

Abstract
We investigated here the mechanism of cytoprotection of nitric oxide (•NO) in bovine aortic endothelial cells treated with H2O2. NONOates were used as •NO donors that released •NO slowly at a well defined rate in the extracellular and intracellular milieus. H2O2-mediated intracellular dichlorofluorescein fluorescence and apoptosis were enhanced by the transferrin receptor (TfR)-mediated iron uptake. •NO inhibited the TfR-mediated iron uptake, dichlorofluorescein fluorescence, and apoptosis in H2O2-treated cells. •NO increased the proteasomal activity and degradation of nitrated TfR via ubiquitination. Nω-nitro-l-arginine methyl ester, a nonspecific inhibitor of endogenous •NO biosynthesis, decreased the trypsin-like activity of 26S proteasome. •NO, by activating proteolysis, mitigates TfR-dependent iron uptake, dichlorodihydrofluorescein oxidation, and apoptosis in H2O2-treated bovine aortic endothelial cells. The relevance of biological nitration on redox signaling is discussed.
Although numerous studies have focused on the cellular interaction between nitric oxide (•NO) and superoxide anion (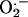 ) (1), relatively little is known about the cellular effects induced by •NO and H2O2 (2–4). These cellular effects include iron homeostasis, oxidative, nitrative, and nitrosative stress, cell proliferation, and apoptosis. The cytoprotective effect of •NO against H2O2-mediated cellular toxicity has been attributed to iron chelation, radical scavenging, and the restoration of mitochondrial respiration (5–9). Recently we reported that H2O2-induced endothelial apoptosis was mediated by the transferrin receptor (TfR)-dependent cellular iron uptake, because blockade of iron uptake by an anti-TfR (IgA class) antibody totally abolished H2O2-induced oxidative stress and apoptosis (10). •NO released from its donor molecules inhibited hydroperoxide-induced apoptosis in endothelial cells (11). Therefore, the antiapoptotic effect of •NO may be related to the regulation of cellular iron signaling.
) (1), relatively little is known about the cellular effects induced by •NO and H2O2 (2–4). These cellular effects include iron homeostasis, oxidative, nitrative, and nitrosative stress, cell proliferation, and apoptosis. The cytoprotective effect of •NO against H2O2-mediated cellular toxicity has been attributed to iron chelation, radical scavenging, and the restoration of mitochondrial respiration (5–9). Recently we reported that H2O2-induced endothelial apoptosis was mediated by the transferrin receptor (TfR)-dependent cellular iron uptake, because blockade of iron uptake by an anti-TfR (IgA class) antibody totally abolished H2O2-induced oxidative stress and apoptosis (10). •NO released from its donor molecules inhibited hydroperoxide-induced apoptosis in endothelial cells (11). Therefore, the antiapoptotic effect of •NO may be related to the regulation of cellular iron signaling.
Endothelial cells acquire iron through TfRs located on the cell surface (12). Cells exposed to hydroperoxides increased the expression of the TfR mRNA resulting from the induction of a 98-kDa, cytosolic, iron-regulatory protein 1 (IRP-1) (13–15). Under these conditions, synthesis of ferritin, an intracellular iron-storage protein, decreased (12). TfR and ferritin syntheses are regulated by iron at the mRNA translation level by the interaction of cytoplasmic IRPs with their respective mRNAs (16). Nitrosothiols have been shown to activate IRPs through nitrosation of cysteines present in IRPs (17, 18). The objective of this study was to investigate the effect of •NO on TfR-mediated iron signaling and apoptosis in endothelial cells exposed to H2O2.
Cells cope with the accumulation of oxidized, nitrated, and nitrosated proteins through increased proteolysis (19, 20). Cellular proteolytic degradation involves posttranslational ubiquitination of modified proteins (21, 22). This study tested two hypotheses: (i) the cytoprotective effects of •NO are due to stimulation of the ubiquitin (Ub)-proteasome pathway, and (ii) •NO regulates oxidant-induced iron signaling, intracellular iron homeostasis, and oxidative stress through increased proteolytic activation. Results show that •NO, both exogenous and endogenous, enhances the proteasomal activity in endothelial cells, and proteasomal inhibitors abrogate •NO-mediated cytoprotection.
Materials and Methods
Materials. N-carbobenzoxyl-l-leucinyl-l-leucinyl-l-norleucinal (MG-132) and epoxomycin (Biomol, Plymouth Meeting, PA), glucose oxidase (GO), clasto-lactacystin-β-lactone (Lac, Sigma), 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Molecular Probes), and diethylenetriamine NONOate (DETA/NO, Cayman Chemical, Ann Arbor, MI) were used as received. Pyrrolidine nonoate methylene acetate (AcOM-PYRRO/NO) was a gift from Larry K. Keefer (National Cancer Institute-Fredrick Cancer Research and Development Center, Bethesda).
Endothelial Cell Experiments. Bovine aortic endothelial cells (BAECs) were obtained from Clonetics (San Diego) and maintained (37°C, 5% CO2) in DMEM containing 10% FBS, l-glutamine (4 mM), penicillin (100 units/ml), and streptomycin (100 μg/ml). Cells used in this study were between passages 4 and 14. On the day of the treatment, the medium was replaced with DMEM containing 2% FBS, which contains ≈25–30 μg of Tf/ml. GO (20 milliunits/ml) was added to cells in the medium containing 25 mM glucose. H2O2 generated from glucose/GO was measured by using a YSI Model 25 oxidase meter (Yellow Springs Instruments). DETA/NO (100 μM) and other proteasome inhibitors, lactacystin-β-lactone (10 μM), MG-132 (10 μM), and epoxomycin (2 μM), were added 2 h before the addition of GO.
