase binds poorly to primer–template DNA compared with either wild-type Pol III or DnaE911 Pol III (ref. 18; Roel Schaaper, personal communication). In contrast to data showing a 3-fold increased rate of adaptive mutation on an F′ episome in the absence of Pol II in nondividing cells (12), elimination of Pol II (polBΔ1) had no measurable effect on the frequencies of either RifR or NalR mutations in dnaE+ or dnaE915 backgrounds (Table 1).
Spontaneous Mutational Spectra of Proofreading-Defective and Wild-Type Pol II.
Based on the 7.8-fold increase in the frequency of RifR mutations in the presence of Pol II exo− (Fig. 1), we asked if there were differences between polB+ and polBex1 strains in types or sites of occurrence of mutations giving the RifR phenotype. Seventy-two and 80 randomly isolated RifR mutants (see Material and Methods) from dnaE915 polB+ and dnaE915 polBex1 cells, respectively, were directly sequenced. Only two clusters within the rpoB gene need to be sequenced to obtain almost all of the single base substitutions that have been observed to give the RifR phenotype (22). However, because of the cell’s absolute requirement for the β-subunit of RNA polymerase, encoded by rpoB, other important classes of mutations, such as frameshifts, are generally not detected in this analysis, [although, one 3-base deletion has been observed in vivo (23)].
In contrast to the polB+ strain, which had a single mutational hot spot, a G → A transition (D516N) at position 1543 (Fig. 2 Upper), polBex1 cells exhibited two major hot spots, the G → A transition at position 1543 and another G → A transition (R529H) at position 1583 (Fig. 2 Lower). The difference at position 1583 is highly significant; 30% (24/80) of the total number of mutants in the polBex1 strain were at this site compared with none (0/72) in the polB+ strain. Position 1543, the largest hot spot, accounted for 63.9% (46/72) of the mutants in polB+ cells but 26% (21/80) of the mutants in polBex1 cells. About 7% (6/80) of the mutations in polBex1 cells were A → T transversions at position 1711 (I572F) compared with none for polB+ (Fig. 2), but this difference in the two spectra may not be significant.
Figure 2.

Forward mutational spectra in the rpoB locus for wild-type and proofreading-defective polB alleles in a dnaE915 antimutator background. (Upper) polB+ mutational spectrum. (Lower) polBex1 mutational spectrum. All strains were mutL::Tn5.
A potentially important difference in the polB+ and polBex1 spectra occurs at position 1544, immediately adjacent to the major hot spot site (Fig. 2). For polB+, we observed A → T transversions (D516V) only, but for polBex1, we found both A → T transversions and A → G transitions (D516G). Although clearly not statistically significant, this result was of interest because while an A → T transversion had been reported at this site by C. Gross and coworkers in a wild-type DNA Pol III background, an A → G transition had not (23).
Pol II-Dependent RifR Mutants Determined by Filter Hybridization.
A filter hybridization method (24) was used to screen 150 RifR mutants from six different genetic backgrounds for G → A transitions at position 1583 and A → G transitions at position 1544. We compared polB+, polBΔ1 and polBex1 strains that were also dnaE915 and dnaE+; all of the strains were in a mutL::Tn5 background.
First we determined the fraction of RifR mutants that had a G → A transition at position 1583. In the dnaE+ background, the fraction was 7% for polB+ and polBex1 cells and 4% for the polBΔ1 cells. In the dnaE915 background, the fraction was 9% for polB+, 4% for polBΔ1, and 32% for polBex1 cells. Therefore, the only strain exhibiting a significant difference in G → A transition mutants at position 1583 was the proofreading-defective polBex1 strain. This result demonstrates that Pol II exo− polymerase copies chromosomal DNA in the presence of DnaE915 antimutator Pol III (Figs. 1 and 2).
Based on the in situ hybridization data, roughly 7 of the 72 RifR mutants sequenced in the polB+ dnaE915 cells should have been G → A transitions at position 1583 (Fig. 2 Upper). It is surprising, therefore, that there were none. We view this as a statistical anomaly because we verified by sequencing that G → A transitions had indeed occurred at position 1583 for each of the RifR mutants identified by in situ hybridization.
