

Abstract
G protein-coupled receptors (GPCRs) mediate the perception of smell, light, taste, and pain. They are involved in signal recognition and cell communication and are some of the most important targets for drug development. Because currently no direct structural information on high-affinity ligands bound to GPCRs is available, rational drug design is limited to computational prediction combined with mutagenesis experiments. Here, we present the conformation of a high-affinity peptide agonist (neurotensin, NT) bound to its GPCR NTS-1, determined by direct structural methods. Functional receptors were expressed in Escherichia coli, purified in milligram amounts by using optimized procedures, and subsequently reconstituted into lipid vesicles. Solid-state NMR experiments were tailored to allow for the unequivocal detection of microgram quantities of 13C,15N-labeled NT(8–13) in complex with functional NTS-1. The NMR data are consistent with a disordered state of the ligand in the absence of receptor. Upon receptor binding, the peptide undergoes a linear rearrangement, adopting a β-strand conformation. Our results provide a viable structural template for further pharmacological investigations.
G protein-coupled receptors (GPCRs) are integral membrane proteins involved in a number of important physiological processes, including sensory transduction, mediation of hormonal activity, and cell-to-cell communication (for reviews, see refs. 1 and 2). More than 1,000 different GPCRs have been identified, and many of them have been implicated as major therapeutic routes to the treatment of human diseases (3). Despite the striking clinical relevance of GPCRs, only one high-resolution structure (rhodopsin) is available (4, 5). The diversity among endogenous GPCR ligands is exceptional. Small molecule ligands such as biogenic amines have been proposed to bind within the hydrophobic core of their respective receptors. In contrast, mutational mapping of ligand-binding sites in peptide receptors indicates that extracellular domains are also involved in ligand recognition (2). To date, direct structural information regarding ligand binding and GPCR activation is very limited (6). Targeted drug design is restricted to computational methods and other ligand design approaches to find and optimize lead compounds (7). The precise knowledge of receptor-bound ligand structures would substantially aid the development of tailor-made medicines.
Neurotensin (NT) is a 13-aa peptide (8) that is involved in a variety of neuromodulatory functions in the central and peripheral nervous system (9). NT binds to its GPCRs, NT type I receptor (NTS-1) and NTS-2 (10–13). The levocabastine-insensitive NTS-1 (10–12) interacts with the agonist NT with high (i.e., subnanomolar) affinity. Similar observations were made for the N-terminally truncated form of rat NTS-1 when expressed as a maltose-binding protein (MBP) fusion in Escherichia coli (14, 15) and purified in the presence of detergents (14, 16). Notably, not only the full-length peptide, but also the C-terminal part of NT, NT(8–13), has been found to interact with NTS-1 with high affinity (see, for example, refs. 11 and 17). The high affinity of the ligand to its receptor has precluded the elucidation of the receptor-bound ligand conformation by solution-state NMR (see, for example, ref. 6). A recent NMR study in the presence of detergent reported NT(8–13) chemical shift changes upon receptor interaction (18, 19). However, the mode of NT binding to its GPCR NTS-1 remains unknown.
Solid-state NMR (ssNMR) spectroscopy provides a means of studying conformational changes of ligands upon binding to high-affinity receptors in a noncrystalline environment. Only the relative size of ligand and natural abundance background (NAB, including receptor, lipids, etc.) determine whether the signal of the bound ligand can be detected unequivocally. For example, simple 13C isotope labeling has been sufficient for structural investigations of membrane proteins (20–22) or peptide/protein complexes (23) where ligand/NAB >10–2.
Double-quantum filtering (2QF, see, for example, refs. 24 and 25) permits the extension of this size limit to ligand/NAB >10–4 and hence allows for the study of NT(8–13) (1 kDa)/NTS-1 (101 kDa) binding in a detergent/lipid environment. As shown previously by ssNMR experiments and ab initio calculations (26), the resulting resonance frequencies are not only diagnostic for each individual peptide residue (27, 28), but they are also very sensitive to polypeptide backbone conformation (27, 29–34). As we will show below, extending 2QF spectroscopy into two spectral dimensions and the study of a uniformly labeled version of NT(8–13) enables the derivation of the backbone structure from a single NMR experiment.
Our study hence involves (i) the optimization of 2D 2QF (13C,13C) correlation spectroscopy and (ii) a subsequent NMR chemical shift analysis. The structure of free, immobilized NT, and the conformational rearrangements associated with receptor binding are studied at atomic resolution, providing unambiguous information about a high-affinity ligand in receptor-bound form.
