

Abstract
The ability to predict the effects of point mutations on the interaction of α-helices within membranes would represent a significant step toward understanding the folding and stability of membrane proteins. We use structure-based empirical parameters representing steric clashes, favorable van der Waals interactions, and restrictions of side-chain rotamer freedom to explain the relative dimerization propensities of 105 hydrophobic single-point mutants of the glycophorin A (GpA) transmembrane domain. Although the structure at the dimer interface is critical to our model, changes in side-chain hydrophobicity are uncorrelated with dimer stability, indicating that the hydrophobic effect does not influence transmembrane helix–helix association. Our model provides insights into the compensatory effects of multiple mutations and shows that helix–helix interactions dominate the formation of specific structures.
How the primary amino acid sequence of a polypeptide determines its three-dimensional structure remains a fundamental question of both practical and intellectual interest in the field of molecular biology. In the field of protein folding and stability, membrane proteins have been less extensively studied than have their soluble counterparts because of the experimental difficulties encountered in handling membrane protein systems. High-resolution structures are available for perhaps a dozen membrane proteins, and calorimetric studies of membrane protein folding are similarly rare (1), but new biochemical methods or tools are expanding the list of accessible experimental questions (2–4). Interest in membrane protein folding is also being driven by the recognition that the environmental constraints placed on membrane proteins may make their folding and stability easier to understand than that of soluble proteins (5).
The success of prediction methods designed to identify α-helical membrane-spanning stretches from protein primary sequence provides strong evidence that membrane protein folding and stability are governed by rather different rules than those of soluble protein folding. It is well established that the hydrophobicity of a stretch of 20 residues is an excellent predictor of whether that sequence will be located within a membrane (6). For soluble proteins, on the other hand, not only does hydrophobicity correlate very poorly with helicity but no other single predictor serves to reliably identify regions of secondary structure (7–9). Atomic details of membrane protein structure will probably prove more complicated to predict than helicity or topology (5), but the success of these approaches to membrane protein structure and organization hints that other simple rules for membrane protein folding might be identified.
A framework for considering the formation of single membrane-spanning helices and the association of these helices into bundles has been provided by the two-stage model for membrane protein folding (10). In the first stage of this model, the insertion of hydrophobic helices into lipid bilayers generates independently stable transmembrane helices. These are proposed to behave as autonomous domains that are unable to unfold or to leave the bilayer because of the high energy penalties associated with breaking hydrogen bonds or exposing hydrophobic side chains to water. The second stage of the model consists of the lateral association of these helices. Specific interactions between the intramembranous portions of these helices are proposed to be responsible for the resulting tertiary and quaternary structures, although lipids, ligands, or extramembranous loops can influence this process. The set of possible interactions is constrained by the membrane environment and by the topological disposition of the polypeptide as determined by the biological insertion machinery (5, 11).
Investigations of the thermodynamics of membrane protein folding can utilize the conceptual framework provided by the two-stage model. For example, the model can greatly simplify the estimation of the influence of amino acid substitutions on the thermodynamic stability of homodimeric helix–helix interactions within a bilayer. Sequence changes will alter the absolute free energies of the species depicted in Fig. 1: the water-solvated unfolded state (A), the monomeric transmembrane helix (B), and the dimeric helical state (C) each might be stabilized or destabilized relative to the wild-type sequence. Estimating the free-energy change of a sequence substitution for state A is particularly challenging because of the difficulties in predicting the ensemble of conformational states sampled by an unfolded polypeptide and in accurately and precisely calculating water–protein solvation energies. However, the two-stage model proposes that the hydrophobicity of the transmembrane peptide presents a sufficiently large thermodynamic barrier to prevent it from leaving the membrane. Because this means that the protein cannot interconvert with the unfolded, water-soluble form, the effects of point mutations on the stability of helix–helix interactions within membranes can be determined by using a thermodynamic cycle that includes only membrane-bound states, as illustrated in Fig. 2. The change in free energy of dimerization because of an amino acid substitution, ΔΔGmut, can be obtained by determining either (ΔG2 − ΔG1) or (ΔG4 − ΔG3). The former expression can be evaluated from experimental results, whereas the latter may prove accessible by calculation, particularly if the solvation of helices by lipid molecules can be easily computed or estimated.