Measurement of Oxidative Stress. After treatment of BAECs with glucose/GO, the medium was aspirated and cells were washed with Dulbecco's PBS (DPBS) and incubated in 2 ml of fresh culture medium without FBS. DCFH-DA was added at a final concentration of 10 μM and incubated for 20 min. Fluorescence was monitored by using a Nikon fluorescence microscope (excitation 488 nm, emission 610 nm) equipped with an FITC filter. Reduced glutathione (GSH) was measured by HPLC as the o-phthalaldehyde adduct. Protein carbonyls were measured as reported (23).
Measurement of 55Fe Uptake. BAECs were grown in DMEM containing 10% FBS until confluence. On the day of treatment the medium was replaced with DMEM containing 2% FBS and the cells were allowed to adjust to the medium conditions. Ferric chloride [0.2 μCi of 55Fe (1 Ci = 37 GBq)] was added to the medium, and its levels were measured after 4 h. Cells were washed with DPBS, lysed with PBS containing 0.1% Triton X-100, and counted in a beta counter.
Electrophoretic Mobility-Shift Assay. Binding of IRP to iron-responsive element (IRE) was measured by electrophoretic mobility-shift assay. 32P-labeled IRE mRNA for the RNA band-shift assay was prepared by using as a template a 1,000-bp rat l-ferritin pseudogene that contains the conserved IRE sequence. The plasmid (p66-L gene) containing this insert [generously provided by Elizabeth Leibold, University of Utah, Salt Lake City (24)] was linearized with SmaI (Invitrogen Life Technologies, Carlsbad, CA) and used for in vitro transcription of IRE mRNA. Transcription was carried out with Sp6 RNA polymerase by using a Promega Riboprobe transcription system.
Proteasome Function Assays. 26S proteasome. Proteasome function was measured as reported (19, 25). Briefly, cells were washed with buffer I (50 mM Tris, pH 7.4/2 mM DTT/5 mM MgCl2/2 mM ATP) and homogenized with buffer I containing 250 mM sucrose. Twenty micrograms of 10,000 × g supernatant were diluted with buffer I to a final volume of 900 μl. The fluorogenic proteasome substrates SucLLVY-AMC (chymotrypsin-like) and Z-Leu-Leu-Lys-AMC (trypsin-like) were added in a final concentration of 80 μM. Proteolytic activity was measured by monitoring the release of the fluorescent group 7-amido-4-methylcoumarin (excitation 380 nm, emission 460 nm).
20S proteasome activity. Activity of the 20S proteasome was determined according to Grune et al. (26). Cells were lysed in PBS containing 0.1% Triton X-100 and 0.5 mM DTT. The assay mixture contained 50 μl of buffer (50 mM Tris·HCl pH 7.8/20 mM KCl/5 mM MgCl2/0.1 mM DTT), 250 μM sLLVY-MCA, and 50 μl of cell lysate (15 μg of protein). After 30 min at 37°C, the reaction was stopped by adding 1 ml of 0.2 M glycine buffer, pH 10, and the fluorescence of the liberated 7-amido-4-methylcoumarin was measured by using excitation and emission wavelengths at 365 and 460 nm, respectively.
Western Analysis. BAECs were washed with ice-cold PBS and resuspended in 100 μl of radioimmunoprecipitation assay buffer (20 mM Tris·HCl, pH 7.4/2.5 mM EDTA/1% Triton X-100/1% sodium deoxycholate/1% SDS/100 mM NaCl/100 mM sodium fluoride) containing 1 mM sodium vanadate and a mixture of protease inhibitors. The lysate was centrifuged at 750 × g for 10 min at 4°C to pellet out the nuclei. The remaining supernatant was centrifuged for 30 min at 12,000 × g. Proteins were resolved on 8% for TfR, nitrated TfR (nitrotyrosine), and protein carbonyls and 12% for total ubiquitinated proteins by SDS/PAGE gels and blotted onto nitrocellulose membranes. Membranes were probed with mouse anti-human TfR monoclonal antibody (Zymed), monoclonal mouse anti-nitrotyrosine antibody (Cayman Chemical), rabbit anti-2,4-dinitrophenol antibody for protein carbonyls (Zymed), mouse anti-cytochrome c antibody (PharMingen), or an affinity-purified polyclonal rabbit anti-Ub antibody and then incubated with either horseradish peroxidase-conjugated rabbit anti-mouse IgG or horseradish peroxidase-conjugated goat anti-rabbit IgG. Protein bands were detected by using the ECL method (Amersham Pharmacia).
Immunoprecipitation of TfR. After treatment, cell lysates (100 μg of protein) were incubated with anti-TfR antibody in a total volume of 500 μl at 4°C overnight, and 30 μl of immobilized protein A was added and incubated further for 2 h. Beads then were washed with DPBS. The immunoprecipitated proteins were boiled for 15 min in Laemmli sample buffer containing 4% of 2-mercaptoethanol and resolved on SDS/8% PAGE. They then were blotted onto nitrocellulose membranes and probed with either a polyclonal anti-Ub antibody or a monoclonal anti-nitrotyrosine antibody to detect Ub-conjugated TfR or nitrated TfR by the ECL method.
Statistical Analysis. Results were analyzed by a one-way analysis of variance (ANOVA), and differences estimated by a Student's t test were considered to be statistically significant at P < 0.05.