Materials and Methods
Preparation of 13C,15N-Labeled NT(8–13). The uniformly 13C,15N-labeled hexapeptide Ac-Arg-Arg-Pro-Tyr-Ile-Leu-OH ([13C,15N]-NT(8–13)) (Fig. 1) was prepared via solid-phase 9-fluorenylmethoxycarbonyl (Fmoc) chemistry on a Wang resin (74 μmol scale) with benzotriazol-1-yloxytris(dimethylamino) phosphonium hexafluorophosphate/1-hydroxybenzotriazole activation by using an ABI 433A (Applied Biosystems/Perkin–Elmer) peptide synthesizer (for a review on Fmoc chemistry, see e.g., ref. 35). All isotope-labeled amino acid derivatives were purchased from Cambridge Isotope Laboratories (Andover, MA). Loading of Fmoc-[U-13C,15N]-Leu-OH (1.9 eq) on the resin was accomplished manually by means of N,N′-diisopropylcarbodiimide/4-dimethylaminopyridine-assisted coupling. Small or no excess of the amino acid building blocks was used in the peptide coupling steps, i.e., 1.9 eq Fmoc-[U-13C,15N]-Ile-OH, 2 eq Fmoc-[U-13C,15N]-Pro-OH, 1.6 eq Fmoc-[U-13C,15N]-Tyr(OtBu)-OH, and 1 eq Fmoc-[U-13C,15N]-Arg(Pmc)-OH. The coupling time was 2 h. The peptide was cleaved from the resin with the following reagents: 120 μl of 1,2-ethanedithiol, 120 μl of triisopropylsilane, 80 μl of H2O, 80 mg of phenol, and 9.6 ml of trifluoroacetic acid (TFA) (for general TFA-cleavage protocol see ref. 36). The crude peptide was purified with RP-HPLC on an Alltima C18 column (10 × 250 mm, 5-μm particles, AllTech Associates) applying a gradient of acetonitrile in 0.1% aqueous TFA. The lyophilized 13C,15N-labeled hexapeptide was obtained in 40% yield (34.3 mg). In addition, the 13C,15N-labeled pentamer Ac-Arg-Pro-Tyr-Ile-Leu-OH [NT(9–13)], which resulted from incomplete coupling of the last amino acid, could be readily isolated by RP-HPLC in 14% yield (9.1 mg). For NMR experiments with solid-phase [13C,15N]-NT(9–13), 2 mg of the lyophilized peptide was loaded into a 4-mm magic angle spinning (MAS) rotor. To reduce sample heterogeneity, 1 μl of distilled water was added. For NMR experiments with solid-phase [13C,15N]-NT(8–13), 0.1 mg of the peptide was dissolved in detergent-containing buffer [25 mM Tris·HCl, pH 7.4/5% glycerol/1 mM EDTA/50 mM NaCl/0.1% n-dodecyl-β-d-maltoside/0.2% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate/0.04% cholesteryl hemisuccinate] and placed into the MAS rotor.
Fig. 1.

(Upper) Sequences of NT, NT(8–13), and NT(9–13) in single-letter code. (Lower) Schematic representation of the NT receptor fusion proteins. Amino acids in single-letter notation are given in gray; subscripts stand for repetition.
Purified, Detergent-Solubilized NTR Sample (NTS-1A). The NTS-1 fusion protein MBP-rT43NTR-thioredoxin (TrxA)-H10 (Fig. 1) consisted of the E. coli MBP, followed by the N-terminally truncated rat NTS-1 (rT43NTR), the E. coli TrxA, and a decahistidine tail (H10) (37). The receptor fusion protein was expressed in functional form in E. coli (37) (50 liters) and purified at large scale (two purifications) in the presence of the detergents n-dodecyl-β-d-maltoside and 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate, and cholesteryl hemisuccinate, by immobilized metal affinity chromatography (60-ml Ni-NTA column), followed by a 10-ml NT column and anion exchange chromatography (5-ml HiTrap Q-Sepharose column), as described for the human NTS-1 fusion protein MBP-huNTR-TrxA-H10 (38). The Q-Sepharose eluate was concentrated by using a Centriprep-30 device (Amicon, 2.9 mg of functional receptor fusion protein available after concentration) and stored in liquid nitrogen. In preparation for the NMR experiments, the concentrated NTS-1 fusion protein was diluted with detergent-containing buffer to give final concentrations of 25 mM Tris·HCl (pH 7.4), 5% glycerol, 1 mM EDTA, and 50 mM NaCl. The receptor was concentrated at 3°C by using Ultrafree-15 Millipore MWCO 50K and Microcon YM-30 (Amicon) devices. Protein determination (39) and [3H]NT binding analysis (16) of the concentrated sample gave a value of 7,563 pmol/mg (565 μl at 3.68 mg/ml). A theoretical value of 10,361 pmol/mg is calculated for MBP-rT43NTR-TrxA-H10 (molecular mass of 96.5 kDa), assuming one ligand binding site per receptor molecule. Seventy three percent of the concentrated receptor preparation hence binds agonist. 13C,15N-labeled NT(8–13) (molecular mass of 973 Da) (2.3 μg) was then added to the receptor preparation (0.37 mg) [agonist/receptor ratio of 0.6 (M/M), based on total protein content] and incubated for 2 h on ice. The [13C,15N]-NT(8–13)/receptor sample (100 μl) was placed into a 4-mm MAS rotor, frozen on dry ice, and subjected to NMR analysis. To titrate the amount of bound agonist, an additional 10 μg of [13C,15N]-NT(8–13) was added to the receptor preparation and analyzed. Sample NT/NTS-1A contains in total 0.37 mg of receptor protein and 12.3 μg of [13C,15N]-NT(8–13) [agonist/receptor ratio of 3.3 (M/M), based on total protein content]. Consideration of the ligand binding data gives an agonist/functional receptor ratio of 4.5 (M/M) in the sample NT/NTS-1A.
