

Abstract
Learning and memory are exquisitely sensitive to behavioral stress, but the underlying mechanisms are still poorly understood. Because activity-dependent persistent changes in synaptic strength are believed to mediate memory processes in brain areas such as the hippocampus we have examined the means by which stress affects synaptic plasticity in the CA1 region of the hippocampus of anesthetized rats. Inescapable behavioral stress (placement on an elevated platform for 30 min) switched the direction of plasticity, favoring low frequency stimulation-induced decreases in synaptic transmission (long-term depression, LTD), and opposing the induction of long-term potentiation by high frequency stimulation. We have discovered that glucocorticoid receptor activation mediates these effects of stress on LTD and long-term potentiation in a protein synthesis-dependent manner because they were prevented by the glucocorticoid receptor antagonist RU 38486 and the protein synthesis inhibitor emetine. Consistent with this, the ability of exogenously applied corticosterone in non-stressed rats to mimic the effects of stress on synaptic plasticity was also blocked by these agents. The enablement of low frequency stimulation-induced LTD by both stress and exogenous corticosterone was also blocked by the transcription inhibitor actinomycin D. Thus, naturally occurring synaptic plasticity is liable to be reversed in stressful situations via glucocorticoid receptor activation and mechanisms dependent on the synthesis of new protein and RNA. This indicates that the modulation of hippocampus-mediated learning by acute inescapable stress requires glucocorticoid receptor-dependent initiation of transcription and translation.
Keywords: long-term potentiation, long-term depression, memory storage
The profound neurobiological effects of stress are believed to be the basis of many cognitive and affective changes in health and disease. In particular, learning and memory is dramatically modified by the presence of stress (1–4). In the case of hippocampus-dependent memory and learning, which is believed to mediated by persistent adjustments of synaptic weights (5–9), stress can facilitate or block the acquisition, consolidation, and/or recall of such tasks depending on experimental conditions (10–14).
Although stress is known to affect hippocampal synaptic plasticity, little is known about how it does so. A brief experience of acute inescapable stress can produce a dramatic change in both the susceptibility to, and the direction of, plasticity without affecting baseline transmission (15–16). Stress blocks high frequency stimulation-induced persistent increases in synaptic efficacy (long-term potentiation, LTP) in the CA1 area of the hippocampus (16–18). In contrast, low frequency stimulation-induced long-term depression (LTD) (16, 19), is known to be facilitated by such stress. The effects of stress in freely behaving rats were found to be relatively short lasting as acclimatization to, or removal from, the aversive conditions led to a rapid loss of the ability to induce LTD and recovery of the ability to induce LTP (16). However, the time window was greatly prolonged by inducing anesthesia immediately after the stress. Indeed, the block of LTP and the facilitation of LTD by stress was even observed in hippocampal slices from animals that had previously been stressed (19).
Behavioral stress leads to the activation of a wide variety of neurotransmitter and neuroendocrine systems that can potentially affect the induction of synaptic plasticity (1–4, 20). We focused on the possible involvement of corticosteroid-dependent mechanisms in stress modification of plasticity, (i) because effective stressors raise plasma corticosterone levels (16–18) and (ii) because stress levels of corticosterone or selective glucocorticoid receptor (type II) agonists can block the induction of LTP and facilitate the induction of LTD (21–29). We investigated the effects of antagonists/inhibitors of glucocorticoid receptors (RU 38486) (20, 26, 30–31), transcription (actinomycin D, ref. 32) and translation (emetine, ref. 32) on the effects of inescapable stress on synaptic plasticity. The stress consisted of placing rats on an elevated platform for 30 min immediately prior to anesthesia (16, 33). We also examined the effects of these agents on the ability of corticosterone to mimic the effects of stress in nonstressed animals.
MATERIALS AND METHODS
Animals.
Experiments were carried out on male Wistar rats (inbred strain, BioResources Unit, Trinity College, Dublin), weighing 250–300 g. Animals were group-housed with free access to water and food in an established animal house having a 12 h:12 h light/dark cycle and thermoregulated environment. The animal care and experimental protocol was licensed by the Department of Health, Ireland.
Electrophysiology and Surgery.
