

Abstract
Mutations arise in a single individual and at a single point in time and space. The geographic distribution of mutations reflects both historical population size and frequency of migration. We employ coalescence-based methods to coestimate effective population size, frequency of migration, and level of recombination compatible with observed genealogical relationships in sequence data from nine nuclear genes in wild barley (Hordeum vulgare ssp. spontaneum), a highly self-fertilizing grass species. In self-fertilizing plants, gamete dispersal is severely limited; dissemination occurs primarily through seed dispersal. Also, heterozygosity is greatly reduced, which renders recombination less effective at randomizing genetic variation and causes larger portions of the genome to trace a similar history. Despite these predicted effects of this mating system, the majority of loci show evidence of recombination. Levels of nucleotide variation and the patterns of geographic distribution of mutations in wild barley are highly heterogeneous across loci. Two of the nine sampled loci maintain highly diverged, geographic region-specific suites of mutations. Two additional loci include region-specific haplotypes with a much shallower coalescence. Despite inbreeding, sessile growth habit, and the observation of geographic structure at almost half of sampled loci, parametric estimates of migration suggest that seed dispersal is sufficient for migration across the ≈3,500-km range of the species. Recurrent migration is also evident based on the geographic distribution of mutational variation at some loci. At one locus a single haplotype has spread rapidly enough to occur, unmodified by mutation, across the range of the species.
Mutations arise in a single individual at a single point in time and space, but they may slowly spread across a species range owing to dispersal, random genetic drift, and possibly selection (1). Individual organisms, or their gametes, spores, or seeds, migrate each generation. When amassed over thousands of generations, migration may lead to homogeneity in allele frequencies over substantial portions of a species' range. Because individual genes do not migrate in isolation but rather as part of a complete genome, we might expect geographic patterns of allelic differentiation to be homogeneous across the genome and estimates of migration rates to be correlated across different loci. However, not all evolutionary forces affect all parts of the genome in a homogeneous fashion. In particular, selection tends to act on very limited regions of the genome and often on changes at single nucleotide sites. The impact of selection will be limited to those regions that are correlated in transmission with the selected site. In addition, individual mutations arise at different points in time, and if demographic and migration forces are not consistent over time, heterogeneous patterns across loci may result.
The coalescent approach of population genetics provides a powerful means to investigate the spatiotemporal process of genetic change. Moreover, when applied to genes from across the genome, it provides a means of dissecting the gene-specific impact of various evolutionary forces. The coalescent approach looks backward in time by attempting to deconstruct the history of an observed gene genealogy (2, 3); it is this time-reversed inferential framework that allows us to trace historical spatial processes. Put differently, the coalescent process traces backward across both time and space to the most recent common ancestor of a sample. Each segment of the genome is a replicate of this coalescent process, and, given that dispersal and mutation are stochastic processes, comparisons across multiple loci are more likely to accurately reflect population history (3). The standard coalescent process can be expanded to two or more populations and to the estimation of migration between them (4–6) and can be adapted to include other forces such as population growth and recombination (3). Analytical methods that make use of observed mutations to estimate the gene genealogy at a locus have the potential to provide better estimates of population parameters than methods that use only summary statistics such as the number of segregating sites or the number of pairwise differences between samples (7, 8).
For the majority of problems in population genetics, the actual gene genealogy cannot be observed, and statistical methods must be used that account for uncertainties in the genealogy, provide a method of accepting or rejecting genealogies conditional on the data, and focus sampling on the most relevant subset of all possible genealogies (8–10). Given such a method, population parameters such as long-term effective population size, rate of migration for portions of a species range, and level of recombination can be estimated jointly (8–13). Our goal in this article is to apply this inferential framework to DNA sequence data from nine loci from samples that span the geographic range of wild barley (Hordeum vulgare ssp. spontaneum), a predominantly self-fertilizing species.
The potential for the structuring of genetic variation is greatest when the mating system or physical barriers to dispersal limit potential matings or the exchange of migrants. Self-fertilizing (selfing) plants, as inbreeding, sessile organisms, represent one extreme in the degree of genetic structuring, because gametes and adult organisms travel very short distances, if at all. Migration is primarily limited to seed dispersal. However, selfing species have a considerable advantage when undergoing dispersal (14). For outcrossing organisms, successful migration requires reaching a suitable habitat and then finding mating opportunities in a new population, within the lifespan of the migrant. For selfing species, only a suitable habitat is required (14). Individual migrants can found new populations or persist in an existing population without interbreeding and still can contribute to the future genetic diversity in the population or region.
