

Abstract
The freshwater turtle Trachemys scripta is among the most anoxia tolerant of vertebrates, a true facultative anaerobe able to survive without oxygen for days at room temperature to weeks or months during winter hibernation. Our good friend and colleague Peter Lutz devoted nearly 25 years to the study of the physiology of anoxia tolerance in these and other model organisms, promoting not just the basic science but also the idea that understanding the physiology and molecular mechanisms behind anoxia tolerance provides insights into critical survival pathways that may be applicable to the hypoxic/ischemic mammalian brain. Work by Peter and his colleagues focused on the factors which enable the turtle to enter a deep hypometabolic state, including decreases in ion flux (“channel arrest”), increases in inhibitory neuromodulators like adenosine and GABA, and the maintenance of low extracellular levels of excitatory compounds such as dopamine and glutamate. Our attention has recently turned to molecular mechanisms of anoxia tolerance, including the upregulation of such protective factors as heat shock proteins (Hsp 72, Hsc73), the reversible downregulation of voltage gated potassium channels, and the modulation of MAP kinase pathways. In this review we discuss three phases of anoxia tolerance, including the initial metabolic downregulation over the first several hours, the long-term maintenance of neuronal function over days to weeks of anoxia, and finally recovery upon reoxygenation, with necessary defenses against reactive oxygen stress.
Keywords: anoxia, antioxidants, channel arrest, hypometabolism, neurotransmitter, reactive oxygen species, Trachemys scripta
Introduction
Oxygen is considered critical to nearly all life on earth, as the end electron acceptor that makes mitochondrial oxidative phosphorylation possible. Acute hypoxia or cerebral ischemia causes neuronal death due to the failure of ATP-driven ion transporters; the breakdown of membrane potential is followed by the release of excitatory amino acids (EAA) such as aspartate and glutamate, with glutamate binding to postsynaptic receptors that regulate calcium channels. The resulting Ca2+ influx activates proteases, lipases, and endonucleases, which in turn destroy cellular integrity. Additional neuronal damage occurs during reperfusion, thought to be caused by the post-ischemic release of oxygen radicals, the synthesis of nitric oxide, inflammation, and an imbalance between the excitatory and inhibitory neurotransmitter systems (Berger et al., 2002). When temperature differences are taken into account these catastrophic effects of hypoxia ischemia are characteristic of the brains of nearly all vertebrates ranging from fish to mammals (Lutz et al., 2003). Neurons are generally viewed as among the most anoxia sensitive of all cells, though recent studies have shown a wide variation in the capacity of neurons to tolerate hypoxia, reflective of function and the degree of hypoxia normally encountered.
Not all animals, however, share the mammalian susceptibility to hypoxia, or even full anoxia. Freshwater turtles of the genera Trachemys and Chrysemys are true facultative anaerobes, able to survive from up to 48 hrs at room temperature to months (during winter hibernation) in the total absence of oxygen (Jackson, 2000). Trachemys scripta has been the subject of extensive research into the adaptations that permit neuronal survival without oxygen; the turtle is able to decrease its metabolic rate to approximately 10–15% of basal, such that energy utilization is matched to anaerobic energy production (Lutz et al. 2003). By preventing an energy deficit, the turtle brain avoids the catastrophic drop in ATP levels which in mammalian neurons results in the breakdown of cellular ion homeostasis, release of excitatory neurotransmitters, and excitotoxic cellular death (Lutz et al. 2003). To decrease neuronal energy requirements, Trachemys decreases membrane ion permeability (“channel arrest”) (Chih et al., 1989), inhibits the release of excitatory neurotransmitters such as dopamine (Milton and Lutz, 1998) and glutamate (Milton et al. 2002), increases the release of inhibitory compounds such as adenosine (Nilsson and Lutz, 1992) and GABA (Nilsson and Lutz, 1991), and decreases electrical activity (Fernandes et al. 1997). Thus at no time do turtle neurons experience an energy deficit that would otherwise constitute a trigger for catastrophic cell death. Such extended anoxic survival time is not a matter of ectothermy, as other reptiles survive only 20–30 minutes without oxygen (Belkin 1963) and do not exhibit the same neurological adaptations that permit true anoxic tolerance (Nilsson et al. 1991).
A very extensive body of knowledge concerning the mechanisms of anoxia tolerance was contributed by our good friend and colleague, Peter Lutz, who was a pioneer and leader in this field. Peter authored more than 50 articles in his lifetime on anoxia tolerance alone, within a career of interests ranging from Nigerian trematodes (Lutz and Siddiqi, 1967), to the platypus (Lutz et al. 1989). Moving to South Florida in 1976, he soon began a series of studies on the diving physiology of sea turtles; their remarkable ability to dive for long periods of time soon piqued Peter’s interest in brain survival during hypoxia, and this in turn led him to begin investigating the best of facultative anaerobes, the freshwater turtle. This interest continued and expanded from his first Trachemys paper in 1980 until his death in 2005. This review will cover many of those significant findings, along with some directions we expect to more fully investigate in the future, building on the legacy of scientific inquiry among the many friends, students, and colleagues who continue working in this field.
It became clear in recent years that while a great deal of effort has gone into describing the events leading from normoxic physiology to anoxic hypometabolism, there are three significant phases of anoxic survival, each with its own challenges. To survive bouts of anoxia the turtle brain must be capable of (1) tolerating an early transition to anoxia (the first 1–2 hours), (2) a more prolonged deep hypometabolic state where neuronal network integrity must be maintained (to permit recovery at a later time when oxygen becomes available) and finally (3) a re-oxygenation phase where the turtle brain must contend with the potential for massive levels of reactive oxygen species while simultaneously restoring normal brain function.
1. The early transition to anoxia and channel arrest
The early response of the turtle brain to anoxia (within the first 1 – 2 hours) is a drastic decrease in ATP demand such that the energy requirements can be fully met by anaerobic glycolysis. Energy consumption decreases by 70–80 per cent during this time. A significant but temporary decrease (20%) is seen in brain ATP levels during the anoxic transition, after which energy costs decline sufficiently for anaerobic glycolysis to once more restore ATP levels (Lutz et al., 1984).
Because greater than 50% of the energy consuming processes of the normoxic neuron can be accounted for by ion pumping, a decrease in membrane ion leakage can contribute in a major way to energy savings. There is extensive evidence that channel arrest occurs in the anoxic turtle brain. This is reflected for example in a decreased K+ flux (Pek and Lutz, 1998) and a reduction in the density of voltage-gated Na+ channels (Perez-Pinzon et al., 1992). NMDA receptor activity is decreased in turtle brain during anoxia together with an associated decrease in Ca2+ permeability (Bickler et al., 2000; Bickler, 1998). The decreased ion permeability reported in turtle brain is also paralleled by a dramatic down-regulation of EEG (Fernandes et al., 1997). Because channel arrest is an important strategy for energy saving, the mechanisms underlying this decreased neuronal activity continue to be a central theme for ongoing investigation both in our laboratory and by others (e.g. Shin et al., 2005).
During the first hour of anoxia the turtle experiences a temporary energy crisis as energy demand initially outstrips supply, causing a limited but significant drop in ATP (Lutz et al., 1984). This temporary decline in ATP has two important effects: a concomitant increase in adenosine, and the opening of KATP channels. Both factors contribute significantly to metabolic downregulation, including channel arrest and the modulation of neurotransmitter release (see below). Extracellular adenosine levels begin to rise almost immediately when the turtle is ventilated on nitrogen; levels peak at about 100 mins anoxia and decline thereafter to near basal levels by 300 mins (Nilsson and Lutz, 1992). The neuroprotection afforded by adenosine is so significant that it has been termed a “retaliatory metabolite” for the mammalian heart and brain; it is produced during periods of energy insufficiency and acts in turn to reduce energy consumption while increasing supply. In the turtle brain, adenosine temporarily increases brain blood flow (Hylland et al., 1994), modulates early decreases in dopamine (Milton and Lutz, 2005) and glutamate (Milton et al., 2002), and plays a critical role in the anoxia induced decrease in turtle brain NMDA receptor activity (Buck and Bickler, 1998) and in potassium channel arrest (Pek and Lutz, 1997).
Ion channels
In early anoxia, the reduced efflux of K+ is also mediated in part by opening of ATP-dependent potassium channels (KATP) (Pek and Lutz, 1998). In the mammalian brain ATP dependent potassium channels are known to play a protective role in the early stages of ischemia. Upon energy imbalance these channels are activated by a drop in cellular ATP levels and the resulting increase in K+ conductance causes hyperpolarization (Krnjevic, 1993). Hyperpolarization can reduce the duration of the action potential and decrease the anoxia induced depolarization, as well as decrease the release of excitatory neurotransmitters (Tanaka et al., 1995). In the long term KATP channel activation in mammals is associated with increases in extracellular K and a gradual depolarization. The KATP channels are also a route for the rapid K+ efflux found during anoxic depolarization (Xia and Haddad, 1991). Thus during ischemia/anoxia mammalian KATP channels play an important but short term protective role. In the anoxic turtle brain the KATP channel is activated by the initial entry into energy imbalance and these channels in turn activate processes that result in a decrease in K+ efflux (Pek and Lutz, 1998). This decrease in K+ efflux is not achieved through membrane hyperpolarization, as the turtle maintains membrane potential, but instead through mechanisms that may involve an elevated adenosine A1 receptor response (Pek and Lutz, 1998). Open KATP channels are also involved in the initial decreased release of excitotoxic neurotransmitters (see below). Once the ATP demand processes have been down-regulated sufficiently to match the reduced ATP supply, the KATP channels are once more inactivated. In the hypometabolic turtle brain however, membrane ion permeability remains depressed.
A second important contributor to the switch for channel arrest is adenosine, which plays a key role in decreasing K+ efflux as well as in down-regulating Ca permeability (Pek and Lutz, 1998; Buck and Bickler, 1998). In mammalian myocardium the KATP channel acts synergistically with adenosine A1 receptors in tissue protection during preconditioning, the process whereby an early mild ischemic event elicits protection against a later lethal ischemia (Yao et al., 1997). Both KATP channel and adenosine are also involved in preconditioning in the mammalian brain (Yoshida et al., 2004). The role of adenosine in K+ efflux has been investigated in the in situ anoxic turtle brain. Extracellular K+ in the normoxic and anoxic brain, upon inhibition of the Na+K+ATPase by ouabain, follows a three-phase pattern similar to that seen in the mammal of: (1) an initial slow increase in K+ until threshold followed by (2) an abrupt elevation in extracellular K+ to (3) an elevated plateau (Pek and Lutz, 1997). The time to reach full depolarization (K+ plateau) is 3 times longer in the anoxic brain than in normoxic controls and the initial rate of K+ leakage is reduced by 70%. Administration of the adenosine A1R blocker 8CPT significantly shortened the time to full depolarization and increased the rate of K+ efflux (Pek and Lutz, 1997). Interestingly adenosine receptor blockers did not fully disinhibit the anoxia induced reduction in K+ leakage, pointing to a role for additional factors in producing channel arrest.