Figure 1.

Possible states for a peptide containing a stretch of approximately 18 hydrophobic residues. Water-solvated, unfolded molecules (A) may be inserted in the membrane as α-helices (B) that in turn may associate to form higher-order structures (C).
Figure 2.

Thermodynamic cycle for point mutation of a dimerizing transmembrane α-helix. Red arrows indicate the change in association state (monomer to dimer), and blue arrows indicate that one residue is being mutated (triangle to circle). The change in free energy of dimerization caused by a mutation, ΔΔGmut, may be obtained from experimental data (ΔG2 − ΔG1) or by computation (ΔG4 − ΔG3).
This simplification of the membrane protein-folding problem makes the two-stage model an appealing tool for examining the basis of the stability and specificity of helix–helix interactions. Here, we use the two-stage model to analyze the detergent-resistant homodimerization of the glycophorin A (GpA) transmembrane helix. A previously published mutagenesis study determined the relative dimerization propensities of more than 160 single-point mutants of the glycophorin A transmembrane domain in a detergent environment (12). Based on the average disruption caused by the 105 hydrophobic substitutions at each site along the transmembrane domain, the authors identified a seven-residue motif as responsible for dimerization (12, 13). These residues (Leu-75, Ile-76, Gly-79, Val-80, Gly-83, Val-84, Thr-87) were subsequently shown by the solution NMR structure of the GpA transmembrane peptide dimer to be located at the dimer interface (14).
Given that the average disruptive effects of single-point mutants accurately identified the residues at the dimerization interface, we hypothesize that the details of the geometry of the interface as seen in the wild-type structure may be used to predict the oligomerization states of each of the individual point mutants. We assume that any stable dimers seen in the mutagenesis experiments will form the same interface as in the wild-type case. Making no changes in the main chain atom positions, we use a rotamer library to artificially create “structures” for sequence variants based on the NMR structure of the wild-type sequence. We represent ΔΔGmut using three empirical parameters designed to describe differences in helix–helix interactions and protein side-chain conformational entropy between mutant and wild-type sequences. Regression analysis identifies combinations of these parameters that best fit the reported experimental dimerization propensities. Our results indicate that predicted changes in steric clashes, side-chain rotamer conformational freedom, and van der Waals contacts within the dimer are able to reproduce the effects of specific point mutations on GpA helix dimerization.
METHODS
Our approach begins with building structures for each sequence variant. The starting coordinates of nonhydrogen atoms are taken from the NMR structure of the wild-type GpA dimer (Protein Data Bank reference 1afo). Side-chain “mutations” are made by using a library of rotamers corresponding to the most commonly observed conformations of side chains in α-helical regions of proteins whose structures have been determined to high resolution (15–17). For residues with more than one rotamer in the library, the rotamer having minimal clashes or the largest number of intermonomer van der Waals contacts is chosen. In cases where one side chain interacts across the dimer interface with another side chain and both have multiple allowed rotamers, all possible combinations of rotamers at each site are tried. Variations about the ideal dihedral angle of up to ±15° for χ1 and χ2 of aliphatic residues, and up to ±30° for χ2 of aromatic residues, are permitted to alleviate small clashes. No energy minimization of the “mutated” structures is performed.
Definitions of Empirical Parameters.
Each structure is scored for three parameters: dsrot, a measure of the gain of side-chain conformational freedom associated with a substitution; vdw, the increase in the number of favorable intermonomer van der Waals contacts associated with making a substitution; and clsh, the extent to which the substitution introduces a steric clash.
The parameter dsrot represents the side-chain rotamer entropy change of a given substitution using the formula:
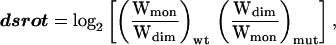 |
1 |
where Wmon, the number of states available to a side chain in a monomeric helix, is simply the number of rotamer library entries for that residue. The number of states available in the dimeric structure, Wdim, is determined by building each of these rotamers into the structure—following the same rules as for building the best structure for each mutant—and eliminating any rotamers that make intermonomer steric clashes. Values for dsrot range from −2 to +2 for the GpA data set.
