

Abstract
The aryl hydrocarbon receptor (AHR) mediates the toxicity of a variety of environmental chemicals. Although little is known about the physiological role of the AHR, studies suggest that it plays an important role in regulating ovulation because Ahr deficient (AhRKO) mice have a reduced number of ovulations compared to wild-type (WT) mice. The reasons for the reduced ability of AhRKO mice to ovulate are unknown. Normal ovulation, however, requires estrous cyclicity, appropriate luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels, and LH and FSH responsiveness. Thus, the purpose of this study was to test the hypothesis that Ahr deletion regulates ovulation by altering cyclicity, FSH and LH levels, follicle-stimulating hormone receptor (Fshr) and luteinizing hormone receptor (Lhcgr) levels, and/or gonadotropin responsiveness. The data indicate that AhRKO and WT mice have similar levels of FSH and LH, but AhRKO mice have reduced Fshr and Lhcgr mRNA levels compared to WT mice. Further, AhRKO ovaries contain fewer corpora lutea compared to WT ovaries after 5 IU equine chorionic gonadotropin (eCG) treatment. Lastly, both AhRKO and WT mice ovulate a similar number of eggs in response to 5 IU human chorionic gonadotropin (hCG), but AhRKO mice ovulate fewer eggs than WT mice in response to 2.5 IU and 1.25 IU hCG. Collectively, these data indicate that AhRKO follicles have a reduced capacity to ovulate compared to WT follicles and that this is due to reduced responsiveness to gonadotropins. Thus, in addition to mediating toxicity of environmental chemicals, the Ahr is required for normal ovulation.
Keywords: AhR, ovary, mouse, gonadotropins
INTRODUCTION
Since the discovery of the aryl hydrocarbon receptor (AHR), studies have determined that it binds numerous xenobiotics and naturally occurring exogenous biological compounds (Bock, 1994; Mukai and Tischkau, 2007; Safe et al., 1998). The binding of xenobiotics and other compounds to the AHR initiates a cascade of events (including gene transcription) that often leads to toxicity. Despite extensive research, however, the AHR is still considered to be an orphan receptor because a strictly physiological, high-affinity ligand for this receptor has not yet been discovered. Although investigators have not identified the endogenous ligand for the AHR, it is still thought to have an endogenous ligand(s) because several studies show that it can be activated in the absence of exogenous ligands (Ma and Whitlock, 1997; Sadek and Allen-Hoffmann, 1994; Wang et al., 1998). The AHR is also thought to play an endogenous role in female reproduction because it is present in all cell types in the ovary (Baldridge and Hutz, 2007; Robles et al., 2000).
Studies using Ahr deficient (AhRKO) mice have provided further evidence of an endogenous role of the Ahr (Abbott et al., 1999; Benedict et al., 2000; Benedict et al., 2003; Fernandez-Salguero et al., 1995; Fernandez-Salguero et al., 1997). Initial studies using AhRKO mice revealed impairment of the digestive and immune systems, hepatic fibrosis, and lesions in the skin, heart, liver, spleen, and uterus (Fernandez-Salguero et al., 1995; Fernandez-Salguero et al., 1997). Later studies using AhRKO mice showed that the Ahr plays a role in regulating normal female reproduction (Abbott et al., 1999; Benedict et al., 2000; Benedict et al., 2003). Specifically, Abbott et al. 1999 showed that AhRKO mice have difficulty maintaining conceptuses during pregnancy, surviving pregnancy and lactation, and rearing pups to weaning. Benedict et al. 2000 found that AhRKO mice have fewer antral follicles compared to WT mice, suggesting that the Ahr may play a role in the regulation of antral follicle numbers. Further, Benedict et al. 2003 found that AhRKO ovaries have a reduced number of corpora lutea compared to WT ovaries, suggesting that AhRKO mice have a reduced number of ovulations compared to WT mice.
The reasons for the reduced ovulations in AhRKO mice compared to WT mice are unknown. Normal ovulation in rodents, however, requires regular estrous cyclicity, the normal release of gonadotropins (FSH and LH), and responsiveness to gonadotropins by their corresponding receptors, FSH receptor (Fshr) and LH receptor (Lhcgr) (Hirshfield, 1991; Richards et al., 2002). Therefore, the purpose of the present study was to test the hypothesis that Ahr deletion affects ovulation by altering estrous cyclicity, FSH and LH levels, Fshr and Lhcgr levels, and/or gonadotropin responsiveness.
To test this hypothesis, we first compared estrous cyclicity in AhRKO and WT mice. In addition, we investigated whether the effect of Ahr deletion on ovulation is due to abnormal levels of FSH and LH in AhRKO mice compared to WT mice. Further, we compared Fshr and Lhcgr mRNA levels in AhRKO and WT mice. To test whether AhRKO mice are less responsive to gonadotropins than WT mice, we compared ovarian weight and the number of corpora lutea in AhRKO and WT mice after treatment with equine chorionic gonadotropin (eCG). Lastly, we conducted superovulation experiments to determine whether Ahr deletion alters the number of ovulated eggs in response to gonadotropin treatment.
METHODS
Animals
AhRKO mice were generated as described by Schmidt et al. 1996 and breeding pairs were generously provided by Dr. Richard Peterson (University of Wisconsin). AhRKO and WT animals were housed in the University of Maryland School of Medicine and the University of Illinois Central Animal Facilities and provided food and water ad libitum. All mice were housed in 12 hour (hr) light: 12 hr dark cycles in a temperature controlled room (24 ± 1°C) with 35 ± 4% relative humidity. The University of Maryland School of Medicine and the University of Illinois Institutional Animal Use and Care Committees approved all procedures involving animal care, euthanasia, and tissue collection.