The decrease in potassium ion flux is an important factor underlying early anoxic survival in the turtle brain. Voltage gated ion channels which mediate this reduced K+ efflux (Kv) play a key role in setting the resting potential and in determining the rate of membrane repolarization and the rate of neuronal firing (Meir et al., 1999). In mammalian neurons certain Kv channels act as oxygen sensors; and acute hypoxia is responsible for blocking a number of Kv channels including Kv1.5 and Kv1.2. We have investigated the involvement of regulated expression of Kv channel transcripts as part of the turtle’s defense strategy of reducing brain metabolic demands. At 4 hours anoxia turtle brains show a substantial down-regulation in Kv1 channel transcription to less than 20% of normoxic levels (Prentice et al., 2003). It is likely that this decrease in Kv1 transcription is accompanied by a decrease in channel protein as Kv1 channels are known to have a rapid turnover rate. The down-regulation of Kv1 channel transcripts indicates an important mechanism that matches brain activity to oxygen supply. Signals that underlie this decreased Kv1 transcription could be directly through an oxygen sensor (HIF-1, NADPH oxidase or mitochondrial) or indirectly through altered levels of metabolites, neurotransmitters or cytokines.
It has been proposed by Sick et al. (1982), that an overall decrease in membrane ion permeability (termed channel arrest) could come about by two processes: (1) Leakage arrest where ion leakage is prevented when neurons are electrically inactive and (2) Spike arrest where channels involved in action potentials are inhibited. There is a 42% decrease in the density of voltage sensitive Na+ channels in anoxic turtle cerebellum (Perez-Pinzon et al., 1992), which is a likely cause of the observed elevation in action potential threshold (Sick et al., 1993). This decreased sodium channel density would therefore result in a down-regulation in sodium ion flux and reduced neuronal activity through the process of spike arrest.
Calcium influx is the ubiquitous signal for neurotransmitter release. In turtle brain in the first 1–8 minutes of anoxia NMDA receptor activity is decreased by 50–60%. This reduction confers an immediate protection to the turtle brain against uncontrolled glutamate activated Ca2+ influx (Bickler et al., 2000). NMDA receptors are silenced in early anoxia by a rapid dephosphorylation controlled by activation of phosphatase 1 or 2A (Bickler and Donohoe, 2002). This initial decrease in receptor activity is not associated with an increase in intracellular calcium. Subsequently suppression of NMDA receptors is predicted by a calcium and calmodulin dependent control mechanism acting during the first 2 hours of anoxia (Bickler et al., 2000). Interestingly adenosine also plays a role in decreased NMDA receptor activity (Buck and Bickler, 1998). In long term anoxia NMDA receptor activity is further reduced through processes involving receptor renewal and internalization (Bickler et al., 2000). These decreases in NMDA receptor activity are accompanied by anoxia-induced decreases in the release of glutamate, which is also mediated by both adenosine and KATP channels (see below).
Neurotransmitters
In mammalian brain, excitotoxic cell death occurs within minutes of exposure to anoxic conditions. Excitatory neurotransmitters, primarily glutamate and dopamine, are rapidly released, causing an acceleration in energy use and calcium influx. The elevation in extracellular transmitter levels comes about as the result of a decrease in reuptake as well as an increase in both vesicular and non-vesicular release (Dawson et al., 2000).
In contrast to the mammalian brain, in the early anoxic turtle extracellular glutamate release is decreased by 44%. Thus it’s likely that the decrease in NMDA receptor activity at this time is not necessary to counteract excitotoxicity, since glutamate levels remain low, but instead NMDA receptor channel inactivation may be a function of the generalized down regulation of neuronal activity. Levels of glutamate in early anoxia appear to be a function of both reduced glutamate release and continued reuptake, which would help maintain neuronal tone and facilitate recovery of the neuronal network after long term anoxia (Milton et al., 2002).
In the hypoxic mammalian brain glutamate and dopamine release are inhibited by mechanisms involving the KATP channels, which may in turn be regulated through adenosine signaling pathways. For example, in rat hippocampal slices a KATP channel blocker abrogated the effects of preconditioning in focal ischemia, as did blockade of adenosine receptors (Yoshida et al., 2004). The mechanisms of decreased glutamate release in anoxic turtle brain also depends on regulation by KATP channels and adenosine signaling pathways. Either pathway is capable of mediating the suppression of glutamate release and only simultaneous blocking both KATP and adenosine pathways prevents the anoxia-induced decrease in release (Milton et al., 2002). As the uptake of glutamate is energetically expensive (Swanson and Duan, 1999), it is likely that activation of adenosine and/or KATP pathways serves to conserve energy by reducing glutamate release and thus also reducing glutamate uptake requirements.
Very similar observations have been made for dopamine release and reuptake in the anoxic turtle brain. Low extracellular levels of dopamine are also maintained in the turtle brain in anoxia (Milton and Lutz, 1998; Milton and Lutz, 2005). As is the case with glutamate, dopamine release is decreased but not abolished and a balance is maintained by the continued function of reuptake transporters during long-term anoxia (Milton and Lutz, 1998). Blocking dopamine reuptake in the first hour of anoxia in turtle brain does not increase intracellular dopamine levels, however, thus in early anoxia reuptake mechanisms do not contribute to the decreased level of dopamine. In common with the regulatory mechanisms for early anoxic glutamate, external dopamine balance during the transition to hypometabolism is maintained by activation of both KATP channels and adenosine pathways, as blocking either pathway increased extracellular dopamine (Milton and Lutz, 2005).
Protective signaling in early anoxia
Heat Shock Proteins
As molecular chaperones that protect proteins against denaturation and help reform mis-folded proteins, heat shock proteins of the HSP70 family are of critical importance in the mammalian brain in increasing tolerance to ischemic damage (Sun et al., 2005). HSPs, clearly provide only limited protection, however, as the brain dies within minutes of an ischemic episode. One member, Hsc73, is regarded as constitutive because it is present in non-stressed tissues and is only slightly induced by stress (Snoeckx et al., 2001). The inducible heat shock protein Hsp72 is at minimal levels in the normoxic mammalian brain and is highly elevated in ischemia. Hsp72 is also an important mediator of brain preconditioning whereby a short ischemic episode results in induction of key protective pathways that protect against a subsequent more severe ischemia (for review, see Latchman, 2004). In marked contrast to mammalian brain, basal Hsp72 and Hsc73 are found at high levels in normoxic turtle brain, consistent with the likely possibility that the turtle is “constitutively preconditioned” prior to anoxic stress (Prentice et al., 2003).
MAP Kinases
There are at least three major pathways of the mitogen activated protein kinase family (ERK, p38, and JNK) and each of these pathways transmits particular signals from stressors such as inflammatory cytokines, heat shock or ischemia. Early responses to ischemia in the mammal may involve immediate early genes which are in part activated through the action of JNK, which phosphorylates the transcription factor c-jun, a major AP-1 complex component. In hypoxia/ischemia JNK may also target members of the Bcl-2 family and in this way modify apoptosis related mechanisms (Kharbanda et al., 2000). Of the MAPK pathways ERK is distinguished by its predominantly pro-survival effects in models of brain ischemia (Xia et al., 1995; Kharbanda et al., 2000). MAP kinases, and especially ERK also play a role in brain ischemia preconditioning. In early anoxia in the turtle brain our preliminary data indicate that there is high level expression of phosphorylated ERK within the first hour of anoxia.
2. Basal Maintenance Phase: Hypometabolism
Following the initial hours of metabolic suppression, the turtle must maintain, over days to weeks, the integrity of the brain in a state of deeply depressed metabolism at an order of magnitude lower than in normoxia. This cannot be a matter of simply “shutting off” the brain, however, as the function and integrity of the neural network must be maintained and in a state of readiness for recovery. This makes the turtle an interesting model in which to determine the minimal processes fundamental to survival, and the adaptations that prevent cell death in the face of continued degradative processes. Comparative studies indicate that such processes are widespread and of general relevance, although these can be difficult to study in the exquisitely anoxia-sensitive mammalian model (Lutz and Milton, 2004).
Neuromodulators and neurotransmitters
During the first hour of anoxia the turtle experiences a temporary energy crisis as energy demand initially outstrips supply, causing a limited but significant drop in ATP (Lutz et al., 1984). As metabolic processes are suppressed ATP levels are restored and the adenylate energy charge (AEC) stabilizes (Kelly and Storey, 1988). With ATP levels now maintained by anaerobic metabolism, energy can be expended to maintain basal functions, including the tightly balanced release and reuptake of neurotransmitters.
We have shown that both dopamine and glutamate continue to be released and taken back up into the cell during long term anoxia such that the turtle maintains basal extracellular levels (Milton and Lutz, 2005; Milton et al. 2002). By contrast, even mild (14%) hypoxia significantly increases extracellular dopamine in the mammal brain (Huang et al., 1994), even prior to depolarization.
The reuptake of both dopamine and glutamate is dependent on membrane ion gradients and thus ultimately on the functioning of Na+/K+ ATPase: the dopamine transporter (DAT) co-transports 2 Na+ and 1 Cl− ion with its substrate (Torres et al. 2003), while glutamate reuptake is Na+ and K+ dependent (Swanson and Duan, 1999). With an estimated reuptake expenditure as high as 1.5 ATP per glutamate anion (Swanson and Duan, 1999) the reuptake of neurotransmitters is energetically expensive, implying a critical role in maintaining these processes despite the energy cost. Hints of this also appear in the mammalian literature, where much research on potential therapeutic interventions in stroke has focused on blocking the release or ultimate effects of glutamate. While promising in laboratory studies, such therapeutic interventions have been complete failures in clinical trials (Hoyte et al., 2004), suggesting a critical role for at least a minimum of glutamate cycling to maintain normal brain function. Indeed, a recent study by Biegon et al. (2004) reported significant attenuation of neural deficits and restored cognitive performance following glutamate receptor stimulation for 24 and 48 hrs after traumatic head injury in mice.
One potential role of neurotransmitter release and reuptake could be the continued stimulation of both autoreceptors and the transporters themselves. Stimulation of dopamine autoreceptors on the pre-synaptic membrane, for example, keeps synaptic levels low by decreasing dopamine release, decreasing synthesis, and increasing the rate of dopamine uptake by DATs (for review, see Sibley, 1999). As these functions clearly maintain normal synaptic function and prevent overstimulation of postsynaptic receptors under normoxic conditions, it perhaps should not have been unexpected to find that the turtle maintains homeostatic function under anoxic conditions as well, albeit at reduced rates of energy consumption.
Recent studies also highlight the primary role of monoamine transporters not only in the regulation of extracellular concentrations but also in the homeostatic maintenance of presynaptic function (Torres et al. 2003). Gainetdinov et al. (1998), for example, reported significant alteration of the homeostasis of the nigrostriatal dopamine system in the absence of the DAT in mice, particularly regarding the control of synthesis and storage mechanisms. Thus a long-term suppression of neurotransmitter release over weeks of anoxia could have profound effects on the continued functioning of the neural network and post-anoxic recovery.
As anoxia progresses, glutamate cycling continues, albeit at a reduced rate, but in contrast to the initial hours of anoxia this reduction is modulated by adenosine and GABAA receptors (Thompson and Lutz, 2001). By several hours into anoxia, KATP channels have closed with the restoration of ATP levels, and no longer affect either neurotransmitter balance or the downregulation of ion channels (Milton and Lutz, 2005; Milton et al., 2002; Pek and Lutz, 1998).