The parameter vdw is calculated by taking the difference between the number of favorable van der Waals contacts in the mutant structure and in the wild-type structure. A “favorable van der Waals contact” refers to any pair of atoms from different monomers whose interatomic separation produces significant attractive interactions, that is, from 0.4 Å less than the ideal van der Waals separation (18) for those atom types to 0.8 Å more than the ideal van der Waals separation. Values for the parameter vdw range from −14 to +9 for the GpA data set.
The parameter clsh is set to 0 for structures showing no intermonomer van der Waals clashes, to 1 for structures having clashes of less than 0.4 Å, and to 2 for structures having clashes greater than 0.4 Å. “Clashes” refers to interatomic separations of nonbonded atoms that are smaller than the closest approaches expected for those atom types because of electron cloud repulsion (18). The 0.4 Å cut-off corresponds to the r.m.s.d. of the backbone atoms of the members of the family of NMR structures (14). Mutants that receive a clsh score of 2 have their vdw and dsrot scores set to 0.
Each mutant is also scored for the differences in hydrophobicity (ges) and side-chain volume (vol) between the mutant and the wild-type sequence. Neither of these parameters required any information or assumptions about the GpA structure. The hydrophobicity scale for the ges term (6) gives scores ranging from −4.4 to +3.8 kcal mol−1 for the GpA data set. The term vol is based on the volumes of the amino acids (given in number of methylene units) and results in scores ranging from −6 to +9 for the GpA data set.
Regression Analysis and Statistical Methods.
The experimental GpA dimerization propensities from SDS/PAGE reported by Lemmon and coworkers (12) are represented in this analysis as integers 0 through 3, with the score of 3 corresponding to a dimerization level “as wild type” and zero corresponding to “no dimer.” Coefficients of different combinations of the described parameters are least-squares fit to these data by using the program mathematica (Wolfram Research, Champaign, IL). The square of the regression coefficient, R2, is reported, as are standard deviations (σ) for the best-fit coefficients; in cases where the 2σ confidence limit of a given coefficient overlaps zero, the corresponding parameter is excluded and the regression is repeated. Regressions employing linear terms are reported here because inclusion of second-order terms does not alter the coefficients of the linear terms within error.
The regression analysis treats the dimerization phenotypes as real numbers, but they should be considered integers when evaluating the predictive properties of a given model. Accordingly, the best-fit coefficients of each model are used to generate calculated stabilities for each mutant; these real numbers are rounded to the nearest integer, scored against the experimental data, and arranged in a 4×4 correlation table (Fig. 3). The extent of agreement between the model-derived stabilities and the experimental stabilities, Rgrp, is calculated by using the formula (19):
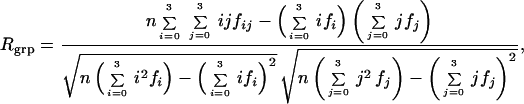 |
2 |
where fi represents the frequency of experimental class i, fj represents the frequency of model class j, and fij represents the frequency of cell (i, j) in the correlation table (Fig. 3).
Figure 3.

Correlation table for model III: calculated and experimental dimerization propensities. Binning of the data by experimental (i) and calculated (j) stability enables direct comparison of the scores as integers, where 0 corresponds to no dimerization and 3 corresponds to wild-type dimerization. Model III scores 75 of 105 mutants correctly; the clustering of the data at or near the diagonal is reflected in the Rgrp2 of 0.760 (see Eq. 2).
Probability Estimates.
We judge the usefulness of each regression model by comparing its prediction success rate with the success rate achieved by using either of two simplistic approaches to predicting stabilities of sequence variants of GpA.