Results
Effect of •NO on H2O2-Induced Intracellular Oxidation of DCFH and Apoptosis. BAECs were treated with H2O2 (1 μM/min) generated from glucose/GO. Before the addition of DCFH-DA, cells were washed free of glucose/GO. Intracellular oxidation of the active probe carboxy-DCFH to dichlorofluorescein (DCF), a green fluorescent, two-electron oxidation product, was measured (Fig. 1A). The DCF fluorescence intensity was nearly 1,000-fold greater in BAECs than in the control after a 4-h treatment with glucose/GO (Fig. 1B). H2O2-induced intracellular DCF f luorescence was almost totally abrogated by DETA/NO that slowly released •NO (7.2 nM/min) (Fig. 1 A and B). BAECs treated with H2O2 and DETA/NO were washed extensively before the addition of DCFH-DA. Fig. 1C shows that DETA/NO treatment inhibits the release of cytochrome c from the mitochondria into the cytosol in BAECs treated with glucose/GO for 8 h. After treatment of BAECs with H2O2 for 8 h (compare with Fig. 1 A), the caspase-3 proteolytic activity increased by nearly 5-fold as compared with the control (Fig. 1D). DETA/NO greatly decreased caspase-3 activation from cells treated with H2O2, which is consistent with the interpretation that the cytochrome c release into cytosol is a prerequisite for caspase-3 activation. Apoptosis was further confirmed by using the terminal deoxynucleotidyltransferase-mediated dUTP end-labeling (TUNEL) technique (10, 11). Results showed that incubation with DETA/NO (50–100 μM) substantially decreased the fraction of TUNEL-positive BAECs after treatment with glucose/GO for 8 h (data not shown).
Fig. 1.

Effect of •NO on H2O2-induced oxidation of DCFH, cytochrome c release, and caspase-3 activity in BAECs. (A) BAECs treated with glucose (Glu)/GO for 4 h in the presence and absence of •NO donor DETA/NO (10–100 μM). Cells were washed with DPBS and incubated with DCFH-DA (10 μM) for 20 min. Cells were washed twice with DPBS and kept in a culture medium. Green fluorescence due to DCF was monitored. In one experiment, cells were treated with AcOM-PYRRO/NO (1 μM), an esterase-specific intracellular NO donor. (B) Average fluorescence intensity (n = 3) of photographs obtained in A. (C) BAECs were treated with glucose/GO for 8 h with and without DETA/NO (100 μM), and the release of cytochrome c from the mitochondria into the cytosol was measured by Western blot analysis. (D) BAECs were treated with glucose/GO (20 milliunits) for 8 h with and without DETA/NO (100 μM), and caspase-3 activity was measured. Data aremean ± SD for three experiments.
The addition of DETA/NO did not affect H2O2 released from glucose/GO (data not shown). Additional evidence for the inhibitory effect of •NO on H2O2-dependent oxidative damage was obtained by using an intracellular •NO donor. Because extracellular •NO is predominantly oxidized to 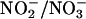 , relatively higher concentrations (50–100 μM) of DETA/NO were required. In contrast, the cell-permeable esterase-specific pro-NO donor AcOM-PYRRO/NO releases •NO intracellularly (27). Thus, lower concentrations (1 μM) of AcOM-PYRRO/NO inhibited H2O2-induced oxidative stress (Fig. 1). These results show that •NO mitigates H2O2-dependent intracellular oxidant formation and apoptosis in endothelial cells.
, relatively higher concentrations (50–100 μM) of DETA/NO were required. In contrast, the cell-permeable esterase-specific pro-NO donor AcOM-PYRRO/NO releases •NO intracellularly (27). Thus, lower concentrations (1 μM) of AcOM-PYRRO/NO inhibited H2O2-induced oxidative stress (Fig. 1). These results show that •NO mitigates H2O2-dependent intracellular oxidant formation and apoptosis in endothelial cells.
Effect of •NO on H2O2-Induced Changes in GSH, Aconitase, and IRP-1 Activities and TfR Expression. Because •NO strongly inhibited H2O2-induced oxidant stress, we assessed the role of •NO on intracellular GSH levels and total aconitase activity. BAECs were treated with glucose/GO for 4 h with and without DETA/NO. Fig. 2A shows that GSH levels were decreased by >50% after a 4-h treatment with H2O2. However, the addition of DETA/NO (50 and 100 μM) increased GSH levels from 2.3 nmol/mg protein to 4 and 5 nmol/mg protein in H2O2-treated cells (Fig. 2A). DETA/NO alone did not affect GSH levels significantly (Fig. 2A). The aconitase activity (28) was measured in cytosol and mitochondria of BAECs treated with glucose/GO in the presence and absence of DETA/NO. •NO markedly inhibited the H2O2-induced inactivation of the mitochondrial but not the cytosolic aconitase activity (Fig. 2 B and C).
Fig. 2.

Effect of •NO on H2O2-induced changes in GSH, aconitase activity, TfR, and IRP-1 levels in BAECs. (A) BAECs were treated with glucose (Glu)/GO (20 milliunits) for 4 h with and without DETA/NO (50 and 100 μM), and GSH levels were measured. (B and C) Cells were treated with glucose/GO (20 milliunits) for 4 h with and without DETA/NO (100 μM), and aconitase activity was measured in the cytosolic (B) and mitochondrial (C) fractions. (D) BAECs were treated with glucose/GO for 4 h with and without DETA/NO, and TfR protein expression was measured by using 20 μg of protein of the 12,000 × g supernatant. (E) BAECs were treated as described for B, and cytoplasmic extracts were analyzed by gel-shift assay with and without 2-mercaptoethanol (2-ME) to measure IRP/IRE binding. (F) Cells were treated as described for D by using 12 μg of protein of the 12,000 × g supernatant except that the effect of lactacystin (10 μM) on TfR protein expression in glucose/GO-treated cells with and without DETA/NO was determined. *, P < 0.05 vs. control.
