

Abstract
A complex mechanism may be operational for dendritic cell (DC) maturation, wherein Toll-like receptor and other signaling pathways may be coordinated differently depending on the nature of the pathogens, in order for DC maturation to be most effective to a given threat. Here, we show that IFN-α/β signaling is selectively required for the maturation of DCs induced by double-stranded RNA or viral infection in vitro. Interestingly, the maturation is still observed in the absence of either of the two target genes of IFN-α/β, TLR3 and PKR (double-stranded-RNA-dependent protein kinase R), indicating the complexity of the IFN-α/β-induced transcriptional program in DCs. We also show that the DCs stimulated in vivo by these agents can migrate into the T cell zone of the spleen but fail to mature without the IFN signal. The immune system may have acquired the selective utilization of this cytokine system, which is essential for innate antiviral immunity, to effectively couple with the induction of adaptive immunity.
Conserved microbial structures, termed pathogen-associated molecular patterns (PAMPs) (1), are recognized by innate immunity receptors such as Toll-like receptors (TLRs) (2, 3). The activation of TLRs results in the induction of the genes involved in antimicrobial activity as well as the maturation of antigen-presenting cells (APCs), typically dendritic cells (DCs), that is central to the induction of adaptive immunity (2, 4). Whereas the cardinal features of signaling molecules and transcription factors functioning downstream of each TLR have been documented well, there is increasing evidence that the repertoire of TLRs for detecting a given pathogen may coordinate a response tailored for defense against a given pathogen, and that molecules other than TLRs also participate in the response in collaboration or in parallel with TLRs (5, 6).
The adaptive immune response against viruses depends on the CTL response and antibody production (7, 8), and DC maturation triggered by virus-associated molecular patterns through TLRs is considered to be central to the induction of virus-specific T cell responses (9). Notably, it has been shown that the induction of CTL response against certain viruses depends on IFN-α/β signaling (7, 10). Moreover, IFN-α/β has been shown to potently enhance antibody response through DC stimulation (11). On the other hand, IFN-β is also induced rapidly following exposure to a wide variety of nonviral infectious agents such as lipopolysaccharide (LPS) (12, 13) or unmethylated DNA (CpG) (14), raising the issue of how IFN-α/β signaling contributes to DC functions in response to each PAMP or viral infection. In the present study, we aimed at determining the role of IFN-α/β signaling in the regulation of DC functions, i.e., their maturation and migration, induced by stimulation with distinct PAMPs or infection by an RNA virus.
Materials and Methods
Mice. IFNAR1–/– mice (10) were purchased from B & K Universal (Hull, U.K.). PKR–/– mice (15) were provided by C. Weissmann (University of Zurich, Zurich). All of the mice were maintained under specific pathogen-free conditions.
Reagents. poly(I:C) and LPS from Salmonella minnesota Re-595 were purchased from Amersham Biosciences and Sigma, respectively. CpG was purchased from Hokkaido System Science (16). We confirmed that treatment of the poly(I:C) preparation with RNaseA (Sigma) abolished poly(I:C) stimulatory activity for DCs. Recombinant murine IFN-β was kindly provided by Toray Industries (Tokyo).
RNA Analysis. RNA extraction and reverse-transcription reaction were performed as described (17, 18). Quantitative real-time RT-PCR analysis was performed by using a LightCycler and SYBRGreen system (Roche Applied Science), and data were normalized by the β-actin expression level for each individual sample. Primers for β-actin, IFN-α4, -nonα4, -β, TNF-α, and IL-6 have been described (18). The following primers specific for IL-12 p35 and IL-12 p40 were used: IL-12 p35, 5′-CCTGCACTGCTGAAGACATC-3′ (sense) and 5′-GAAGCAGGATGCAGAGCTTC-3′ (antisense); IL-12 p40, 5′-GACACGCCTGAAGAAGATGAC-3′ (sense) and 5′-TAGTCCCTTTGGTCCAGTGTG-3′ (antisense).