In species with a history of inbreeding, larger portions of chromosomes are expected to trace the same or a highly correlated history owing to larger domains of linkage disequilibria (15). With perpetual self-fertilization, the maternal and paternal parent are one and the same, so two identical chromosomes are transmitted to their progeny. Reduced heterozygosity renders recombination less effective at randomizing mutations at different chromosomal sites and expands the imprint of selective sweeps or background selection against deleterious mutations (16).
Wild barley (H. vulgare ssp. spontaneum) is an annual, diploid grass species with chromosome number = 7 and an estimated rate of self-fertilization of 98% (17) and is the progenitor of cultivated barley (Hordeum vulgare ssp. vulgare). The natural distribution of wild barley ranges from the Mediterranean portion of the Middle East, across the Zagros Mountains, and into adjacent southwest Asia, a distance east to west of ≈3,500 km (see Fig. 1). The eastern and western portions of the species range are relatively low-elevation regions; wild barley has limited cold tolerance and is rare above a 1,500-m elevation (18). Wild barley populations are abundant in the western portion of the range, but the species is rare at higher elevations (e.g., parts of the Zagros and the continental plateau in Turkey and Iran) and sporadic in the Eastern portion of the range (18). The Zagros Mountains, trending northwest to southeast, roughly bisect the range with a series of smaller mountain ranges, including many peaks above 3,000 m and a tallest single peak of 4,500 m. Thus they represent a significant disruption of the natural range of the species and a possible barrier to the movement of animals that serve as potential seed dispersers. Spikelets of wild barley have long, barbed lemma awns, well suited for attachment to animal fur (19); a number of large animal species occur natively within the range of wild barley, including goats, boars, deer, and gazelles. Because of genomic resources developed for domesticated barley and previous studies of genetic diversity in the species (e.g., 17, 20–23), wild barley is an excellent candidate for the study of species-level migration patterns and for measuring the degree of geographic structure present at individual loci.
Fig. 1.

The geographic distribution of major haplotype variants from Lin et al. (22) at Adh3 is shown.
Sequence Diversity in Wild Barley. In a previous study of sequence diversity at the Adh3 locus among 25 accessions of wild barley, Lin et al. (22) identified two deeply diverged lineages in eastern and western portions of the range (Fig. 1). Samples from the eastern and western regions differed by 2.2% sequence divergence at Adh3, whereas three accessions from the Zagros region seemed to be recombinants between the eastern and western lineages (22). The geographic distribution of haplotypes and level of divergence at the other two independently segregating Adh loci bore little resemblance to that identified at Adh3, even though the same 25 individuals composed the sample (23). Indeed, sampled haplotypes at Adh1 and Adh2 seemed to be distributed almost at random across the range of the species, suggesting that the rate of migration was at least of the order of the temporal history of the coalescent process (23). Thus, although the distribution of haplotype diversity at Adh3 implies a barrier to gene flow across the range of wild barley, the broad geographic distribution of some haplotypes at Adh1 and Adh2 implies migration sufficient to distribute all haplotypes across the range (23).
This study was designed to further investigate the strength of migration across the geographic range of wild barley and to investigate heterogeneities in spatial patterns of genetic diversity. Accordingly, sequence data were obtained from six additional loci by using the same sample of 25 accessions used for the Adh genes (Table 5, which is published as supporting information on the PNAS web site, www.pnas.org). In allocating fixed experimental resources, we opted to increase our sample of loci while keeping the sample of accessions fixed at 25, because previous work had suggested that a sample size of 25 was sufficient to detect broad geographic patterns. However, sample size limits geographic resolution to broad-scale spatial heterogeneities. Thus, we focus on the Western, Zagros, and Eastern regions as defined above. The analyses employ a maximum-likelihood, coalescence-based approach (24) to estimate the level of genetic diversity in each population, reported here as ΘL, which, under a strict drift-mutation process, is equal to 4Neμ, where Ne is the effective population size and μ is the mutation rate (25). The level of migration (ℳ) among the three major portions of the species range and the samplewide extent of recombination (reported here as rL) are coestimated. The results reveal large heterogeneities across loci in diversity statistics, migration rates that are sufficient to produce spatial homogeneity in the absence of local selection, and some residual geographic patterns at two loci in addition to a strong geographic pattern similar to that of Adh3 at a third locus.