In situ hybridization also revealed that A → G mutations occurred at position 1544 at roughly similar frequencies for polB+ and polBex1 alleles in the presence of wild-type Pol III. Therefore, these data cannot be used either to demonstrate or to exclude the possibility that Pol II may be active in copying chromosomal DNA in the presence of wild-type Pol III. The reason that no A → G mutations were detected at position 1544 in the experiments of C. Gross and coworkers (22, 23) may be because these mutations are particularly well corrected by methyl-directed mismatch repair.
Episomal Lac− → Lac+ Mutations Induced by DNA Pol II in Dividing Cells.
We measured the effect of Pol II on base substitution and frameshift mutations using a series of F′(lac) episomes that carry mutant lacZ alleles, each of which reverts to Lac+ phenotype by a specific mutational event (13, 14). These experiments employed genetic backgrounds similar to those used to measure the effect of Pol II on chromosomal Rifr mutations—i.e., polB+ and polBex1 alleles combined with dnaE+ and dnaE915 alleles. The strains were also mutL::Tn5.
Base substitution mutations.
Compared with polB+, the presence of the polBex1 allele resulted in significant increases in the frequency of Lac+ revertants due to G·C → A·T transitions in the presence of both dnaE+ and dnaE915 alleles (4.2-fold and 9.8-fold compared with polB+, respectively) (Table 2). G·C → A·T mutations have been shown to be reduced by dnaE915 (17), as confirmed by the data in Table 2. The results suggest that both Pol II and Pol III are able to copy the same DNA template position—i.e., the G·C → A·T reversion site on F′(lac).
Table 2.
Spontaneous reversion frequencies of episomal lacZ alleles in various polB backgrounds
| Reversion event | Lac+ revertants/108 cells plated*
|
|||
|---|---|---|---|---|
|
dnaE+
|
dnaE915
|
|||
| polBex1 | polB+ | polBex1 | polB+ | |
| Base substitutions | ||||
| G·C → A·T | 64.5 ± 2.1 | 14 ± 2.6 | 22.2 ± 0.3 | 2.8 ± 0.8 |
| G·C → C·G | 0.5 ± 0.1 | 0.02 ± 0.02 | 7 ± 1.2 | 0.2 ± 0.1 |
| G·C → T·A | 1.7 ± 0.3 | 0.6 ± 0.2 | 6.2 ± 0.7 | 0.3 ± 0.1 |
| A·T → T·A | 0.8 ± 0.2 | 0.2 ± 0.1 | 2.1 ± 0.3 | 0.2 ± 0.1 |
| A·T → G·C | 20.3 ± 2.8 | 13.3 ± 1.9 | 2.8 ± 0.3 | 17.0 ± 2.1 |
| A·T → C·G | ND | ND | ND | ND |
| Frameshifts | ||||
| +1 A | 175 ± 6 | 35 ± 3 | 1497 ± 71 | 96 ± 3 |
| −1 A | 663 ± 76 | 564 ± 50 | 1114 ± 68 | 258 ± 19 |
| −1 G | 4196 ± 177 | 2074 ± 305 | 7687 ± 187 | 4428 ± 79 |
| +1 G | 7241 ± 245 | 6952 ± 342 | 3092 ± 96 | 3287 ± 174 |
| −2 (CG) | 638 ± 25 | 846 ± 53 | 530 ± 37 | 605 ± 34 |
All strains were mutL::Tn5. ND, not detected.
Average of five or more independent isolates ± SEM.
Pol II exo− also appeared to increase G·C → T·A and A·T → T·A transversions (Table 2). Because of the low number of Lac+ revertants, the estimation of the mutational frequencies in the polB+ background are unreliable. However, the reversion frequencies in polBex1 cells were consistently elevated. In the dnaE+ background, Pol II exo− appeared to stimulate G·C → T·A mutations about 4-fold, and A·T → T·A mutations about 3-fold (Table 2). In the dnaE915 background, Pol II exo− appeared to stimulate G·C → T·A mutations about 20-fold, and A·T → T·A mutations about 10-fold. (Table 2). We note that Pol II exo−-induced A·T → T·A revertants were reproducibly detected at frequencies of about 2 × 10−8, whereas revertants were barely detectable in the presence of wild-type Pol II (Table 2). Thus, it is quite likely that Pol II exo− is stimulating both G·C → T·A and A·T → T·A transversions in the dnaE915 background.