Preparation and Analysis of DCs. Immature DCs were generated from mouse bone marrow (BM) as described (19). Immature DCs were collected and further cultured with or without 100 μg/ml poly(I:C), 100 ng/ml LPS, or 0.1 μM CpG in a fresh medium. Infection of DCs with viruses was carried out as described (17). DCs were stained with FITC-conjugated CD11c and PE-conjugated antibodies against CD40, CD80, CD86, or MHC class I or II (Pharmingen). Flow cytometric analysis was performed by using FACSCalibur with cellquest software (BD Biosciences). For mixed lymphocyte reaction assay, CD4+ or CD8+ T cells purified with MACS (Miltenyi Biotec, Auburn, CA) from a BALB/c spleen were used as responder cells (5 × 104). The responder cells were cultured with irradiated (30 Gy) DCs as stimulator cells. The cultures were pulsed with 1 μCi (1 Ci = 37 GBq) of [3H]thymidine at the final 15 h. [3H]Thymidine incorporation was measured by β-scintillation counting.
Electrophoretic Mobility-Shift Assay. After stimulation, the cell extract was prepared from DCs and analyzed by electrophoretic mobility-shift assay using an oligonucleotide probe containing the NF-κB-binding site of the IFN-β promoter or the IFN-stimulated responsive element (ISRE) of the 2,5-oligoadenylate synthetase promoter as described (20). We performed supershift with anti-RelA antibody (Santa Cruz Biotechnology).
Immunohistochemistry. Spleens were embedded in an OCT compound and snap-frozen in liquid nitrogen. Cryostat sections (10-μm-thick) were fixed with acetone and stained with FITC-conjugated anti-B220 and biotin-conjugated anti-CD11c or anti-CD86 antibodies (Pharmingen), and subsequently with streptavidin-Alexa Fluor 546 (Molecular Probes). Sections were examined by using a fluorescence microscope (Nikon).
Results
Selective Involvement of IFN-α/β Signaling in Double-Stranded RNA (ds-RNA)-Induced DC Maturation. To examine the contribution of IFN-α/β signaling to DC maturation, BM-derived WT DCs or mutant DCs from mice lacking the IFN-α/β receptor component, IFNAR1 (IFNAR1–/– DC) (10), were stimulated with poly(I:C), LPS, or CpG and subjected to flow cytometric analysis for the induction of MHC and costimulatory molecules. Both IFN-α and -β genes were induced by poly(I:C), whereas only the IFN-β gene is induced by LPS or CpG in WT DCs (data not shown and refs. 12–14). As shown in Fig. 1a, all these molecules were induced in WT DCs in response to the three stimuli. In contrast, a different observation was made with IFNAR1–/– DCs. Most notably, the poly(I:C)-mediated induction was abolished for all molecules. The induction of these molecules by LPS or CpG was also suppressed in IFNAR1–/– DCs; however, the suppression was not as severe as that of the poly(I:C)-dependent induction except for the MHC class I induction, which was impaired in IFNAR1–/– DCs in response to all three stimuli.
Fig. 1.

Selective requirement of IFN-α/β signaling in DC maturation in response to ds-RNA. (a) WT or IFNAR1–/– DCs were stimulated with poly(I:C), LPS, or CpG for 24 h and analyzed for cell-surface expression of the indicated molecules by flow cytometry. (b) WT DCs were stimulated with poly(I:C) or 1,000 units/ml IFN-β and analyzed by flow cytometry. (c) To measure the allostimulatory activity of DCs, poly(I:C)-, LPS-, or CpG-stimulated or unstimulated WT or IFNAR1–/– DCs were irradiated and incubated with allogeneic BALB/c CD4+ or CD8+ T cells at various cell concentrations as indicated. The percent of control [3H]thymidine incorporation was calculated as follows: (cpm obtained in the presence of stimulated DCs)/(cpm obtained in the presence of unstimulated DCs). Data are expressed as mean ± SD of triplicate samples. A representative of two experiments is shown.
Because the ds-RNA induces a large amount of IFN-α/β compared with the other PAMPs examined here (13), we next examined whether IFN alone could elicit the signal for full DC maturation. As shown in Fig. 1b, the DCs stimulated by a high IFN-β dose (1,000 units/ml) showed the induction of the MHC and costimulatory molecules, but it was considerably lower than that in the case of poly(I:C)-stimulated DCs. These results therefore suggest that IFN-α/β signaling per se is not sufficient for full DC maturation.