In contrast to the low extracellular levels of excitatory neurotransmitters, tissue and extracellular levels of GABA both increase significantly in the anoxic turtle. In the brain, GABA tissue concentrations increase 45–60% over the first 2–4 hr anoxia, and 127% after 13 hr at 25°C (Hitzig et al., 1985; Nilsson et al., 1990). Such increases are possible as the conversion of glutamate to GABA is anaerobic and can continue during anoxia, while the breakdown of GABA is oxygen-dependent. Increased tissue levels are reflected extracellularly; at about 100 mins of anoxia there begins a large and sustained increase in GABA, with extracellular levels rising from 0.3 to 27 μM over 2–3 hr (Nilsson and Lutz, 1991). Interestingly, this massive GABA increase begins about the time the initial adenosine peak has begun to decline (Nilsson and Lutz, 1992). The rise in GABA is also accompanied by increases in GABAA receptor density, which continues to increase for at least 24 hr anoxia (Lutz and Leone-Kabler, 1995) and is likely to increase the effectiveness of GABA inhibition during extended anoxic periods.
A functional link between glutamate, GABA, and Hsp73 has been proposed by Jin et al. (2003) in the mammalian brain, such that the heat shock cognate provides a structural link facilitating the synthesis of GABA from glutamate and aiding in vesicular packaging. Aiding in the packaging and release of GABA may thus provide a functional role for the long-term increases in Hsp73 levels we have reported (below). The turtle brain also has detectable levels of glutamate acid decarboxylase (GAD), another critical component of this complex (Prentice and Milton, unpublished results).
Electrical activity
The depression of ion channel activity, coupled to decreases in excitatory neurotransmitter release and increased inhibition, results in a dramatic suppression of electrical activity in the anoxic turtle brain, and thus important energy savings. While evoked potentials and electroencephalogram (EEG) activity become fully suppressed within seconds in the anoxic mammalian brain, integrated electrical activity apparently continues in the turtle brain, albeit at reduced levels. During the initial stages of anoxia, EEG activity progresses from irregular, low amplitude waves to a state dominated by low-amplitude, slow-waves (3–12 Hz), with the total EEG power spectra decreased by about one order of magnitude across all frequencies (Fernandes et al., 1997). This depressed basal EEG is interrupted at 0.5 – 2.0 min intervals by high amplitude, mixed frequency burst activity. Such alternating activity cycles have also been reported in hibernating mammals, with the slow-wave sleep-like EEG patterns of deep hibernation interrupted periodically by short bursts of low-amplitude activity (Krilowicz et al., 1988). We can speculate that such activity bursts may thus be critical factors to maintain the brain as an integrated unit in a state of deeply depressed metabolism, and could be functionally linked to the continued release and reuptake of neurotransmitters noted during long-term anoxia. Upon reoxygenation, there is a steep rise in EEG amplitude, with full recovery in less than 2 hrs (Fernandes et al., 1997).
Molecular changes
Heat shock proteins
While detectable levels of heat shock proteins Hsp72 and Hsc73 under normoxic conditions suggest constitutive preconditioning in the turtle, additional increases occur during long-term anoxia. In the turtle brain, Hsp72 is induced early in anoxia, increasing for up to 8 hrs before returning to normoxic levels by 12 hr anoxia. By contrast, Hsc73, generally reported as only slightly inducible in mammals, increases progressively over 12 hr anoxia (Prentice et al., 2004). The differential expression of these heat shock proteins suggests different roles in brain anoxia, with Hsp72 playing a key protective role during the initial transition to the anoxic state, and Hsc73 perhaps more important for long-term maintenance (e.g. GABA packaging and release, see above).
NF-κB
Hsc73 is not the only molecular component that changes preferentially during long term anoxia. In the turtle brain, NF-κB shows maximal DNA binding at 6 hr anoxia (Lutz and Prentice, 2002); such late translocation to the nucleus suggests a role in either long-term maintenance of cellular processes or perhaps as a defense mechanism against presumptive ROS damage and/or apoptosis upon reoxygenation (Lutz and Milton, 2004). NF-κB activity increases in mammalian neurons and glia after ischemia, and is thought to induce genes related to immune function, unflammation, apoptosis, and ROS protection (Haddad, 2002; Martindale and Holbrook, 2002).
Neuroglobin
A recently discovered heme protein of the CNS, neuroglobin is a highly conserved oxygen-binding protein reported in mammals (Burmester et al., 2000), fish, amphibians, and birds (Burmester and Hankelm, 2004). It’s function, however, is still a matter of debate, with suggested roles as a “neuronal myoglobin” transferring oxygen to the mitochondrial respiratory chain (Burmester and Hankelm, 2004), as a terminal oxidase that regenerates NAD+ under anaerobic conditions (Sowa et al., 1998), as an antioxidant (Herold et al., 2004), and as a sensor to detect cellular oxygen concentrations (Kriegl et al., 2002). Increased levels of neuroglobin have been shown to protect against hypoxic-ischemic injury in both cultured neurons (Sun et al., 2001) and in an experimental stroke model in rats (Sun et al., 2003), but investigation of the regulation and role of neuroglobin is difficult in the mammalian brain due to its exquisite sensitivity to severe hypoxia/anoxia. In the turtle brain, neuroglobin mRNA expression as measured by RT-PCR increased by 3.5 fold after 4 hours hypoxia, whereas a more modest but progressive 2.0 fold increase occurred over 4 h anoxia (Milton et al., 2006). On re-oxygenation following 4 h anoxia, even higher levels of expression (4.7 fold) were detected. The greater degree of induction in hypoxia or reoxygenation compared to anoxia implies that induction is not a rapid process in response to low oxygen levels, and that a functional role for neuroglobin is more likely in the presence of oxygen than in anoxia (e.g. ROS detoxification vs. the anoxic regeneration of NAD+).
MAP Kinases
While Greenway and Storey (2000) reported no increases in ERK, JNK, or p38MAPK activity in the brains of one to 20 hr anoxic submerged turtles held at 7°C, we have found significant changes in MAPK levels in early and long-term anoxic animals ventilated on nitrogen at room temperature.
Overall, ERK levels increased nearly three-fold over 1 hr anoxia, and were still two-fold higher in 4 hr anoxic turtle brains compared with control. Phospho-activated ERK shows a more striking pattern of changes, with an approximate 6-fold increase in the initial hour of anoxia that returns to basal by 4 hr anoxia (Milton et al, 2005). We have also found significant increases in whole–brain JNK and phospho-JNK, as well as p38MAPK, at 1 and 4 hr anoxia (Milton, unpublished data).
Changes that promote anoxic survival may also shape ROS protection in the turtle: it has been demonstrated in mammalian models of preconditioning that mitochondrial KATP channels mediate ERK 1/2 activation by generating ROS, and thus confer a protected phenotype on cardiomyocytes (Abas et al., 2000; Gong et al. 2004; Xu et al., 2004) and kidney cells (Park et al. 2001). The neuroprotective effects of adenosine have also been linked to downstream ERK activation (Trincavelli et al. 2002). In mammalian cardiomyocytes, the protective effects of preconditioning triggered by adenosine receptor stimulation are blocked by ERK inhibition (Reid et al. 2005), while A2A agonists increase ERK activation and promote cell survival in sympathetic neurons (Ramirez et al. 2004). As both ATP-regulated potassium channels and adenosine contribute significantly to anoxic survival, either of these signals may affect MAPK pathways.
3. Recovery upon Reoxygenation
The recovery phase begins when oxygen is restored to the system (although, intriguingly, a turtle hibernating underwater may have to upregulate from deep hypometabolsim prior to the restoration of pulmonary oxygen, in order to surface and breathe!). Upon the restoration of oxygen, blood PO2 recovers to normoxic levels within 10 mins (Fernandes et al. 1997). Accompanying this is the rapid restoration of electrical activity; when air respiration was restored following 6 hrs anoxia, a rapid rise in EEG amplitude recovered total EEG power in turtles within 2 hr (Fernandes et al. 1997). Recovery, however, requires more than the addition of oxygen. Processes that were initially downregulated and maintained at minimal levels must be upregulated in a coordinated manner, while the turtle must also avoid toxicity resulting from an overproduction of reactive oxygen free radicals (ROS). Recovery upon reoxygenation has been a relatively neglected area of study in anoxia tolerant organisms. A first priority to re-establish normal brain activity would presumably be the reactivation of ion channels. This is seen, for example, with the upregulation of Kv transcription within 4 h of normoxia following nitrogen respiration (Prentice et al., 2003).
Reactive oxygen species
Anoxia/ischemia followed by reoxygenation/reperfusion in mammals is known to result in a rapid transient increase in ROS (Halliwell and Gutteridge, 1997; Hashimoto et al. 2003; Schild and Reiser, 2005), formed by the incomplete reduction of O2 to superoxide (.O2-) or hydrogen peroxide (H2O2). H2O2 in turn can lead to the production of the highly reactive hydroxyl radicals (.OH) that damage cells by the oxidization of essential cellular lipids, proteins, and nucleic acids (Oliver et al. 1990). While there is evidence for the production of ROS during hypoxia and under normoxic conditions, when it may act as a signaling molecule, significant ROS overproduction occurs during reoxygenation because hypoxia causes an accumulation of reducing equivalents (NADH, FADH2) in the mitochondria (Zuo and Clanton, 2005). Oxidative damage and mutations are particularly common in mitochondrial DNA (mtDNA) (Kang and Hamasaki, 2005), and elevated levels of mtDNA mutations appear to increase apoptosis, cardiomyopathies, neurodegenerative disease, and premature aging (for review, see Stuart and Brown, 2006). Oxidative stress on cellular components has been implicated in the ageing process (Sohal et al., 2002), while comparative studies indicate that the rate of endogenous ROS generation explains at least part of the different rates of aging in animals (Barja, 2002). The mammalian brain is particularly vulnerable to oxidative damage because of its high levels of unsaturated fatty acids and iron, coupled to relatively poor antioxidant defenses (Floyd and Carney, 1992).
After hours to weeks of anoxia, the turtle brain should be primed to experience a massive ROS insult upon reoxygenation. Trachemys scripta, however, is clearly able to survive repeated anoxia-reoxygenation stress, both in vivo and in vitro; primary mixed brain cultures cultivated in our lab are able to withstand 2 days anoxia/24 hr reoxygenation with less than 3% cell death (Lutz et al. 2003). There are a number of possible levels at which turtles could possess adaptations to combat ROS stress, as depicted in Fig 1.
Figure 1.
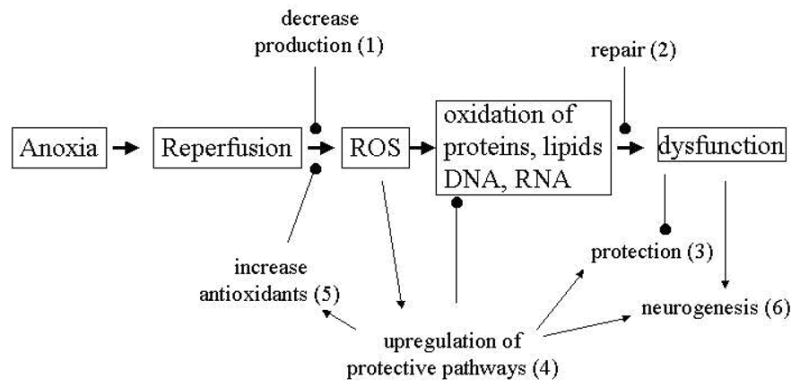
Potential sites where T. scripta may be adapted to reduce or prevent reactive oxygen stress upon reoxygenation following anoxia. Anoxia/reperfusion in mammals results in massive ROS increases that damage proteins, lipids, and nucleotides, resulting in cellular dysfunction and eventual death. Freshwater turtles are clearly able to survive repeated anoxia/reoxygenation, and may have increased resistance to ROS stress.