Genetic Screening of Mice
The genetic screening protocol was performed as previously described (Benedict et al., 2000; Benedict et al., 2003). Briefly, ear punches were lysed in proteinase K buffer at room temperature for 30 minutes (min). Lysates were incubated at 100°C for 3 min, and then subjected to polymerase chain reaction (PCR) assays using the primers previously described (Benedict et al., 2000; Benedict et al., 2003). The PCR products were sized by agarose gel (1.8%) electrophoresis. The WT (Ahr+/+) mice were identified by the presence of a 670 base pair (bp) product. Homozygous AhRKO (Ahr−/−) mice were identified by the presence of a 580 bp product. Heterozygous (Ahr+/−) mice were identified by the presence of both the 580 bp and 670 bp products. Only homozygous AhRKO and WT mice were used in these experiments.
Measurement of Cyclicity
The estrous cycles of adult AhRKO and WT mice were monitored daily for 20 days via vaginal swabs as previously described (Cooper et al., 1993). Timing elapsed in each stage of the estrous cycle was then compared in AhRKO and WT mice.
Measurement of Gonadotropin Levels
Blood samples were collected from adult AhRKO and WT mice and subjected to measurements of FSH and LH as previously described (Tomic et al., 2004). Samples were collected between 9–9:30 AM for proestrus AM, estrus, and diestrus and between 9–9:15 PM for proestrus PM. Briefly, serum FSH and LH levels were measured by radioimmunoassay (RIA) using reagents from the National Hormone and Pituitary Distribution Program. For both FSH and LH, a standard curve was prepared and cold standards and samples (100 μl) were added to labeled tubes along with primary antibody (FSH at 1:1400 dilution and LH at 1:500 dilution) and iodinated FSH or LH. Samples were stored at 4°C overnight. On day 2, secondary antibody was added (1:10 dilution) along with 2% normal rabbit serum (Sigma Aldrich, St. Louis, MO) and incubated at room temperature for 5 min. The tubes were centrifuged for 15 min at 3000 rpm, the supernatant was decanted, and pellets were counted in a gamma counter for 1 min each. All samples were run in duplicate. Sensitivity for the FSH assay was 200 pg/ml, with inter- and intra-assay coefficients of variation of 2.7% and 6.7%, respectively. Sensitivity for the LH assay was 86 pg/ml, with inter- and intra-assay coefficients of variation of 5.3% and 2.5%, respectively.
Real Time PCR Analysis
Levels of Fshr and Lhcgr mRNA expression in AhRKO and WT follicles were compared using real-time PCR as described (Tomic et al., 2004) with minor modifications. Specifically, antral follicles were collected from AhRKO and WT ovaries on postnatal days (PD) 32–35 and immediately stored at −70°C until RNA extraction. Follicles were isolated on PD32–35 because it is a time when both WT and AhRKO mice are sexually mature, WT ovaries exhibit normal follicle growth and a normal number of corpora lutea, and AhRKO ovaries exhibit slow follicle growth and a reduced number of ovulations (Barnett et al., 2007; Benedict et al., 2003). Total RNA was extracted from the follicles using the RNeasy Mini Kit (QIAGEN Inc., Valencia, CA) according to the manufacturer protocol. The quality of RNA was checked using a spectrophotometer and only RNA with readings between 1.8–2.0 was used in experiments. RNA (0.5–1 μg) was reverse transcribed using an Omniscript reverse transcriptase kit (QIAGEN) with random primers according to the manufacturer protocol. Real-time PCR was conducted using a MJ Research (OPTICON) PCR machine and accompanying software according to the manufacturer instructions. The OPTICON quantifies the amount of PCR product generated by measuring the dye (SYBR green) that fluoresces when bound to double-stranded DNA. A standard curve was generated from five serial dilutions of purified PCR product. For each primer sequence described below, a melting curve was performed. Real-time PCR amplification of the individual genes was performed using 3 μl of cDNA. Specific primer sequences for each gene were as follows: Fshr = (Forward) 5′-AGCAAGTTTGGCTGTTATGAGG-3′ (Reverse) 5′-GTTCTGGACTGAATGATTTAGAGG -3′ (Babu et al., 2001). Lhcgr = (Forward) 5′-TCTCTCAGAGTGATTCCCTG-3′ (Reverse) 5′-AGCGTCTGAATGGACTCCAG-3′ (Pubmed Accession # NM_013582). β-actin mRNA was measured in each sample as an internal control using published primer sequences (Weihua et al., 2000).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed as described (Kazi et al., 2005) with minor modifications. Specifically, whole ovaries were collected from adult WT mice on PD 54 and placed in a 1% formaldehyde solution containing 270 μl of 37% formalin and incubated at room temperature for 15 min. Ovaries were collected at this time point because it is a time when WT mice have a normal number of ovulations and AhRKO mice exhibit a reduced number of ovulations (Benedict et al., 2003). The ovaries were then centrifuged at 14,000 RPM for 3 min. The media was aspirated and the ovaries were washed three times with an ice-cold 1X PBS/protease inhibitor (PBS+PI) mix (Complete Mini EDTA-free Protease Inhibitor Cocktail, 1 tablet/10 ml, Roche Applied Science, Indianapolis, IN). After the last supernatant was discarded, PBS+PI mix was added to the ovaries and the ovaries were homogenized. Homogenates were centrifuged at 14,000 RPM for 5 min at 4°C, and the supernatants were removed and discarded. Pellets were re-suspended in lysis buffer (1:5 of PBS+PI:SDS buffer) and incubated on ice for 10 min. Samples were sonicated on ice using a Microson Ultrasonic Cell Disruptor (Misonix, Farmingdale, NY) at a 2.5 power level for three 10 sec cycles, with 20 sec pauses between each. The resulting samples (i.e., soluble chromatin-protein complexes) were then divided into 100 μl aliquots. One sample labeled INPUT (whole ovary positive control) was stored at −80°C until further use.
Sonicated sample aliquots were diluted 1:10 with dilution buffer (20 mM Tris-HCl, pH 8.1, 150 mM NaCl, 2 mM EDTA, 1% triton X-100, and PI). To reduce nonspecific background, the diluted samples were pre-cleared by adding salmon sperm DNA/protein A agarose and incubated on a shaker for 30 min at 4°C. Samples were then centrifuged at 14,000 RPM at 4°C for 3 min. Supernatants were collected and incubated overnight at 4°C with 10 μg mouse monoclonal anti-Rabbit AHR antibody (Ab-10; NeoMarkers/Lab Vision, Fremont, CA).