T Cell Activation by DCs Stimulated by PAMPs. To further assess the status of the DC maturation induced by these PAMPs, we next examined the T cell stimulatory activities of the WT and IFNAR1–/– DCs, stimulated by either of the three PAMPs, using a mixed lymphocyte reaction assay for CD4+ or CD8+ T cells. As shown in Fig. 1c, the stimulatory activity was impaired for both classes of T cell in the poly(I:C)-stimulated IFNAR1–/– DCs. As expected from the results in Fig. 1a, the stimulatory activity for CD8+ T cells is also impaired in the LPS- or CpG-stimulated IFNAR1–/– DCs, but the impairment was not as severe as that observed in the poly(I:C)-stimulated IFNAR1–/– DCs. In addition, IFNAR1–/– DCs also failed to activate the 2C-transgenic CD8+ T cells (21) when stimulated with poly(I:C), whereas the same T cells could be activated when stimulated with LPS (Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). These results further indicate the selective contribution of IFN-α/β signaling to ds-RNA-dependent DC maturation.
Amplification of DC Signaling by IFN-α/β in Response to poly(I:C). To gain more insight into the maturation defect in IFNAR1–/– DCs, we studied the mRNA induction kinetics for inflammatory cytokines in DCs stimulated with poly(I:C) or LPS by real-time RT-PCR (Fig. 2a). Abnormal mRNA induction profiles of TNF-α, IL-6, IL-12 p40, and IL-12 p35 were observed when IFNAR1–/– DCs were stimulated with poly(I:C), but not when they were stimulated with LPS. Most notably, the induction of these cytokine mRNAs occurs at an early phase of the stimulation but is not sustained at a later phase in IFNAR1–/– DCs. The kinetics of TNF-α protein induction in the culture supernatant determined by ELISA was consistent with the kinetics of mRNA induction (Fig. 7, which is published as supporting information on the PNAS web site).
Fig. 2.

IFN-α/β signaling in gene induction events and NF-κB activation in DCs. DCs were stimulated for the indicated time with poly(I:C) or LPS, and the kinetics of cytokine (a) and chemokine receptor (b) mRNA levels was determined by real-time RT-PCR. (c) NF-κB DNA binding activity in poly(I:C)- or LPS-stimulated DCs was measured by electrophoretic mobility-shift assay. The arrowhead indicates the NF-κB complex. The specificity of the NF-κB complex was determined by adding 100-fold molar excess of a specific competitor or a specific antibody to RelA. The same cell lysates were also analyzed by immunoblotting (IB) with the anti-RelA antibody to monitor protein expression level.
We next studied mRNA expression for chemokine receptors, because DCs undergo concomitant down-regulation of inflammatory chemokine receptors (such as CCR1) and induction of CCR7 during their maturation (22). As shown in Fig. 2b, the expression profiles of these mRNAs, namely CCR1 mRNA down-regulation and CCR7 mRNA induction, remained the same between the poly(I:C)-stimulated WT and IFNAR1–/– DCs. Taken together with the above results, we hypothesized the following mechanism of biphasic DC signaling by ds-RNA: In the early phase of DC signaling, DCs are stimulated by ds-RNA to induce the mRNAs for IFN-α/β, inflammatory cytokines, and CCR7, and the IFN signal, in turn, acts on the amplification of the DC signaling to sustain cytokine expression and to induce effective DC maturation in the late phase.
The stimulation by ds-RNA or other PAMPs commonly results in the activation of the NF-κB family of transcription factors. The above observations prompted us to examine the NF-κB activation during the DC stimulation by poly(I:C) or LPS. RelA activation reached a peak 6 h after poly(I:C) stimulation, whereas its activation by LPS occurred faster, peaking 2 h after stimulation (Fig. 2c). Interestingly, the RelA activation by poly(I:C) was suppressed in IFNAR1–/– DCs, whereas the LPS-induced activation occurred almost normally in these cells. Thus, RelA activity is also selectively dependent on IFN signaling during the poly(I:C)-induced DC maturation. It is worth noting that the poly(I:C)-induced RelA activation was not significantly suppressed at an early phase, i.e., 2 h after stimulation, but it is not enhanced at the later stage (Fig. 2c). Therefore, it is likely that the IFN signal is not required for the early phase of the RelA activation but necessary for enhancing its activation in the late phase. In view of the scarce evidence that the IFN signal directly activates RelA, it is likely that the IFN signal enhances RelA activation indirectly by inducing a molecule integrated into the poly(I:C) signaling.