Previous results indicate that H2O2-dependent oxidation of DCFH is due to an increased expression of TfR levels and TfR-dependent iron uptake (29). Fig. 2D shows that TfR expression significantly increases in BAECs treated with glucose/GO, whereas in the presence of DETA/NO, TfR expression is inhibited. Because TfR synthesis is regulated by IRP binding to IRE and the IRP-1 activity is stimulated by H2O2 (30), we assessed the effect of DETA/NO on IRP-1 activity in BAECs treated with glucose/GO. To determine whether the increase in IRP-1 activity with H2O2 and DETA/NO treatment was due to an increase in total IRP-1 (Fig. 2E), lysates were treated with 1% 2-mercaptoethanol, which activates IRP-1 to the high-affinity RNA-binding form (31). Under these conditions, major differences in IRP-1 binding to IRE were not observed between the control and other treatment groups (Fig. 2E). The lack of inhibition of •NO on IRP-1/IRE binding in H2O2-treated cells is confounding, because •NO markedly decreased the expression of TfR in H2O2-treated BAECs (Fig. 2D). A likely explanation is that the decrease in TfR expression in DETA/NO-H2O2 is due to increased degradation of TfR by the proteasomal system. Therefore, cells were preincubated with Lac, a proteasome-specific protease inhibitor that covalently modifies two β-subunits of the proteasome (32). In the presence of Lac, TfR expression in cells treated with H2O2 and DETA/NO was enhanced (Fig. 2F). The increase in TfR expression by the proteasome inhibitor, Lac, suggests that TfR likely undergoes proteolytic degradation in the presence of •NO and H2O2. These results suggest that •NO may modulate H2O2-mediated iron signaling and oxidant stress by stimulating the proteasomal activity.
Effect of Proteasomal Inhibitors on •NO-Mediated Decrease in Cellular Oxidative Stress. The next step was to address whether proteasomal inhibitors could increase iron uptake and DCFH oxidation in cells treated with H2O2. Endothelial cells were pretreated with various proteasome inhibitors (10 μM Lac, 10 μM MG-132, and 2 μM epoxomycin) (32, 33). Proteasomal inhibitors alone, at the concentrations used, did not induce any significant oxidative stress or apoptosis in BAECs. DCFH oxidation increased in cells pretreated with the proteasomal inhibitors for 2 h and exposed to glucose/GO and DETA/NO (Fig. 3A). However, DCF fluorescence was eliminated completely in cells treated with the anti-TfR antibody (IgA class) that specifically binds to the extracellular domain of TfR and inhibits the receptor endocytosis (Fig. 3A).
Fig. 3.

Effect of proteasomal inhibitors on DCFH oxidation, iron uptake, and apoptosis in BAECs treated with H2O2 and •NO. (A) BAECs were treated with glucose (Glu)/GO with or without DETA/NO (100 μM), proteasome inhibitors, lactacystin (10 μM), epoxomycin (2 μM) and MG-132 (10 μM), and anti-TfR antibody (IgA class, 42/6) (12 μg/ml). Cells were pretreated with the proteasome inhibitors and DETA/NO for 2 h before treatment with H2O2 for 4 h. Cells then were washed in DPBS and incubated with DCFH-DA (10 μM) for 20 min. Green fluorescence due to DCF was monitored with time. (B) 55Fe uptake into cells was monitored under the same conditions as described for A and C. Caspase-3 activity was measured under the same conditions described for A except that cells were treated with H2O2 for 8 h. 1, control; 2, glucose/GO; 3, DETA/NO alone; 4, glucose/GO + DETA/NO; 5, Lac alone; 6, Lac + glucose/GO; 7, Lac + DETA/NO; 8, Lac + glucose/GO + DETA/NO; 9, glucose/GO + AcOM-PYRRO/NO; 10, Lac + glucose/GO + AcOM-PYRRO/NO. (D) Cells were treated as described for B, and 5 μg of the derivatized total protein was resolved on an SDS/8% PAGE gel and probed with a monoclonal anti-2,4-dinitrophenol antibody to measure protein carbonyl levels by Western blot analysis. (E) BAECs were treated as described for A, and the cell lysate (100 μg of protein) was immunoprecipitated with 6 μg of anti-TfR antibody, resolved on an SDS/8% PAGE, and probed for nitrated TfR with anti-nitrotyrosine antibody by Western blot analysis. *, P < 0.05 vs. control.
To probe the involvement of iron, the uptake of labeled iron (55Fe) in the presence of Lac was measured. 55Fe uptake was nearly 2.3-fold greater in H2O2-treated cells than in control cells (Fig. 3B). There was no increase in uptake of 55Fe with DETA/NO (Fig. 3B). This finding is consistent with the decreased TfR levels observed in BAECs treated with H2O2 and DETA/NO. Lac treatment counteracted the effect of DETA/NO, as evidenced by the increase in 55Fe uptake in Lac-treated cells (Fig. 3B). Proteasome inhibition with Lac also reversed the antiapoptotic effect of •NO. Fig. 3C shows that Lac caused an increase in caspase-3 activity in cells treated with H2O2 and •NO. These results suggest that the cytoprotective and antioxidative effect of DETA/NO probably is due to stimulation of the proteasome.