Materials and Methods
Sampled Loci and Sequencing Methods. New sequence data reported here derive from both coding and noncoding portions (introns and flanking regions) of α-amy1, Dhn5, Dhn9, G3pdh, Pepc, and Waxy (see Table 6, which is published as supporting information on the PNAS web site). The enzymatic Adh, α-amy1, G3pdh, and Pepc loci are common to many eukaryotic organisms. Waxy encodes granule-bound starch synthase (26). The Dhn loci are functional, nonenzymatic genes: Dhn5 expression is induced by exposure to low temperatures (e.g., 5°C), and Dhn9 is induced by dehydration (27). Initial amplification primers for all loci were designed based on sequence from the nr or EST databases in GenBank (www.ncbi.nlm.nih.gov/blast) and from quality-trimmed EST data available through the program harvest (28). Wild barley is predominately self-fertilizing; thus, sampling individuals is essentially equivalent to sampling gametes, and purified PCR products often can be sequenced directly. Sequence fragments were assembled by using phred/phrap/consed (University of Washington, Seattle), and, when present, vector sequence was screened out by using cross match (University of Washington, Seattle) (29–31). Assemblies of consensus sequence used a minimum quality criterion of a phred score ≥20 on both forward and reverse strands. polyphred (32) was used to screen for potentially polymorphic sites within sequences from individual accessions. PCR products that could not be sequenced directly (e.g., heterozygotes) were cloned, and at least three clones of each haplotype were sequenced. Singletons (nucleotide variants found only once in the sample) were reamplified and resequenced from both the forward and reverse strand. Amplification conditions and primers used for all loci are available from the authors on request.
Data Analysis and Statistics. Estimates of the number of segregating sites and sequence diversity statistics, including Watterson's θ (25) (denoted here as Θw), Tajima's π (33), and comparative statistics such as Tajima's D (34) (denoted here as T to distinguish this test from the conventional linkage disequilibrium statistic), from both coding and noncoding regions were calculated by using dnasp v. 3.53 (35). Haplotype trees for each locus were constructed by using statistical parsimony (36) as implemented by tcs v. 1.13 (37). Migration among portions of the range of wild barley was estimated by using the lamarc v. 1.1 package (24). The estimate of migration in lamarc, when scaled relative to the coestimated value of ΘL for each recipient population, is an estimate of the average number of migrants entering the population per generation. lamarc uses a Metropolis–Hastings Monte Carlo Markov chain algorithm to search for values of parameters compatible with the genealogical relationships estimated from the observed sample of sequences (11, 12). All analyses used the Felsenstein 1984 nucleotide substitution model (38, 39) and empirical base frequencies and transition/transversion ratios. To assure adequate extension of searches, final values for ΘL, ℳ (unscaled migration values), and rL from an initial analysis using Watterson's estimate of Θw and default settings of the program for ℳ and rL were plugged in as the starting values of a second round of analysis that used 20 initial chains of 1,000 and four final chains of 20,000 genealogies with 2,000 genealogies discarded per chain. These settings were used for three replicate searches, and “heating” was used to search for additional compatible genealogies. Heating used temperatures of 1, 1.2, 1.5, and 4; when swapping was very limited, temperatures were set to 1, 1.1, 1.3, and 1.8. Finally, a single replicate search using the same chain length and number of chains as above, with start parameters drawn from the final values from the second-round analysis, was used to confirm results. Reported results are from the third analysis. Only ΘL and ℳ were estimated for loci where DNA sequence showed no evidence of recombination. lamarc analyses were performed by using single nodes of the Linux Beowulf computer Lupin at the Institute of Geophysics and Planetary Physics (IGPP) and Linux computers at the Bioinformatics Core facility at the University of California, Riverside.
Coalescent simulations were performed by using the program ms (40) with 10,000 replicates per simulation. An initial estimate used two parameters, the mean Θw per locus from all sampled loci and a sample size of 25 individuals. A second simulation used a division into samples of 10, 7, and 8 individuals (sample sizes for the Western, Zagros, and Eastern regions), with the relative diversity of each sample, migration between portions of the range, and level of recombination based on the means across loci from lamarc estimates. For scaling of the recombination parameter ρ = 4N0r, where N0 is the initial population size in the simulation, we used a mutation rate of 5 × 10–9 per site and an estimate of species effective population size based on Θw at synonymous sites.
Results
Nucleotide Sequence Polymorphism. Total length of aligned sequence for the six new sequence data sets reported here is 7,351 bp, including 4,302 bp of coding sequence and 3,049 bp of sequence from noncoding regions. Together with sequence from the three Adh loci, total aligned sequence length is 12,566 bp. We were unable to obtain complete sequences from two individuals at the Dhn5 locus and one individual at the G3pdh locus; thus, data from these loci include 23 and 24 individuals, respectively. Diversity estimates for the nine loci are shown in Table 1. Levels of diversity among the loci are heterogeneous (P > 0.001, χ2 = 28.63, df = 8) (41) with mean Θw of 1.15 (×10–3) for Pepc, an order of magnitude below that found at Adh3, where Θw = 15.42. Sampled loci also differed in the minimum number of recombination events detected, with no recombination evident at α-amy1 or Pepc, whereas a minimum of five and six recombination events is apparent in Dhn5 and Waxy, respectively (Table 1).