Salmon sperm DNA/protein A slurry (40 μl) was then added and incubated for 1 hr at 4°C. The samples were then centrifuged at 1,000 RPM at 4°C for 1 min. The supernatant was removed and discarded. The beads were washed sequentially with 1 ml of low salt immune complex wash buffer, high salt immune complex wash buffer, and LiCl immune complex wash buffer, respectively, and then twice with 1 ml of TE buffer at room temperature. Each wash was for 5 min each at 4°C. The complexes were then eluted twice by adding 250 μl of elution buffer (1% SDS and 0.1M NAHCO3) to the pellet for 15 min at room temperature with rotation. NaCl (5 M) was added to the pooled eluted samples as well as the INPUT sample. All samples were incubated at 65°C overnight to separate cross-linked immunoprecipitated protein and DNA from each other. The DNA was purified using the Qiaquick PCR Purification kit (QIAGEN) according to the manufacturer protocol.
The purified DNA was amplified by PCR using primers specific for the Fshr and Lhcgr promoters. The specific sequences were as follows: Fshr = Site no. 1 (Forward) 5′ GCCTCTCATCAGCTCCCAGC 3′, (Reverse) 5′ GCAGTGCAGACTGAGGTCCC 3′; Site no. 2 (Forward) 5′ TCCACACACTGTCCGGTAAG 3′, (Reverse) 5′ CTTGAAGGATAAGACAGGTG 3′. Lhcgr = Site no. 1 (Forward) 5′ GAGAACAGGGACAGGCGGTG 3′, (Reverse) 5′ CGCGACCCTGACAACTCTGG 3′. These sites were selected because they contain AHR binding sites according to the Transcription Element Search System (TESS) website (http://www.cbil.upenn.edu/tess/). PCR products were then subjected to agarose gel (1.8%) electrophoresis.
Measurements of Gonadotropin Responsiveness
Three different approaches were used to measure gonadotropin responsiveness in AhRKO and WT mice. For the first approach, the ability of eCG to increase ovarian weight was compared in AhRKO and WT mice. Specifically, sexually immature AhRKO and WT mice of similar body weights were dosed on PD 25–28 with 0, 5, or 15 IU eCG, a gonadotropin with both FSH- and LH-like properties. The mice were euthanized 48 hr later and the ovaries were removed, cleared of fat, oviduct, and bursa, and weighed.
For the second approach, the ability of follicles to naturally ovulate if forced to grow to the antral stage with eCG treatment was compared in AhRKO and WT mice. Specifically, sexually immature AhRKO and WT mice (PD 25–28) of similar body weights were dosed with eCG (5 IU or 15 IU). After 72 hr, their ovaries were removed, fixed in Kahle solution, and processed for histological evaluation of the number of corpora lutea as described (Benedict et al., 2003). Ovarian sections were used to count the number of corpora lutea without knowledge of genotype. To avoid double counting, each corpus luteum was followed through consecutive sections to ensure that it was only counted once.
For the third approach, the ability of follicles to ovulate in response to eCG followed by human chorionic gonadotropin (hCG) treatment was compared in AhRKO and WT mice. Specifically, sexually immature AhRKO and WT mice were injected with a single subcutaneous (sc) injection of 5 IU eCG on PD 25–28. Exactly 48 hr later, the mice were injected with 1.25, 2.5 or 5 IU of hCG per mouse. After 18 hr, the ovaries and oviducts were removed, the oviducts were flushed, and the number of ovulated eggs was counted in AhRKO and WT mice.
Statistical Analysis
Data were analyzed using SPSS statistical software (SPSS, Inc., Chicago). An independent sample t-test was used to compare mean differences between AhRKO and WT samples. Analysis of variance followed by Scheffe post-hoc test was used to compare differences between hormone treatment groups. Data are presented as means ± standard errors of the means (SEM). A p value less than or equal to 0.05 was considered statistically significant.
RESULTS
The Effect of Ahr Deletion on Cyclicity
When cyclicity was compared in WT and AhRKO mice, no differences were observed between WT and AhRKO mice in the amount of time spent in each stage of the cycle over a 20 day period. WT mice spent 5.6 ± 0.5 days in estrus, 3.9 ± 0.3 days in metestrus, 4.3 ± 0.4 days in diestrus, and 6.0 ± 0.8 days in proestrus over a 20 day period. Similarly, AhRKO mice spent 5.7 ± 0.3 days in estrus, 3.7 ± 1.2 days in metestrus, 5.3 ± 0.3 days in diestrus, and 5.3 ± 1.2 days in proestrus over a 20 day period (n = 10 for WT; n = 3 for AhRKO; p = 0.95 for WT vs. AhRKO in estrus, p = 0.78 for WT vs. AhRKO in metestrus, p = 0.17 for WT vs. AhRKO in diestrus, p = 0.68 for WT vs. AhRKO in proestrus).
The Effect of Ahr Deletion on FSH and LH Levels
To determine if gonadotropin levels were altered in AhRKO mice compared to WT mice, serum levels of FSH and LH were compared in WT and AhRKO mice. The results indicate that AhRKO and WT mice have similar levels of FSH at all stages of the estrous cycle (proestrus AM: WT = 4.84 ± 0.96 ng/ml; AhRKO = 5.46 ± 1.6 ng/ml; n = 3 – 5; p = 0.74; proestrus PM: WT = 8.50 ± 1.58 ng/ml; AhRKO: 8.60 ± 1.06 ng/ml; n = 3; p = 0.98; estrus: WT = 5.84 ± 1.40 ng/ml; AhRKO = 6.21 ± 0.30 ng/ml; n = 3 – 4; p = 0.83; diestrus: WT = 3.03 ± 0.32 ng/ml; AhRKO = 3.34 ± 0.38 ng/ml; n = 4; p = 0.33).