We then surmised that if •NO protects against oxidative stress in H2O2-treated cells through activation of intracellular proteolysis, cellular accumulation of oxidized protein should be inhibited. Protein carbonyl levels were significantly more in glucose/GO-treated cells than in untreated cells (Fig. 3D). However, protein carbonyl levels were decreased drastically in the presence of DETA/NO (Fig. 3D). In the presence of Lac, protein carbonyls were increased (Fig. 3D). Collectively, these results indicate a previously uncharacterized antioxidant role for •NO as a stimulator of proteasomal function.
Recently, an increase in protein nitration was detected in the presence of •NO, H2O2, and iron (34). The decrease in TfR levels coupled with an increase in proteasomal activity in cells treated with DETA/NO and H2O2 (Fig. 2D) prompted us to examine whether TfR is modified through nitration. TfR nitration was assessed by immunoprecipitating TfR and probing it with a monoclonal anti-nitrotyrosine antibody. Nitration of TfR increased in BAECs treated with H2O2 and DETA/NO for 4 h (Fig. 3E). Nitrated TfR levels, however, decreased during prolonged incubation time (>6 h) (data not shown). This probably is due to •NO stimulation of proteasomal activity.
Effect of •NO on 20S and 26S Proteasomal Activities: Degradation of Nitrated TfR by Ubiquitination. Degradation of oxidatively modified or nitrated protein by proteasomes could occur by a Ub-dependent or Ub-independent mechanism (19, 20, 35). Therefore, peptidase activities of the 26S and 20S proteasome in cells treated with DETA/NO alone and in the presence of H2O2 were measured. Treatment of BAECs for 6 h with glucose/GO almost completely nullified both the chymotrypsin-like and trypsin-like activities of the 26S proteasome (Fig. 4 A and B). DETA/NO alone significantly increased the chymotrypsin-like activity in cells. The enzyme activities remained elevated in cells during the combined treatment of H2O2 and DETA/NO (Fig. 4 A and B), suggesting that •NO enhances the proteolytic activities in endothelial cells treated with H2O2. The 26S and 20S proteasomal activities were measured in the presence or absence of Lac and MG-132. Both Lac and MG-132 inhibited •NO-stimulated proteasomal activities (Fig. 4 A and B). These results indicate that •NO activates the proteasomal activity in endothelial cells treated with H2O2.
Fig. 4.

Effect of •NO on proteasomal activity in BAECs: degradation of ubiquitinated proteins. (A and B) BAECs were pretreated with DETA/NO (100 μM) and proteasome inhibitors (10 μM) for 2 h before the addition of glucose (Glu)/GO (20 milliunits) for 6 h. The chymotrypsin-like (A) and trypsin-like (B) activities of 26S proteasome were measured as described in Materials and Methods. (C) Treatment conditions were the same as described for A and B, but the 20S proteasome activity was measured as a function of time from 0 to 6 h by using the fluorogenic peptide, sLLVY-MCA, as the substrate. (D) Total ubiquitination of proteins measured in BAECs treated with glucose/GO for 6 h and resolved on SDS/12% PAGE followed by Western blot analysis with a polyclonal anti-Ub antibody. (E) Densitometric analysis of the data shown in D. (F) BAECs pretreated with DETA/NO and/or proteasome inhibitors for 2 h were treated with glucose/GO for 6 h and immunoprecipitated with anti-TfR antibody, resolved on SDS/8% PAGE, and probed with anti-Ub antibody by Western blot analysis. (G) BAECs were pretreated with l-NAME for 8 h, and the trypsin-like activity of 26S proteasome was measured. *, P < 0.05 vs. control.
The total ubiquitination of cellular proteins and their degradation in the presence of H2O2 and DETA/NO were then investigated. BAECs were treated with glucose/GO with and without DETA/NO for 6 h. Fig. 4D shows that glucose/GO treatment did not alter ubiquitinated proteins compared with control cells, suggesting that ubiquitination per se is not affected by H2O2 when cells were treated for 6 h under these experimental conditions. Ubiquitination of proteins was lowered drastically in cells treated with glucose/GO-DETA/NO (Fig. 4 D and E), which is consistent with an increase in the 26S proteasomal activity, leading to an enhanced rate of degradation of ubiquitinated proteins.
Next we investigated whether TfR degradation depends on the ubiquitination of this protein (Fig. 4F). Cells were treated with glucose/GO for 6 h with and without DETA/NO, and the cell extracts were immunoprecipitated with a monoclonal anti-TfR antibody, resolved on an SDS/8% PAGE, and probed with a polyclonal anti-Ub antibody. Fig. 4F shows that TfR undergoes Ub-dependent degradation, and the ubiquitinated TfR (Ub-TfR) increases in H2O2-treated cells and decreases in cells treated with H2O2 and DETA/NO (Fig. 4F). This is consistent with the enhanced proteasomal activities in the presence of H2O2 and DETA/NO. Finally, we investigated the effects of Nω-nitro-l-arginine methyl ester (l-NAME) and its inactive structural analog (Nω-nitro-d-arginine methyl ester) on the 26S proteasomal activity. l-NAME, a nonspecific inhibitor of NO synthase (NOS) activity, decreased the trypsin-like activity in a dose-dependent manner (Fig. 4G). This suggests that the endogenous •NO generated by the endothelial NOS is essential for maintaining the inherent proteasomal activity.