Recordings of field excitatory postsynaptic potentials (EPSPs) were made from the CA1 stratum radiatum of the hippocampus in response to ipsilateral stimulation of the Schaffer collateral/commissural pathway by using techniques similar to those described (16, 34). Experiments were carried out under pentobarbitone sodium (60 mg/kg, i.p.) anesthesia and core temperature was maintained at 37 ± 0.5°C. Electrode implantation sites were identified by using stereotaxic coordinates. Three stainless steel screws (1.5 mm diameter) were inserted into the skull through a drill hole without piercing the dura. One served as a ground electrode (7 mm posterior to bregma and 5 mm left of the midline), another acted as an anchor (opposite the ground screw, 7 mm posterior to bregma and 5 mm right of the midline) and the third served as the reference electrode (8 mm anterior to bregma and 1 mm left of the midline). Recording and stimulating electrodes were made by gluing together a pair of twisted Teflon-coated 90% platinum/10% iridium wires (50 μm inner diameter, 75 μm outer diameter). The recording electrode was inserted 3.4 mm posterior to bregma and 2.5 mm right of the midline and the stimulating electrode was inserted 4.2 mm posterior to bregma and 3.8 mm right of the midline. In some animals a second stimulating electrode was placed ipsilaterally (3.4 mm posterior to bregma and 3.4 mm right of the midline) to stimulate a separate, independent pathway. The optimal depth of the wire electrodes in the stratum radiatum of the CA1 region of the dorsal hippocampus was determined by using electrophysiological criteria and was verified by post mortem examination. In all experiments test EPSPs were evoked by stimulating with a square wave constant current pulse of 0.2 ms duration, at a frequency of 0.033 Hz and at a stimulation intensity adjusted to give an EPSP amplitude of 50% of maximum. For the two pathway experiments, lack of paired-pulse interaction (interval 40 ms) with responses evoked in the both pathways was used as a criterion of independence. None of the drugs studied here affected baseline transmission at the doses tested (data not shown).
Low frequency conditioning stimulation consisted of 900 pulses at 3 Hz. The high frequency stimulation protocol for inducing LTP consisted of 10 trains of 20 stimuli, interstimulus interval 5 ms (200 Hz), intertrain interval 2 s. The electroencephalogram was simultaneously monitored (from the hippocampal recording electrode) during all experiments so as to ensure that no abnormal activity was evoked by the conditioning stimulation. LTP/LTD was measured 30 min after the conditioning stimulation.
A guide cannula was implanted in the lateral cerebral ventricle (coordinates: 0.5 mm anterior to bregma and 1.0 mm right of midline) just prior to electrode implantation. Intracerebroventricular (i.c.v.) injections of volumes of 12 μl were made over a 6-min period through an internal cannula.
Statistical comparisons were made by using either Student’s t test or ANOVA. Values are expressed as the mean % of the baseline field EPSP amplitude ± SEM over at least a 30-min baseline period. Similar results were obtained when EPSP slope was measured.
Stress Protocol.
Behavioral stress was evoked by taking the rat out of its home cage in which it was group-housed and placing it on an elevated platform made of clear perspex (21×20 cm2 and ≈90 cm above ground level) in the middle of a brightly lit room for 30 min. During this period the animals show behavioral (“freezing” immobility, piloerection, urination, and defecation) and endocrine (elevated serum corticosterone levels measured from cardiac samples taken immediately after anesthesia, 41 ± 7 μg/dl vs. 1 ± 0.3 μg/dl in nonstressed; ref. 16) signs of stress. The animals were anesthetized immediately after the stress.