Table 1. Estimates of nucleotide sequence diversity.
| Gene | Length, bp | ΘW per gene | ΘW, all sites | ΘW, synonymous sites | ΘW, nonsynonymous sites | T | Rm | rL |
|---|---|---|---|---|---|---|---|---|
| Adh1 | 1,362 | 3.71 | 2.73 (± 1.11) | 3.14 | 2.20 | -0.926 | 0 | — |
| Adh2 | 1,980 | 9.53 | 4.84 (± 1.72) | 5.72 | 3.31 | -1.289 | 2 | 0.253 (0.137, 0.487) |
| Adh3 | 1,873 | 27.81 | 15.42 (± 5.11) | 19.44 | 8.90 | 1.790 | 2 | 0.294 (0.178, 0.365) |
| α-amy1 | 856 | 2.65 | 3.10 (± 1.36) | 6.54 | 0.54 | -1.948* | 0 | — |
| Dhn5 | 1,061 | 11.11 | 10.59 (± 3.77) | 16.77 | 8.24 | -0.130 | 5 | 0.404 (0.296, 0.557) |
| Dhn9 | 1,011 | 4.77 | 4.90 (± 1.91) | 7.51 | 0.00 | -0.725 | 1 | 0.511 (0.318, 0.768) |
| G3pdh | 2,010 | 15.80 | 7.93 (± 2.64) | 10.84 | 0.48 | 0.823 | 1 | 0.094 (0.046, 0.268) |
| Pepc | 1,154 | 1.32 | 1.15 (± 0.61) | 2.13 | 0.00 | -0.023 | 0 | — |
| Waxy | 1,232 | 11.42 | 9.43 (± 1.05) | 17.25 | 0.88 | -0.615 | 6 | 0.457 (0.413, —) |
Values shown (× 10-3 for per-site measures) are for a common set of 25 samples at nine different loci in wild barley. For ΘW, all sites, SD is shown, based on no recombination. For the coalescence-based estimate of recombination rL, 95% confidence intervals are shown. ΘW, Watterson's estimate; T, Tajima's D test; Rm, minimum number of recombination events, rL, ratio between per-site recombination and per-site mutation rates. *, 0.01 < P < 0.05.
Five of the six additional loci reported here have very low levels of nonsynonymous nucleotide polymorphism (Table 1). At both Dhn9 and Pepc no nonsynonymous polymorphisms were found; α-amy1 and G3pdh included a single nonsynonymous change, whereas Waxy included two amino acid-encoding changes occurring only once and twice in the sample. However, in coding portions of Dhn5, 56% of nucleotide changes are nonsynonymous. In Dhn5, replacement substitutions are relatively common, as is evident from π = 7.54 (and Θw = 8.24) at nonsynonymous sites versus an average at all other loci of π = 1.63 (Table 7, which is published as supporting information on the PNAS web site).
With samples partitioned into Western, Zagros, and Eastern regions, average Θw across loci suggests a larger Ne for the Western region (Table 2). This difference is evident at eight of the nine loci. At Pepc, estimates of Θw are similar across the three regions (see Table 8, which is published as supporting information on the PNAS web site). Average Θw also suggests that Ne is greater in the Zagros than in the Eastern region. Estimates of Θw are inflated when the sample includes deeply divergent lineages, as is evident at Adh3 and G3pdh. Samples from the Zagros region included both Adh3 lineages and recombinants between them. The Western region also includes a single sample with the second of the two divergent Adh3 lineages, greatly increasing the number of segregating sites in the sample (see Table 8). The Eastern region includes one of the two major Adh3 lineages. At G3pdh, the Zagros region includes only one of the two divergent lineages and thus has much lower estimated Θw than do the Eastern and Western regions. With Adh3 and G3pdh excluded, average Θw is still larger in the Western region but very similar in the Eastern and Zagros regions.
Table 2. Estimates of θ (× 10-3) among geographic regions.
| Region | ΘW | π | ΘL |
|---|---|---|---|
| Western | 7.15 | 6.08 | 10.14 |
| Zagros | 4.47 | 5.34 | 2.10 |
| Eastern | 4.04 | 4.11 | 1.80 |
Values are based on number of segregating sites in the sample (ΘW) and the distribution of pairwise differences between sequences (π) and conditional on the underlying genealogy (ΘL).