Lipid-Reconstituted NTR Sample (NTS-1B). The NTS-1 fusion protein MBP-N10-Tev-T43NTR-N5G3S-Tev-G3S-TrxA-H10 is shown schematically in Fig. 1. The receptor fusion protein was expressed in functional form in E. coli (90 liters) and purified (50-ml Ni-NTA column, followed by a 10-ml NT column; 5.2 mg of purified protein from three purifications). For reconstitution into lipid vesicles, receptors (final concentration of 0.13 mg/ml) were incubated with n-dodecyl-β-d-maltoside-saturated brain polar lipids [Avanti Polar Lipids, final concentration of 0.4 mg/ml, lipid-to-receptor ratio of ≈ 400 (M/M)] for 4–5 h at 4°C in a volume of 13 ml, followed by the addition of Bio-beads SM-2 (BioRad, 1.5 gram). The Bio-beads were exchanged four times over a time period of 3 days. Proteoliposomes were recovered by ultracentrifugation and resuspended in 50 mM Tris·HCl, pH 7.4/1 mM EDTA at a protein concentration of 2–2.5 mg/ml. We estimated from [3H]NT binding assays (40) that 66% of the reconstituted receptors are accessible to ligand, with the remaining receptors either not binding agonist or having their ligand binding sites facing inside the lipid vesicles (data not shown). For the NMR experiments, 22 μg of [13C,15N]-NT(8–13) was added to 3.8 mg of reconstituted receptor fusion protein (molecular mass of 101 kDa) [agonist/receptor ratio of 0.6 (M/M), based on total protein content]. After incubation (4 h, 4°C), the sample (NT/NTS-1B) was centrifuged at 40,000 rpm (70 Ti rotor) for 30 min and loaded as a pellet into a 4-mm MAS rotor. Consideration of the ligand binding data gives an agonist/functional receptor ratio of 0.9 (M/M) in the sample NT/NTS-1B.
ssNMR Experiments and Analysis. All NMR experiments were performed on a wide-bore, 600-MHz (1H resonance frequency, Bruker Biospin, Rheinstetten, Germany) spectrometer by using double (1H,13C) or triple (1H,13C,15N) resonance MAS (41) probe heads. Experiments on solid-phase [13C,15N]-NT(9–13) samples were conducted at 5°C, whereas experiments involving buffer/protein solutions and reconstituted sample NT/NTS-1B were performed at –80°C or –85°C. An MAS spinning rate of 7 kHz was used with tppm decoupling (42) at 110-kHz radio-frequency amplitude during free evolution and detection periods. Cross polarization (43, 44) experiments involved amplitude modulated radio-frequency fields (45). 2QF experiments were performed by using the POST-C7 (46) dipolar recoupling scheme with 32-step phase cycling scheme to suppress unwanted single-quantum (1Q) signals (see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). These phase cycles were tested by performing 2QF experiments in the side-chain region of the natural abundance tri-peptide Ala-Gly-Gly. Results of the 1D experiments with the four preparations (see below) containing uniformly 13C,15N-labeled NT peptides are given in Figs. 6 and 7, which are published as supporting information on the PNAS web site. Additional test experiments on mixtures of labeled and unlabeled amino acids confirmed the suppression of 1Q signals by at least a factor of 100 in the 2Q spectra in line with the theoretical expectation. As a result, microgram peptide quantities can be reliably detected in the presence of a large background signal. Details for the presented 2D correlations experiments are given in the figure legends and Fig. 5. Further information regarding the discussed data interpretation is given in Fig. 8, which is published as supporting information on the PNAS web site.
For the construction of the backbone model, the talos (47) package was used. As recently demonstrated in the context of fibrous peptides (30, 33, 34), talos analyses NMR chemical shifts assignments of three consecutive peptide residues by using a statistical homology search to arrive at an estimation of the dihedral angles of the peptide of interest. Within this framework, backbone dihedral angles of NT(8–13) can be obtained for residues Arg-9–Ile-12 (see Table 1, which is published as supporting information on the PNAS web site). For each residue, >80% of the predicted dihedral angles sets are consistent with an elongated peptide conformation. Additional information regarding ψ of Arg-8 and φ of Leu-13 is accessible from considering an extended sequence Pro-7-NT(8–13)-X, where Pro-7 corresponds to the residue of the full-length agonist NT and X is varied. This approach predicts ψ (Arg-8) = –34 ± 15° and φ (Leu-13) = –104 ± 16° for X = Ala, Pro, Thr.
Results
Four preparations containing uniformly 13C,15N-labeled NT peptides (see Fig. 1) were analyzed by ssNMR: (i) solid-phase NT (9–13) as a spectroscopic reference [this pentapeptide, in contrast to NT(8–13), binds to NTS-1 with low affinity only (17)], (ii) NT(8–13) in detergent-containing buffer to investigate the structure of the free ligand, (iii) NT(8–13) in complex with the purified, detergent solubilized receptor (sample NT/NTS-1A), and (iv) in complex with lipid-reconstituted NTS-1 (sample NT/NTS-1B). The latter samples allow us to determine the conformation of NT(8–13) when bound to its GPCR with high affinity. To do so, NMR signals arising from microgram quantities of peptide ligand must be detected in the presence of large background signals (NAB) resulting from buffer components (glycerol) and detergents (sample NT/NTS-1A) or lipid (sample NT/NTS-1B). Because ligand/NAB <10–2, conventional 1Q experiments are not suitable to probe ligand binding in the current context (see Fig. 6). Instead, 2QF (see, for example, refs. 24 and 25) methods that select for pairs of nearby 13C-13C nuclei (e.g., directly bonded) are mandatory. Measurements on the purified NTS-1 fusion protein (0.27 mg of functional receptor) in the presence of increasing amounts of NT(8–13) in microgram quantities gave 2Q signals that changed upon agonist addition (data not shown), reflecting adequate sensitivity of our 2QF experiments. For the spectral identification of the individual peptide residues and the subsequent side-chain 13C resonance assignment, 2D 2QF correlation experiments are compulsory and are discussed below.