The results also show that AhRKO and WT mice have similar levels of LH in the morning and evening of proestrus and during estrus (proestrus AM: WT = 3.03 ± 0.21 ng/ml; AhRKO = 2.86 ± 0.47 ng/ml; n = 3–5; p = 0.71; proestrus PM: WT = 5.77 ± 0.23 ng/ml; AhRKO = 5.45 ± 0.20 ng/ml; n = 3 – 5; p = 0.35; estrus: WT = 2.32 ± 0.27 ng/ml; AhRKO = 2.28 ± 0.22 ng/ml; n = 3; p = 0.89).
The Effect of Ahr Deletion on Fshr and Lhcgr
To determine if AhRKO and WT follicles have a different capacity to respond to gonadotropins, Fshr and Lhcgr mRNA levels were compared in AhRKO and WT follicles (Fig. 1). The results indicate that isolated AhRKO follicles express significantly lower levels of Fshr compared to WT follicles (WT = 1.24 ± 0.03 genomic equivalents (ge); AhRKO = 0.80 ± 0.09 ge; n = 3; p ≤ 0.01). AhRKO follicles also express significantly lower levels of Lhcgr mRNA compared to WT follicles (WT = 1.16 ± 0.32 ge; AhRKO = 0.16 ± 0.05 ge; n = 3; p ≤ 0.04).
Figure 1. Effect of Ahr Deletion on Hormone Receptor Levels.

Antral follicles were isolated from WT and AhRKO ovaries and subjected to real time PCR analysis for Fshr and Lhcgr. All data were normalized to β-actin. Each bar represents the mean ± SEM. Asterisks indicate significant differences between WT and AhRKO follicles (n = 3; p ≤ 0.01 for Fshr; p ≤ 0.04 for Lhcgr).
Interaction of AHR with Fshr and Lhcgr Promoter
Since Ahr deletion reduced expression of Fshr and Lhcgr, ChIP assays were conducted to determine whether the AHR directly interacts with the promoter regions of the Fshr and/or Lhcgr. No interactions were observed between the AHR and site no. 1 of the Fshr promoter region or between the AHR and the Lhcgr promoter region as indicated by the lack of a band in the sample lane, but the presence of a band in the positive control lane (data not shown). In contrast, interaction was observed between the AHR and promoter region of the Fshr at site no. 2 as indicated by the presence of a 250 bp band in sample lane 4, the presence of a 250 bp band in the positive control lane 5, and lack of bands in the negative control lanes 2, 6, and 7 (Fig. 2).
Figure 2. Interaction of Ahr with the Fshr Promoter.
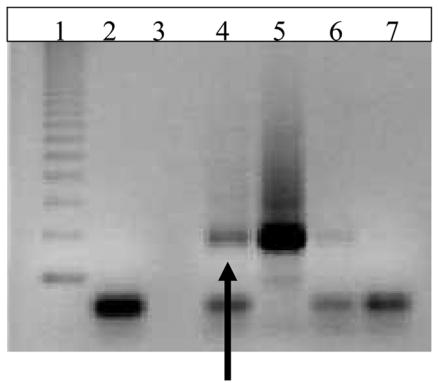
ChIP assays were conducted using a chromatin immunoprecipitation kit (Upstate Biotechnology), ovarian tissue, anti-AHR antibody (Zymed), and primers to the promoter region of the Fshr (site 2) as described in the methods. Lane 1 = ladder; lane 2 = PCR negative control; lane 3 = blank; lane 4 =ovarian DNA with anti-AHR antibody (sample lane), lane 5 = ovarian DNA input before immunoprecipitation with antibody (positive control lane); lane 6 = negative control (no antibody); lane 7 = negative control (no DNA). The arrow indicates AHR binding to Fshr promoter in ovarian sample (n =3).
The Effect of Ahr Deletion on Gonadotropin Responsiveness
As an indicator of gonadotropin responsiveness, ovarian weight in response to eCG treatment was compared in WT and AhRKO mice (Fig. 3A). WT and AhRKO ovaries had similar ovarian weights before eCG treatment (WT = 0.0029 ± 0.0006 g; AhRKO = 0.0028 ± 0.0004 g; n = 6 for WT, n = 3 for AhRKO; p = 0.92). After treatment with the low dose of eCG (5 IU), however, AhRKO ovarian weight was significantly less than WT ovarian weight (WT = 0.0068 ± 0.0012 g; AhRKO = 0.0028 ± 0.0006 g; n = 8 for WT, n = 4 for AhRKO; p ≤ 0.013). After treatment with the high dose of eCG (15 IU), ovarian weights in AhRKO and WT mice increased to a similar size (WT = 0.0070 ± 0.0005 g; AhRKO = 0.0057 ± 0.0004 g; n = 3; p = 0.13).
Figure 3. The Effect of Ahr Deletion on Ovarian Weight and the Number of Corpora Lutea in Response to Gonadotropin Treatment.

Panel A: AhRKO and WT mice were injected with eCG (5 IU and 15 IU) on postnatal days 25–28 and ovarian weight was measured as described in methods. Each bar represents the mean ± SEM. Bars with different letters are significantly different from each other (n = 3 – 8, p = 0.92 for 0 IU eCG in WT vs. 0 IU eCG in AhRKO; p ≤ 0.013 for 5 IU eCG in WT vs. 5 IU eCG in AhRKO; p = 0.13 for 15 IU eCG in WT vs. 15 IU eCG in AhRKO). Panel B: AhRKO and WT mice were injected with eCG (5 IU and 15 IU) on postnatal days 25–28 and then the number of corpora lutea were counted in each ovary. Each bar represents the mean ± SEM. Asterisks indicate statistically significant differences between genotypes (n = 3 – 8, p ≤ 0.02 for 5 IU eCG in WT vs. 5 IU eCG in AhRKO; p = 0.68 for 15 IU eCG in WT vs. 15 IU eCG in AhRKO).