Statistical parsimony analysis produces a unique distribution of genealogies at each locus, including the Adh1 and Adh2 loci that are closely linked on barley chromosome 4 (23). At several loci, region-specific haplotypes are observed. At α-amy1, four different haplotypes were detected among Western samples, one of which also predominates among the Eastern and Zagros samples (Fig. 2), whereas only one Zagros sample carries an additional single nucleotide change (see Fig. 4, which is published as supporting information on the PNAS web site). No amino acid variant was observed that differentiated this high-frequency haplotype. At Dhn5, 16 unique haplotypes were sampled, and there is evidence of recombination among haplotypes (see Fig. 5, which is published as supporting information on the PNAS web site). No geographic structure is apparent at the locus. At Dhn9, only two haplotypes, differentiated by 13 mutational steps, are found in the Eastern samples (see Fig. 6, which is published as supporting information on the PNAS web site). These same two haplotypes, and two additional types two and five mutational steps from them, are the only haplotypes present in the Zagros samples. Each of the Western samples contained a unique haplotype, and there is evidence of recombination among them. The two divergent lineages at G3pdh differ by a minimum of 42 nucleotide changes (and four insertion/deletion events) or 2.2% sequence divergence (see Fig. 7, which is published as supporting information on the PNAS web site). Only one amino acid-encoding polymorphism is present in the data set, an Ile/Val polymorphism that differs between the two major lineages. Half of the samples in the Western region carry the rarer of the two major haplotypes, and one individual from the Eastern region also has this haplotype. Nucleotide sequence diversity in the Zagros region includes only two segregating sites. There is evidence of recombination within the more common of the divergent lineages at G3pdh, but not among them. At Pepc there are six observed haplotypes. Two of these six predominate, occurring in 19 of the 25 samples (see Fig. 8, which is published as supporting information on the PNAS web site). One of these two principal haplotypes was not found in any Western samples. At Waxy, there are 22 haplotypes (see Fig. 9, which is published as supporting information on the PNAS web site). There is little evidence of geographic structure; neighboring haplotypes on the tree are often from different geographic regions. A minimum of six recombination events are inferred at the Waxy locus (Table 1).
Fig. 2.

The statistical parsimony gene genealogy of α-amy1 is shown.
Coalescent Estimation of Population Parameters. The coalescence-based estimates of migration show a wide range of values, with the frequency of migration for a per-locus, per-region pair ranging from 0 to 21.27 (see Table 9, which is published as supporting information on the PNAS web site). Across loci, average exchange of migrants between regions is close to one migrant per generation (Table 3). The one exception is migration from Eastern into Western regions, which is estimated at 3.54 migrants per generation. This value is inflated by an unusually large estimate of migration (21.27) from the Eastern into the Western region at Adh2; without this value, the mean across the other eight loci is 1.32, much closer to other estimates (Table 9).
Table 3. Bidirectional estimates of migration at all loci for three regions.
| Western | Zagros | Eastern | |
|---|---|---|---|
| Western | — | 1.77 | 3.54 |
| Zagros | 1.11 | — | 1.31 |
| Eastern | 1.46 | 0.90 | — |
Values reported are estimated from the region along the x axis into the region along the y axis. Reported values are 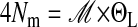 of the recipient population.
of the recipient population.
Estimates of θ = 4Neμ (from Θw, π, and ΘL) for each region and each locus are shown in Table 2. Coalescence-based estimates also suggest a larger effective population size for the Western portion of the sample, with the average ΘL for the Western region roughly five times greater than that from both Eastern and Zagros regions. As was the case for Θw, average ΘL was slightly higher in the Zagros than in the Eastern sample. There is heterogeneity among estimates of ΘL at individual loci; in the Western samples, for example, ΘL has a range from 0.81 at Pepc to 21.03 at Adh1, with an average value of 10.14. The ΘL values among Eastern samples show even greater variation, with Dhn9 at 0.04 and Waxy at 6.07.
Intralocus recombination, in units of recombination events per mutation per site per generation, ranges from rL = 0.094 at G3pdh to rL = 0.511 at Dhn9. Estimates of recombination as inferred from the coalescent estimator are roughly consistent with the observed ratio of the minimum number of recombination events at a locus [by using the method of Hudson and Kaplan (42)] and the number of segregating sites at a locus.
Coalescent Simulations. An initial simulation using only average Θw per locus and a sample size of 25 chromosomes produced an estimated 95% confidence interval of Θw per site of 3.0 and 13.1. Three loci, Adh1, α-amy1, and Pepc, are below the 95% confidence interval, and only Adh3 was above the interval. A more complex simulation (described above) produces a 95% confidence interval of 4.2 and 15.0 (Fig. 3).
Fig. 3.

Distribution of Θw per site (×10–3) from a 10,000-replicate coalescent simulation based on estimates of diversity, migration, and recombination from lamarc. The 2.5% and 97.5% confidence intervals are shown in hatched shading. Actual values of Θw per site for nine sample loci are shown as dotted lines.