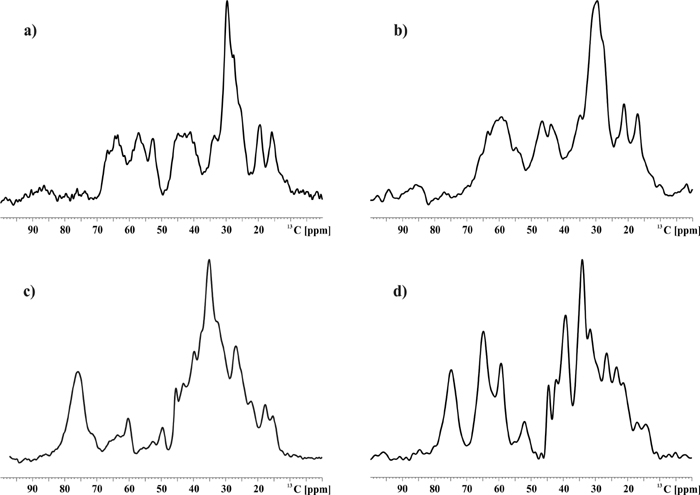
Immobilized NT in the Absence of Receptor. Fig. 2 contains results of a 2D 13C-13C 2QF correlation experiment for free NT(9–13) and NT(8–13) in detergent solution. The observed 2D patterns agree well with standard correlation maps (32, 48) expected from the NT amino acid sequence. The observed (1Q) line width varies between 1 and 2.5 ppm and amounts to ≈1.5 ppm for the majority of the observed correlations. For sensitivity reasons, results of Figs. 2b and 3 (see below) were obtained by using a smaller number of t1 increments. As a result, the 2Q line width is enlarged but does not affect the spectral analysis presented here. If rare-spin polarization transfer is used, interand intraresidue correlation experiments are in general necessary to unambiguously assign the NMRs of larger polypeptides under MAS conditions (48).
Fig. 2.

2D 13C-13C (2Q,1Q)-correlation experiments for 2 mg of solid-phase [13C,15N]-NT(9–13) (a) and 0.1 mg of frozen [13C,15N]-NT(8–13) in detergent solution (b). In a and b, 128 and 44 t1 experiments, respectively, were recorded by using tppm (42) decoupling (at a radio-frequency amplitude of 110 kHz) in evolution and detection periods. Residue assignments are given in single-letter notation; intraresidue side-chain correlations are indicated in a. POST-C7 recoupling was used for 2Q excitation and reconversion. Data shown result from 256 (a) and 1,536 (b) scans. Below each 2D correlation spectrum, secondary chemical shifts Δδ as obtained from Table 1 are given. (b) Arg-8 and Arg-9 Δδ values are shaded to indicate that secondary chemical shifts here are only tentatively assigned (see also Fig. 3).
Fig. 3.

2D 13C-13C (2Q,1Q) correlation experiments for the two [13C,15N]-NT(8–13)/receptor preparations NT/NTS-1A (a) and NT/NTS-1B (b, in red). In both cases, 32 t1 experiments were recorded by using tppm (42) decoupling in evolution and detection periods. Residue assignments are given in single-letter notation. Similar to Fig. 2, POST-C7 recoupling was used for 2Q excitation and reconversion. (a) A total of 13,568 scans were accumulated for each t1 experiment. (b) A total of 11,136 scans were coadded in t2. Below each 2D correlation spectrum, secondary chemical shifts Δδ as obtained from Table 1 are given. Arg-8 and Arg-9 Δδ values are shaded to indicate that secondary chemical shifts here are only tentatively assigned. (b) A 1D slice along the t1 axis (54 ppm, indicated in red) and results from frozen [13C,15N]-NT(8–13) (see Fig. 2b) are included in black for reference. Additional information is given in Fig. 9, which is published as supporting information on the PNAS web site.
In the (2Q,1Q) correlation experiment considered here, resonance frequencies of two dipolar coupled spins detected in the 1Q dimension (1QC) must resonate at the sum frequency along the ω1 (2QC) axis. Hence, knowledge of the characteristic 13C chemical shifts of the five [NT(9–13)] or six [NT(8–13)] involved residues and construction of the corresponding standard 2D 2Q correlation map (48) is sufficient to assign resonances for all residues in the spectrum (see also Fig. 8). In particular, all backbone [(δ(Cα) + δ(Cβ), δ(Cα,β)] correlations are easily identified (see Table 1). Knowledge of the isotropic chemical-shift values can be used to interpret the observed spectra in terms of a 3D backbone conformation of NT(9–13) and NT(8–13). For this purpose, we can calculate the parameter Δδ that reflects the conformation-dependent chemical shift of Cα and Cβ resonances (32) and relates the experimentally observed carbon chemical shifts under MAS conditions to standard, isotropic random coil values (see BioMagResBank, a repository for data from NMR spectroscopy on proteins, www.bmrb.wisc.edu/index.html). These parameters are plotted in Fig. 2 for the C-terminal residues of NT. In particular, negative values of Δδ are consistent with large positive values of the backbone dihedral angle ψ, whereas Δδ > 0 is indicative of helical (i.e., ψ < 0) peptide backbone conformations (32).