A similar conclusion can be made for the G·C → C·G transversions. A clear qualitative increase in frequency of Lac+ revertants was found for Pol II exo− in the dnaE915 background (Table 2). The 35-fold increase is a very rough estimate because the frequency of polB+ dnaE915 revertants was too small to obtain a reliable measurement (Table 2). These low frequencies of spontaneous revertants for several of the base substitution markers agrees with previously published data (13).
It was surprising that in dnaE915 cells the presence polBex1 resulted in a 6-fold reduction in the frequency of A·T → G·C mutations compared with polB+ (Table 2). Although we cannot offer a mechanistic explanation for this result, we have previously shown that mutations in the conserved exonuclease domain of Pol II (3, 4) also reduce the specific activity of the polymerase function by about a factor of 3 (26, 27). Perhaps there are alterations in the specificity of nucleotide incorporation accompanying the reduction in polymerase activity, allowing for more accurate insertions in certain sequence contexts. In dnaE+ cells, there was no significant difference between polBex1 and polB+ cells in A·T → G·C mutation frequencies (Table 2).
Frameshift mutations.
A large increase was observed in the frequency of +1 A frameshift mutations in polBex1 cells (Table 2). The increase was 5-fold in dnaE+ cells and 15-fold in the dnaE915 cells (Table 2). The polBex1 also resulted in a smaller (≈2-fold), yet significant, increase in −1 G frameshift mutations in both dnaE and dnaE915 backgrounds (Table 2). It is interesting to note that the +1 A and −1 G frameshifts occurred at higher frequencies in the dnaE915 strains, a result reminiscent of the bacteriophage T4 DNA polymerase antimutator tsL141 which also showed a decreased frequency of base substitutions (28) with a concomitant increased frequency of frameshift mutations (29). A similar observation was made for the dnaE915 and dnaE911 antimutator strains (17). For the remaining frameshift events listed in Table 2, there were no significant differences in the frequency of Lac+ revertants between polB+ and polBex1 alleles in either dnaE+ or dnaE915 cells.
Based on the data in Table 2, we conclude that the proofreading-defective Pol II exo− polymerase causes significant increases in the frequencies of base substitution and frameshift mutations on episomes. In contrast to the increase in chromosomal mutations, which was significant only in the dnaE915 background, the increase in episomal mutations occurred in both wild-type and antimutator Pol III backgrounds.
DISCUSSION
Previous attempts to define a role for DNA Pol II in E. coli have been hampered by the absence of phenotypes associated with polB mutants. It has been established that dinA(polB), the structural gene coding for Pol II, is repressed by LexA as part of the SOS regulon, and that the cellular levels of Pol II are increased by a factor of 7 following induction of the SOS regulon (2–4). In spite of data suggesting that Pol II could be involved in repair of UV-induced damage (5), null mutants of polB do not appear to exhibit an increased sensitivity to UV light (6).
In an effort to demonstrate a role for Pol II in DNA synthesis in vivo, we replaced the wild-type polB gene on the chromosome with a proofreading-defective polB derivative (polBex1) and examined whether there was a concomitant increase in mutagenesis on the E. coli chromosome and on F′ episomes. To maximize detection of Pol II-induced mutations and to ensure that the mutational events were an unbiased sample of replication errors, all of the mutagenesis data were obtained in the absence of methyl-directed mismatch repair. Mutations to RifR and NalR (mutations in the rpoB and gyrA genes), and the reversion of specific mutant alleles of lacZ, were used to measure mutation frequencies on the E. coli chromosome and on F′(lac) episomes, respectively. Each of these loci have been extremely well characterized (13, 14, 22, 23). It was of particular interest to study the effects Pol II exo− on chromosomal and episomal mutations in dividing cells in light of our recent observation that adaptive mutation, occurring on F′(lac) episomes in nondividing E. coli, is significantly enhanced in both polBex1 and polBΔ1 backgrounds (12).
Pol II Replicates Chromosomal DNA in Dividing E. coli.