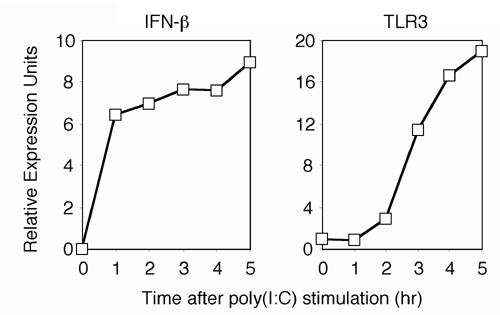
Role of TLR3 or PKR in poly(I:C)-Induced DC Maturation. What is the target of IFN-α/β functioning downstream of its signaling pathway to induce DC maturation? The recently identified TLR3 is shown to be selectively involved in ds-RNA recognition (23), and it has also been reported that TLR3 mRNA is induced by IFN-α/β (13, 24, 25). The above observations, together with those in previous reports, suggest that the IFN-mediated induction of the TLR3 gene and subsequent enhancement of TLR3 signaling are critical to the ds-RNA-induced DC maturation at the late phase. Congruent with this notion is the observation that the TLR3 mRNA induction is followed by the induction of IFN-β mRNA during the poly(I:C)-induced maturation of DCs (Fig. 8, which is published as supporting information on the PNAS web site).
To examine the effect of TLR3 gene deficiency on the ds-RNA-induced DC maturation, we generated TLR3-deficient mice, using a strategy essentially similar to that of a previous report (ref. 16; Fig. 9, which is published as supporting information on the PNAS web site), and analyzed BM-derived DCs (TLR3–/– DCs). Consistent with a previous report (23), the poly(I:C)-induced expression of costimulatory molecules and MHC class II was significantly suppressed in TLR3–/– DCs (Fig. 3a). The induction of IFN-β, TNF-α, and IL-6 mRNAs was also severely impaired in TLR3–/– DCs (Fig. 3b). These results indicate that TLR3 is indeed critical for the full DC response to ds-RNA. On the other hand, the poly(I:C) response is not abolished in TLR3–/– DCs. The induction of MHC class I is almost intact, and CD86 and CD40 are both induced, albeit at lower levels relative to those in WT DCs (Fig. 3a). Furthermore, the induction of IFN-α mRNAs was also detectable in the poly(I:C) stimulated TLR3–/– DCs (Fig. 3b).
Fig. 3.

Role of TLR3 or PKR induced by IFN-α/β in poly(I:C)-stimulated DCs. (a) WT, TLR3–/–, or PKR–/– DCs were stimulated with poly(I:C) or LPS for 24 h and analyzed as in Fig. 1a.(b) Real-time RT-PCR analysis of indicated cytokine mRNAs after poly(I:C) stimulation in WT, TLR3–/–, or PKR–/– DCs.
The above results indicate that a molecule other than TLR3 is also involved in ds-RNA signaling. In this regard, PKR is activated through direct interaction with ds-RNA and its gene induction is also dependent on IFN-α/β (26). To determine whether PKR is also involved in ds-RNA-induced DC maturation, we analyzed DCs from PKR-deficient mice (PKR–/– DCs) (15). As shown in Fig. 3, PKR–/– DCs exhibited a normal induction of T cell stimulatory molecules and cytokine mRNAs, except that the induction of IFN-α/β mRNAs was slightly suppressed. Thus, although TLR3 induction by IFN-α/β may be a critical event missing in the IFNAR1–/– DC maturation induced by ds-RNA, PKR is seemingly not involved in this process.