Decreased ROS production (1)
Preliminary evidence from our lab using MitoTracker Red® to follow mitochondrial ROS production (Imoto et al., 2006) in neuronally enriched turtle cell cultures has shown that while ROS are apparent during normoxia and upon recovery (and disappear over 4 hr anoxia), the number and intensity of fluorescent cells upon reoxygenation of cultures following 4 hr anoxia is not significantly different from basal normoxic conditions (Fig 2). The actual production of ROS in vivo or in vitro in the turtle brain has not been previously measured; whether the apparent low production of ROS during recovery is due to true reduced production or to highly efficient scavenging (see (5), below) has yet to be determined. It could be speculated, however, that the turtle may be able to avoid a mammalian-like overproduction of ROS upon reoxygenation in part by some of the very mechanisms that permit anoxic survival, including the upregulation of MAPK pathways, adenosine increases, and the ability to prevent excitotoxin release.
Figure 2.

Representative digital images showing mitochondrial oxidation of rhodamine dye in neuronally-enriched turtle primary cultures utilizing Mitotracker Red®. Cultures were photographed using a BioRad Radiance 2000 Scanning Laser Confocal Microscopemicroscope at 10X magnification. a) normoxic controls b) 4 hr anoxia c) upon reoxygenation following 4 hr anoxia.
The overactivation of glutamate receptors (Sharp et al. 2005; Kahlert et al. 2005) and elevated intracellular calcium (Sharikabad et al. 2004) are both associated with free-radical formation, and the direct activation of NMDA receptors results in a massive release of .OH (Lancelot et al. 1998; Laplanche et al. 2003). By keeping extracellular glutamate (Milton et al., 2002) and intracellular calcium levels low (Bickler, 1992), and through an anoxia-induced reduction of NMDA receptor activity (Buck and Bickler, 1998; Bickler et al. 2000), the turtle brain would be positioned to avoid ROS increases associated with NMDA receptor overstimulation. Similarly, significant increases in extracellular dopamine result even from mild hypoxia in the mammalian brain (Huang et al. 1994), and dopamine increases are correlated with ROS formation. Ferger et al. (1999), for example, demonstrated that the malonate-induced release of dopamine was paralleled by hydroxyl radical generation. Dopamine levels, however, are maintained by the turtle over 4 hr anoxia (above). While excitatory neurotransmitter levels are kept low, however, neuroprotective compounds like GABA (Nilsson and Lutz, 1991) and adenosine (Nilsson and Lutz, 1992) increase in the extracellular space during anoxia. Recent data from mammalian systems has shown that adenosine given at reperfusion can significantly protect both the heart and brain against ischemia/reperfusion damage (Xu et al., 2005, Xu et al., 2006).
Robust repair mechanisms (2)
Superoxides are known to damage proteins, lipids, and nucleotides; an organism could theoretically avoid cumulative ROS damage by the possession of robust repair mechanisms. Measurements of lipid peroxidation damage products, including conjugated dienes, lipid hydroperoxides, and thiobarbuturic acid reactive substances (TBARS) showed minimal changes in turtle tissues following anoxic submergence and recovery (Willmore and Storey, 1997a), however in these studies it cannot be determined solely from post-anoxic measurements whether damage is prevented or instead repaired. Little work has been done to investigate the role of repair enzymes in turtle anoxia and reoxygenation survival, though such mechanisms are highly conserved and can be presumed to exist. Mermet et al. (2002) reported the presence of Kin protein, involved in nuclear DNA repair, in several regions of the Trachemys scripta brain, though activity levels were not determined. And an early report by Woodhead et al. (1980) found no difference in excision repair capacity in cultured cardiac cells from long-lived turtles (Carolina box turtle, Terrapene carolina) compared to shorter-lived fish species; the study was carried out, however, only 24 hr after uv irradiation, which in ectotherms may have been too early a time point for differences to become apparent.
One compound known to act as both an antioxidant and to function in the repair of oxidatively damaged proteins is the enzyme methionine sulfoxide reductase (MsrA) (Weissbach et al. 2002). Under mild oxidizing conditions, methionine (Met), both free and in proteins, can be converted to methionine sulfoxide [Met(O)], resulting in the inactivation of a variety of proteins; MsrA and MsrB catalyze the reduction of Met(O) epimers in proteins back to methionine, and can thus restore protein function. Because of its role as a repair enzyme, it has also been suggested that the reversible oxidation of Met residues in proteins provides an ROS sink, resulting in the destruction of ROS and reactive nitrogen species (RNI) at the expense of NADPH (Levine et al. 1996). MsrA has been has been shown to protect mammalian neurons against brief hypoxia/reoxygenation (Yermolaieva et al., 2004). Preliminary evidence from our labs indicates a greater than four-fold induction of MsrA transcription in whole brain tissues by 4 hr anoxia, however no upregulation is seen following anoxia-reoxygenation compared to basal levels (Milton and Prentice, 2006). In the turtle, brain, therefore, this could imply a critical role for MsrA during anoxia itself, perhaps in repairing proteins damaged during the initial critical events of the transition phase, or alternatively that the enzyme is “banked” in reserve against future reoxygenation stress.
Protection of structure and function (3)
Another potential adaptation to avoid anoxia-reoxygenation dysfunction would be to increase the stability of nucleotide, protein and lipid structures in the face of oxidative stressors, thus ameliorating damage from excess ROS production that may occur without having to upregulate repair functions simultaneously with the restoration of normal function. While numerous studies have demonstrated the reversible down-regulation of a number of proteins during anoxia (Fraser et al., 2001; for review see Storey, this issue), there have been no studies to our knowledge of actual protein or lipid stability in turtle tissues in anoxia or reoxygenation (as there have been, for example, in investigations of proteins stability under thermal stress or high pressure). However, the significant upregulation of heat shock proteins reported in turtle tissues could aid in stabilizing structures during both anoxia itself and during the ensuing reoxygenation.
Upregulation of protective pathways (4)
While much attention has focused on the physiology of metabolic downregulation at the cellular and biochemical level, more recently attention has turned to the molecular adaptations underlying the physiology of hypometabolism; some of the same factors which permit anoxic survival may also be critical to surviving ROS stress. Putative protective mechanisms in mammalian systems that have been investigated in turtles include members of the mitogen-activated protein kinase family: p38-MAPK, extracellular regulated kinase (ERK-1/2), and c-jun kinase (JNK), as well as the heat shock proteins (above). JNK and p38MAPK are both activated by ischemia-reperfusion in intact mammalian hearts, but neither has been shown to be exclusively pro- or anti-apoptotic (Dougherty et al. 2002). ERK-1/2 has been shown to alter ROS production in the mammalian brain, and ERK upregulation is thought to be protective in several tissues (Solenkova et al., 2006; Conde de la Rosa et al. 2006). ERK activation is protective against applied ROS stress in mouse proximal tubule cells (Arany et al. 2005), as well as in hepatocytes (Conde de al Rosa, 2006). Other studies link ERK to ROS production and neuronal cell damage, thus studies of anoxia-adapted turtles may allow us to distinguish between robust survival pathways and the mixed pathological/adaptive responses that occur in mammalian cells (Lutz et al. 2003).
Work by Greenway and Storey (2000) with one to 20 hr anoxic submerged turtles held at 7°C, showed increased JNK activity from 1.5- to approximately 4-fold between 5 and 20 hr in the liver, heart, and kidney, and an increase in phosphorylated ERK of 2.8-fold in the spleen. We have reported a significant 6-fold increase in phospho-ERK in the 1 hr anoxic brain at room temperature, a clear upregulation of this enzyme, though this initial upregulation decreased to near basal by 4 hr anoxia (Milton et al., 2005). Phospho-ERK would thus play a more likely role in surviving the initial anoxic stress than as a defense against reoxidative stress many hours later. Other MAPK pathways are currently under investigation in our lab both in vivo and using in vitro cell culture approaches.
Increased antioxidants (5)
The most straightforward way to combat ROS stress would be to increase the levels of tissue antioxidants. T. scripta maintains high constitutive levels of catalase, superoxide dismutase (SOD), and alkyl hydroperoxide reductase (Willmore and Storey, 1997a; 1997b), while cortex levels of ascorbic acid are 2–3 times greater than in mammalian brains (Rice et al., 1995). Glutathione peroxidase (GPOX) and SOD activities in turtle liver are actually comparable to mammalian enzyme activity (Willmore and Storey, 1997b) despite much lower overall metabolic rates. Array screening of anoxic Chrysemys picta marginata hatchlings also revealed the upregulation of antioxidant genes in the heart and liver, including isozymes of SOD, glutathione peroxidase, glutathione-S-transferase (GST), and peroxiredoxin (Storey, 2005; 2006). Of note is the finding that liver GST in T. scripta is apparently modified during anoxia to a more stable form, resulting in a 50% reduction in specific activity, coupled to a change in substrate preference (Willmore and Storey, 2005).
In addition to the upregulation of the potential antioxidant MsrA (see above), preliminary results in our lab have also revealed an increase in SOD transcription, which in contrast to MsrA occurs during reoxygenation. While no upregulation occurs over 4 hr anoxia, by 4 hr recovery there is a 5-fold increase in SOD-1 transcription (Milton and Prentice, 2006).
Neurogenesis (6)
Despite the hypometabolism of anoxia and hypothermia of winter hibernation, a number of turtles still fail to survive the season, implying that despite their tolerance, months of anoxia can result in severe damage. The replacement of irreversibly damaged neurons following recovery would allow turtles to survive repeated winters even with considerable damage, depending on the extent and location of damaged cells. Much recent research supports the concept that neurons and glial cells in certain parts of the CNS continue to be produced throughout life (Gross, 2000; Gould and Gross, 2002). Studies concerned with the occurrence of immature proliferating cells in the CNS of vertebrates have focused primarily on birds and mammals, and a temperature-related response has been reported in adult lizards (Ramirez et al., 1997; Penafiel et al., 2001). Recent work has expanded these findings to turtles, demonstrating the presence of recently divided cells positive for neuronal markers in the spinal cord of juvenile Chrysemys d’orbigny (Fernandez et al. 2002; Russo et al. 2004). In the brain these cells proliferate more strongly under warm conditions than at colder temperatures (Radmilovich et al. 2003), providing a potential means to replace anoxia-damaged cells during the warmer months.