The first of these approaches sets the predicted stability of all sequences equal to the mean of the experimental stabilities. This choice minimizes the sum of the square of the error associated with the “predicted” values. Because regression analysis also yields models whose squared errors have been minimized, this approach is equivalent to performing the regression analysis with a constant term only. Any improvement in prediction by a more complex model over the single term therefore may be attributed to the additional terms introduced.
A second approach sets the predicted stability of all sequences equal to the mode of the experimental data, thus maximizing the possible score for a model consisting of a single constant term. This method constitutes the “best guess” that can be made assuming perfect knowledge of the bias in the data set.
The statistical significance of a success rate may be estimated by calculating the probability of randomly equaling or exceeding that number of successful predictions. Assuming a success rate of ¼ for random predictions, this probability is given by:
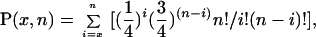 |
3 |
where x represents the number of successful predictions made by the model out of n attempts.
RESULTS
Table 1 presents four models obtained by fits to different subsets of the mutagenesis data, together with statistical measures of the quality of the fit and the predictive power of these models. The estimated coefficients, standard errors, and squared correlation coefficients derived by regression analysis are presented on the left side of the table. The rightmost portion of the table reports the agreement between the experimental and calculated stabilities after the latter have been rounded to the nearest integer. The number of correct predictions achieved by the regression model (calc) should be compared with the scores attained by setting the stability equal to either the average (mean) or the most common (mode) value of the experimental data. In parentheses below each score is the probability of reaching that score purely by chance.
Table 1.
Models obtained by regression analysis to single-point mutants of GpA
| Model | Data (n) | Model coefficients
|
R2 | Accuracy
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| k (σ) | clsh (σ) | dsrot (σ) | vdw (σ) | ges (σ) | vol (σ) | Rgrp2 | calc (Pcalc) | Mean (Pmean) | Mode (Pmode) | |||
| I | All | 2.26 | −1.29 | +0.28 | +0.10 | −0.36 | −0.06 | 0.535 | 0.496 | 79 | 45 | 59 |
| (165) | (0.08) | (0.13) | (0.11) | (0.02) | (0.04) | (0.02) | (2⋅10−10) | (0.28) | (1⋅10−3) | |||
| II | Polar | 1.67 | −0.83 | –* | –* | −0.33 | –* | 0.312 | 0.240 | 24 | 14 | 24 |
| (60) | (0.19) | (0.19) | (0.09) | (7⋅10−3) | (0.67) | (7⋅10−3) | ||||||
| III | Apolar | 2.72 | −1.62 | +0.54 | +0.19 | –* | –* | 0.743 | 0.760 | 75 | 26 | 56 |
| (105) | (0.06) | (0.11) | (0.08) | (0.02) | (2⋅10−23) | (0.56) | (2⋅10−9) | |||||
| IV | All | 1.91 | –† | –† | –† | −0.34 | −0.11 | 0.209 | 0.265 | 54 | 45 | 59 |
| (165) | (0.08) | (0.06) | (0.03) | (0.02) | (0.28) | (1⋅10−3) | ||||||
The sizes (n) of the “polar” (Gly, Ser, Thr, Tyr) and “apolar” (Ala, Cys, Val, Ile, Leu, Met, Phe, Trp) subsets are given in parentheses. Standard deviations (σ) are given in parentheses below the coefficients. R2 is the square of the regression coefficient. Accuracy: number of mutant phenotypes correctly predicted by the regression model (calc), the mean of the data (mean), or the mode of the data (mode). The probability of equaling or exceeding these scores by chance, as calculated using Eq. 2, is given in parentheses below the accuracy scores.
Excluded from regression: 2σ confidence limits for the coefficient overlap zero.
Excluded from regression to test a hypothesis.
Regression Analysis Reveals Correlations.
Model I uses coefficients for the parameters clsh, dsrot, vdw, ges, and vol, plus a constant term k, to fit all 165 pieces of experimental data. The R2 and Rgrp2 of ≈0.5 show that a correlation exists between the model and the data, but this model correctly predicts only 45% of the mutant stabilities and incorrectly predicts wild-type stability to be 2 rather than 3.