Replacing wild-type Pol II with a proofreading-deficient Pol II mutant resulted in a 7.8-fold increase in mutation frequencies to RifR and a 9.3-fold increase in mutation frequencies to NalR in a dnaE915 antimutator background (Fig. 1). These increases are strong evidence that Pol II can copy chromosomal DNA in dividing cells. However, Pol II exo− did not have this effect in either dnaE+ or dnaE911 cells. This result suggests that Pol II can compete effectively with the DnaE915 antimutator DNA polymerase, but not nearly as well with either the wild-type or the DnaE911 polymerases.
Schaaper and coworkers (ref. 30; Roel Schaaper, personal communication) suggested that the DnaE915 polymerase is an antimutator because it has difficulty extending from a primer with a mismatch at the 3′ terminus. Thus, in comparison to DnaE+, DnaE915 may dissociate much more readily from a mismatched terminus, and, when the same or another polymerase rebinds to the terminus, DnaE915 may be more likely to excise the mismatch rather than extend from it. In contrast to DnaE915, the DnaE911 antimutator polymerase may bind much more avidly to primer 3′ termini. The high fidelity of DnaE911 is presumed to be due to an enhanced proofreading-to-polymerase ratio compared with wild-type Pol III, which would favor excision of rather than extension from a mismatch at the 3′ terminus (31–33). If these suppositions are valid, then Pol II may be able to compete more successfully for primer 3′ termini against DnaE915 than against either DnaE911 or DnaE+.
A comparison of the spectra of RifR mutations in polBex1 and polB+ backgrounds revealed a mutational hot spot at position 1583 that is unique to the proofreading-defective polymerase (Fig. 2 Lower). As the overall frequency of RifR mutants was 3-fold higher in polBex1 than in polB+ cells (based on the colony hybridization data), the G → A transitions at position 1583, which represent about 30% of all the RifR mutants in polBex1 dnaE915 cells, must occur at a greatly enhanced frequency in the presence of Pol II exo− (Fig. 2). It is important to note that because RNA polymerase is absolutely required for viability, the RifR mutational spectra consists only of base substitutions. In an in vitro gap-filling assay, however, Pol II exo− catalyzes a variety of simple and complex deletions (27). These mutational events would not be scored in the RifR screening assay and, therefore, the RifR spectrum may not reflect the most common mutational events produced by the proofreading-defective Pol II.
Pol II Copies F′ Episomal DNA in Dividing E. coli.
Replacement of polB+ with polBex1 on the chromosome resulted in significant increases in the spontaneous reversion frequencies of several different lacZ alleles carried on F′ episomes (Table 2). In contrast to chromosomal mutations, the increase in episomal mutations occurred in the presence of either the wild-type Pol III or the DnaE915 polymerase. The frequencies of four of the six base substitutions (G·C → A·T, G·C → C·G, G·C → T·A, and A·T → T·A) and three of the five frameshifts (+1 A, −1 A, and −1 G) were increased significantly in the presence of Pol II exo− (Table 2).
Surprisingly, polBex1 had an antimutator phenotype for 1 of the 11 lacZ− alleles. In the dnaE915 background, the A·T → G·C reversion frequency was 6-fold higher in polB+ compared with polBex1 cells (Table 2). This unexpected result might relate to our observation that the mutations introduced in the polB Exo I domain (27), which reduce 3′-exonuclease activity by ≈1000-fold, also cause a 3-fold reduction in the specific activity of nucleotide insertion (26, 27). Thus, the Pol II exo− polymerase may have a decrease in the efficiency of either dGMP·T or dCMP·A misinsertion that over-compensates for the absence of proofreading. Note also that the +1A frameshift reverts at a significantly higher frequency in the dnaE915 compared with dnaE+ background. Schaaper (17) has reported that for dnaE915 and dnaE911, both A·T → G·C and G·C → A·T mutations are reduced in a mutL background, but not transversions and frameshifts. An increase in frameshift mutagenesis in an antimutator background has also been observed in bacteriophage T4, where the temperature-sensitive L141 antimutator DNA polymerase reduces A·T → G·C mutations (28) but increases frameshift mutations (29).