As a second test of gonadotropin responsiveness, the number of corpora lutea formed in response to eCG treatment were compared in WT and AhRKO mice (Fig. 3B). After treatment with the low dose of eCG (5 IU), AhRKO ovaries contained fewer corpora lutea compared to WT ovaries (WT = 7.0 ± 0.6; AhRKO = 3.2 ± 0.9; n = 3 for WT and n = 5 for AhRKO; p ≤ 0.02). After treatment with the high dose of eCG (15 IU), however, the number of corpora lutea in AhRKO and WT ovaries was similar (WT = 4.0 ± 0.0; AhRKO = 3.5 ± 0.6; n = 3; p = 0.2).
As a third test of gonadotropin responsiveness, the ability to ovulate in response to exogenous gonadotropins (eCG + hCG) was compared in WT and AhRKO mice (Fig. 4). After treatment with eCG followed by the two lowest doses of hCG (1.25 IU and 2.5 IU hCG), significantly fewer oocytes were collected from AhRKO mice compared to WT mice. Specifically, after 1.25 IU hCG treatment, only 12.0 ± 2.8 oocytes were collected from AhRKO mice, while 22.4 ± 1.9 oocytes were collected from WT mice (n = 5 for WT and n = 3 for AhRKO; p ≤ 0.02). After 2.5 IU hCG treatment, only 18.2 ± 1.3 oocytes were collected from AhRKO mice, whereas 25.8 ± 2.0 oocytes were collected from WT mice (n = 5 for WT and n = 4 for AhRKO; p ≤ 0.02). After treatment with 5 IU eCG followed by the highest dose of hCG (5 IU), however, no significant differences were observed in the average number of oocytes collected from AhRKO and WT mice (WT = 52.0 ± 7.6 oocytes, n = 7; AhRKO = 54.0 ± 14.0 oocytes; n = 4; p = 0.9).
Figure 4. Effect of Ahr Deletion on the Number of Ovulated Eggs.

WT and AhRKO mice were injected with eCG (5 IU), and 48 hr later, the mice were injected with hCG (1.25, 2.5, and 5 IU). After 18 hr, the ovaries and oviducts were removed, and the number of eggs in each oviduct was counted. Each bar represents the mean ± SEM. Asterisks indicate statistically significant differences between genotypes (n = 3 – 7; p ≤ 0.02 for 1.25 IU hCG in WT vs. 1.25 IU in AhRKO; p ≤ 0.02 for 2.5 IU hCG in WT vs. 2.5 IU hCG in AhRKO; p = 0.9 for 5 IU hCG in WT vs. 5 IU hCG in AhRKO).
DISCUSSION
Previous studies have shown that deletion of the Ahr has negative effects on the ovary (Benedict et al., 2000; Benedict et al., 2003). Specifically, these studies indicate that AhRKO mice have a decreased number of antral follicles (Benedict et al., 2000), slower follicular growth (Barnett et al., 2007), and a reduced number of corpora lutea compared to WT mice (Benedict et al., 2003). The reduced number of corpora lutea in AhRKO mice indicates that AhRKO mice have a reduced capacity to ovulate compared to controls. Therefore, the purpose of this study was to investigate the mechanism by which Ahr deletion reduces ovulation in the mouse. Collectively, the data obtained from this study show that Ahr deletion does not affect ovulation by disrupting estrous cyclicity or altering serum levels of gonadotropins. Instead, Ahr deletion directly affects ovulation in the mouse by reducing the levels of gonadotropin receptors in the ovary and thus, decreasing gonadotropin responsiveness.
Our findings that Ahr deletion does not affect gonadotropin levels are consistent with those from studies using a 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), which is a potent AHR ligand and known inhibitor of ovulation (Chaffin et al., 1997, Franczak et al., 2006; Gao et al., 2000; Mizuyachi et al., 2002; Petroff et al., 2001; Petroff et al., 2003; Shi et al., 2007). For example, Chaffin et al. 1997 showed that in utero and lactational exposure to a single oral dose of 1 μg TCDD/kg does not affect serum FSH or LH concentrations in female rat pups. Similarly, Shi et al. (2007) showed that chronic exposure to 50 and 200 ng TCDD/kg does not affect serum FSH and LH profiles and Franczak et al. 2006 showed that chronic exposure to TCDD (50 and 200 ng/kg) from fetal life to 8 months of age does not affect diestrous concentrations of LH. Further, Gao et al. 2000 showed that LH and FSH surges occur in TCDD-treated rats in response to gonadotropin releasing hormone (GnRH), but are not sufficient to restore ovulation. These data suggest that TCDD treatment and Ahr deletion may affect ovulation through similar mechanisms and that the mechanism is unlikely to involve changes in the levels of gonadotropins synthesized/secreted by the anterior pituitary. This hypothesis is supported by semi-quantitative RT-PCR studies indicating that TCDD treatment downregulates expression of the Ahr in the rat ovary (Son et al., 1999). It is also supported by western blot and immunohistochemical studies indicating that TCDD treatment significantly reduces AHR levels in liver cells (Pollenz, 1996) and male reproductive organs such as the testes, seminal vesicles, and prostate (Roman et al., 1998). In addition, the hypothesis is supported by a study indicating that TCDD treatment induces the degradation of the AHR in liver cells by a ubiquitin-proteasome pathway (Ma and Baldwin, 2000).