To see if these parameters could better predict the behavior of subsets of the data, we divide the data set into “apolar” and “polar” groups. Hydrophobicity has been previously noted to affect the stability of GpA mutants: charged or strongly polar substitutions at any site invariably disrupt the dimer, whereas hydrophobic substitutions show a pattern of disruption that corresponds to a helical repeat (12).
Polar Substitutions Are Weakly Predicted.
Model II, which is fit to the data for the slightly polar substitutions (Gly, Ser, Thr, Tyr), shows poorer R2 and Rgrp2 values than model I. Both clsh and ges show negative correlations with stability; the other terms do not contribute to the fit. Although this model predicts stabilities better than using the mean of the data, the mode of the data performs just as well (Table 1). We conclude that the relative stabilities of slightly polar substitutions are not well explained by these variables.
Hydrophobic Substitutions Are Well Predicted.
In contrast, model III, which is fit to the 105 apolar substitutions (Ala, Cys, Val, Leu, Ile, Met, Phe, Trp), has excellent R2 and Rgrp2 values and remarkably high prediction accuracy. The wild-type stability and 75 mutant phenotypes are correctly calculated with this equation; as shown in Table 1, the probability of achieving this prediction rate by chance is essentially nil. Model III makes 49 more correct predictions than are obtained by setting the calculated stability to the mean of the apolar data subset: this difference can be attributed to the three parameters clsh, vdw, and dsrot. The data set is strongly biased to the score of 3 by mutations away from the interface, which usually score as wild type. Nevertheless, model III makes 19 more correct predictions than are obtained by setting the predicted stability to 3. In fact, assigning the stabilities of substitutions at nonmotif sites to 3 while setting motif sites to 1 (the mean score of these sites) results in only 62 correct predictions, or 13 fewer than are achieved by model III. Clearly, our model has significant predictive value and does more than simply differentiate between motif and nonmotif sites.
The correlation table in Fig. 3 demonstrates that even the incorrectly predicted dimerization propensities of model III are strongly biased toward the experimental score. Each mutant is classified into one cell of a 4×4 table according to its experimental (i) and calculated (j) stability. The majority of mutants lie on the diagonal whereas the rest fall close to it, suggesting that the parameters employed in the models are useful for all mutants. However, more than half of the mutants whose experimental dimerization propensities are scored as “2” are calculated by model III to be as stable as wild type. Examination of these mutants reveals that most occur away from the interface and thus receive scores of zero for clsh, vdw, and dsrot. The slightly lowered stabilities of these mutants cannot be explained by using the chosen empirical parameters, suggesting that another physical mechanism, perhaps interactions with detergent, might be involved.
Coefficients Provide Insight into Stability.
Limiting the coefficients of model III to a single decimal place, the calculated stability (calc) can be expressed:
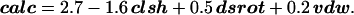 |
4 |
The best-fit constant term of 2.7 is near the wild-type value of 3. This is quite satisfying, because the empirical parameters were all defined as deviations from the wild-type structure. The relative importance of steric clashes, side-chain rotamer entropy, and van der Waals contacts to the stability of GpA transmembrane helix–helix interactions can be inferred from the products of the coefficients and ranges of each parameter. For the apolar data subset, clsh ranges from 0 to 2, and so the second term on the right-hand side of Eq. 4 can reduce calc by as much as 3.2. Because dsrot ranges from −2 to +2, the third term can affect calc by 1.0 in either the positive or negative direction, whereas the vdw range of −12 to +9 means that the final term can raise calc by up to 1.8 and lower it by as much as 2.4. Any of the three empirical parameters therefore can significantly alter the predicted stability. The three components contributing to calc are depicted graphically in Fig. 4 for an instructive set of mutations. Because both vdw and dsrot have the capacity to increase the calculated stability, our model has the potential to predict compensations or even outright increases in dimer stability.
Figure 4.