Relationship of Pol II to the DNA Polymerase from the Archaeon Methanococcus jannaschii.
E. coli Pol II is a group B polymerase sharing five domains with eucaryotic Pol α and bacteriophage T4 Pol (3, 4). The entire genome from the Archaeon M. jannaschii has been recently sequenced (34) and found to contain a single DNA polymerase gene which conforms to group B polymerases (35, 36). A comparison of the M. jannaschii and E. coli Pol II shows that domains IV, II, III, I, and V (36) exhibit amino acid identities of 40%, 57%, 27%, 45%, and 86%, respectively; allowing for conservative amino acid substitutions, the identities are 53%, 71%, 39%, 64%, and 86%, respectively. The signature sequences for the motifs A (domain II), B (domain III), and C (domain I) (37) are also highly conserved. Because there may be just one DNA polymerase in M. jannaschii, this group B enzyme may be able to catalyze both replicative and repair DNA synthesis.
E. coli has three DNA polymerases, and specific roles for Pol II have proven difficult to define. The observation that both Pol II and Pol III are converted from relatively nonprocessive to highly processive polymerases in the presence of the β clamp (8–10), the replication accessory protein that confers processive DNA synthesis, suggests that Pol II might share overlapping functions with Pol III in vivo. We have shown here that Pol II is involved in both chromosomal and episomal DNA synthesis in dividing cells. These observations, coupled with the finding that Pol II is involved in DNA synthesis in nondividing E. coli (12), provides a strong rationale to intensify efforts to identify precise roles for Pol II in pathways of DNA replication and repair.
Acknowledgments
We are grateful to Dr. Roel M. Schaaper (National Institute on Environmental Health Sciences) for providing dnaE antimutator bacterial strains, to Dr. Thomas A. Cebula (U.S. Food and Drug Administration) for his valuable advice concerning the filter hybridization analysis, and to Dr. Miriam Suskind (University of Southern California) for providing excellent advice on all aspects of this study. This work was supported by National Institutes of Health Grant GM42554, National Science Foundation Grant MCB-9214137, and by an National Institutes of Health–National Institute on Aging Predoctoral Training Grant Fellowship T32 AG00093 to Z.Q.
Footnotes
Abbreviations: Pol II, E. coli DNA polymerase II; Pol III, E. coli DNA polymerase III; LB, Luria–Bertani; RifR, rifampicin resistance; NalR, nalidixic acid resistance.
References
- 1.Knippers R. Nature (London) 1970;228:1050–1053. doi: 10.1038/2281050a0. [DOI] [PubMed] [Google Scholar]
- 2.Bonner C A, Randall S K, Rayssiguier C, Radman M, Eritja R, Kaplan B E, McEntee K, Goodman M F. J Biol Chem. 1988;263:18946–18952. [PubMed] [Google Scholar]
- 3.Bonner C A, Hays S, McEntee K, Goodman M F. Proc Natl Acad Sci USA. 1990;87:7663–7667. doi: 10.1073/pnas.87.19.7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwasaki H, Nakata A, Walker G, Shinagawa H. J Bacteriol. 1990;172:6268–6273. doi: 10.1128/jb.172.11.6268-6273.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masker W, Hanawalt P, Shizuya H. Nature (London) New Biol. 1973;244:242–243. doi: 10.1038/newbio244242a0. [DOI] [PubMed] [Google Scholar]
- 6.Escarcellar M, Hicks J, Gudmundsson G, Trump G, Touati D, Lovett S, Foster P, McEntee K, Goodman M F. J Bacteriol. 1994;176:6221–6228. doi: 10.1128/jb.176.20.6221-6228.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tessman I, Kennedy M A. Genetics. 1993;136:439–448. doi: 10.1093/genetics/136.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wickner S, Hurwitz J. Proc Natl Acad Sci USA. 1974;71:4120–4124. doi: 10.1073/pnas.71.10.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hughes A J, Bryan S K, Chen H, Moses R E, McHenry C S. J Biol Chem. 1991;266:4568–4573. [PubMed] [Google Scholar]