IFN-α/β and TLR3 Signaling in DC Maturation by Virus. We next examined whether IFN-α/β and TLR3 signaling is also essential for DC maturation in response to infection by a live RNA virus. Because a suitable assay system has not been described previously with which to analyze the virus-induced DC maturation in vitro, we first attempted to establish such a system. We infected DCs with several viruses, such as lymphocytic choriomeningitis virus, encephalomyocarditis virus, rotavirus, vesicular stomatitis virus (VSV), and Newcastle disease virus (NDV), and found that NDV induces DC maturation most effectively (Fig. 10, which is published as supporting information on the PNAS web site). Because the UV-inactivated NDV fails to induce DC maturation in this system, viral infection and replication are considered to be necessary for the maturation (Fig. 10).
Similar to the response to poly(I:C), a high-level expression of MHCs and costimulatory molecules was induced in the BM-derived DCs from WT mice on NDV infection, whereas the induction of these molecules was again abolished in the IFNAR1–/– DCs (Fig. 4a). The induction of TNF-α, IL-12 p40, and IL-6 mRNAs was also severely impaired in the IFNAR1–/– DCs (Fig. 4b). These results indicate that IFN signaling is also essential for the NDV-induced DC maturation. To investigate to what extent TLR3 contributes to this process, TLR3–/– DCs were subjected to this assay. In contrast to the IFNAR1–/– DCs, maturation occurred normally in the TLR3–/– DCs, as revealed by the induction of MHCs and costimulatory molecules on NDV infection (Fig. 4a). The induction of IFN-α/β, TNF-α, IL-12, and IL-6 mRNAs was also normal in the TLR3–/– DCs (Fig. 4c). In addition, essentially the same observations were made in the PKR–/– DCs (Fig. 4 a and c), except that IL-12 p40 production further increased in the PKR–/– DCs, for reasons that are not yet clear at present (Fig. 4c). These results point to the complexity of the virus-induced DC maturation process (see Discussion).
Fig. 4.

Role of IFN-α/β signaling in live-virus-mediated DC maturation. (a) WT, TLR3–/–, PKR–/–, and IFNAR1–/– DCs were infected with NDV (0.5 hemagglutinating units) for 18 h, and the indicated molecules were analyzed by flow cytometry. (b and c) The induction of mRNAs for the indicated cytokines at the indicated time after NDV infection of WT, IFNAR1–/–, PKR–/–, or TLR3–/– DCs was monitored by real-time RT-PCR.
Enhancement of DC Activation by IFN-α/β Signaling on Viral Infection. To further characterize the maturation defect in IFNAR1–/– DCs during NDV infection, we studied the kinetics of CD86 expression by DCs (Fig. 5a). CD86 became up-regulated in the WT DCs 3 h after NDV infection and its expression level increased until 18 h, at which time it reached a plateau. Interestingly, the early induction of CD86 in the IFNAR1–/– DCs, i.e., 3 h after NDV infection, occurred similarly to WT DCs, but its expression level did not increase at 18 h (Fig. 5a). Essentially the same result was observed in response to poly(I:C) (data not shown). Furthermore, the initial induction of TNF-α in the IFNAR1–/– DCs (≈6 h) was normal, but in the later phase, it could not reach the levels of that in the WT DCs (Fig. 7). The RelA activation occurred normally until 2 h in IFNAR1–/– DCs on NDV infection. This activity was further increased until 12 h after viral infection in the WT DCs, but it was not observed in the IFNAR1–/– DCs after 6 h (Fig. 5b). Together with the observation of the activation of the ISGF3 complex after 6 h in the WT DCs by NDV (Fig. 5b), our data suggest that the mechanism of the IFN-dependent positive feedback regulation of DC maturation is operational and critical for the late phase of DC maturation in response to NDV infection.
Fig. 5.