Components contributing to calc for six mutants of GpA, with weightings given by Eq. 4, and comparison with experiment. Values for expt are reported relative to the best-fit constant term, 2.7, so that the difference between calc and expt accurately reflects the model error. The series of mutations at Thr-87 shows that one, two, or all three parameters may contribute to reductions in stability. Terms may also offset one another. Substitution Val-80 → Leu results in superior packing (vdw) for one of the available leucine rotamers, but because the other rotamers clash, this substitution incurs a large cost in side-chain rotamer freedom (dsrot). By contrast, the parameter dsrot has a stabilizing effect on the mutant Leu-75 → Ala because the wild-type residue loses rotamer freedom upon dimerization, whereas the mutant side chain does not. This partially offsets the loss of favorable packing (vdw) resulting from this sequence change. Mutation Ile-76 → Leu achieves superior packing to the wild-type interface by rearrangement of the side chain of Leu-75, but this gain is offset by the cost of excluding a side-chain rotamer. The agreement of calc with expt is representative of the fit of the model; the mean squared error is 0.27 for model III and 0.25 for these six examples.
Calculated Propensities Correlate with Experimental Dissociation Free Energies.
The recent determination of the dissociation constants for a GpA fusion protein and two point mutants by equilibrium ultracentrifugation in nonionic detergent (20) provides the opportunity to compare the predicted dimerization propensities with experimental helix–helix interaction free energies. The strong correlation between calc and the dissociation free energies (Fig. 5) suggests that model III might be placed on an energy scale. From the slope of the line in Fig. 5, each calc unit corresponds to one kcal mol−1 of dissociation free energy. With this scale, the terms of Eq. 4 might be viewed as representing the energetic impacts of single-point mutations on GpA transmembrane helix homodimerization, in units of kcal mol−1. More data will be needed to assess the precision and accuracy of this energy scale, but we can compare the empirically derived scale with theoretical estimates of side-chain rotamer entropy. The entropic cost of restricting, for two GpA monomers, a given side chain from two rotamer states to one may be estimated by counting the reduction in the states of the system (21); the corresponding free-energy change is given by −TΔS = 2⋅RTln2, or 0.84 kcal mol−1. The coefficient of dsrot from model III indicates that restricting a single side-chain rotamer from two states to one costs 0.54 (±0.08) kcal per mole of dimer (Table 1). This agrees well with theory, especially because nonequivalent population of the rotamers could reduce the theoretical estimate. Simple methods such as those we present here may have potential for quantitative estimation of helix–helix interaction free energies.
Figure 5.

Correlation of calculated dimerization propensity and free energy of dissociation for wild-type GpA and two point mutants. Values for calc, using Eq. 4, are strongly correlated with the free energies of dissociation, ΔGdiss, reported in ref. 20. The squared correlation coefficient, R2, is 0.997 and the slope is 1.04 (±0.03) kcal−1.
Multiple Mutations Are Well Predicted.
If the agreement between calc and the experimental stabilities described above reflects the presence of chemical information in the empirical parameters, then the effects of multiple sequence changes might also be predicted by this model. A series of single and multiple alanine insertion mutations of GpA generated by von Heijne and coworkers (22, 23) provides the opportunity to test this idea. We analyze the single alanine insertion variants (22) as well as all sequence variants in ref. 23 having up to four residues inserted. The data include 15 insertions of a single Ala, a double-point mutant (Leu-75 → Val/Ile-76 → Ala), 1 insertion of AlaAla, 4 insertions of AlaAlaAla (with various point mutations), and 12 insertions of AlaAlaAlaAla (with various point mutations). To score these insertion mutations for the empirical parameters, the sequences are aligned with wild type to minimize changes to the motif, usually with Thr-87 as a reference point (Fig. 6). Changes to residues in the motif-aligned positions are treated as multiple-point mutations. To obtain experimental scores for these mutants, we compute an apparent free energy of dissociation, ΔGapp, from the monomer and dimer band concentrations reported in refs. 22 and 23 by using the equation:
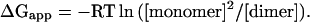 |
5 |
Given that insertions within the motif must rotate and translate the residues that participate in the wild-type interface relative to one another, this class of mutants would not be expected to dimerize (22, 23). In agreement with these expectations, our model suggests that insertion of one or two alanines after position 81 disrupts dimerization because of steric clashes (Fig. 6, constructs 1A and 2A). More significantly, our model also explains the unexpected finding that insertion of three or four alanines supports dimerization (23). The model shows how sequence changes that disrupt the GpA dimer as single-point mutations can be structurally compatible with the GpA interface as part of a multiple substitution [Fig. 6, constructs 3A, 4A, and 4A(G79L)]. Fig. 7 shows the excellent correlation between calc and the apparent free energy of dissociation, ΔGapp, for wild type and all 33 mutants reported by von Heijne and coworkers that bear up to four inserted residues (22, 23). Like the examples in Fig. 6, which are plotted in Fig. 7 with triangles, many of these constructs dimerize despite containing insertions within the seven-residue GpA motif that place individually disruptive sequence changes at motif positions. With the exception of a single point, the dimerization behavior of all these constructs is well explained by our model. We conclude that the model is able to account for compensatory interactions between multiple-point mutations.