- 10.Bonner C A, Stukenberg P T, Rajagopalan M, Eritja R, O’Donnell M, McEntee K, Echols H, Goodman M F. J Biol Chem. 1992;267:11431–11438. [PubMed] [Google Scholar]
- 11.Maki H, Kornberg A. J Biol Chem. 1988;263:6561–6569. [PubMed] [Google Scholar]
- 12.Foster P L, Gudmundsson G, Trimarchi J M, Cai H, Goodman M F. Proc Natl Acad Sci USA. 1995;92:7951–7955. doi: 10.1073/pnas.92.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cupples C G, Miller J H. Proc Natl Acad Sci USA. 1989;86:5345–5349. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cupples C G, Cabrera M, Cruz C, Miller J H. Genetics. 1990;125:275–280. doi: 10.1093/genetics/125.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cairns J, Foster P L. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller J H. A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. [Google Scholar]
- 17.Schaaper R M. Genetics. 1993;134:1031–1038. doi: 10.1093/genetics/134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fijalkowska I J, Schaaper R M. J Bacteriol. 1995;177:5979–5986. doi: 10.1128/jb.177.20.5979-5986.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schaaper R M. J Biol Chem. 1993;268:23762–23765. [PubMed] [Google Scholar]
- 20.Koziol J A. Mutat Res. 1991;249:275–280. doi: 10.1016/0027-5107(91)90154-g. [DOI] [PubMed] [Google Scholar]
- 21.Stewart F M. Genetics. 1994;137:1139–1146. doi: 10.1093/genetics/137.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin D J, Gross C A. J Mol Biol. 1988;202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 23.Jin D J, Gross C A. J Bacteriol. 1989;171:5229–5231. doi: 10.1128/jb.171.9.5229-5231.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cebula T, Koch W. Mutat Res. 1990;229:79–87. doi: 10.1016/0027-5107(90)90010-2. [DOI] [PubMed] [Google Scholar]
- 25.Modrich P. Annu Rev Biochem. 1987;56:435–466. doi: 10.1146/annurev.bi.56.070187.002251. [DOI] [PubMed] [Google Scholar]
- 26.Cai H, Yu H, McEntee K, Goodman M F. Methods Enzymol. 1995;262:13–22. doi: 10.1016/0076-6879(95)62004-4. [DOI] [PubMed] [Google Scholar]
- 27.Cai H, Yu H, McEntee K, Kunkel T A, Goodman M F. J Biol Chem. 1995;270:15327–15335. doi: 10.1074/jbc.270.25.15327. [DOI] [PubMed] [Google Scholar]
- 28.Drake J W, Allen E F. Cold Spring Harbor Symp Quant Biol. 1968;33:339–344. doi: 10.1101/sqb.1968.033.01.039. [DOI] [PubMed] [Google Scholar]
- 29.Ripley L S, Shoemaker N B. Genetics. 1983;103:353–366. doi: 10.1093/genetics/103.3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fijalkowska I J, Dunn R L, Schaaper R M. Genetics. 1993;134:1023–1030. doi: 10.1093/genetics/134.4.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muzyczka N, Poland R L, Bessman M J. J Biol Chem. 1972;247:7116–7122. [PubMed] [Google Scholar]
- 32.Bessman M J, Muzyczka N, Goodman M F, Schnaar R L. J Mol Biol. 1974;88:409–421. doi: 10.1016/0022-2836(74)90491-4. [DOI] [PubMed] [Google Scholar]
- 33.Clayton L K, Goodman M F, Branscomb E W, Galas D J. J Biol Chem. 1979;254:1902–1912. [PubMed] [Google Scholar]
- 34.Bult C J, White O W, Olsen G J, Zhou L, Fleischmann R D, et al. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 35.Jung G H, Leavitt M C, Hsieh J C, Ito J. Proc Natl Acad Sci USA. 1987;84:8287–8291. doi: 10.1073/pnas.84.23.8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang T S, Wong S W, Korn D. FASEB J. 1989;3:14–21. doi: 10.1096/fasebj.3.1.2642867. [DOI] [PubMed] [Google Scholar]
- 37.Delarue M, Poch O, Tordo N, Moras D, Argos P P. Protein Eng. 1990;3:461–467. doi: 10.1093/protein/3.6.461. [DOI] [PubMed] [Google Scholar]