RESULTS AND DISCUSSION
The first set of experiments determined the possible role of glucocorticoid receptor activation in mediating the effects of stress on hippocampal plasticity by using the glucocorticoid receptor antagonist RU 38486. Stressing rats by placing them on an elevated platform for 30 min immediately prior to anesthesia-enabled low frequency stimulation (900 pulses at 3 Hz) to induce homosynaptic LTD (n = 5; 78.9 ± 4.7% of baseline 30 min later; P < 0.05) and blocked the induction of LTP by high frequency stimulation (200 Hz) (92.4 ± 8.3% of baseline 30 min later) in the CA1 area of the dorsal hippocampus (Fig. 1 A–C). These effects of stress were completely prevented by the administration of RU 38486 (5–20 mg/kg, s.c.). LTD was no longer induced and reliable homosynaptic LTP was elicited in stressed rats that received an injection of the antagonist 30 min before they were put on the platform (n = 7, 107.3 ± 6.0% of baseline 30 min after low frequency stimulation; 139.7 ± 9.7% of baseline 30 min after high frequency stimulation, P < 0.05 compared with baseline; Fig. 1 D and E). Although this was the same as that observed in nonstressed rats (n = 5; 101.8 ± 4.6% of baseline 30 min after low frequency stimulation; 139.3 ± 3.9% of baseline 30 min after high frequency stimulation, P < 0.01 compared with baseline; Fig. 1A), the change in the inducibility of LTD and LTP was not due to a reduction in the affective response to the aversive environment because the drug was also active even when it was given immediately after the stress, at the time of anesthesia (n = 8; 101.3 ± 4.5% of baseline at 30 min after low frequency stimulation; 130.6 ± 3.8% of baseline at 30 min after high frequency stimulation, P < 0.05; Fig. 1F).
Figure 1.
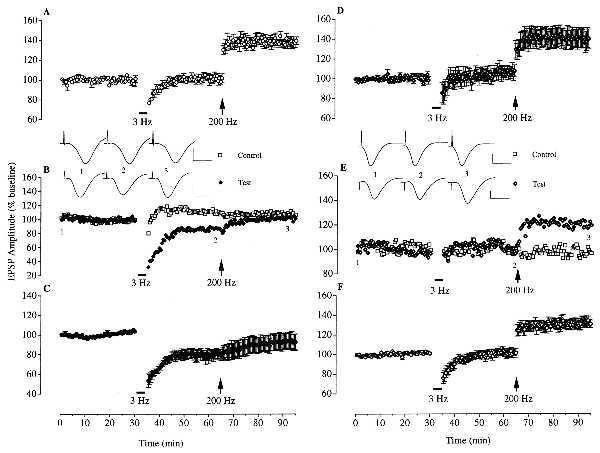
Activation of glucocorticoid receptors mediates the facilitation of low frequency stimulation-induced LTD and the block of high frequency stimulation-induced LTP by behavioral stress. (A) Low frequency stimulation (3 Hz; bar) failed to induce LTD of the field EPSP amplitude in the CA1 area of anesthetized nonstressed rats (n = 5). Subsequent high frequency stimulation (200 Hz, arrow) induced reliable LTP in these animals. (B and C) Reliable LTD was induced by low frequency stimulation in animals that had been stressed immediately before anesthesia (n = 5). These animals received an injection of polyethylene glycol (vehicle for RU 38486, 0.2 ml, s.c.) at the time of anesthesia and the conditioning stimulation was applied ≈150 min later. High frequency stimulation failed to induce LTP. An example of a two-pathway experiment is shown in B. Homosynaptic LTD was induced in the test pathway without affecting the responses in the control pathway. (D and E) In animals injected with the glucocorticoid receptor antagonist RU 38486 (5–20 mg/kg, s.c.) 30 min before the stress, low frequency stimulation failed to induce LTD (n = 7). Subsequent high frequency stimulation induced reliable LTP in these animals that was not significantly different from that observed in the nonstressed controls. An example of a two-pathway experiment is shown in E. Homosynaptic LTP was induced in the test pathway without affecting the responses in the control pathway. (F) Similar results were obtained when the RU 38486 was injected immediately after the stress (n = 8). (Insets) Representative traces of field potentials (average of 6 consecutive sweeps) at the times indicated by the numbers. Horizontal bar = 10 ms. Vertical bar = 1 mV.