Discussion
We have used a coalescence-based approach to simultaneously estimate levels of diversity, migration, and recombination using nucleotide sequence data from nine loci from 25 samples that span the native range of wild barley. The mean frequency of migration across loci is one to three migrations per generation among the three major portions of the species range. This rate seems sufficient to ensure geographic homogeneity of diversity. Levels of diversity at individual loci, as inferred by Θw, π, and ΘL, are heterogeneous, with a 13-fold difference in Θw among loci. Estimated levels of diversity differ dramatically among the three major geographic regions occupied by wild barley, apparently reflecting major differences in Ne among regions. The set of samples from the Western portion of the range has consistently higher diversity based on the distribution of segregating sites (Θw and π); the difference is much more pronounced based on estimates of ΘL, which are conditional on the genealogy relating haplotypes. Diversity at two of the nine sampled loci (Adh3 and G3pdh) is divided into two deeply divergent lineages largely specific to either the Western or Eastern region. Finally, intralocus recombination is apparent at six of the nine sampled loci despite the high frequency of self-fertilization in wild barley. Only the three loci with the lowest levels of diversity show no evidence of recombination.
Under the simplest scenario (i.e., that of a drift/mutation/migration equilibrium), gene genealogies across the genome are expected to be replicates of the same stochastic process. Instead we find that the patterns of diversity fall into distinct categories, reminiscent of those described by Lewontin and Hubby (43) in their classic paper on isozyme variation. Thus, we can summarize observed nucleotide sequence diversity at sampled loci, using six qualitative categories (Table 4).
Table 4. Comparison of levels of nucleotide diversity and extent of geographic structuring of haplotype variation at nine loci.
| Diversity | Geographic structure | No geographic structure |
|---|---|---|
| Low | α-amy1 | Adh1, Pepc |
| Medium | Dhn9 | Adh2 |
| High | Adh3, G3pdh | Dhn5, Waxy |
Low Polymorphism Throughout the Species Range, No Geographic Structure. Adh1 and Pepc demonstrate this pattern. The number of observed haplotypes is limited (11 and 6), and haplotypes that are observed differ by a small number of mutational steps, owing to the small number of segregating sites (Fig. 8 and figure 1 in ref. 21). Identical haplotypes are frequently found in samples from very distant localities, a result consistent with the parametric estimates of migration. The pattern is consistent with limited Ne, perhaps resulting from a selective sweep, in combination with relatively high rates of migration. Most amino acid replacements are found as singletons in the sample and may result from a selection/mutation balance in small local populations.
Low Polymorphism Throughout the Species Range, with Geographic Structure. The α-amy1 locus most clearly demonstrates this pattern. Twenty-one of 25 samples carry an identical haplotype, and samples from very distant locations have the same haplotype (Fig. 2). Almost all haplotype diversity is found in the Western portion of the species' range. However, diversity is extremely limited throughout the geographic range of the species. This pattern is most consistent with limited Ne in combination with recurrent migration and/or a selective sweep at the locus.
Moderate Polymorphism Throughout the Species Range, No Geographic Structure. The Adh2 locus is most representative of this class. The observed number of haplotypes is much greater than in the two categories described above, but the number of segregating sites is sufficiently small that mutational steps between haplotypes are limited, and observed haplotypes often form small clusters separated by only one or two mutational steps. This pattern is most consistent with intermediate Ne in populations at equilibrium between drift, migration, and mutation.
Moderate Polymorphism, with Geographic Structure. This pattern is most evident at Dhn9, where the Eastern and Zagros regions are dominated by two haplotypes (Fig. 6). A much more diverse array of haplotypes, including recombinant types, is evident in the Western region. The pattern is consistent with drift/ migration/mutation equilibrium, likely in combination with a much larger Ne for one portion of the species range.
High Diversity, No Geographic Structure. Dhn5 and Waxy are most representative of this class. Almost every sampled individual carries a unique haplotype, and many inferred haplotypes are not found in the sample; i.e., there are few clusters of related haplotypes, and observed haplotypes often differ by a large number of mutational steps (Figs. 5 and 9). Parametric, bidirectional estimates of migration for Dhn5 and Waxy are relatively large for all pairs of populations (Table 9). Both loci in this class have a relatively high inferred number of recombination events, consistent with estimates of rL. The patterns observed are consistent with a large Ne and migration.
High Diversity at the Locus, Strong Pattern of Geographic Structure. Adh3 and G3pdh fall into this class. At both loci, deeply divergent lineages are restricted almost entirely to one of the major portions of the species range. The majority of segregating sites at these loci are accounted for by differences between the two major haplotype groups: these differences result in estimates of Θw per site at Adh3 and G3pdh for the entire sample of 14.9 and 7.93, respectively. However, within-lineage diversity is relatively limited, with Θw of 5.11 and 2.34 and 0.44 and 2.18 for each of the two major lineages at Adh3 and G3pdh, respectively. Possible causes of this pattern include restricted migration between regions or selection for preservation of the divergent haplotypes based on differential patterns of environmental adaptation.