From Fig. 2, we find that secondary chemical shifts for free NT(9–13) and NT(8–13) exposed to detergent-containing buffer are in general small and vary in sign. Significant changes in Δδ between both preparations are detected for Pro-10, Ile-12, and Leu-13. From the homonuclear correlation experiment in Fig. 2b we cannot, a priori, assign the Arg side-chain correlations to either Arg-8 or Arg-9. For this reason, Arg-8 and Arg-9 Δδ values are shaded in Fig. 2b. Notably, the Arg assignment obtained for NT(9–13) matches one set of (Cα,Cβ) Arg chemical shifts detected in all other considered preparations (see below). Therefore, the corresponding correlations in Figs. 2b and 3 were tentatively assigned to Arg-9. Irrespective of this ambiguity, we conclude from the data presented in Fig. 2 that NT(9–13) and NT(8–13) remain largely unstructured in the absence of the receptor.
NT/NTR Complex. Next, we investigated the interaction of NT(8–13) with purified NTS-1 in detergent solution (sample NT/NTS-1A) (Fig. 3a). The ratio of agonist/functional receptor in sample NT/NTS-1A is 4.5 (M/M). Under these experimental conditions, we expect signal contributions from both free and receptor-bound NT(8–13). In line with 1D results (see Figs. 6 and 7), we observe in the 2D 2QF experiment broad signals ≈60 and 30 ppm in ω1 and ω2, respectively. These signals most likely stem from natural abundance 2Q contributions of the detergents and other buffer components. On the other hand, well-separated side-chain resonances and the (Cα,Cβ) correlations of interest lie outside this range and can be identified (Table 1). Again, these resonance assignments do not permit discriminating between Arg-8 and Arg-9 but they can be used to define conformation-dependent chemical shifts Δδ summarized in Fig. 3a. Except for Leu-13, all residues are now described by negative values of Δδ.
The interpretation of Fig. 3a in terms of secondary chemical shifts is not only influenced by the occurrence of strong detergent and buffer signals, but also by a relatively low NT(8–13) signal intensity as a consequence of the limited amount of purified receptor in the NMR rotor. To increase the amount of functional receptor and hence the amount of bound NT(8–13) for ssNMR measurements, and to reduce the noise contributions from detergent and buffer components, the NTS-1 fusion protein was reconstituted into lipid, which allows for packing of the receptor at higher density (2.5 mg of functional receptors) into the NMR rotor. As a result, the amount of NT(8–13) (22 μg) could be significantly increased (sample NT/NTS-1B). At a peptide/receptor molar ratio of 0.9, we can assume that the ssNMR signals must predominantly result from bound NT(8–13) molecules. Fig. 3b (red) shows a 2Q correlation pattern of the sample NT/NTS-1B. Consistent with additional 1D 2QF (see Fig. 7) experiments, the major part of the spectrum is free of lipid correlations and the general (2Q,1Q) correlation pattern for NT(8–13) can be readily identified. In particular, the detection of virtually all side-chain resonances allows for an unambiguous identification of Ile-12 and Leu-13 residues. The resulting (Cα and Cβ) chemical shift assignments are given in Fig. 3b (see Table 1).
For reference, results of NT(8–13) in detergent buffer are included in black in Fig. 3b and reveal that the side-chain resonances do not significantly change upon receptor binding. In contrast, one observes that the spectral separation between Cα and Cβ resonances diminishes considerably upon receptor binding. Correspondingly, the conformation-dependent chemical shifts are strongly negative (see Fig. 3b and Table 1). Remarkably, results of Fig. 3 lead to similar secondary chemical shifts, suggesting that both preparations reflect similar ligand–receptor interactions.
Discussion
In the following, we discuss the structural implications of our ssNMR study. In the presence of its receptor NTS-1, the agonist NT(8–13) changes from a disordered state into a defined β-strand conformation. Our structural model of the receptor-bound ligand could represent a viable template for 3D pharmacophore studies (7).