In contrast, a few studies indicate that TCDD exposure alters the levels of gonadotropins (Franczak et al., 2006; Petroff et al., 2003). Specifically, Petroff et al. 2003 have shown that TCDD (8 or 32 μg/kg via oral administration) causes a significant premature increase in serum FSH and LH concentrations in immature female rats. Similarly, Franczak et al. 2006 showed that chronic exposure to TCDD (50 ng/kg) from fetal life to 8 months of age significantly elevates serum FSH levels in mature female rats. The reasons for differences in the effects of TCDD on gonadotropin levels are unclear. It is possible that they stem from differences in doses and route of administration of TCDD. Further, it is possible that they are due to differences in the timing of TCDD exposure and gonadoptropin measurements. Perhaps, in some dosing regimens and age groups, TCDD directly targets the ovary and reduces ovarian estrogen synthesis/secretion. In turn, the reduced estrogen levels may be insufficient to exert negative feedback at the level of the hypothalamus or anterior pituitary, resulting in increased gonadotropin synthesis/secretion. This possibility is supported by studies indicating that TCDD reduces estradiol concentrations in rats (Shi et al., 2007) and in cultured rat ovaries (Chaffin et al., 1997). Interestingly, we recently reported that Ahr deletion reduces serum estradiol levels in mice and it reduces the synthesis/secretion of estradiol by mouse antral follicles in vitro (Barnett et al., 2007). Thus, it is possible that some TCDD treatments and Ahr deletion have similar effects on estradiol concentrations, and that TCDD treatment is affecting estradiol levels by downregulating expression of the Ahr in the ovary.
Our previous finding that Ahr deletion reduces estradiol levels (Barnett et al., 2007) is consistent with the data reported here. Since LH and FSH responsiveness are important for estradiol synthesis, the reduced gonadotropin responsiveness observed in AhRKO mice could lead to the reduced estradiol levels observed in AhRKO mice. Perhaps, AhRKO follicles have reduced responsiveness to FSH and this is turn leads to their slow follicle growth and reduced estradiol synthesis. The reduced estradiol synthesis may then exacerbate the slow follicular growth.
We next examined whether AhRKO mice could have reduced responsiveness to gonadotropins due to low gonadotropin receptor expression levels in the ovary. Our results show that AhRKO follicles have significantly lower Fshr and Lhcgr mRNA levels compared to WT follicles. Our results are consistent with studies by Roby et al. (2001), which showed that FSH and LH binding sites are reduced in TCDD-treated rats. Further, our results are consistent with a study showing that TCDD reduces Lhcgr mRNA expression and stability in cultured rat granulosa cells (Minegishi et al., 2003). Thus, while TCDD and Ahr deletion do not affect gonadotropin levels, they both reduce the levels of gonadotropin receptors. Thus, it is possible that TCDD decreases expression of the Ahr in ovarian cells, and in turn, this leads to reduced expression of gonadotropin receptors in the ovary.
Since Ahr deletion reduced the expression of Fshr and Lhcgr, we hypothesized that the Ahr directly regulates gene expression by interacting with the promoter regions of the Fshr and Lhcgr genes. We determined that the promoter region of the mouse Fshr gene contains two AHR response elements and that the mouse Lhcgr gene contains one AHR response element. Thus, these regions were selected for evaluation of the interaction of the AHR with the promoters of gonadotropin receptors via ChIP assays. Our results show that the AHR directly interacts with one of the AHR response elements present in the promoter of the Fshr gene. In contrast, our results show no interaction between the AHR and the Lhcgr promoter region. Thus, our data suggest that the AHR may directly regulate Fshr expression, but not Lhcgr expression. The ability of the AHR to regulate Fshr expression is consistent with findings from other studies indicating that the AHR may regulate other genes involved in ovulation. For example, the nuclear receptor interacting protein 1 (Nrip1) gene has been proven essential for oocyte release during ovulation (Steel et al., 2005), and this gene has been shown to be up-regulated upon activation the AHR signaling pathway (Augereau et al., 2006). Another gene, prostaglandin-endoperoxide synthase 2 (Ptgs2), is reduced upon AHR activation by TCDD in the ovary (Mizuyachi et al., 2002), and deletion of Ptgs2 has been shown to result in a block in ovulation (Lim et al., 1997).
The mechanism by which Ahr deletion indirectly leads to reduced Lhcgr expression in antral follicles is unknown. A previous study by Chen et al. (1994) indicates that FSH up-regulates Lhcgr in mouse cumulus cells. Thus, it is possible that in the presence of the Ahr, AHR is present and able to bind to the promoter region of the Fshr, increasing its expression. This may lead to normal FSH responsiveness, which is required for normal Lhcgr expression and LH responsiveness. In the absence of the Ahr, however, there is no AHR to bind to the promoter region of the Fshr. This may lead to reduced Fshr expression and thus, reduced responsiveness to FSH. In turn, the reduced FSH responsiveness may lead to reduced expression of Lhcgr and thus, reduced LH responsiveness
The reduced levels of gonadotropin receptors in AhRKO ovaries likely led to the reduced ability of AhRKO ovaries to respond to exogenous gonadotropins (eCG, hCG). At low doses of eCG or hCG, the AhRKO ovaries were not as responsive as WT ovaries. At high doses of exogenous gonadotropins, however, AhRKO ovaries responded similarly to WT ovaries, indicating that gonadotropin responsiveness can be restored in AhRKO mice under super-physiological levels of exogenous gonadotropins. Our data are consistent with those showing that TCDD-treated rats exhibit a reduced number of ovulated ova and corpora lutea, and reduced ovarian weight gain after eCG treatment compared to control rats (Mizuyachi et al., 2022; Roby, 2001).
In conclusion, the present study shows that the reduced ovulation in AhRKO mice is due to a reduced capacity of AhRKO follicles to respond to gonadotropins. Further, the reduced responsiveness to gonadotropin treatment in AhRKO mice is due to reduced levels of gonadotropin receptors in AhRKO ovaries compared to WT ovaries. Thus, in addition to mediating the toxicity of environmental chemicals (Bock, 1994; Mukai and Tischkau, 2007; Safe et al., 1998), the Ahr is required for normal ovulation and gonadotropin responsiveness.