Emerging themes in anoxia tolerance and cerebral ischemic protection
A number of major themes in neuroprotection emerge from the studies on turtle brain anoxia tolerance; in many cases these offer unique insights into the molecular pathways that underlie physiological survival mechanisms. The anoxia tolerant turtle brain has many adaptations that share common pathways with those described in the mammalian literature of brain ischemic preconditioning (IP), including 1) maintaining ATP levels (Zhang et al. 2002), 2) opening of ATP-dependent potassium channels (Milton et al. 2002) 3) the increased release of GABA (Grabb et al. 2002) and upregulation of GABAA receptors (Lutz and Leone-Kabler, 1995), 4) decreased glutamate release regulated by KATP and adenosine (Milton et al. 2002), and 5) increased adenosine release coupled to an increase in adenosine receptor (ADR) affinity (Lutz and Manuel, 1999). However, the vulnerability of mammalian cells to hypoxic/ischemic stress can make it difficult to distinguish between adaptive and injurious responses, and indeed some molecular pathways are thought to induce both; critical pathways may thus be identified by considering the turtle as a “constitutively preconditioned” model. In IP, a brief, sublethal exposure to ischemia or anoxia induces changes that result in a more ischemia-tolerant phenotype; key pathways hypoxic insult can provide protection against a later, more severe event that would otherwise be lethal. Preconditioning protection creates two distinct periods of protection against an index ischemic insult; an initial window of protection that disappears within 60–120 min, and a second, longer window of protection appearing 24 hrs later (Bernaudin 2002). This second window of protection (SWOP) is of intense clinical interest, as elucidation of the molecules that confer the protected, preconditioned phenotype could conceivably enable therapeutic exploitation of these endogenous protective mechanisms against the effects of various diseases including stroke and myocardial infarction. The shift to the defensive phenotype is the result of a complex cascade of molecular events; in the mammalian myocardium, nitric oxide (Qui et al. 1997; Bolli et al. 1997), reactive oxygen species (ROS) (Zhou et al. 1996; Sun et al. 1996), bradykinin (Ebrahim et al. 2001; Kositprapa et al 2001), and adenosine (Baxter et al. 1994) have all been shown to contribute to delayed IP. Triggers to the protected phenotype then affect downstream molecular pathways, which may block pro-apoptotic changes that lead to mitochondrial damage, cytochrome c release, and activation of caspases. Numerous reports document changes in kinase signaling pathways in ischemia and redox-stressed cells; many focus on mitogen-activated protein kinase/extracellular-signal-regulated protein kinase (MAPK/ERK), and the stress activated protein kinases c-JunN-terminal kinase (JNK), and p38 MAPK, as the balance of these may determine cell fate (Xia et al. 1995). Current research indicates that IP results from stimulation of key survival mechanisms that may involve nitric oxide; NO in turn activates Ras and then ERK (the downstream MAPK) (Nandagopal et al., 2001). In a number of cases ERK activation arises from the activation of adenosine A1 receptors and KATP channels (Heurteaux et al., 1995; Dawson and Dawson, 2000; Solenkova et al., 2006). Transduction of signals for cardiac as well as brain preconditioning are heavily dependent upon protein kinase C-epsilon activation and in brain this activation occurs in response to signals from NMDA and adenosine A1 receptors (Lange-Asschenfeldt et al., 2004). Data on PKC-epsilon activation in mammalian brain are leading investigators to consider PKC-epsilon as a target for developing protective drug therapies that mimic brain preconditioning (Chou and Messing, 2005). In turtle brain, a number of the same pathways: adenosine A1 receptors, KATP channels and MAP kinase pathways are central to anoxic survival. A critical role for adenosine receptor stimulation has been shown to underlie the switch to a hypometabolic state, and adenosine receptors, in concert with KATP channels, modulate excitatory neurotransmitter levels that enable turtle neurons to continue functioning at basal levels of excitability under prolonged anoxia. Phosphorylated ERK is elevated in early anoxia and is therefore a likely contributor to the development of concerted survival responses in this transition phase.
The down-regulated release of excitatory neurotransmitters with continued reuptake mechanisms in the anoxic turtle brain contrasts markedly with the uncontrolled release of excitatory neurotransmitters seen in the ischemic mammalian brain. This strategy for continued basal function and maintaining neural network integrity in the anoxia tolerant brain matches closely recent experimental observations on the therapeutic potential in mammalian brain of maintaining an operational glutaminergic system following cerebral injury
Major protective kinase pathways implicated in mammalian neuronal preconditioning include the MAP kinases and the Akt pathway. The NMDA receptor and NO may signal in neuronal preconditioning through ERK activation (Gonzalez-Zulueta et al., 2000). Although JNK is generally thought of as a pro-death signal, in cardiac myocytes subjected to hypoxia/re-oxygenation (Waetzig and Herdegen, 2005), JNK is protective if adequate ATP is available (Dougherty et al., 2002). Akt has the potential to mediate neuronal ischemic protection though activation of eNOS (Gonzalez-Zulueta et al., 2000; Huang, 2004) or through the phosphorylation and inactivation of BAD, a pro-apoptotic signal (Wang et al., 1999). In anoxic turtle neurons the observed elevations in phospho-JNK, phospho-ERK and phospho-p38 may differentially relay downstream signals to alternative targets resulting in a net outcome that is pro-survival, and in which the activation of pro-apoptotic cell targets is prevented. Phosphorylated Akt is also elevated in anoxic neurons and this protective kinase may therefore target aspects of the apoptotic machinery to usurp a potential death signal.
A number of recent therapeutic interventions for stroke have targeted apoptotic pathway components including caspases and Bcl-2 family members (Antonawich et al., 1999; Tanaka et al., 2004). Bcl-2 and Bcl-xl are induced in cerebral ischemia (Shimizu et al., 2001) and recent studies point to increased Bcl-2 expression in brain following preconditioning. Reports of an induction of the antioxidant thioredoxin (TRX) in brain preconditioning (Chiueh et al., 2005) indicate that this occurs upstream of a protective Bcl-2 activation. Interestingly a traditional apoptosis effector, caspase 3, is up-regulated in preconditioning but held in check by heat shock proteins of the HSP70 family (McLaughlin et al., 2003). In anoxic turtle brain we have reported an induction of Bcl-2 transcription as well as elevated Hsc73 protein expression, which may point to the presence of tight controls over both pro-and anti-apoptotic processes during prolonged anoxia.
Recent studies on mechanisms of brain repair in stroke and in neurodegenerative diseases point to a key role for neurogenesis mediated by increases in expression of growth factors such as vascular endothelial growth factor (VEGF) and brain-derived neurotrophic factor (BDNF) which may then signal through survival pathways involving NO and Akt (Kokaia et al., 1995; Jin et al., 2002). In the turtle there is potential for neurogenesis; recent reports point to substantial neuronal cell division in the turtle central nervous system.
Peter
Peter Lutz’s phenomenal contributions to science have greatly influenced the thought processes of scientists including many of us who knew him well. Peter opened our eyes to the critical importance and the reality of integrated approaches to biology. Peter’s humanity, creativity and sense of fun have given us tremendous inspiration for our own lifetimes in science.
Acknowledgments
This work was funded in part by NIH AREA grant No. 1 R15 NS048909-01 and American Heart Association Florida/Puerto Rico Affiliate Grant in Aid of Research No. 0455318B to S.L.M., Principal Investigator, and the FAU Foundation. This is contribution number P200614 from the Florida Center of Excellence in Biomedical and Marine Biotechnology.
Footnotes
Footnote for cover page: This paper derives from a presentation at a Memorial Symposium in honor of Dr. Peter Lutz held at Florida Atlantic University on September 23rd, 2005.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abas L, Bogoyevitch MA, Guppy M. Mitochondrial ATP production is necessary for activation of the extracellular-signal-regulated kinases during ischaemia/reperfusion in rat myocyte-derived H9c2 cells. Biochem J. 2000;349:119–126. doi: 10.1042/0264-6021:3490119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonawich FJ, Federoff HJ, Davis JN. BCL-2 transduction, using a herpes simplex virus amplicon, protects hippocampal neurons from transient global ischemia. Exp Neurol. 1999;156:130–7. doi: 10.1006/exnr.1998.7004. [DOI] [PubMed] [Google Scholar]
- Arany I, Megyesi JK, Reusch JE, Safirstein RL. CREB mediates ERK-induced survival of mouse renal tubular cells after oxidant stress. Kidney Int. 2005;68:1573–82. doi: 10.1111/j.1523-1755.2005.00569.x. [DOI] [PubMed] [Google Scholar]
- Barja G. Rate of generation of oxidative stress-related damage and animal longevity. Free Radic Biol Med. 2002;33:1167–72. doi: 10.1016/s0891-5849(02)00910-3. [DOI] [PubMed] [Google Scholar]
- Baxter GF, Marber MS, Patel VC, Yellon DM. Adenosine receptor involvement in a delayed phase pf myocardial protection 24 h after ischemic preconditioning. Circulation. 1994;90:2993–3000. doi: 10.1161/01.cir.90.6.2993. [DOI] [PubMed] [Google Scholar]
- Belkin DA. Anoxia: tolerance in reptiles. Science. 1963;139:492–93. doi: 10.1126/science.139.3554.492. [DOI] [PubMed] [Google Scholar]
- Berger R, Garnier Y, Jensen A. Perinatal brain damage: underlying mechanisms and neuroprotective strategies. J Soc Gynecol Investig. 2002;9:319–28. [PubMed] [Google Scholar]
- Bernaudin M, Tang Y, Reilly M, Petit E, Sharp FR. Brain genomic response following hypoxia and re-oxygenation in the neonatal rat. Identification of genes that might contribute to hypoxia-induced ischemic tolerance. J Biol Chem. 2002;277:3972–38. doi: 10.1074/jbc.M204619200. [DOI] [PubMed] [Google Scholar]