Comparison of Figs. 2 and 3 allows for a qualitative structural interpretation of the observed chemical shift variations of NT(8–13). The size and the sign changes of Δδ imply that solid-phase NT(9–13) and NT(8–13) immobilized in detergent buffer remain largely unstructured. Our ssNMR data are hence in qualitative agreement with previous solution-state NMR studies of NT in aqueous, methanol, and SDS solutions (49, 50), which indicated an inherent conformational flexibility with no discernible elements of secondary structure in water and methanol. Notably, the observed chemical changes with respect to the random coil are largest for the Arg-8–Arg-9–Pro-10 segment in buffer-detergent solution, consistent with previously postulated charge–charge interactions of the peptide with surrounding detergent molecules (49). In the same manner, the observed NMR correlations can be analyzed for NT(8–13) in the presence of functional NTS-1 (Fig. 3). For both samples NT/NTS-1A and NT/NTS-1B, comparable chemical shift changes are observed. Except for Leu-13, both signal sets indicate negative values of Δδ. Because of the higher concentration of NT(8–13) and therefore higher signal-to-noise ratio in our NMR studies on NT/NTS-1B (Fig. 3b), we conclude that the peptide conformation (i.e., also Leu-13) is described by negative values for secondary chemical shifts Δδ. Knowledge of Δδ and the peptide amino acid sequence allow for an estimation of the backbone dihedral angles of NT(8–13) in the receptor-bound form. For this purpose, the chemical shift assignments of Fig. 3 were used as entry parameters within the talos (47) prediction routine. Similar to recent ssNMR studies on fibrous proteins (33, 34), the resulting dihedral angles can be used to construct a 3D model of the backbone conformation of receptor-bound NT(8–13), which is shown in Fig. 4. Our data suggest a β-strand conformation of the agonist bound to NTS-1. The current analysis does not yet permit the refinement of the side-chain conformations of NT(8–13). For this reason, all side chains in Fig. 4 are shaded and indicated only for reference.
Fig. 4.

Backbone model of NT(8–13) when bound to NTS-1. Backbone dihedral angle constraints as obtained from talos (see Table 1) and standard potentials were used to sample the allowed conformational space within cns (56). A representative backbone structure together with a transparent hose-shaped object reflecting the ensemble distribution of a set of 100 structures is shown. The current analysis does not permit refinement of the side-chain conformations. Hence, all side chains are shown by using thin lines and are indicated only for reference. The figure was prepared by using molmol (57).
Previously published molecular modeling studies (51, 52) of NT bound to NTS-1 resulted in conflicting information about the presumed structure of the bound agonist. Pang et al. (51) predicted a compact conformation of NT(8–13), with a proline type I turn for the segment Arg-9–Pro-10–Tyr-11–Ile-12. The corresponding backbone angles (ϕ, ψ) for Pro-10 and Tyr-11 would be given by (–60°, –30°) and (–90°, 0°), respectively, contradicting the ssNMR data presented here. More recently, mutagenesis and structure-activity studies combined with modeling techniques were used to predict the receptor binding site and the conformation of bound NT(8–13) (52, 53). In this model, NT(8–13) adopts a linear backbone conformation in qualitative agreement with our ssNMR results. The receptor-NT(8–13) side chain contacts proposed by Barroso et al. (52) will be investigated by additional ssNMR experiments that measure interatomic distances in the NT(8–13)-receptor complex.
To date, no structural information at the molecular level has been reported on NT bound to its high-affinity receptor. We have used 2D ssNMR correlation experiments to elucidate the interaction of NT(8–13) with the rat NTS-1. The observed chemical shifts not only allow for a comparative study of the NT conformation in different chemical environments, but also enable a direct interpretation of the NMR signals in terms of the local backbone conformation of the neuropeptide.
Our results on solid-phase NT(9–13) and NT(8–13) immobilized in a detergent environment indicate that the peptide remains largely unstructured in the absence of the receptor. When bound to NTS-1, the secondary chemical shifts observed in the solid state considerably change for most of the amino acid residues of NT(8–13). The corresponding backbone torsional angles ψ are consistent with a β–strand arrangement of the agonist in complex with its receptor, both purified and reconstituted into lipid. Our selected NMR approach, using chemical shift information only, was largely dictated by signal-to-noise considerations. The accuracy of the reported backbone dihedral angles could be further refined by dipolar double-quantum dephasing experiments (see, for example, ref. 54) or chemical shift-selective distance measurements. These experiments along with additional 15N-13C and CHHC (55) correlation experiments are ongoing.
The presented ssNMR data provide direct experimental evidence for a distinct conformation of a neuropeptide bound with high affinity to its GPCR. We have analyzed microgram ligand quantities in the presence of milligram receptor quantities and demonstrate that ssNMR experiments are suitable for structural studies on membrane protein systems for which structural information at the atomic level is currently lacking.
Supplementary Material
Acknowledgments
Technical assistance during the course of the NMR experiments by H. Förster (Bruker) and L. Sonnenberg is gratefully acknowledged. We thank K. Seidel for performing the structure calculations within the cns software. We are indebted to R. Tycko and S. Becker for comments on the manuscript. We thank R. Henderson and C. Griesinger for contin-uous support of this project. The work of A.K.S. and R.G. on sample NT/NTS-1A was supported by GlaxoWellcome, AstraZeneca, and the Medical Research Council LINK program. The work of J.F.W. and R.G. on sample NT/NTS-1B was supported by the intramural program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Abbreviations: Fmoc, 9-fluorenylmethoxycarbonyl; GPCR, G protein-coupled receptor; MAS, magic angle spinning; MBP, maltose-binding protein; NAB, natural abundance background; NT, neurotensin; NTS-1, NT type I receptor; ssNMR, solid-state NMR; 1Q, single quantum; 2QF, double-quantum filtering.