Discussion
This study found that •NO released slowly from •NO donors inhibits TfR-mediated iron signaling, intracellular DCFH oxidation, and apoptosis in H2O2-treated endothelial cells. •NO enhanced nitration of TfR and proteasomal activity. NOS inhibition decreased the proteasomal activity. We suggest that, by stimulating proteolysis, •NO inhibits TfR-mediated iron uptake, oxidative stress, and apoptotic signaling in H2O2-treated endothelial cells.
Hydrogen Peroxide-Induced Iron Signaling, Oxidant Stress, and Apoptosis Involvement of TfR. Reports indicate that the cellular damage caused by reactive oxygen and nitrogen species is critically controlled by intracellular iron homeostasis (36, 37). Exposure of murine fibroblasts to H2O2 enhances the expression of TfR mRNA and DCF fluorescence, implicating a potential link between oxidative stress and TfR-mediated iron uptake (13, 14). Recently we showed that the exogenous addition of bolus or continuously generated H2O2 to endothelial cells causes an enhanced oxidation of DCFH to DCF that is regulated by TfR-mediated uptake of Tf iron (29). Blockade of iron uptake by anti-TfR antibody (42/6, IgA class) that specifically binds to the extracellular domain of TfR abolishes H2O2-induced DCF fluorescence. This intriguing finding suggests that intracellular oxidation of DCFH is catalyzed by both H2O2 and iron transported through TfR. Here we show that •NO generated from DETA/NO down-regulates TfR expression in H2O2-treated cells and inhibits TfR-mediated iron uptake. Consequently, intracellular DCFH oxidation decreases. In cells treated with Lac, TfR levels were up-regulated, which led to enhanced iron uptake and DCFH oxidation (Fig. 3A).
Our results demonstrate that H2O2-induced caspase activation is accompanied by an increase in TfR-mediated uptake of iron (Fig. 2). •NO released from DETA/NO inhibits H2O2-induced apoptosis and TfR-mediated iron uptake. Thus, TfR acts as an effective “gatekeeper” and modulator of H2O2-induced endothelial apoptosis. •NO mitigates H2O2-induced oxidative stress and endothelial apoptosis by inhibiting iron signaling.
Degradation of Proteins by the Ub-Proteasome Pathway-Modulatory Effect of •NO. The Ub-proteasome pathway plays a major role in the breakdown of oxidatively modified proteins that otherwise would disrupt normal cellular function. The protein targeted for degradation is covalently attached to several molecules of Ub, a 76-aa protein (38). Nitration typically targets the modified protein for ubiquitination and rapid degradation by 26S proteasomes (39). Ubiquitinated protein is subsequently escorted to the 26S proteasome where it undergoes degradation via a pathway involving a series of enzymes known as E1, E2, and E3 (21). The proteolytic core of the 26S complex, the 20S proteasome, contains multiple peptidase activities including the chymotrypsin-like activity (a serine endopeptidase that hydrolyzes peptide bonds present in the carboxyl group of hydrophobic amino acids such as phenylalanine) and trypsin-like activity (peptidase-mediated cleavage after basic side chains). The chymotrypsin-like activity was thought to be crucial for removing nitrated proteins in plasma (20). Nitrated proteins (e.g., glyceraldehyde-3-phosphate dehydrogenase) were shown to induce the Ub-proteasome pathway (40).
NO donors induced protein nitration via a mechanism involving redox-active iron and H2O2 (34). Similarly, TfR protein was nitrated in the presence of iron, H2O2, and •NO (Fig. 3E). The nitrated TfR was subsequently targeted for proteasomal degradation. Previous work showed that the activity of Ub-conjugating enzymes (E1 and E2) is reversibly inhibited during oxidative stress (21). The present data show that both 20S and 26S proteasome systems are inactivated by glucose/GO, which effectively compromised the removal of oxidized proteins. As a result, the protein carbonyls were higher in cells treated with H2O2. However, the protein carbonyls were considerably diminished in the presence of DETA/NO (Fig. 4D). Although a recent report suggests that Ub conjugation is not required for degradation of oxidized proteins by proteasome (35), the present data indicate that both 20S and 26S proteasomes are involved in degradation of nitrated and oxidized proteins, because both 20S and 26S proteasome activities were down-regulated with H2O2 and up-regulated with H2O2 and DETA/NO treatment.
Nitration of Receptor Proteins and Redox Signaling. Protein tyrosyl nitration has been reported to modify the function of the N-methyl-d-aspartate (NMDA) receptor causing alterations in cell signaling (41). Hypoxia-induced nitration of the NMDA receptor subunits increased Ca2+ influx in the newborn piglet brain (41). Neuronal NOS which is colocalized with NMDA receptors is activated by Ca2+ influx, leading to increased •NO production. NOS inhibition prevents both NMDA receptor nitration and receptor-mediated Ca2+ influx. Therefore, nitration is a viable mechanism by which a redox-sensitive receptor can acquire a gain or loss of function. In this study we postulate that TfR nitration can alter the TfR-dependent iron uptake. The crystal structure of the homodimeric TfR ectodomain has a helical domain (residues 607–760) that binds to Tf (42, 43). There are six tyrosines (residues 611, 614, 628, 643, 683, and 689) in the Tf-binding domain. Nitration of tyrosines present in the helical domain will likely inhibit Tf binding due to steric hindrance and conformational changes. This tentative proposal explains how TfR nitration can inhibit H2O2-induced iron uptake.