IFN signal-dependent amplification of maturation signaling on viral infection. (a) The kinetics of CD86 expression on NDV-stimulated WT or IFNAR1–/– DCs was analyzed by flow cytometry. The numbers indicate the mean fluorescent intensity of the cells. (b) NF-κB or IFN-stimulated gene factor 3 (ISGF3) activation at different time points was analyzed by electrophoretic mobility-shift assay. The filled and open arrowheads indicate the NF-κB and ISGF3 complex with the probe DNA, respectively. Cell lysates were also subjected to immunoblotting to analyze total RelA level. (c) The kinetics of chemokine receptor mRNA levels in NDV-stimulated DCs was determined by real-time RT-PCR. (d) Immunohistochemical analyses of spleens from WT and IFNAR1–/– mice were performed after i.p. injection with control PBS, poly(I:C) (100 μg per body), NDV (250 hemagglutinating units per body), or LPS (200 ng per body). Spleen sections were doubly stained with antibodies against B220 (FITC; green) and CD11c or CD86 (Alexa Fluor 546; red). It is worth noting that CD11c– CD86high cells are detected in the marginal zone of the spleen of both virus-infected WT and IFNAR1–/– mice, but the nature of these cells remains unknown.
Role IFN-α/β in DC Migration and Maturation in Vivo. Finally, we examined the role of IFN-α/β signaling in DC migration and maturation in vivo in response to LPS, poly(I:C), or NDV. The migration and maturation of DCs in the T cell zone of the spleen were examined by immunohistochemical analysis using anti-CD11c and anti-CD86 antibodies. As shown in Fig. 5d, CD11c+ cells migrated into the T cell zone in both the WT and IFNAR1–/– mice on poly(I:C) injection or NDV infection. On the other hand, the CD86 expression was found to be notably weak in the DCs that migrated in the IFNAR1–/– mice. These results indicate that the loss of IFN-α/β signaling affects the maturation but not migration of DCs in vivo. In fact, these observations are consistent with the results shown in Figs. 2b and 5c, showing that the induction of CCR7 mRNA occurs normally in the poly(I:C)- or NDV-stimulated IFNAR1–/– DCs. These in vivo results suggest a unique dissociation between the migration and maturation events of DCs in response to ds-RNA stimulation or viral infection, the latter of which requires IFN-α/β signaling.
Discussion
In this study, we presented evidence that IFN-α/β plays a selective role in the DC maturation induced by ds-RNA or an RNA virus. We also found that ds-RNA- or virus-mediated DC activation can be divided into two distinct phases, wherein IFN-α/β signaling plays a critical role in the later phase. In the IFNAR1–/– DCs, both the initial induction of inflammatory cytokines and activation of NF-κB occur, but they are not sustained in the late phase. In view of the facts that the induction of IFN-α/β mRNAs by poly(I:C) or NDV occurs in the early phase of DC maturation (Figs. 3b, 4c, and 8) and that a high dose of recombinant IFN-β cannot mimic the effect of poly(I:C) (Fig. 1b), we infer that the IFN-α/β signal contributes to this signal amplification not directly but indirectly by inducing the molecules involved in ds-RNA- or virus-mediated activation.
Our results are consistent with previous reports (13, 23) in that TLR3 is critical for the IFN-α/β-dependent DC maturation by extracellularly delivered ds-RNA, whereas PKR is dispensable in these events. Our results also suggest that the signaling mechanism by ds-RNA is complex, because the poly(I:C)-dependent maturation and cytokine induction are not abolished in TLR3–/– DCs. It is therefore important to study the nature of another molecule mediating the ds-RNA response.
After completion of this work, it was reported that PKR is required for intracellular ds-RNA recognition (27). Although the issue of how this PKR-mediated DC activation contributes to its maturation was not addressed in this report, it is possible that DC maturation is induced by intracellular ds-RNA, in which IFN-α/β signaling, but not TLR3 signaling, will be involved (27). Considering the fact that both extra- and intracellular ds-RNAs may be produced during viral infection, it may be advantageous for the host to possess distinct extra- and intracellular sensing mechanisms, which may be called “the double check mechanism,” for viral invasions. In view of our results that the DC maturation by NDV does not depend on either PKR or TLR3, the DC activation mechanisms may be differentially dependent on the nature of the viruses. We infer that DCs may specifically use several different pattern recognition receptors and other signaling molecules to detect several features of a virus simultaneously.