References
- 1.Gether, U. & Kobilka, B. K. (1998) J. Biol. Chem. 273, 17979–17982. [DOI] [PubMed] [Google Scholar]
- 2.Ji, T. H., Grossmann, M. & Ji, I. (1998) J. Biol. Chem. 273, 17299–17302. [DOI] [PubMed] [Google Scholar]
- 3.Wilson, S., Bergsma, D. J., Chambers, J. K., Muir, A. I., Fantom, K. G., Ellis, C., Murdock, P. R., Herrity, N. C. & Stadel, J. M. (1998) Br. J. Pharmacol. 125, 1387–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., Le Trong, I., Teller, D. C., Okada, T., Stenkamp, R. E., et al. (2000) Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- 5.Okada, T., Fujiyoshi, Y., Silow, M., Navarro, J., Landau, E. M. & Shichida, Y. (2002) Proc. Natl. Acad. Sci. USA 99, 5982–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inooka, H., Ohtaki, T., Kitahara, O., Ikegami, T., Endo, S., Kitada, C., Ogi, K., Onda, H., Fujino, M. & Shirakawa, M. (2001) Nat. Struct. Biol. 8, 161–165. [DOI] [PubMed] [Google Scholar]
- 7.Klabunde, T. & Hessler, G. (2002) Chembiochem 3, 928–944. [DOI] [PubMed] [Google Scholar]
- 8.Carraway, R. & Leeman, S. E. (1973) J. Biol. Chem. 248, 6854–6861. [PubMed] [Google Scholar]
- 9.Martin, S., Botto, J.-M., Vincent, J.-P. & Mazella, J. (1999) Mol. Pharmacol. 55, 210–215. [DOI] [PubMed] [Google Scholar]
- 10.Vita, N., Laurent, P., Lefort, S., Chalon, P., Dumont, X., Kaghad, M., Gully, D., Le Fur, G., Ferrara, P. & Caput, D. (1993) FEBS Lett. 317, 139–142. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka, K., Masu, M. & Nakanishi, S. (1990) Neuron 4, 847–854. [DOI] [PubMed] [Google Scholar]
- 12.Watson, M., Isackson, P. J., Makker, M., Yamada, M. S., Yamada, M., Cusack, B. & Richelson, E. (1993) Mayo Clin. Proc. 68, 1043–1048. [DOI] [PubMed] [Google Scholar]
- 13.Chalon, P., Vita, N., Kaghad, M., Guillemot, M., Bonnin, J., Delpech, B., Le Fur, G., Ferrara, P. & Caput, D. (1996) FEBS Lett. 386, 91–94. [DOI] [PubMed] [Google Scholar]
- 14.Tucker, J. & Grisshammer, R. (1996) Biochem. J. 317, 891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grisshammer, R., Duckworth, R. & Henderson, R. (1993) Biochem. J. 295, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grisshammer, R., Averbeck, P. & Sohal, A. K. (1999) Biochem. Soc. Trans. 27, 899–903. [DOI] [PubMed] [Google Scholar]
- 17.Goedert, M. (1989) Methods Enzymol. 168, 462–481. [DOI] [PubMed] [Google Scholar]
- 18.Williamson, P. T. F., Bains, S., Chung, C., Cooke, R., Meier, B. H. & Watts, A. (2001) in Focus on Structural Biology: Perspectives on Solid State NMR in Biology, eds. Kiihne, S. & de Groot, H. J. M. (Kluwer, Dordrecht, The Netherlands), Vol. 1, pp. 191–201. [Google Scholar]
- 19.Williamson, P. T. F., Bains, S., Chung, C., Cooke, R. & Watts, A. (2002) FEBS Lett. 518, 111–115. [DOI] [PubMed] [Google Scholar]
- 20.Creuzet, F., McDermott, A., Gebhard, R., Vanderhoef, K., Spijkerassink, M. B., Herzfeld, J., Lugtenburg, J., Levitt, M. H. & Griffin, R. G. (1991) Science 251, 783–786. [DOI] [PubMed] [Google Scholar]
- 21.Smith, S. O., Aschheim, K. & Groesbeek, M. (1996) Q. Rev. Biophys. 29, 395–449. [DOI] [PubMed] [Google Scholar]
- 22.Creemers, A. F. L., Kiihne, S., Bovee-Geurts, P. H. M., DeGrip, W. J., Lugtenburg, J. & de Groot, H. J. M. (2002) Proc. Natl. Acad. Sci. USA 99, 9101–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weliky, D. P., Bennett, A. E., Zvi, A., Anglister, J., Steinbach, P. J. & Tycko, R. (1999) Nat. Struct. Biol. 6, 141–145. [DOI] [PubMed] [Google Scholar]
- 24.Bax, A., Freeman, R. & Kempsell, S. P. (1980) J. Am. Chem. Soc. 102, 4849–4851. [Google Scholar]
- 25.Munowitz, M. & Pines, A. (1987) Adv. Chem. Phys. 66, 1–152. [Google Scholar]
- 26.Dedios, A. C., Pearson, J. G. & Oldfield, E. (1993) Science 260, 1491–1496. [DOI] [PubMed] [Google Scholar]
- 27.Saito, H. (1986) Magn. Reson. Chem. 24, 835–852. [Google Scholar]
- 28.Cavanagh, J., Fairbrother, W. J., Palmer, A. G., Skelton, N. J. (1996) Protein NMR Spectroscopy, Principles, and Practice (Academic, San Diego).