Figure 6.

Sequences, inferred structures, and calculated and experimental dimer stabilities for insertion mutants of GpA. Sequences of GpA fusion proteins from ref. 23 are aligned by using Thr-87. Inserted or mutated residues are in red; residues aligned with the motif are in large type. Numbering of residues in the panels corresponds to their positions in the motif alignment, not to numbering in the construct. Values for calc and ΔGapp values are computed by using Eq. 4 and 5, respectively. The six panels show the packing at the interface resulting from building these sequences into the GpA wild-type backbone geometry. Molecular surfaces are displayed for wild type, 3A, 4A, and 4A(G79L), where one monomer approaches within 1.5 Å of the other and serve to depict the close packing across the interface. For sequences 1A and 2A, clashes are created upon building in the residues, and the surfaces serve to highlight these steric collisions. (wildtype) The wild-type residues Leu-75, Ile-76, Gly-79, and Val-80 exhibit excellent intermonomer packing. (1A) Introducing Val at position 79 of the motif generates a steric clash. (2A) Either Met at position 79 or Ile at position 75 will clash with any rotamer of the Phe at position 76. (3A) Four weakly disruptive single mutations are placed into motif positions, but our model indicates that these should have compensatory interactions with one another. The Ala at position 79 would clash slightly in the wild-type sequence as described previously (14), but having Ala replace Val at position 80 eliminates this clash and generates good packing. The Phe side chain of position 75 would need to swing away from the interface to avoid a clash in a wild-type context, but the space afforded by the Gly at position 76 enables close packing between the Phe side chain and the backbone of the opposite monomer. (4A) Interactions between Ala residues at 79 and 80 are favorable, as for 3A. Placing Val in position 76 reintroduces the favorable interactions obtained in the wild type by having a β-branched residue at the dyad. (4A(G79L)) As 4A, except that a (wild-type) Leu is placed at position 75 in a new geometry. The rank order of the calculated and predicted dimerization propensities compare well, showing that the stabilities of these mutants can be predicted by using the same rules as were used for the single-point mutants.
Figure 7.

Correlation of calculated dimerization propensity and apparent free energy of dissociation for wild type and 33 multiple mutants of GpA. Values for calc, using Eq. 4, are shown to be strongly correlated with the apparent free energies of dissociation, ΔGapp, computed by using Eq. 5 and data from refs. 22 and 23). Excluding the outlier, the squared correlation coefficient, R2, is 0.845 and the slope is 0.85 (±0.07) kcal−1. Triangles indicate data points corresponding to sequences depicted in Fig. 6.
On the other hand, the outlier in Fig. 7, mutation 4A(G79L, A2W), is predicted by our model to be nondimeric due to an inferred clash, but this sequence actually forms a large amount of dimer. Similarly, several more extensive insertion mutations reported in ref. 23 are not well described by our model; for instance, the stabilities of 6 of 11 insertions of five residues are poorly predicted by calc. We conclude that the assumptions in our approach are no longer valid for this class of mutations: either our method fails to account for the effects of such extensive changes to the GpA motif or the peptides associate in a fashion that is not analogous to wild-type GpA dimerization.