We also demonstrated here a unique dissociation of DC migration and maturation in response to poly(I:C) stimulation or virus infection. In fact, CCR7 mRNA induction requires only the IFN-independent early phase of the activation. The mechanism in which DC migration precedes the maturation may be an important aspect in that the DCs activated by antigens (such as viruses) in the periphery first migrate into the T cell zone of the lymphoid tissues and fully mature there so as to effectively activate T cells. In this context, it is interesting to note that a special type of cells, termed IFN-producing cells, were identified; these cells localize in peripheral lymphoid tissues and are capable of producing large amounts of IFN-α/β following viral infection (28, 29). It is therefore possible that the IFN-α/β signaling in DCs operates in both autocrine and paracrine mechanisms in vivo on viral infection. The acquisition of the signal enhancement by IFN-α/β in the late phase of DC maturation must have been critical for the evolution of the antiviral adaptive immune systems.
Supplementary Material
Acknowledgments
We thank Drs. H. Iba, S. Ohka, R. Miura, and T. Mizutani for technical assistance, and Drs. E. Barsoumian, R. A. Flavell, and K. Takeda for invaluable discussion and advice. The work was supported by special grant for Advanced Research on Cancer 11182101 and Grant-In-Aid 14021017 for Scientific Research on Priority Area from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by grants from the Virtual Research Institute of Aging of Nippon Boehringer Ingelheim.
Abbreviations: DC, dendritic cell; ds-RNA, double-stranded RNA; LPS, lipopolysaccharide; NDV, Newcastle disease virus; PAMP, pathogen-associated molecular pattern; PKR, ds-RNA dependent protein kinase R; TLR, Toll-like receptor.
References
- 1.Medzhitov, R. & Janeway, C. A., Jr. (1997) Curr. Opin. Immunol. 9, 4–9. [DOI] [PubMed] [Google Scholar]
- 2.Akira, S., Takeda, K. & Kaisho, T. (2001) Nat. Immunol. 2, 675–680. [DOI] [PubMed] [Google Scholar]
- 3.Barton, G. M. & Medzhitov, R. (2003) Science 300, 1524–1525. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau, J. & Steinman, R. M. (1998) Nature 392, 245–252. [DOI] [PubMed] [Google Scholar]
- 5.Balachandran, S., Roberts, P. C., Brown, L. E., Truong, H., Pattnaik, A. K., Archer, D. R. & Barber, G. N. (2000) Immunity 13, 129–141. [DOI] [PubMed] [Google Scholar]
- 6.Roger, T., David, J., Glauser, M. P. & Calandra, T. (2001) Nature 414, 920–924. [DOI] [PubMed] [Google Scholar]
- 7.van den Broek, M. F., Muller, U., Huang, S., Zinkernagel, R. M. & Aguet, M. (1995) Immunol. Rev. 148, 5–18. [DOI] [PubMed] [Google Scholar]
- 8.Guidotti, L. G. & Chisari, F. V. (2001) Annu. Rev. Immunol. 19, 65–91. [DOI] [PubMed] [Google Scholar]
- 9.Fonteneau, J. F., Larsson, M. & Bhardwaj, N. (2002) Curr. Opin. Immunol. 14, 471–477. [DOI] [PubMed] [Google Scholar]
- 10.Muller, U., Steinhoff, U., Reis, L. F., Hemmi, S., Pavlovic, J., Zinkernagel, R. M. & Aguet, M. (1994) Science 264, 1918–1921. [DOI] [PubMed] [Google Scholar]
- 11.Le Bon, A., Schiavoni, G., D'Agostino, G., Gresser, I., Belardelli, F. & Tough, D. F. (2001) Immunity 14, 461–470. [DOI] [PubMed] [Google Scholar]