- Bickler PE. Cerebral anoxia tolerance in turtles, regulation of intracellular calcium and pH. Am J Physiol. 1992;263:R1298–R1302. doi: 10.1152/ajpregu.1992.263.6.R1298. [DOI] [PubMed] [Google Scholar]
- Bickler PE. Reduction of NMDA receptor activity in cerebrocortex of turtles (Chrysemys picta) during 6 wk of anoxia. Am J Physiol. 1998;275:R86–R91. doi: 10.1152/ajpregu.1998.275.1.R86. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Donohoe PH. Adaptive responses of vertebrate neurons to hypoxia. J Exp Biol. 2002;205:3579–3586. doi: 10.1242/jeb.205.23.3579. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Donohoe PH, Buck LT. Hypoxia-induced silencing of NMDA receptors in turtle neurons. J Neurosci. 2000;20:3522–3528. doi: 10.1523/JNEUROSCI.20-10-03522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegon A, Fry PA, Paden CM, Alexandrovich A, Tsenter J, Shomani E. Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: Implications for treatment of neurological and cognitive deficits. Proc Natl Acad Sci USA. 2004;101:5117–22. doi: 10.1073/pnas.0305741101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli R, Manchikalapudi S, Tang XL, Takano H, Qui Y, Guo Y, Zhang Q, Jadoon AK. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase. Evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circ Res. 1997;81:1094–1107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- Buck LT, Bickler PE. Adenosine and anoxia reduce N-methly-D-aspartate receptor open probabilities in the turtle cerebrocortex. J Exp Biol. 1998;201:289–297. doi: 10.1242/jeb.201.2.289. [DOI] [PubMed] [Google Scholar]
- Burmester T, Hankeln T. Neuroglobin: A respiratory protein of the nervous system. News Physiol Sci. 2004;19:110–113. doi: 10.1152/nips.01513.2003. [DOI] [PubMed] [Google Scholar]
- Burmester T, Weich B, Reinhardt S, Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- Chih CP, Rosenthal M, Sick TJ. Ion leakage is reduced during anoxia in turtle brain: a potential survival strategy. Am J Physiol. 1989;257:R1562–R1564. doi: 10.1152/ajpregu.1989.257.6.R1562. [DOI] [PubMed] [Google Scholar]
- Chiueh CC, Andoh T, Chock PB. Induction of thioredoxin and mitochondrial survival proteins mediates preconditioning-induced cardioprotection and neuroprotection. Ann N Y Acad Sci. 2005;1042:403–18. doi: 10.1196/annals.1338.034. [DOI] [PubMed] [Google Scholar]
- Chou WH, Messing RO. Protein kinase C isozymes in stroke. Trends Cardiovasc Med. 2005;15:47–51. doi: 10.1016/j.tcm.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Conde de la Rosa L, Shoemaker MH, Vrenken TE, Buist-Homan M, Havinga R, Jansen PL, Moshage H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: Involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44:918–29. doi: 10.1016/j.jhep.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Neuronal ischaemic preconditioning. Trends Pharmacol Sci. 2000;21:423–4. doi: 10.1016/s0165-6147(00)01560-1. [DOI] [PubMed] [Google Scholar]
- Dawson LA, Djali S, Gonzales C, Vinegra MA, Zaleska MM. Characterization of transient focal ischemia-induced increases in extracellular glutamate and aspartate in spontaneously hypertensive rats. Brain Res Bull. 2000;53:767–776. doi: 10.1016/s0361-9230(00)00363-4. [DOI] [PubMed] [Google Scholar]
- Dougherty CJ, Kubasiak LA, Prentice H, Andreka P, Bishopric NH, Webster KA. Activation of c-Jun N-terminal kinase promotes survival of cardiac myocytes after oxidative stress. Biochem J. 2002;362(Pt 3):561–71. doi: 10.1042/0264-6021:3620561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty CJ, Kubasiak LA, Frazier DP, Li H, Xiong WC, Bishopric NH, Webster KA. Mitochondrial signals initiate the activation of c-Jun N-terminal kinase (JNK) by hypoxia-reoxygenation. FASEB J. 2004;18:1060–70. doi: 10.1096/fj.04-1505com. [DOI] [PubMed] [Google Scholar]
- Ebrahim Z, Yellon DM, Baxter GF. Bradykinin elicits ‘second window’ myocardial protection in rat heart through an NO-dependent mechanism. Am J Physiol. 2001;281:H1458–H1466. doi: 10.1152/ajpheart.2001.281.3.H1458. [DOI] [PubMed] [Google Scholar]
- Ferger B, Eberhardt O, Teismann P, de Groote C, Schulz JB. Malonate-induced generation of reactive oxygen species in rat striatum depends on dopamine release but not on NMDA receptor activation. J Neurochem. 1999;73:1329–32. doi: 10.1046/j.1471-4159.1999.0731329.x. [DOI] [PubMed] [Google Scholar]
- Fernandes JA, Lutz PL, Tannenbaum A, Todorov AT, Liebovitch L, Vertes R. Electroencephalogram activity in the anoxic turtle brain. Am J Physiol. 1997;273:R911–R919. doi: 10.1152/ajpregu.1997.273.3.R911. [DOI] [PubMed] [Google Scholar]
- Fernandez A, Radmilovich M, Trujillo-Cenoz O. Neurogenesis and gliogenesis in the spinal cord of turtles. J Comp Neuro. 2002;453:131–144. doi: 10.1002/cne.10388. [DOI] [PubMed] [Google Scholar]
- Floyd RA, Carney JM. Free radical damage to protein and DNA: mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann Neurol. 1992;32(Suppl):S22–S27. doi: 10.1002/ana.410320706. [DOI] [PubMed] [Google Scholar]
- Fraser KPP, Houlihan DF, Lutz PL, Leone-Kabler S, Manuel L, Brechin JG. Complete suppression of protein synthesis during anoxia with no post-anoxia protein synthesis debt in the red-eared slider turtle Trachemys scripta elegans. J Exp Biol. 2001;204:4353–4360. doi: 10.1242/jeb.204.24.4353. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Jones SR, Fumagalli F, Wightman RM, Caron MG. Re-evaluation of the role of the dopamine transporter in dopamine system homeostasis. Brain Res Brain Res Rev. 1998;26:148–53. doi: 10.1016/s0165-0173(97)00063-5. [DOI] [PubMed] [Google Scholar]
- Gong KZ, Zhang ZG, Li LH, Huang AF, Bu P, Dong F, Liu J. ROS-mediated ERK activation in delayed protection from anoxic preconditioning in neonatal rat cardiomyocytes. Chin Med J (Engl) 2004;117:395–400. [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci U S A. 2000;97:436–41. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabb MC, Lobner D, Turetsky DM, Choi DW. Preconditioned resistance to oxygen glucose deprivation-induced cortical neuronal death: alterations in vesicular GABA and glutamate release. Neurosci. 2002;115:173–183. doi: 10.1016/s0306-4522(02)00370-6. [DOI] [PubMed] [Google Scholar]
- Greenway SC, Storey KB. Mitogen-activated protein kinases and anoxia tolerance in turtles. J Exp Zool. 2000;287:477–484. doi: 10.1002/1097-010x(20001201)287:7<477::aid-jez3>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Gross CG. Neurogenesis in the adult brain: death of a dogma. Nat Rev Neurosci. 2000;1:67–73. doi: 10.1038/35036235. [DOI] [PubMed] [Google Scholar]
- Gould E, Gross CG. Neurogenesis in adult mammals: some progress and problems. J Neurosci. 2002;22:619–623. doi: 10.1523/JNEUROSCI.22-03-00619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad JJ. Oxygen-sensing mechanisms and the regulation of redox-responsive transcription factors in development and pathophysiology. Respir Res. 2002;22:1–26. doi: 10.1186/rr190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. Lipid peroxidation in brain homogenates: the role of iron and hydroxyl radicals. J Neurochem. 1997;69:1330–1. doi: 10.1046/j.1471-4159.1997.69031330.x. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Yonetani M, Nakamura H. Selective brain hypothermia protects against hypoxia-ischemic injury in newborn rats by reducing hydroxyl radical production. Kobe J Med Sci. 2003;49:83–91. [PubMed] [Google Scholar]
- Herold S, Fago A, Weber RE, Dewilde S, Moens L. Reactivity studies of the Fe(III) and Fe(II)NO forms of human neuroglobin reveal a potential role against oxidative stress. J, Biol, Chem. 2004;279:22841–22847. doi: 10.1074/jbc.M313732200. [DOI] [PubMed] [Google Scholar]
- Heurteaux C, Lauritzen I, Widmann C, Lazdunski M. Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc Natl Acad Sci U S A. 1995;92:4666–70. doi: 10.1073/pnas.92.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitzig BM, Kneussl MP, Shih V, Brandstetter RD, Kazemi H. Brain amino acid concentrations during diving and acid-base stress in turtles. J Appl Physiol. 1985;58:1751–1754. doi: 10.1152/jappl.1985.58.6.1751. [DOI] [PubMed] [Google Scholar]
- Hoyte L, Barber PA, Buchan AM, Hill MD. The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol Med. 2004;4:131–136. doi: 10.2174/1566524043479248. [DOI] [PubMed] [Google Scholar]
- Huang PL. Nitric oxide and cerebral ischemic preconditioning. Cell Calcium. 2004;36:323–9. doi: 10.1016/j.ceca.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Huang CC, Lajevardi NS, Tammela O, Pastuszko A, Delivoria-Papadopoulos M, Wilson DF. Relationship of extracellular dopamine in striatum of newborn piglets to cortical oxygen pressure. Neurochem Res. 1994;19:649–655. doi: 10.1007/BF00967702. [DOI] [PubMed] [Google Scholar]
- Hylland P, Nilsson GE, Lutz PL. Time course of anoxia induced increase of cerebral blood flow rate in turtles: evidence for a role of adenosine. J Cereb Blood Flow Metab. 1994;16:290–295. doi: 10.1038/jcbfm.1994.110. [DOI] [PubMed] [Google Scholar]
- Imoto K, Kukidome D, Nishikawa T, Matsuhisa T, Sonoda K, Fujisawa K, Yano M, Motoshima H, Taguchi T, Tsuruzoe K, Matsumura T, Ichijo H, Araki E. Impact of mitochondrial reactive oxygen species and apoptosis signal-regulating kinase 1 on insulin signaling. Diabetes. 2006;55:1197–204. doi: 10.2337/db05-1187. [DOI] [PubMed] [Google Scholar]
- Jackson DC. Living without oxygen: lessons from the freshwater turtle. Comp Biochem Physiol A. 2000;125:299–315. doi: 10.1016/s1095-6433(00)00160-4. [DOI] [PubMed] [Google Scholar]
- Jin H, Wu G, Osterhaus J, Wei K, Davis D, Sha E, Floor C, Hsu R, Kopke D, Wu J. Demonstration of functional coupling between γ-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc Natl Acad Sci USA. 2003;100:4293–4298. doi: 10.1073/pnas.0730698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlert S, Zundorf G, Reiser G. Glutamate-mediated influx of extracellular Ca2+ is coupled with reactive oxygen species generation in cultured hippocampal neurons but not in astrocytes. J Neurosci Res. 2005;79:262–71. doi: 10.1002/jnr.20322. [DOI] [PubMed] [Google Scholar]
- Kang D, Hamasaki N. Alterations of mitochondrial DNA in common diseases and disease states: aging, neurodegeneration, heart failure, diabetes, and cancer. Curr Med Chem. 2005;12:429–41. doi: 10.2174/0929867053363081. [DOI] [PubMed] [Google Scholar]