The discovery that stress needs to activate glucocorticoid receptors to switch the direction of hippocampal synaptic plasticity led us to examine the involvement of protein and RNA synthesis in mediating the effects of stress because glucocorticoid receptors act as nuclear transcription factors throughout the brain, including hippocampal pyramidal cells (20, 30, 35–36). Treatment with the protein synthesis inhibitor emetine (240 μg, injected i.c.v. ≈40 min after the stress) completely prevented the facilitation of LTD (n = 4, 103.3 ± 2.8% of baseline 30 min after low frequency stimulation) and block of LTP induction by stress (147.1 ± 9.0% of baseline 30 min later, P < 0.05; Fig. 2A). This effect of emetine was not due to a direct interference with LTD induction mechanisms because LTD was induced reliably when the drug was injected just prior to the low frequency stimulation (n = 4, 83.5 ± 3.6% of baseline 30 min later, P < 0.05; Fig. 2B). Thus, protein synthesis is required for stress to affect both the induction of LTD and LTP. Consistent with a role for RNA synthesis in mediating the facilitation of LTD induction by stress, the transcription inhibitor actinomycin D (60 μg, injected i.c.v. ≈40 min after the stress) blocked the response to low frequency stimulation (n = 6, 106.9 ± 6.9% of baseline 30 min later; Fig. 2C). However, high frequency stimulation did not induce significant LTP in these animals (117.4 ± 8.2% of baseline 30 min later). The lack of LTP was not due to an inhibitory effect of actinomycin D on the induction of LTP because the drug did not affect the response to high frequency stimulation in nonstressed animals (n = 5, 140.9 ± 8.7% of baseline 30 min later, P < 0.05; Fig. 2D).
Figure 2.
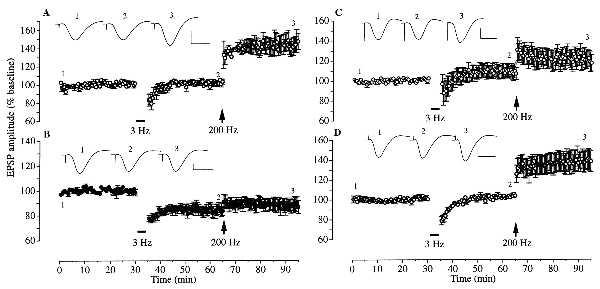
Blocking effects of RNA and protein synthesis inhibitors on the redirection of synaptic plasticity by behavioral stress. (A) Injection with the translation inhibitor emetine (240 μg, i.c.v.) 40 min after anesthesia (i.e., about 120 min before low frequency stimulation) blocked the effect of stress on both LTD and LTP induction (n = 4). Low frequency stimulation failed to induce LTD. Subsequent high frequency stimulation induced significant LTP, a magnitude not significantly different from that observed in nonstressed controls. (B) The same dose of emetine given 15–30 min before low frequency stimulation (≈130 min after stress) failed to affect the effect of stress on synaptic plasticity (n = 4). LTD was induced by low frequency stimulation and subsequent high frequency stimulation failed to induce LTP. (C and D) Actinomycin D (60 μg, i.c.v.), a transcription inhibitor, was given 40 min after anesthesia (i.e., about 120 min before low frequency stimulation). In stressed animals (C) low frequency stimulation failed to induce LTD. Subsequent high frequency stimulation did not induce significant LTP. In the nonstressed rats (D) low frequency stimulation failed to induce LTD and subsequent high frequency stimulation resulted in significant LTP (n = 5). (Insets) Representative traces of field potentials at the times indicated by the numbers on the graph. Horizontal bar = 10 ms. Vertical bar = 1 mV.
The finding that the effect of stress on LTP induction was not significantly affected by actinomycin D at a dose level that produces ≈95% inhibition of transcription (32) and at a time when it blocked the effect of stress on LTD induction can be interpreted as evidence that there is a differential requirement for transcription mechanisms in the effects of stress on the induction of LTD and LTP. Alternatively, because the actinomycin D was administered 40 min after the stress, it is possible that its inability to restore LTP was due to sufficient new RNA being synthesized during this interval. To help distinguish between these interpretations and to confirm the role of corticosteroid-dependent transcription in mediating the enablement of LTD by stress, we examined the effect of administering actinomycin D prior to exogenously applied corticosterone. Injection of corticosterone (5 mg/kg, i.p., 30 min before the low-frequency stimulation) in nonstressed rats mimicked the effect of stress on synaptic plasticity, enabling the induction of LTD by low-frequency stimulation (n = 5, 85.2 ± 2.1% of baseline 30 min later, P < 0.01) and blocking the induction of LTP by high frequency stimulation (97.3 ± 8.4% of baseline 30 min later; Fig. 3A). Pretreatment with actinomycin D prevented the facilitation of LTD (n = 5, 99.5 ± 3.1% of baseline) without significantly affecting the block of LTP by corticosterone (112.9 ± 5.7% of baseline after 30 min, P > 0.05; Fig. 3B). The further finding that emetine prevented the block of LTP by corticosterone even in the presence of actinomycin D (n = 3, 146.7 ± 6.7% of baseline at 30 min after high frequency stimulation, P < 0.05; Fig. 3C) shows that the lack of effect of actinomycin D on the corticosterone-mediated block of LTP was not due to an inability to induce LTP. It also supports the proposal that stress-mediated block of LTP requires the synthesis of new protein.