Several features of these data are largely consistent across the sampled loci. First, the Western region, thought to be the geographic center of barley domestication, has much higher levels of diversity at almost all loci. Second, all loci with at least moderate levels of diversity show clear evidence of intralocus recombination, despite the extreme restriction in recombination associated with a mating system of ≈98% self-fertilization. Third, estimates of migration across loci are broadly consistent. Are moderate to high levels of migration plausible for wild barley? As we have noted, wild barley has long, barbed awns that promote dispersal of disarticulated spikelets through adherence to animal fur. Many large and small mammal species are native to the region. Moreover, it is possible that hunter–gatherer peoples could have transported wild barley as they moved from region to region. So moderate migration rates are consistent with the population biology of wild barley.
The evident heterogeneity in diversity statistics is hard to reconcile with the assumption that the genealogies estimated in this study are replicates of the same stochastic process. Even when detailed population parameters are incorporated in the coalescent simulations, replicates of the neutral evolutionary process can result in very different levels of sequence diversity at a locus (Fig. 3). However, heterogeneity at sampled loci is greater still than that expected based on replicates of the same neutral process. We must therefore conclude that the genome of wild barley is a mosaic of different histories generated by different evolutionary processes. It is difficult to escape the conclusion that selection has played a role in molding the geographic structure of genetic diversity at some of the loci in this sample. Those loci with very little diversity and homogeneous geographic distributions may have experienced a selective sweep (Adh1, Pepc), whereas those loci that have maintained strong geographic patterns of diversity despite ample migration may represent locally adapted types (Adh3, G3pdh). Although these patterns are suggestive, much larger samples need to be assayed to establish the generality of our conclusions. If, however, we take the data at face value, four of nine loci may show some signature of selection. At a minimum, two of nine loci must be affected by selection because it does not seem possible to simultaneously reconcile the strongly divergent geographic and genealogical patterns represented by the Adh1, Pepc class with the Adh3, G3pdh class. So it seems that somewhere between 20% and 50% of the loci that constitute the sample show some imprint of selection over the time period and geographic range spanned by the estimated genealogies.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Robert Wayne Allard (1919–2003). Professor Allard was an exceptional researcher and teacher who played the central role in the development of experimental plant population genetics. We thank P. Beerli, M. K. Kuhner, and J. Yamato for assistance with data analysis using the lamarc package; T. J. Close for helpful discussion; and H. Chen, M. L. Durbin, T. Girke, A. M. Montalvo, and P. Robinson for technical assistance. P. Beerli, A. H. D. Brown, A. G. Clark, B. S. Gaut, C. Vogl, and D. E. Wolf provided helpful comments on an earlier version of the manuscript. This research was partially supported by National Science Foundation Grant DEB-0129247.
Data deposition: The sequences reported in this article have been deposited in the GenBank database (accession nos. AY349195–AY349349).
References
- 1.Barton, N. H. & Wilson, I. (1995) Philos. Trans. R. Soc. London B 349, 49–59. [DOI] [PubMed] [Google Scholar]
- 2.Kingman, J. F. C. (1982) Stochastic Processes: Formalism Appl. Prc. Winter Sch. 13, 235–248. [Google Scholar]
- 3.Hudson, R. R. (1990) in Oxford Survey of Evolutionary Biology, eds. Dawkins, R. & Ridley, M. (Oxford Univ. Press, Oxford), Vol. 7, pp. 1–44. [Google Scholar]
- 4.Notohara, M. (1990) J. Math. Biol. 29, 59–75. [DOI] [PubMed] [Google Scholar]
- 5.Takahata, N. (1988) Genet. Res. 52, 213–222. [DOI] [PubMed] [Google Scholar]
- 6.Takahata, N. & Slatkin, M. (1990) Theor. Popul. Biol. 38, 331–350. [DOI] [PubMed] [Google Scholar]
- 7.Felsenstein, J. (1992) Genet. Res. 59, 139–147. [DOI] [PubMed] [Google Scholar]
- 8.Stephens, M. & Donnelly, P. (2000) J. R. Stat. Soc. B 62, 605–635. [Google Scholar]
- 9.Griffiths, R. C. & Tavare, S. (1994) Philos. Trans. R. Soc. London B 344, 403–410. [DOI] [PubMed] [Google Scholar]
- 10.Kuhner, M. K., Yamato, J. & Felsenstein, J. (1995) Genetics 140, 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beerli, P. & Felsenstein, J. (2001) Proc. Natl. Acad. Sci. USA 98, 4563–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beerli, P. & Felsenstein, J. (1999) Genetics 152, 763–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhner, M. K., Yamato, J. & Felsenstein, J. (2000) Genetics 156, 1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker, H. G. (1955) Evolution 9, 347–348. [Google Scholar]
- 15.Nordborg, M., Borevitz, J. O., Bergelson, J., Berry, C. C., Chory, J., Hagenblad, J., Kreitman, M., Maloof, J. N., Noyes, T., Oefner, P. J., et al. (2002) Nat. Genet. 30, 190–193. [DOI] [PubMed] [Google Scholar]
- 16.Charlesworth, B., Morgan, M. T. & Charlesworth, D. (1993) Genetics 134, 1289–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown, A. H. D., Zohary, D. & Nevo, E. (1978) Heredity 41, 49–62. [Google Scholar]
- 18.Zohary, D. & Hopf, M. (1994) Domestication of Plants in the Old World: The Origin and Spread of Cultivated Plants in West Asia, Europe, and the Nile Valley (Oxford Univ. Press, New York).