- Kelly DA, Storey KB. Organ-specific control of glycoysis in anoxic turtles. Am J Physiol. 1988;255:R774–779. doi: 10.1152/ajpregu.1988.255.5.R774. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, Yuan ZM, Narula J, Weichselbaum R, Nalin C, Kufe D. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-xl in response to DNA damage. J Biol Chem. 2000;275:322–327. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- Kokaia Z, Zhao Q, Kokaia M, Elmer E, Metsis M, Smith ML, Siesjo BK, Lindvall O. Regulation of brain-derived neurotrophic factor gene expression after transient middle cerebral artery occlusion with and without brain damage. Exp Neurol. 1995;136:73–88. doi: 10.1006/exnr.1995.1085. [DOI] [PubMed] [Google Scholar]
- Kositprapa C, Ockaili RA, Kukreja RC. Bradykinin B2 receptor is involved in the late phase of preconditioning in rabbit heart. J Mol Cell Cardiol. 2001;33:1355–1362. doi: 10.1006/jmcc.2000.1396. [DOI] [PubMed] [Google Scholar]
- Kriegl JM, Bhattacharyya AJ, Nienhaus K, Deng P, Minkow O, Nienhaus GU. Ligand binding and protein dynamics in neuroglobin. Proc Natl Acad Sci. 2002;99:7992–7997. doi: 10.1073/pnas.082244399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krilowicz BL, Glotzbach SF, Heller HC. Neuronal activity during sleep and complete bouts of hibernation. Am J Physiol. 1988;255:R1008–19. doi: 10.1152/ajpregu.1988.255.6.R1008. [DOI] [PubMed] [Google Scholar]
- Krnjevic K. Membrane current activation during hypoxia in hippocampal neurons. In: Hochachka PW, Lutz PL, Sick T, Rosenthal M, van den Thilart G, editors. Surviving Hypoxia: Mechanisms of Control and Adaptation. Boca Raton, FL: CRC Press; 1993. pp. 365–388. [Google Scholar]
- Lancelot E, Revaud ML, Boulu RG, Plotkine M, Callebert J. A microdialysis study investigating the mechanisms of hydroxyl radical formation in rat striatum exposed to glutamate. Brain Res. 1998;809:294–6. doi: 10.1016/s0006-8993(98)00942-1. [DOI] [PubMed] [Google Scholar]
- Lange-Asschenfeldt C, Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. J Cereb Blood Flow Metab. 2004;24:636–45. doi: 10.1097/01.WCB.0000121235.42748.BF. [DOI] [PubMed] [Google Scholar]
- Laplanche L, Michaud M, Kamenka JM, Barbanel G. Hydroxyl radicals release in rat striatum involves metabotropic glutamate receptors. Brain Res Bull. 2003;61:453–7. doi: 10.1016/s0361-9230(03)00171-0. [DOI] [PubMed] [Google Scholar]
- Latchman DS. Protective effect of heat shock proteins in the nervous system. Curr Neurovasc Res. 2004 2004 Jan;1(1):21–7. doi: 10.2174/1567202043480206. Erratum in: Curr Neurovasc Res. 2004 Apr;1, 191. [DOI] [PubMed] [Google Scholar]
- Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci USA. 1996;93:15036–40. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz PL, Siddiqi AH. Comparison of hemoglobins of Fasciola gigantica (Trematoda: Digenea) and its host. Exp Parisit. 1967;20:83–87. doi: 10.1016/0014-4894(67)90025-2. [DOI] [PubMed] [Google Scholar]
- Lutz PL, Leone-Kabler SA. Upregulation of GABAA receptor during anoxia in the turtle brain. Am J Physiol. 1995;37:R1332–R1335. doi: 10.1152/ajpregu.1995.268.5.R1332. [DOI] [PubMed] [Google Scholar]
- Lutz PL, Manuel L. Maintenance of adenosine A1 receptor function during long term anoxia in the turtle brain. Am J Physiol. 1999;276:R633–R636. doi: 10.1152/ajpregu.1999.276.3.R633. [DOI] [PubMed] [Google Scholar]
- Lutz PL, Prentice H. Sensing and responding to hypoxia, molecular and physiological mechanisms. Integ Comp Biol. 2002;42:436–468. doi: 10.1093/icb/42.3.463. [DOI] [PubMed] [Google Scholar]
- Lutz PL, Milton SL. Negotiating brain anoxia survival in the turtle. J Exp Biol. 2004;207:3141–3147. doi: 10.1242/jeb.01056. [DOI] [PubMed] [Google Scholar]
- Lutz PL, McMahon P, Rosenthal M, Sick TJ. Relationships between aerobic and anaerobic energy production in turtle brain in situ. Am J Physiol. 1984;247:R740–R744. doi: 10.1152/ajpregu.1984.247.4.R740. [DOI] [PubMed] [Google Scholar]
- Lutz PL, Edwards R, McMahon P. GABA concentrations are maintained in the anoxic turtles brain. Am J Physiol. 1985;249:R372–R374. doi: 10.1152/ajpregu.1985.249.3.R372. [DOI] [PubMed] [Google Scholar]
- Lutz PL, Dawson TJ, Bonnet E, Fanning D. Oxygen affinity of monotreme blood: hypoxic adaptations. J Exp Zool. 1989;251:285–289. [Google Scholar]
- Lutz PL, Nilsson GE, Prentice H. The Brain without Oxygen: Causes of Failure Molecular and Physiological Mechanisms for Survival. 3. Dordrecht: Kluwers Press; 2003a. [Google Scholar]
- Lutz PL, Prentice H, Milton SL. Is turtle longevity linked to enhanced mechanisms for surviving brain anoxia and reoxygenation? J Exp Gerontol. 2003b;38:797 –800. doi: 10.1016/s0531-5565(03)00111-6. [DOI] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci USA. 2003;100:715–20. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir A, Ginsburg S, Butkevic A, Kachalsky SG, Kaiserman I, Ahdut R, Demirgoren S, Rahamimof R. Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol Rev. 1999;79:1019–1088. doi: 10.1152/physrev.1999.79.3.1019. [DOI] [PubMed] [Google Scholar]
- Mermet N, Angulo J, Medina M, Reperant J, Ward R, Araneda S. Expression of Kin, a nuclear protein binding to curved DNA, in the brain of the frog (Rana esculenta), turtle (Trachemys scripta), quail (Coturnix coturnix) and mouse (Mus musculus) Anat Embryol (Berl) 2002;205:37–51. doi: 10.1007/s00429-001-0224-7. [DOI] [PubMed] [Google Scholar]
- Milton SL, Lutz PL. Low extracellular dopamine levels are maintained in the anoxic turtle brain. J Cereb Blood Flow Metab. 1998;18:803–807. doi: 10.1097/00004647-199807000-00010. [DOI] [PubMed] [Google Scholar]
- Milton SL, Lutz PL. Adenosine and ATP sensitive potassium channels modulate dopamine release in the anoxic turtle (Trachemys scripta) striatum. Am J Physiol. 2005;289:R77–R83. doi: 10.1152/ajpregu.00647.2004. [DOI] [PubMed] [Google Scholar]
- Milton SL, Prentice HM. Anoxic and reoxygenation survival in the turtle brain. Comp Biochem Physiol, SEB 2006 Abstracts. 2006:A9.5. [Google Scholar]
- Milton SL, Thompson JW, Lutz PL. Mechanisms for maintaining extracellular glutamate in the anoxic turtle striatum. Am J Physiol. 2002;282:R1317 – R1323. doi: 10.1152/ajpregu.00484.2001. [DOI] [PubMed] [Google Scholar]
- Milton SL, Prentice HM, Lutz PL. Molecular mechanisms of ROS defense in the turtle Trachemys scripta. Comp Biochem Physiol SEB 2005 Abstracts. 2005:A10.38. [Google Scholar]
- Milton SL, Nayak G, Lutz PL, Prentice HM. The regulation of neuroglobin gene transcription in hypoxia and anoxia in the brain of the anoxia-tolerant turtle Trachemys scripta. J Biomed Sci. 2006;13:509–14. doi: 10.1007/s11373-006-9084-8. [DOI] [PubMed] [Google Scholar]
- Nandagopal K, Dawson TM, Dawson VL. Critical role for nitric oxide signaling in cardiac and neuronal ischemic preconditioning and tolerance. J Pharmacol Exp Ther. 2001;297:474–8. [PubMed] [Google Scholar]
- Nilsson GE, Lutz PL. Release of inhibitory neurotransmitters in response to anoxia in turtle brain. Am J Physiol. 1991;261:R32–R37. doi: 10.1152/ajpregu.1991.261.1.R32. [DOI] [PubMed] [Google Scholar]
- Nilsson GE, Lutz PL. Adenosine release in the anoxic turtle brain: a possible mechanism for anoxic survival. J Exp Biol. 1992;162:345–351. [Google Scholar]
- Nilsson GN, Alfaro AA, Lutz PL. Changes in turtle brain neurotransmitters and related substances during anoxia. Am J Physiol. 1990;259:R376–R384. doi: 10.1152/ajpregu.1990.259.2.R376. [DOI] [PubMed] [Google Scholar]
- Nilsson GN, Lutz PL, Jackson TL. Neurotransmitters and anoxic survival in the brain: A comparison of anoxia-tolerant and anoxia-intolerant vertebrates. Physiol Zool. 1991;64:638–652. [Google Scholar]
- Oliver CN, Starke-Reed PE, Stadtman ER. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury in gerbil brain. Proc Natl Acad Sci USA. 1990;87:5144–5147. doi: 10.1073/pnas.87.13.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KM, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J Biol Chem. 2001;276:11870–6. doi: 10.1074/jbc.M007518200. [DOI] [PubMed] [Google Scholar]
- Pek M, Lutz PL. Role for adenosine in "channel arrest" in the anoxic turtle brain. J Exp Biol. 1997;200:1913–1917. doi: 10.1242/jeb.200.13.1913. [DOI] [PubMed] [Google Scholar]
- Pek M, Lutz PL. K+ATP channel activation provides transient protection in anoxic turtle brain. Am J Physiol. 1998;44:R2023–R2027. doi: 10.1152/ajpregu.1998.275.6.R2023. [DOI] [PubMed] [Google Scholar]
- Penafiel A, Rivera A, Gutierrez A, Trias S, De La Calle A. Temperature affects adult neurogenesis in the lizard brain. Int J Dev Biol. 2001;45:83–84. [Google Scholar]
- Perez-Pinzon M, Rosenthal M, Sick T, Lutz PL, Marsh D. Down-regulation of sodium channels during anoxia: a putative survival strategy of turtle brain. Am J Physiol. 1992;31:R712–R715. doi: 10.1152/ajpregu.1992.262.4.R712. [DOI] [PubMed] [Google Scholar]
- Prentice HM, Milton SL, Scheurle D, Lutz PL. Gene transcription of brain voltage-gated potassium channels is reversibly regulated by oxygen supply. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1317–R1321. doi: 10.1152/ajpregu.00261.2003. [DOI] [PubMed] [Google Scholar]
- Prentice HM, Milton SL, Scheurle D, Lutz PL. The upregulation of cognate and inducible heat shock proteins in the anoxic turtle brain. J Cereb Blood Flow Metab. 2004;24:826–828. doi: 10.1097/01.WCB.0000126565.27130.79. [DOI] [PubMed] [Google Scholar]
- Qui Y, Rizvi A, Tang KL, Manchikalapudi S, Takano H, Jadoon AK, Wu WJ, Bolli R. Nitric oxide triggers late preconditioning against myocardial infarction in conscious rabbits. Am J Physiol. 1997;273:H2931–H2936. doi: 10.1152/ajpheart.1997.273.6.H2931. [DOI] [PubMed] [Google Scholar]
- Ramirez C, Nacher J, Molowny A, Sanchez-Sanchez F, Irurzun A, Lopez-Garcia C. Photo-period temperature and neuroblast proliferation-migration in lizard cortex. Neuroreport. 1997;7:1257–1260. doi: 10.1097/00001756-199707070-00047. [DOI] [PubMed] [Google Scholar]
- Ramirez SH, Fan S, Maguire CA, Perry S, Hardiek K, Ramkumar V, Gelbard HA, Dewhurst S, Maggiwar SB. Activation of adenosine A2A receptor protects sympathetic neurons against nerve growth factor withdrawal. J Neurosci Res. 2004;77:258–69. doi: 10.1002/jnr.20150. [DOI] [PubMed] [Google Scholar]
- Radmilovich M, Fernandez A, Trujillo-Cenoz O. Environment temperature affects cell proliferation in the spinal cord and brain of juvenile turtles. J Exp Biol. 2003;206:3085–3093. doi: 10.1242/jeb.00515. [DOI] [PubMed] [Google Scholar]
- Reid EA, Kristo G, Yoshimura Y, Ballard-Croft C, Keith BJ, Mentzer RM, Jr, Lasley RD. In vivo adenosine receptor preconditioning reduces myocardial infarct size via subcellular ERK signaling. Am J Physiol Heart Circ Physiol. 2005;288:H2253–9. doi: 10.1152/ajpheart.01009.2004. [DOI] [PubMed] [Google Scholar]