Figure 3.
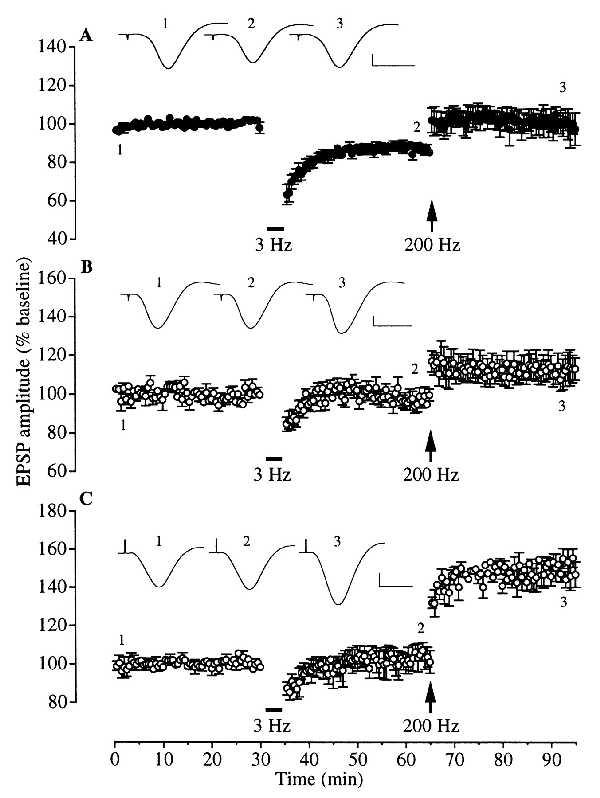
Blocking effects of RNA and protein synthesis inhibitors on the redirection of synaptic plasticity by corticosterone in nonstressed animals. (A) In nonstressed animals injected with corticosterone (5 mg/kg, i.p.), 30 min before low frequency stimulation, stable LTD was induced (n = 5). When high frequency stimulation was applied 30 min after low frequency stimulation, the LTD was reversed only to control baseline levels. (B) The RNA synthesis inhibitor actinomycin D (60 μg, i.c.v.) was given 40 min after anesthesia (i.e., about 120 min before the injection of corticosterone) to nonstressed animals (n = 5). Low frequency stimulation failed to induce LTD. Subsequent high frequency stimulation failed to induce significant LTP. (C) When, in addition to actinomycin D treatment, emetine (240 μg, i.c.v.) was given 30 min before corticosterone (≈120 min after actinomycin D) low frequency stimulation still failed to induce LTD but high frequency stimulation now induced LTP (n = 3). (Insets) Representative traces of field potentials at the times indicated by the numbers on the graph. Horizontal bar = 10 ms. Vertical bar = 1 mV.