Summary. This study described the role of •NO, generated slowly at defined rates from NONOates (44), on oxidant-induced iron signaling. NONOates inhibit DCFH oxidation, TfR iron signaling, and apoptosis in endothelial cells under oxidant stress. Admittedly, the response to •NO donors in cells is varied and often paradoxical (45). Two different sources of •NO donors elicit opposing effects on the same cell function. Whether •NO enhances or suppresses the proteasome activity seems to depend on the proliferative status of a cell (46). •NO-mediated signaling and activation of the proteasomes may play a critical regulatory role in apoptosis and cell survival by modulating TfR-dependent iron signaling in endothelial cells subjected to oxidative and nitrative modification. Most vascular pathologies (diabetes, hyperglycemia, and atherosclerosis) are characterized by a decrease in endothelial •NO production (47). Under these conditions, oxygen radicals and protein degradation are usually enhanced. Thus, this role of •NO (i.e., maintaining the proteasome function) is highly significant from a pathophysiological perspective.
Acknowledgments
This work was supported by National Institutes of Health Grants RR01008, 1PO1HL68769-01, CA77822, HL073056-01, and GM 34009.
Abbreviations: Tf, transferrin; TfR, Tf receptor; IRP, iron-regulatory protein; Ub, ubiquitin; MG-132, N-carbobenzoxyl-l-leucinyl-l-leucinyl-l-norleucinal; GO, glucose oxidase; Lac, clasto-lactacystin-β-lactone; DCFH, dichlorodihydrofluorescein; DA, diacetate; DETA/NO, diethylenetriamine NONOate; AcOM-PYRRO, pyrrolidine nonoate methylene acetate; BAEC, bovine aortic endothelial cell; GSH, glutathione; IRE, iron-responsive element; DCF, dichlorofluorescein; l-NAME, Nω-nitro-l-arginine methyl ester; NOS, NO synthase.
References
- 1.Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A. & Freeman, B. A. (1990) Proc. Natl. Acad. Sci. USA 87, 1620–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paxinou, E., Weisse, M., Chen, Q., Souza, J. M., Hertkorn, C., Selak, M., Daikhin, E., Yudkoff, M., Sowa, G., Sessa, W. C. & Ischiropoulos, H. (2001) Proc. Natl. Acad. Sci. USA 98, 11575–11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas, S. R., Chen, K. & Keaney, J. F., Jr. (2002) J. Biol. Chem. 277, 6017–6602. [DOI] [PubMed] [Google Scholar]
- 4.Drummond, G. R., Cai, H., Davis, M. E., Ramasamy, S. & Harrison, D. G. (2000) Circ. Res. 86, 347–354. [DOI] [PubMed] [Google Scholar]
- 5.Wink, D. A., Hanbauer, I., Krishna, M. C., DeGraff, W., Gamson, J. & Mitchell, J. B. (1993) Proc. Natl. Acad. Sci. USA 90, 9813–9817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beltran, B., Mathur, A., Duchen, M. R., Erusalimsky, J. D. & Moncada, S. (2000) Proc. Natl. Acad. Sci. USA 97, 14602–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joshi, M. S., Pontheir, J. L. & Lancaster, J. R. (1999) Free Radical Biol. Med. 27, 1357–1366. [DOI] [PubMed] [Google Scholar]
- 8.Wang, Y., Vodovotz, Y., Kim, P. K. M., Zamora, R. & Billiar, T. R. (2002) Ann. N.Y. Acad. Sci. 962, 415–422. [DOI] [PubMed] [Google Scholar]
- 9.Rubbo, H., Parthasarathy, S., Barnes, S., Kirk, M., Kalyanaraman, B. & Freeman, B. (1995) Arch. Biochem. Biophys. 324, 15–25. [DOI] [PubMed] [Google Scholar]
- 10.Kotamraju, S., Chitambar, C. R., Kalivendi, S. V., Joseph, J. & Kalyanaraman, B. (2002) J. Biol. Chem. 277, 17179–17187. [DOI] [PubMed] [Google Scholar]
- 11.Kotamraju, S., Hogg, N., Joseph, J., Keefer, L. K. & Kalyanaraman, B. (2001) J. Biol. Chem. 276, 17316–17323. [DOI] [PubMed] [Google Scholar]
- 12.Rouault, T. A. & Klausner, R. D. (1996) Trends Biochem. Sci. 21, 174–177. [PubMed] [Google Scholar]
- 13.Pantopoulos, K. & Hentze, M. W. (1995) EMBO J. 14, 2917–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pantopoulos, K., Mueller, S., Atzberger, A., Ansorge, W., Stremmel, W. & Hentze, M. W. (1997) J. Biol. Chem. 272, 9802–9808. [DOI] [PubMed] [Google Scholar]
- 15.Pantopoulos, K. & Hentze, M. W. (1998) Proc. Natl. Acad. Sci. USA 95, 10559–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Testa, U., Pelosi, E. & Peschle, C. (1993) Crit. Rev. Oncog. 4, 241–276. [PubMed] [Google Scholar]