Acknowledgments
This work was funded by NIH HD 047275 and NIH MARC Predoctoral Fellowship F31 GM072195. The authors thank Drs. Armina Kazi and Robert Koos for their help with the ChIP assays, Dr. Patricia Hoyer and Mr. Sam Marion for their help with the RIAs, and Dr. Chuck Greenfeld for his help with dosing.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to disclose that could inappropriately influence this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott BD, Schmidt JE, Pitt JA, Buckalew AR, Wood CR, Held GA, Diliberto JJ. Adverse reproductive outcomes in the transgenic Ah receptor-deficient mouse. Toxicol Appl Pharmacol. 1999;155:62–70. doi: 10.1006/taap.1998.8601. [DOI] [PubMed] [Google Scholar]
- Augereau P, Badia E, Fuentes M, Rabenoelina F, Corniou M, Derocq D, Balaguer P, Cavailles V. Transcriptional regulation of the human NRIP/RIP gene by estrogen is modulated by dioxin signalling. Mol Pharm. 2006;69:1336–1346. doi: 10.1124/mol.105.017376. [DOI] [PubMed] [Google Scholar]
- Babu PS, Danilovich N, Sairam MR. Hormone-induced receptor gene splicing: enhanced expression of the growth factor type I follicle-stimulating hormone receptor motif in the developing mouse ovary as a new paradigm in growth regulation. Endocrinology. 2001;142:381–389. doi: 10.1210/endo.142.1.7886. [DOI] [PubMed] [Google Scholar]
- Baldridge MG, hutz RJ. Autoradiographic localization of aromatic hydrocarbon receptor (AHR) in rhesus money ovary. Am J Primatol. 2007;69:681–691. doi: 10.1002/ajp.20381. [DOI] [PubMed] [Google Scholar]
- Barnett K, Tomic D, Gupta R, Miller KP, Meachum S, Paulose T, Flaws JA. The aryl hydrocarbon receptor affects mouse ovarian follicle growth via mechanisms involving estradiol regulation and responsiveness. Biol Reprod. 2007 doi: 10.1095/biolreprod.106.057687. in press. [DOI] [PubMed] [Google Scholar]
- Benedict JC, Lin TM, Loeffler IK, Peterson RE, Flaws JA. Physiological role of the aryl hydrocarbon receptor in mouse ovary development. Toxicol Sci. 2000;56:382–388. doi: 10.1093/toxsci/56.2.382. [DOI] [PubMed] [Google Scholar]
- Benedict JC, Miller KP, Lin TM, Greenfeld C, Babus JK, Peterson RE, Flaws JA. Aryl hydrocarbon receptor regulates growth, but not atresia, of mouse preantral and antral follicles. Biol Reprod. 2003;68:1511–1517. doi: 10.1095/biolreprod.102.007492. [DOI] [PubMed] [Google Scholar]
- Bock KW. Aryl hydrocarbon or dioxin receptor: biologic and toxic responses. Rev Physiol Biochem Pharmacol. 1994;125:1–42. doi: 10.1007/BFb0030908. [DOI] [PubMed] [Google Scholar]
- Chaffin CL, Trewin AL, Watanabe G, Taya K, Hutz RJ. Alterations to the pituitary-gonadal axis in the peripubertal female rat exposed in utero and through lactation to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biol Reprod. 1997;56:1498–1502. doi: 10.1095/biolreprod56.6.1498. [DOI] [PubMed] [Google Scholar]
- Chen L, Russell PT, Larsen WJ. Sequential effects of follicle-stimulating hormone and luteinizing hormone on mouse cumulus expansion in vitro. Biol Reprod. 1994;51:290–295. doi: 10.1095/biolreprod51.2.290. [DOI] [PubMed] [Google Scholar]
- Cooper RL, Goldman J, Vandenbergh JG. Monitoring of estrus cyclicity in the laboratory rodent by vaginal lavage. In: Chapin RE, Heindel J, editors. Methods in Toxicology. 3B. Academic Press; Orlando: 1993. pp. 45–56. [Google Scholar]
- Fernandez-Salguero PM, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Ward JM, Sundberg JP, Gonzalez FJ. Lesions of aryl-hydrocarbon receptor-deficient mice. Vet Pathol. 1997;34:605–614. doi: 10.1177/030098589703400609. [DOI] [PubMed] [Google Scholar]
- Franczak A, Nynca A, Valdez KE, Mizinga KM, Petroff BK. Effects of acute and chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin on the transition to reproductive senescence in female Sprague-Dawley rats. Biol Reprod. 2006;74:125–130. doi: 10.1095/biolreprod.105.044396. [DOI] [PubMed] [Google Scholar]
- Gao X, Petroff BK, Rozman KK, Terranova PF. Gonadotropin-releasing hormone (GnRH) partially reverses the inhibitory effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on ovulation in the immature gonadotropin-treated rat. Toxicology. 2000;147:15–22. doi: 10.1016/s0300-483x(00)00161-x. [DOI] [PubMed] [Google Scholar]
- Hirshfield AN. Development of follicles in the mammalian ovary. Int Rev Cytol. 1991;124:43–101. doi: 10.1016/s0074-7696(08)61524-7. [DOI] [PubMed] [Google Scholar]
- Kazi AA, Jones JM, Koos RD. Chromatin immunoprecipitation analysis of gene expression in the rat uterus in vivo: estrogen-induced recruitment of both estrogen receptor alpha and hypoxia-inducible factor 1 to the vascular endothelial growth factor promoter. Mol Endocrinol. 2005;19:2006–2019. doi: 10.1210/me.2004-0388. [DOI] [PubMed] [Google Scholar]
- Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, Dey SK. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell. 1997;91:197–208. doi: 10.1016/s0092-8674(00)80402-x. [DOI] [PubMed] [Google Scholar]
- Ma Q, Baldwin KT. 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. J Biol Chem. 2000;275:8432–8438. doi: 10.1074/jbc.275.12.8432. [DOI] [PubMed] [Google Scholar]
- Ma Q, Whitlock JP., Jr A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem. 1997;272:8878–8884. [PubMed] [Google Scholar]