The present finding that corticosteroid mechanisms mediate the control of hippocampal plasticity by stress appear to conflict with a previous study that found that the effect of stress on LTP induction was only partly blocked in chronically (≥4 weeks) adrenalectomized animals (37). This may be due to the fact that in the present study we acutely blocked glucocorticoid receptors whereas chronic adrenalectomy can have complex adaptive effects, e.g., on both mineralocorticoid and glucocorticoid receptors over time (20, 38–39). As activation of mineralocorticoid receptors facilitates LTP induction (26), any adaptive changes, such as an alteration in the relative proportion of functional mineralocorticoid and glucocorticoid receptors, may complicate the interpretation of the effects of adrenalectomy. Alternatively, because the adrenalectomy experiments examined the effect of restraint/tailshock stress on the induction of LTP of the population spike in vitro and adrenalectomy had large baseline effects on LTP, it is possible that the effect they observed may involve different mechanisms than those described here for elevated-platform stress. Although the present results do not exclude other, noncorticosteroid mechanisms such as N-methyl-d-aspartate (19) or opioid (40) receptor activation, it is likely, given the present findings, that the involvement of such pathways is contingent on glucocorticoid receptor activation. The sequence of events following the activation of these receptors is clearly dependent on the synthesis of new protein and in the case of LTD, RNA synthesis. Given the relatively rapid onset of the facilitatory effect of stress and corticosterone on LTD induction, it is probable that the modulation of immediate early genes is involved (30, 41). One likely candidate protein for the facilitation of LTD by stress is the voltage-gated L-type Ca2+ channel (36, 42). It seems less likely, however, that this channel is involved in the block of LTP by stress because activation of L-type Ca2+ channels facilitates LTP induction (43), although it is possible that excess/inappropriate Ca2+ entry may inhibit LTP induction. The finding that actinomycin D did not significantly affect the block of LTP induction by either stress or exogenous corticosterone is also indicative of possible different mechanisms for the changes in LTP and LTD induction.
The long trains of pulses used here to induce LTD and LTP are based on standard protocols derived from in vitro studies and are not normally found in the intact hippocampus. Hippocampal pyramidal neurons usually fire in brief high frequency bursts of two to seven action potentials (complex spikes). In freely moving animals this burst firing is generally phase-locked to either the positive or negative peaks of background theta (≈3–12 Hz) electroencephalographic activity. Recently, we reported that as few as 5 pulses applied as a single brief burst (200 Hz) on the positive phase of sensory-evoked theta activity reliably elicited LTP in the CA1 area of nonstressed rats (44). Brief burst stimulation on the negative phase of theta waves had no effect on baseline transmission in these animals (i.e., no LTD) but did induce depotentiation of previously potentiated synaptic responses. In contrast, in stressed rats, burst stimulation triggered on the positive phase of theta activity failed to induce LTP whereas such stimulation given on the negative phase of theta activity now elicited LTD. Indeed, a single burst of five pulses was sufficient to induce LTD in stressed animals (45). It seems likely, therefore, that naturally occurring plasticity is subject to similar behavioral control mechanisms to those studied in the present experiments.
The corticosteroid-dependent mechanisms of LTD/LTP regulation described here would be expected to occur naturally in the hippocampus of animals in inescapable stressful situations. Such a glucocorticoid receptor-mediated and genomic/protein synthesis-dependent redirection of hippocampal synaptic plasticity would provide a powerful means for acute stress to affect hippocampus-dependent memory and behavior. Like stress (10–14), depending on experimental conditions, glucocorticoid receptor activation can either block or facilitate learning that engages hippocampal neural networks (46–48). Learning that is blocked by glucocorticoid receptor activation may depend on LTP-like plasticity and conversely, learning that is facilitated may rely on LTD-like plasticity. Indeed, LTP-like mechanisms fulfill many of the requirements as a basis for reinforcement-related learning (e.g., refs. 4 and 49) whereas LTD-like mechanisms are attractive models for learning from mistakes, where incorrect connections are weakened (e.g., ref. 50).
Acknowledgments
This research was supported by the Health Research Board of Ireland, the European Union DGXII, and the Wellcome Trust.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: LTD, long-term depression; LTP, long-term potentiation; EPSP, excitatory postsynaptic potential; i.c.v., intracerebroventricular.