- Rice ME, Lee EJ, Choy Y. High levels of ascorbic acid, not glutathione, in the CNS of anoxia-tolerant reptiles contrasted with levels in anoxia-intolerant species. J Neurochem. 1995;64:1790–1799. doi: 10.1046/j.1471-4159.1995.64041790.x. [DOI] [PubMed] [Google Scholar]
- Russo RE, Fernandez A, Reali C, Radmilovich M, Trujillo-Cenoz O. Functional and molecular clues reveal precursor-like cells and immature neurons in the turtle spinal cord. J Physiol. 2004;560:831–838. doi: 10.1113/jphysiol.2004.072405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild L, Reiser G. Oxidative stress is involved in the permeabilization of the inner membrane of brain mitochondria exposed to hypoxia/reoxygenation and low micromolar Ca. FEBS J. 2005;272:3569–601. doi: 10.1111/j.1742-4658.2005.04781.x. [DOI] [PubMed] [Google Scholar]
- Sharikabad MN, Ostbye KM, Brors O. Effect of hydrogen peroxide on reoxygenation-induced Ca2+ accumulation in rat cardiomyocytes. Free Radic Biol Med. 2004;37:531–538. doi: 10.1016/j.freeradbiomed.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Sharp CD, Houghton J, Elrod JW, Warren A, Jackson TH, 4th, Jawahar A, Nanda A, Minagar A, Alexander JS. N-methyl-D-aspartate receptor activation in human cerebral endothelium promotes intracellular oxidant stress. Am J Physiol Heart Circ Physiol. 2005;288:H1893–9. doi: 10.1152/ajpheart.01110.2003. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Nagayama T, Jin KL, Zhu L, Loeffert JE, Watkins SC, Graham SH, Simon RP. Bcl-2 Antisense treatment prevents induction of tolerance to focal ischemia in the rat brain. J Cereb Blood Flow Metab. 2001;21:233–43. doi: 10.1097/00004647-200103000-00007. [DOI] [PubMed] [Google Scholar]
- Shin DS, Wilkie MP, Pamenter ME, Buck LT. Calcium and protein phosphatase 1/2A attenuate N-methyl-D-aspartate receptor activity in the anoxic turtle cortex. Comp Biochem Physiol A Mol Integr Physiol. 2005;142:50–7. doi: 10.1016/j.cbpa.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Sibley DR. New insights into dopaminergic receptor function using antisense and genetically altered animals. Annu Rev Pharmacol Toxicol. 1999;39:313–41. doi: 10.1146/annurev.pharmtox.39.1.313. [DOI] [PubMed] [Google Scholar]
- Sick TJ, Rosenthal M, LaManna JC, Lutz PL. Brain potassium homeostasis, anoxia, and metabolic inhibition in turtles and rats. Am J Physiol. 1982;243:R281–R288. doi: 10.1152/ajpregu.1982.243.3.R281. [DOI] [PubMed] [Google Scholar]
- Sick TJ, Perez-Pinzon M, Lutz PLRosenthal M. Maintaining coupled metabolism and membrane function in anoxic brain: a comparison between the turtle and rat. In: Hochachka PW, Lutz PL, Sick T, Rosenthal M, van den Thilart G, editors. Surviving Hypoxia. Boca Raton: CRC Press; 1993. pp. 351–364. [Google Scholar]
- Snoeckx LH, Cornelussen EH, Van Nieuwenhoven RN, Reneman FA, Van Der Vusse GJ. Heat shock proteins and cardiovascular pathophysiology. Physiol Rev. 2001;81:1461–1497. doi: 10.1152/physrev.2001.81.4.1461. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Mockett RJ, Orr WC. Mechanisms of aging: an appraisal of the oxidative stress hypothesis. Free Radic Biol Med. 2002;33:575–86. doi: 10.1016/s0891-5849(02)00886-9. [DOI] [PubMed] [Google Scholar]
- Solenkova NV, Solodushko V, Cohen MV, Downey JM. Endogenous adenosine protects preconditioned heart during early minutes of reperfusion by activating Akt. Am J Physiol Heart Circ Physiol. 2006;290:H441–449. doi: 10.1152/ajpheart.00589.2005. [DOI] [PubMed] [Google Scholar]
- Sowa AW, Duff SMG, Guy PA, Hill RD. Altering hemoglobin levels changes energy status in maize cells under hypoxia. PNAS. 1998;95:10317–10321. doi: 10.1073/pnas.95.17.10317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey KB. Gene hunting in hypoxia and exercise. In: Roach RC, Hackett P, Wagner P, editors. Hypoxia and Exercise: Proceedings of the 14th International Hypoxia symposium. Springer Publishers; New York: 2005. [Google Scholar]
- Storey KB. Reptile freeze tolerance: metabolism and gene expression. Cryobiology. 2006;52:1–16. doi: 10.1016/j.cryobiol.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Stuart JA, Brown ME. Mitochondrial DNA maintenance and bioenergetics. Biochim Biophys Acta. 2006;1757:79–89. doi: 10.1016/j.bbabio.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Mao XO, Zhu Y, Greenberg DA. Neuroglobin is up-regulated by and protects neurons from hypoxic-ischemic injury. PNAS USA. 2001;98:15306–15311. doi: 10.1073/pnas.251466698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jin K, Peel A, Mao XO, Xie L, Greenberg DA. Neuroglobin protects the brain from experimental stroke in vivo. Proc Natl Acad Sci USA. 2003;100:3497–3500. doi: 10.1073/pnas.0637726100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ouyang YB, Xu L, Chow AM, Anderson R, Hecker JG, Giffard RG. The carboxyl-terminal domain of inducible Hsp70 protects from ischemic injury in vivo and in vitro. J Cereb Blood Flow Metab. 2005;26:937–50. doi: 10.1038/sj.jcbfm.9600246. [DOI] [PubMed] [Google Scholar]
- Sun JZ, Tang XL, Park SW, Qiu Y, Turrens JF, Bolli R. Evidence for an essential role of reactive oxygen species in the genesis of late preconditioning against myocardial stunning in conscious pigs. J Clin Invest. 1996;97:562–576. doi: 10.1172/JCI118449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Duan S. Regulation of glutamate transporter function. Neuroscience. 1999;5:280–282. [Google Scholar]
- Tanaka T, Yoshida M, Yokoo H, Mizoguchi K, Tanaka M. The role of ATP-sensitive potassium channels in striatal dopamine release: an in vivo microdialysis study. Pharmacol Biochem Behav. 1995;52:831–835. doi: 10.1016/0091-3057(95)00151-l. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, Bennett MV, Zukin RS. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci. 2004;24:2750–9. doi: 10.1523/JNEUROSCI.5475-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson W, Lutz PL. Maintaining glutamate homeostasis in the anoxic turtle brain. FASEB J. 2001;15:A129. [Google Scholar]
- Torres GE, Cameiro A, Seamans K, Fiorentini C, Sweeney A, Yao WD, Caron MG. Oligomerization and trafficking of the human dopamine transporter. Mutational analysis identifies critical domains important for the functional expression of the transporter. J Biol Chem. 2003;278:2731–9. doi: 10.1074/jbc.M201926200. [DOI] [PubMed] [Google Scholar]
- Trincavelli ML, Tuscano D, Marroni M, Klotz KN, Lucacchini A, Martini C. Involvement of mitogen protein kinase cascade in agonist-mediated human A(3) adenosine receptor regulation. Biochim Biophys Acta. 2002;1591:55–62. doi: 10.1016/s0167-4889(02)00248-3. [DOI] [PubMed] [Google Scholar]
- Waetzig V, Herdegen T. Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage. Trends Pharmacol Sci. 2005;26:455–61. doi: 10.1016/j.tips.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–43. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Weissbach H, Etienne F, Hoshi T, Heinemann SH, Lowther WT, Matthews B, St John G, Nathan C, Brot N. Peptide methionine sulfoxide reductase: structure, mechanism of action, and biological function. Arch Biochem Biophys. 2002;397:172–8. doi: 10.1006/abbi.2001.2664. [DOI] [PubMed] [Google Scholar]
- Willmore WG, Storey KB. Antioxidant systems and anoxia tolerance in a freshwater turtle Trachemys scripta elegans. Mol Cell Biochem. 1997a;170:177–185. doi: 10.1023/a:1006817806010. [DOI] [PubMed] [Google Scholar]
- Willmore WG, Storey KB. Glutathione systems and anoxia tolerance in turtles. Am J Physiol. 1997b;273:R219–R225. doi: 10.1152/ajpregu.1997.273.1.R219. [DOI] [PubMed] [Google Scholar]
- Willmore WG, Storey KB. Purification and properties of the glutathione S-transferases from the anoxia-tolerant turtle, Trachemys scripta elegans. FEBS J. 2005;272:3602–3614. doi: 10.1111/j.1742-4658.2005.04783.x. [DOI] [PubMed] [Google Scholar]
- Woodhead AD, Satlow RB, Grist E. DNA repair and longevity in three species of cold-blooded vertebrates. Exp Gerontol. 1980;15:301–4. doi: 10.1016/0531-5565(80)90034-0. [DOI] [PubMed] [Google Scholar]
- Xia Y, Haddad GG. Major differences in CNS sulfonylurea receptor distribution between the rat (newborn, adult) and turtle. J Comp Neuro. 1991;330:363–380. doi: 10.1002/cne.903140206. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effect of ERK and JNK-p38 MAP kinase on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Xu FF, Liu XH, Cai LR. Role of hypoxia-inducible factor-1alpha in the prevention of cardiomyocyte injury induced by hypoxic preconditioning. Sheng Li Xue Bao. 2004;56:609–14. [PubMed] [Google Scholar]
- Xu K, Puchowicz MA, Lust WD, LaManna JC. Adenosine treatment delays postischemic hippocampal CA1 loss after cardiac arrest and resuscitation in rats. Brain Res. 2006;1071:208–17. doi: 10.1016/j.brainres.2005.11.060. [DOI] [PubMed] [Google Scholar]
- Xu Z, Mueller RA, Park SS, Boysen PG, Cohen MV, Downey JM. Cardioprotection with adenosine A2 receptor activation at reperfusion. J Cardiovasc Pharmacol. 2005;46:794–802. doi: 10.1097/01.fjc.0000188161.57018.29. [DOI] [PubMed] [Google Scholar]
- Yao Z, Mizumura T, Mei DA, Gross GJ. KATP channels and memory of ischemic preconditioning in dogs: synergism between adenosine and KATP channels. Am J Physiol. 1997;272:H334–H342. doi: 10.1152/ajpheart.1997.272.1.H334. [DOI] [PubMed] [Google Scholar]
- Yermolaieva O, Xu R, Schinstock C, Brot N, Weissbach H, Heinemann SH, Hoshi T. Methionine sulfoxide reductase A protects neuronal cells against brief hypoxia/reoxygenation. Proc Natl Acad Sci USA. 2004;101:159–64. doi: 10.1073/pnas.0308215100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Nakakimura K, Cui YJ, Matsumoto M, Sakabe T. Adenosine A(1) receptor antagonist and mitochondrial ATP-sensitive potassium channel blocker attenuate the tolerance to focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24:771–9. doi: 10.1097/01.WCB.0000122742.72175.1B. [DOI] [PubMed] [Google Scholar]
- Zhang C, Rosenbaum DM, Shaikh AR, Li Q, Rosenbaum PS, Pelham DJ, Roth S. Ischemic preconditioning attenuates apoptotic cell death in the rat retina. Invest Opth Visual Sci. 2002;43:3059–66. [PubMed] [Google Scholar]
- Zhou X, Zhai X, Ashraf M. Direct evidence that initial oxidative stress triggered by preconditioning contributes to a second window of protection by endogenous antioxidant enzymes in myocytes. Circulation. 1996;93:1177–1184. doi: 10.1161/01.cir.93.6.1177. [DOI] [PubMed] [Google Scholar]
- Zuo L, Clanton TL. Reactive oxygen species formation in the transition to hypoxia in skeletal muscle. Am J Physiol Cell Physiol. 2005;289:C207–16. doi: 10.1152/ajpcell.00449.2004. [DOI] [PubMed] [Google Scholar]