- 17.Pantopoulos, K., Weiss, G. & Hentze, M. W. (1996) Mol. Cell. Biol. 16, 3781–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim, S. & Ponka P. J. (2000) J. Biol. Chem. 275, 6220–6226. [DOI] [PubMed] [Google Scholar]
- 19.Coux, O., Tanaka, K. & Goldberg, A. L. (1996) Annu. Rev. Biochem. 65, 801–847. [DOI] [PubMed] [Google Scholar]
- 20.Gow, A., Duran, D., Malcolm, S. & Ischiropoulos, H. (1996) FEBS Lett. 385, 63–66. [DOI] [PubMed] [Google Scholar]
- 21.Haas, A. L. & Siepmann, T. J. (1997) FASEB J. 11, 1257–1268. [DOI] [PubMed] [Google Scholar]
- 22.Hershko, A. & Ciechanover, A. (1998) Annu. Rev. Biochem. 67, 425–479. [DOI] [PubMed] [Google Scholar]
- 23.Levine, R. L., Williams, J. A., Stadman, E. R. & Shacter, E. (1994) Methods Enzmol. 233, 346–357. [DOI] [PubMed] [Google Scholar]
- 24.Leibold, E. A. & Munro, H. N. (1988) Proc. Natl. Acad. Sci. USA 85, 2171–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pajonk, F., Riess, K., Sommer, A. & McBride, W. H. (2002) Free Radical Biol. Med. 32, 536–543. [DOI] [PubMed] [Google Scholar]
- 26.Grune, T., Reinheckel, T. & Davies, K. J. A. (1996) J. Biol. Chem. 271, 15504–15509. [DOI] [PubMed] [Google Scholar]
- 27.Saavedra, J. E., Shami, P. J., Wang, L. Y., Davies, K. M., Booth, M. N., Ciitro, M. L. & Keefer, L. K. (2000) J. Med. Chem. 43, 261–269. [DOI] [PubMed] [Google Scholar]
- 28.Ruzicka, F. J. & Beinert, H. (1978) J. Biol. Chem. 253, 2514–2517. [PubMed] [Google Scholar]
- 29.Tampo, Y., Kotamraju, S., Chitambar, C. R., Kalivendi, S. V., Keszler, A., Joseph, J. & Kalyanaraman, B. (2003) Circ. Res. 92, 56–63. [DOI] [PubMed] [Google Scholar]
- 30.Mueller, S., Pantopoulos, K., Hubner, C. A., Stremmel, W. & Hentze, M. W. (2001) J. Biol. Chem. 276, 23192–23196. [DOI] [PubMed] [Google Scholar]
- 31.Hentze, M. W., Rouault, T. A., Harford, J. B. & Klausner, R. D. (1989) Science 244, 357–359. [DOI] [PubMed] [Google Scholar]
- 32.Dick, L. R., Cruikshank, A. A., Destree, A. T., Grenier, L., McCormack, T. A., Melandri, F. D., Nunes, S. L., Palombella, V. J., Parent, L. A., Plamondon, L. & Stein, R. L. (1997) J. Biol. Chem. 272, 182–188. [DOI] [PubMed] [Google Scholar]
- 33.Meng, L., Mohan, R., Kwok, B. H., Elofsson, M., Sin, N. & Crews, C. M. (1999) Proc. Natl. Acad. Sci. USA 96, 10403–10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas, D. D., Espey, M. G., Vitek, M. P., Miranda, K. M. & Wink, D. A. (2002) Proc. Natl. Acad. Sci. USA 99, 12691–12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shringarpure, R., Grune, T., Mehlhase, J. & Davies, K. J. A. (2003) J. Biol. Chem. 278, 311–318. [DOI] [PubMed] [Google Scholar]
- 36.Gehring, N. H., Hentze, M. W. & Pantopoulos, K. (1999) J. Biol. Chem. 274, 6219–6225. [DOI] [PubMed] [Google Scholar]
- 37.Martins, E. A., Robalinho, R. L. & Meneghini, R. (1995) Arch. Biochem. Biophys. 316, 128–134. [DOI] [PubMed] [Google Scholar]
- 38.Sze, S. K., Ge, Y., Oh, H. & McLafferty, F. W. (2002) Proc. Natl. Acad. Sci. USA 99, 1774–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Souza, J. M., Choi, I., Chen, Q., Weisse, M., Daikhin, E., Yudkoff, M., Obin, M., Ara, J., Horwitz, J. & Ischiropoulos, H. (2000) Arch. Biochem. Biophys. 380, 360–366. [DOI] [PubMed] [Google Scholar]
- 40.Buchczyk, D. P., Briviba, K., Hartl, F. U. & Sies, H. (2000) Biol. Chem. 381, 121–126. [DOI] [PubMed] [Google Scholar]
- 41.Zanelli, S. A., Ashraf, Q. M. & Mishra, O. P. (2002) Neuroscience 112, 869–877. [DOI] [PubMed] [Google Scholar]
- 42.Bennett, M. J., Lebron, J. A. & Bjorkman, P. J. (2000) Nature 403, 46–53. [DOI] [PubMed] [Google Scholar]
- 43.Lebron, J. A., Bennet, M. J., Vaughn, D. E., Chirino, A. J., Snow, P. M., Mintier, G. A., Feder, J. N. & Bjorkman, P. J. (1998) Cell 93, 111–123. [DOI] [PubMed] [Google Scholar]
- 44.Goss, S. P. A., Hogg, N. & Kalyanaraman, B. (1997) J. Biol. Chem. 272, 21647–21653. [DOI] [PubMed] [Google Scholar]
- 45.Fiorucci, S., Mencarelli, A., Mannucci, R., Distrutti, E., Morelli, A., del Soldato, P. & Moncada, S. (2002) FASEB J. 12, 1645–1647. [DOI] [PubMed] [Google Scholar]
- 46.Glockzin, S., von Knethen, A., Scheffner, M. & Brune, B. (1999) J. Biol. Chem. 274, 19581–19586. [DOI] [PubMed] [Google Scholar]
- 47.Cai, H. & Harrison, D. G. (2000) Circ. Res. 87, 840–844. [DOI] [PubMed] [Google Scholar]