- 12.Toshchakov, V., Jones, B. W., Perera, P. Y., Thomas, K., Cody, M. J., Zhang, S., Williams, B. R., Major, J., Hamilton, T. A., Fenton, M. J. & Vogel, S. N. (2002) Nat. Immunol. 3, 392–398. [DOI] [PubMed] [Google Scholar]
- 13.Doyle, S. E., O'Connell, R., Vaidya, S. A., Chow, E. K., Yee, K. & Cheng, G. (2003) J. Immunol. 170, 3565–3571. [DOI] [PubMed] [Google Scholar]
- 14.Sun, S., Zhang, X., Tough, D. F. & Sprent, J. (1998) J. Exp. Med. 188, 2335–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang, Y. L., Reis, L. F., Pavlovic, J., Aguzzi, A., Schafer, R., Kumar, A., Williams, B. R., Aguet, M. & Weissmann, C. (1995) EMBO J. 14, 6095–6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemmi, H., Takeuchi, O., Kawai, T., Kaisho, T., Sato, S., Sanjo, H., Matsumoto, M., Hoshino, K., Wagner, H., Takeda, K. & Akira, S. (2000) Nature 408, 740–745. [DOI] [PubMed] [Google Scholar]
- 17.Sato, M., Suemori, H., Hata, N., Asagiri, M., Ogasawara, K., Nakao, K., Nakaya, T., Katsuki, M., Noguchi, S., Tanaka, N. & Taniguchi, T. (2000) Immunity 13, 539–548. [DOI] [PubMed] [Google Scholar]
- 18.Sakaguchi, S., Negishi, H., Asagiri, M., Nakajima, C., Mizutani, T., Takaoka, A., Honda, K. & Taniguchi, T. (2003) Biochem. Biophys. Res. Commun. 306, 860–866. [DOI] [PubMed] [Google Scholar]
- 19.Inaba, K., Inaba, M., Romani, N., Aya, H., Deguchi, M., Ikehara, S., Muramatsu, S. & Steinman, R. M. (1992) J. Exp. Med. 176, 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuyama, T., Kimura, T., Kitagawa, M., Pfeffer, K., Kawakami, T., Watanabe, N., Kundig, T. M., Amakawa, R., Kishihara, K., Wakeham, A., et al. (1993) Cell 75, 83–97. [PubMed] [Google Scholar]
- 21.Sha, W. C., Nelson, C. A., Newberry, R. D., Kranz, D. M., Russell, J. H. & Loh, D. Y. (1988) Nature 335, 271–274. [DOI] [PubMed] [Google Scholar]
- 22.Dieu, M. C., Vanbervliet, B., Vicari, A., Bridon, J. M., Oldham, E., Ait-Yahia, S., Briere, F., Zlotnik, A., Lebecque, S. & Caux, C. (1998) J. Exp. Med. 188, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexopoulou, L., Holt, A. C., Medzhitov, R. & Flavell, R. A. (2001) Nature 413, 732–738. [DOI] [PubMed] [Google Scholar]
- 24.Miettinen, M., Sareneva, T., Julkunen, I. & Matikainen, S. (2001) Genes Immun. 2, 349–355. [DOI] [PubMed] [Google Scholar]
- 25.Heinz, S., Haehnel, V., Karaghiosoff, M., Schwarzfischer, L., Muller, M., Krause, S. W. & Rehli, M. (2003) J. Biol. Chem. 278, 21502–21509. [DOI] [PubMed] [Google Scholar]
- 26.Nakaya, T., Sato, M., Hata, N., Asagiri, M., Suemori, H., Noguchi, S., Tanaka, N. & Taniguchi, T. (2001) Biochem. Biophys. Res. Commun. 283, 1150–1156. [DOI] [PubMed] [Google Scholar]
- 27.Diebold, S. S., Montoya, M., Unger, H., Alexopoulou, L., Roy, P., Haswell, L. E., Al-Shamkhani, A., Flavell, R., Borrow, P. & Sousa, C. R. (2003) Nature 424, 324–328. [DOI] [PubMed] [Google Scholar]
- 28.Asselin-Paturel, C., Boonstra, A., Dalod, M., Durand, I., Yessaad, N., Dezutter-Dambuyant, C., Vicari, A., O'Garra, A., Biron, C., Briere, F. & Trinchieri, G. (2001) Nat. Immunol. 2, 1144–1150. [DOI] [PubMed] [Google Scholar]
- 29.Nakano, H., Yanagita, M. & Gunn, M. D. (2001) J. Exp. Med. 194, 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}