- 29.Heller, J., Laws, D. D., Tomaselli, M., King, D. S., Wemmer, D. E., Pines, A., Havlin, R. H. & Oldfield, E. (1997) J. Am. Chem. Soc. 119, 7827–7831. [Google Scholar]
- 30.Balbach, J. J., Ishii, Y., Antzutkin, O. N., Leapman, R. D., Rizzo, N. W., Dyda, F., Reed, J. & Tycko, R. (2000) Biochemistry 39, 13748–13759. [DOI] [PubMed] [Google Scholar]
- 31.Laws, D. D., Bitter, H. M. L., Liu, K., Ball, H. L., Kaneko, K., Wille, H., Cohen, F. E., Prusiner, S. B., Pines, A. & Wemmer, D. E. (2001) Proc. Natl. Acad. Sci. USA 98, 11686–11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luca, S., Filippov, D. V., van Boom, J. H., Oschkinat, H., de Groot, H. J. M. & Baldus, M. (2001) J. Biomol. NMR 20, 325–331. [DOI] [PubMed] [Google Scholar]
- 33.Petkova, A. T., Ishii, Y., Balbach, J. J., Antzutkin, O. N., Leapman, R. D., Delaglio, F. & Tycko, R. (2002) Proc. Natl. Acad. Sci. USA 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaroniec, C. P., MacPhee, C. E., Astrof, N. S., Dobson, C. M. & Griffin, R. G. (2002) Proc. Natl. Acad. Sci. USA 99, 16748–16753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Atherthon, E. & Sheppard, R. C. (1989) Solid-Phase Peptide Synthesis: A Practical Approach (IRL, Oxford).
- 36.Dick, F. (1994) in Methods in Molecular Biology, eds. Pennington, M. W. & Dunn, B. M. (Humana, Totowa, NJ), Vol. 35, pp. 63–72.7894609 [Google Scholar]
- 37.Grisshammer, R. & Tucker, J. (1997) Protein Expression Purif. 11, 53–60. [DOI] [PubMed] [Google Scholar]
- 38.Grisshammer, R., Grunwald, T. & Sohal, A. K. (2002) Protein Expression Purif. 24, 505–512. [DOI] [PubMed] [Google Scholar]
- 39.Schaffner, W. & Weissmann, C. (1973) Anal. Biochem. 56, 502–514. [DOI] [PubMed] [Google Scholar]
- 40.Grisshammer, R. & Hermans, E. (2001) FEBS Lett. 493, 101–105. [DOI] [PubMed] [Google Scholar]
- 41.Andrew, E. R., Bradbury, A. & Eades, R. G. (1958) Nature 182, 1659. [Google Scholar]
- 42.Bennett, A. E., Rienstra, C. M., Auger, M., Lakshmi, K. V. & Griffin, R. G. (1995) J. Chem. Phys. 103, 6951–6958. [Google Scholar]
- 43.Hartmann, S. R. & Hahn, E. L. (1962) Phys. Rev. 128, 2042–2053. [Google Scholar]
- 44.Pines, A., Gibby, M. G. & Waugh, J. S. (1973) J. Chem. Phys. 59, 569–590. [Google Scholar]
- 45.Metz, G., Wu, X. L. & Smith, S. O. (1994) J. Magn. Reson. 110, 219–227. [Google Scholar]
- 46.Hohwy, M., Jakobsen, H. J., Eden, M., Levitt, M. H. & Nielsen, N. C. (1998) J. Chem. Phys. 108, 2686–2694. [Google Scholar]
- 47.Cornilescu, G., Delaglio, F. & Bax, A. (1999) J. Biomol. NMR 13, 289–302. [DOI] [PubMed] [Google Scholar]
- 48.Baldus, M. (2002) Prog. Nucl. Magn. Reson. Spectrosc. 41, 1–47. [Google Scholar]
- 49.Xu, G.-Y. & Deber, C. M. (1991) Int. J. Pept. Protein Res. 37, 528–535. [DOI] [PubMed] [Google Scholar]
- 50.Nieto, J. L., Rico, M., Santoro, J., Herranz, J. & Bermejo, F. J. (1986) Int. J. Peptide Protein Res. 28, 315–323. [DOI] [PubMed] [Google Scholar]
- 51.Pang, Y.-P., Cusack, B., Groshan, K. & Richelson, E. (1996) J. Biol. Chem. 271, 15060–15068. [DOI] [PubMed] [Google Scholar]
- 52.Barroso, S., Richard, F., Nicolas-Ethève, D., Reversat, J.-L., Bernassau, J.-M., Kitabgi, P. & Labbé-Jullié, C. (2000) J. Biol. Chem. 275, 328–336. [DOI] [PubMed] [Google Scholar]
- 53.Richard, F., Barroso, S., Nicolas-Ethève, D., Kitabgi, P. & Labbé-Jullié, C. (2001) Eur. J. Pharmacol. 433, 63–71. [DOI] [PubMed] [Google Scholar]
- 54.Feng, X., Verdegem, P. J. E., Lee, Y. K., Sandstrom, D., Eden, M., Bovee-Geurts, P., deGrip, W. J., Lugtenburg, J., deGroot, H. J. M. & Levitt, M. H. (1997) J. Am. Chem. Soc. 119, 6853–6857. [Google Scholar]
- 55.Lange, A., Luca, S. & Baldus, M. (2002) J. Am. Chem. Soc. 124, 9704–9705. [DOI] [PubMed] [Google Scholar]
- 56.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 57.Koradi, R., Billeter, M. & Wüthrich, K. (1996) J. Mol. Graphics 14, 51–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}