- 19.von Bothmer, R., Jacobsen, N., Baden, C., Jorensen, R. B. & Linde-Laurson, I. (1995) An Ecogeographical Study of the Genus Hordeum (Food Agric. Org. U.N., Rome).
- 20.Brown, A. H. D., Nevo, E., Zohary, D. & Dagan, O. (1978) Genetics 49, 97–108. [Google Scholar]
- 21.Cummings, M. P. & Clegg, M. T. (1998) Proc. Natl. Acad. Sci. USA 95, 5637–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin, J.-Z., Brown, A. H. D. & Clegg, M. T. (2001) Proc. Natl. Acad. Sci. USA 98, 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin, J.-Z., Morrell, P. L. & Clegg, M. T. (2002) Genetics 162, 2007–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuhner, M. K., Beerli, P., Yamato, J. & Felsenstein, J. (2002) lamarc, Likelihood Analysis with Metropolis Algorithm using Random Coalescence (Univ. of Washington, Seattle) Version 1.1.
- 25.Watterson, G. A. (1975) Theor. Popul. Biol. 7, 256–276. [DOI] [PubMed] [Google Scholar]
- 26.Mason-Gamer, R. J., Weil, C. F. & Kellogg, E. A. (1998) Mol. Biol. Evol. 15, 1658–1673. [DOI] [PubMed] [Google Scholar]
- 27.Choi, D. W., Zhu, B. & Close, T. J. (1999) Theor. Appl. Genet. 98, 1234–1247. [Google Scholar]
- 28.Wannamaker, S. & Close, T. (2002) harvest (Univ. of California, Riverside, CA) Version 1.07.
- 29.Ewing, B., Hillier, L., Wendl, M. C. & Green, P. (1998) Genome Res. 8, 175–185. [DOI] [PubMed] [Google Scholar]
- 30.Ewing, B. & Green, P. (1998) Genome Res. 8, 186–194. [PubMed] [Google Scholar]
- 31.Gordon, D., Abajian, C. & Green, P. (1998) Genome Res. 8, 195–202. [DOI] [PubMed] [Google Scholar]
- 32.Nickerson, D. A., Tobe, V. O. & Taylor, S. L. (1997) Nucleic Acids Res. 25, 2745–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tajima, F. (1983) Genetics 105, 437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tajima, F. (1989) Genetics 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rozas, J. & Rozas, R. (1999) Bioinformatics 15, 174–175. [DOI] [PubMed] [Google Scholar]
- 36.Templeton, A. R., Crandall, K. A. & Sing, C. F. (1992) Genetics 132, 619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clement, M., Posada, D. & Crandall, K. A. (2000) Mol. Ecol. 9, 1657–1659. [DOI] [PubMed] [Google Scholar]
- 38.Kishino, H. & Hasegawa, M. (1989) J. Mol. Evol. 29, 170–179. [DOI] [PubMed] [Google Scholar]
- 39.Swofford, D., Olsen, G. L., Waddell, P. J. & Hillis, D. M. (1996) in Molecular Systematics, eds. Hillis, D. M., Moritz, C. & Mable, B. K. (Sinauer Assoc., Inc., Sunderland, MA), pp. 407–514.
- 40.Hudson, R. R. (2002) Bioinformatics 18, 337–338. [DOI] [PubMed] [Google Scholar]
- 41.Elandt-Johnson, R. C. (1971) Probability Models and Statistical Methods in Genetics (Wiley, New York).
- 42.Hudson, R. R. & Kaplan, N. L. (1985) Genetics 111, 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewontin, R. C. & Hubby, J. L. (1966) Genetics 54, 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}