References
- 1.McEwen B, Sapolsky R. Curr Opin Neurobiol. 1995;5:205–212. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 2.Gold P. In: Brain and Memory: Modulation and Mediation of Neural Plasticity. McGaugh J, Weinberger N, Lynch G, editors. New York: Oxford Univ. Press; 1995. pp. 41–74. [Google Scholar]
- 3.McGaugh J L, Cahill L, Roozendaal B. Proc Natl Acad Sci USA. 1996;93:13508–13514. doi: 10.1073/pnas.93.24.13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Izquierdo I, Medina J H. Psychobiology. 1997;25:1–9. [Google Scholar]
- 5.Bliss T, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 6.Maren S, Baudry M. Neurobiol Learn Mem. 1995;63:1–18. doi: 10.1006/nlme.1995.1001. [DOI] [PubMed] [Google Scholar]
- 7.Bear M, Abraham W. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- 8.Gluck M, Myers M. Annu Rev Psychol. 1997;48:481–584. doi: 10.1146/annurev.psych.48.1.481. [DOI] [PubMed] [Google Scholar]
- 9.Jeffery K, Reid I. Am J Psychiatr. 1997;154:156–154. doi: 10.1176/ajp.154.2.156. [DOI] [PubMed] [Google Scholar]
- 10.Rudy J. Behav Neurosci. 1995;110:238–246. doi: 10.1037//0735-7044.110.2.238. [DOI] [PubMed] [Google Scholar]
- 11.Francis D, Zaharia M, Shanks N, Anisman H. Physiol Behav. 1995;58:57–65. doi: 10.1016/0031-9384(95)00009-8. [DOI] [PubMed] [Google Scholar]
- 12.Kirchbaum C, Wolf O, May M, Wippich W, Hellhammer D. Life Sci. 1996;58:1475–1483. doi: 10.1016/0024-3205(96)00118-x. [DOI] [PubMed] [Google Scholar]
- 13.Diamond D M, Fleshner M, Ingersoll N, Rose R M. Behav Neurosci. 1996;110:661–672. doi: 10.1037//0735-7044.110.4.661. [DOI] [PubMed] [Google Scholar]
- 14.Healy D, Drugan R. Psychobiology. 1996;24:110–117. [Google Scholar]
- 15.Shors T, Gallegos R, Breindl A. Synapse. 1997;26:209–217. doi: 10.1002/(SICI)1098-2396(199707)26:3<209::AID-SYN2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 16.Xu L, Anwyl R, Rowan M J. Nature (London) 1997;387:497–500. doi: 10.1038/387497a0. [DOI] [PubMed] [Google Scholar]
- 17.Shors T J, Seib T, Levine S, Thompson R. Science. 1989;244:224–226. doi: 10.1126/science.2704997. [DOI] [PubMed] [Google Scholar]
- 18.Diamond D M, Bennett M C, Stevens K E, Wilson R L, Rose G M. Psychobiology. 1990;18:273–281. [Google Scholar]
- 19.Kim J J, Foy M R, Thompson R F. Proc Natl Acad Sci USA. 1996;93:4750–4753. doi: 10.1073/pnas.93.10.4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Kloet E. Front Neuroendocrinol. 1991;12:95–164. [Google Scholar]
- 21.Filipini D, Gijsbers K, Birmingham M K, Dubrovsky B. J Steroid Biochem Mol Biol. 1991;40:87–92. doi: 10.1016/0960-0760(91)90171-z. [DOI] [PubMed] [Google Scholar]
- 22.Diamond D M, Bennett M C, Fleshner M, Rose G M. Hippocampus. 1992;2:421–430. doi: 10.1002/hipo.450020409. [DOI] [PubMed] [Google Scholar]
- 23.Pavlides C, Watanabe Y, McEwen B S. Hippocampus. 1993;3:183–192. doi: 10.1002/hipo.450030210. [DOI] [PubMed] [Google Scholar]
- 24.Kerr D S, Huggett A, Abraham W. Psychobiology. 1994;22:123–133. [Google Scholar]
- 25.Rey M, Carlier E, Talmi M, Soumireu M B. Neuroendocrinology. 1994;60:36–41. doi: 10.1159/000126717. [DOI] [PubMed] [Google Scholar]
- 26.Pavlides C, Watanabe Y, Magariños A M, McEwen B S. Neuroscience. 1995;68:387–394. doi: 10.1016/0306-4522(95)00151-8. [DOI] [PubMed] [Google Scholar]