- Minegishi T, Hirakawa T, Abe K, Kishi H, Miyamoto K. Effect of IGF-1 and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the expression of LH receptors during cell differentiation in cultured granulosa cells. Mol Cell Endocrinol. 2003;203:123–131. doi: 10.1016/s0303-7207(03)00073-x. [DOI] [PubMed] [Google Scholar]
- Mizuyachi K, Son DS, Rozman KK, Terranova PF. Alteration in ovarian gene expression in response to 2,3,7,8-tetrachlorodibenzo-p-dioxin: reduction of cyclooxygenase-2 in the blockage of ovulation. Reprod Toxicol. 2002;16:299–307. doi: 10.1016/s0890-6238(02)00024-2. [DOI] [PubMed] [Google Scholar]
- Mukai M, Tischkau SA. Effects of tryptophan photoproducts in the circadian timing system: searching for a physiological role for the aryl hydrocarbon receptor. Toxicol Sci. 2007;95:172–181. doi: 10.1093/toxsci/kfl126. [DOI] [PubMed] [Google Scholar]
- Petroff BK, Croutch CR, Hunter DM, Wierman ME, Gao X. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) stimulates gonadotropin secretion in the immature Sprague-Dawley rat through a pentobarbital- and estradiol-sensitive mechanism but does not alter gonadotropin-releasing hormone (GnRH) secretion by immortalized GnRH neurons in vitro. Biol Reprod. 2003;68:2100–2106. doi: 10.1095/biolreprod.102.010439. [DOI] [PubMed] [Google Scholar]
- Petroff BK, Roby KF, Gao X, Son DS, Williams S, Johnson DC, Rozman KK, Terranova PF. A review of mechanisms controlling ovulation with implications for the anovulatory effects of polychlorinated dibenzo-p-dioxins in rodents. Toxicology. 2001;158:91–107. doi: 10.1016/s0300-483x(00)00367-x. [DOI] [PubMed] [Google Scholar]
- Pollenz RS. The aryl-hydrocarbon receptor, but not the aryl-hydrocarbon receptor nuclear translocator protein, is rapidly depleted in hepatic and nonhepatic culture cells exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Mol Pharmacol. 1996;49:391–398. [PubMed] [Google Scholar]
- Richards JS, Russell DL, Ochsner S, Hsieh M, Doyle KH, Falender AE, Lo YK, Sharma SC. Novel signaling pathways that control ovarian follicular development, ovulation, and luteinization. Recent Prog Horm Res. 2002;57:195–220. doi: 10.1210/rp.57.1.195. [DOI] [PubMed] [Google Scholar]
- Robles R, Morita Y, Mann KK, Perez GI, Yang S, Matikainen T, Sherr DH, Tilly JL. The aryl hydrocarbon receptor, a basic helix-loop-helix transcription factor of the PAS gene family, is required for normal ovarian germ cell dynamics in the mouse. Endocrinology. 2000;141:450–453. doi: 10.1210/endo.141.1.7374. [DOI] [PubMed] [Google Scholar]
- Roby KF. Alterations in follicle development, steroidogenesis, and gonadotropin receptor binding in a model of ovulatory blockade. Endocrinology. 2001;142:2328–2335. doi: 10.1210/endo.142.6.7993. [DOI] [PubMed] [Google Scholar]
- Roman BL, Pollenz RS, Peterson RE. Responsiveness of the adult male reproductive tract to 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure: Ah receptor and ARNT expression, CYP1A1 induction, and Ah receptor down-regulation. Toxicol Appl Pharmacol. 1998;150:228–239. doi: 10.1006/taap.1998.8388. [DOI] [PubMed] [Google Scholar]
- Sadek CM, Allen-Hoffmann BL. Suspension-mediated induction of Hepa 1c1c7 Cyp1a-1 expression is dependent on the Ah receptor signal transduction pathway. J Biol Chem. 1994;269:31505–31509. [PubMed] [Google Scholar]
- Safe S, Wang F, Porter W, Duan R, McDougal A. Toxicol Lett. 102–103. 1998. Ah receptor agonists as endocrine disruptors: antiestrogenic activity and mechanisms; pp. 343–347. [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci USA. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Valdez KE, Ting AY, Franczak A, Gum SL, Petroff BK. Ovarian endocrine disruption underlies premature reproductive senescence following environmentally relevant chronic exposure to the aryl hydrocarbon receptor agonist 2,3,7,8,-tetrachlorodibenzo-p-dioxin. Biol Reprod. 2007;76:198–202. doi: 10.1095/biolreprod.106.053991. [DOI] [PubMed] [Google Scholar]
- Son DS, Ushinohama K, Gao X, Taylor CC, Roby KF, Rozman KK, Terranova PF. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) blocks ovulation by a direct action on the ovary without alteration of ovarian steroidogenesis: lack of a direct effects on ovarian granulosa and thecal-interstitial cell steroidogenesis in vitro. Reprod Toxicol. 1999;13:521–530. doi: 10.1016/s0890-6238(99)00048-9. [DOI] [PubMed] [Google Scholar]
- Steel JH, White R, Parker MG. Role of the RIP140 corepressor in ovulation and adipose biology. J Endocrinol. 2005;185:1–9. doi: 10.1677/joe.1.05896. [DOI] [PubMed] [Google Scholar]
- Tomic D, Miller KP, Kenny HA, Woodruff TK, Hoyer P, Flaws JA. Ovarian follicle development requires Smad3. Mol Endocrinol. 2004;18:2224–2240. doi: 10.1210/me.2003-0414. [DOI] [PubMed] [Google Scholar]
- Wang F, Hoivik D, Pollenz R, Safe S. Functional and physical interactions between the estrogen receptor SP1 and nuclear aryl hydrocarbon receptor complexes. Nucleic Acids Research. 1998;26:3044–3052. doi: 10.1093/nar/26.12.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihua Z, Saji S, Makinen S, Cheng G, Jensen EV, Warner M, Gustafsson JA. Estrogen receptor (ER) beta, a modulator of ERalpha in the uterus. Proc Natl Acad Sci. 2000;97:5936–5941. doi: 10.1073/pnas.97.11.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]