- 27.Pavlides C, Kimura A, Magariños A M, McEwen B S. Neuroscience. 1995;68:379–385. doi: 10.1016/0306-4522(95)94332-s. [DOI] [PubMed] [Google Scholar]
- 28.Pavlides C, Ogawa S, Kimura A, McEwen B S. Brain Res. 1996;738:229–235. doi: 10.1016/s0006-8993(96)00776-7. [DOI] [PubMed] [Google Scholar]
- 29.Joëls M. Front Neuroendocrinol. 1997;18:2–48. doi: 10.1006/frne.1996.0144. [DOI] [PubMed] [Google Scholar]
- 30.Funder J. Annu Rev Med. 1997;48:231–240. doi: 10.1146/annurev.med.48.1.231. [DOI] [PubMed] [Google Scholar]
- 31.Cadepond F, Ulman A, Baulieu E-E. Annu Rev Med. 1997;48:129–156. doi: 10.1146/annurev.med.48.1.129. [DOI] [PubMed] [Google Scholar]
- 32.Otani S, Marshall C, Tate W, Goddard G, Abraham W. Neuroscience. 1989;28:519–526. doi: 10.1016/0306-4522(89)90001-8. [DOI] [PubMed] [Google Scholar]
- 33.Balfour D J K, Reid I. Arch Int Pharmacodyn Ther. 1979;237:67–74. [PubMed] [Google Scholar]
- 34.Doyle C, Holscher C, Rowan M J, Anwyl R. J Neurosci. 1996;16:418–424. doi: 10.1523/JNEUROSCI.16-01-00418.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karst H, Joëls M. Neurosci Lett. 1991;130:27–31. doi: 10.1016/0304-3940(91)90219-j. [DOI] [PubMed] [Google Scholar]
- 36.Kerr D, Campbell L, Thibault O, Landfield P. Proc Natl Acad Sci USA. 1992;89:8527–8531. doi: 10.1073/pnas.89.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shors T J, Levine S, Thompson R F. Neuroendocrinology. 1990;51:70–75. doi: 10.1159/000125318. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt T, Meyer A. Receptor. 1994;4:229–257. [PubMed] [Google Scholar]
- 39.Spencer R, Moday H, Miller A. Stress. 1997;2:51–64. doi: 10.3109/10253899709014737. [DOI] [PubMed] [Google Scholar]
- 40.Shors T J, Levine S, Thompson R F. Brain Res. 1990;506:316–318. doi: 10.1016/0006-8993(90)91270-q. [DOI] [PubMed] [Google Scholar]
- 41.Ryabinin A, Melia K, Cole M, Bloom F, Wilson M. J Neurosci. 1995;15:721–730. doi: 10.1523/JNEUROSCI.15-01-00721.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coussens C M, Teyler T J. Synapse. 1996;24:97–103. doi: 10.1002/(SICI)1098-2396(199610)24:2<97::AID-SYN1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 43.Mulkeen D, Anwyl R, Rowan M J. Neurosci Lett. 1987;80:351–355. doi: 10.1016/0304-3940(87)90481-2. [DOI] [PubMed] [Google Scholar]
- 44.Hölscher C, Anwyl R, Rowan M J. J Neurosci. 1997;17:6470–6477. doi: 10.1523/JNEUROSCI.17-16-06470.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hölscher C, Xu L, Anwyl R, Rowan M. Soc Neurosci Abstr. 1997;23:788. [Google Scholar]
- 46.Oitzl M, de Kloet E. Behav Neurosci. 1992;106:62–71. doi: 10.1037//0735-7044.106.1.62. [DOI] [PubMed] [Google Scholar]
- 47.Newcomer J, Craft S, Hershey T, Askins K, Bardgett M. J Neurosci. 1994;14:2047–2053. doi: 10.1523/JNEUROSCI.14-04-02047.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roozendaal B, McGaugh J L. Eur J Neurosci. 1997;9:76–83. doi: 10.1111/j.1460-9568.1997.tb01355.x. [DOI] [PubMed] [Google Scholar]
- 49.Seidenbecher T, Reymann K G, Balschun D. Proc Natl Acad Sci USA. 1997;94:1494–1499. doi: 10.1073/pnas.94.4.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cziko G. Without Miracles. Universal Selection Theory and the Second Darwinian Revolution. Cambridge, MA: MIT Press; 1995. [Google Scholar]