Changes in Hydrophobicity and Volume Are Poor Predictors of Stability.
Although the parameters ges and vol have some small predictive value in the regression analysis of the entire data set (model I) and are included in the initial regression for model III, confidence limits indicate that these two parameters do not contribute significantly to the fit of the “apolar” subset (Table 1). Because changes in side-chain hydrophobicity and volume have proven useful in understanding the relative stabilities of point mutants of soluble proteins (24–26), we now focus on these terms.
We fit model IV to the entire data set, specifically excluding clsh, vdw, and dsrot to test the predictive values of the remaining two terms. The poor R2, Rgrp2, and accuracy (Table 1) conclusively demonstrate the minimal value of these terms as predictors of GpA dimer stability. It is noteworthy that for all regressions presented in Table 1, |vol| and vol were each tried in turn. Because increases and decreases in side-chain volume at the interface should both be detrimental to dimer stability (by causing clashes and packing voids, respectively), it was expected that the term |vol| would provide superior predictive power. Surprisingly, in no model does the more physically reasonable form of the function give an improved fit to the data. Tested subsets (polar, apolar, interfacial, noninterfacial) also showed no significant predictive value for ges, |vol|, or vol, although |vol| does show a weak negative correlation with stability for the interfacial subset.
We note that the lack of correlation between ges and the experimental dimer stabilities in model III is not simply due to the apolar data set exhibiting a small range of hydrophobicity changes. In fact, apolar substitutions such as Gly → Phe (−2.7 kcal mol−1) and Ile → Ala (+1.5 kcal mol−1) result in large transfer free-energy changes and a range of more than 8 kcal per GpA dimer for the term ges. By contrast, the free-energy scale derived for model III shows that the effects of single apolar substitutions actually modulate the dimerization free energy of GpA by no more than 3 kcal mol−1 or so. Because ges has a range more than twice that of the experimentally observed association free energies in the apolar subset, the absence of a statistically significant correlation with dimer stability must indicate that hydrophobicity changes are simply lacking in predictive value.
Polar Substitutions May Destabilize the Helical Monomer in SDS.
Interestingly, the coefficient for the parameter ges exhibits, within error, the same negative value for models I, II, and IV despite the fact that model III rules out changes in transfer free energy as a significant contributor to dimer stability (Table 1). We note that model III is obtained by regression of the apolar data subset only: it would appear that the polar residues included in the data sets for regression models I, II, and IV cause a slight negative correlation between ges and the experimental dimer stability. We suggest that the disruptive effects of hydrophilic residues are a result of the SDS/PAGE system in which the dimerization is assayed.
The introduction of polar side chains must destabilize the association of GpA helices with the hydrophobic interiors of detergent micelles because of the energetic cost of burying polar side chains away from water. A structural alternative available in micelles—but not in membranes—is for the peptide to break the helix near the hydrophilic site and expose the main-chain and side-chain hydrogen bond donors and acceptors to water at the micelle surface. The described changes in monomer gel mobility of strongly polar point mutants of the GpA fusion protein on SDS/PAGE (12) are consistent with alterations in the association of the protein with the detergent micelle. The destabilizing effects of strongly polar substitutions on GpA dimerization independent of position in the transmembrane sequence therefore may be attributed to a decreased stability of the monomeric helix (12) relative to an unfolded monomeric species. This idea is supported by data from model peptides, which exhibit helicity in micellar environments that is correlated with the hydrophobicity of a central uncharged segment of the peptide (27). We propose that the GpA transmembrane domain is uniformly hydrophobic enough to remain associated with the interior of SDS micelles as an α-helix, and that hydrophobic substitutions do not affect this but simply modulate helix–helix interactions. In contrast, the dimer stabilities of polar GpA mutants probably reflect not only helix–helix interaction effects but also reductions of the stability of the helical monomer relative to the unfolded state.
DISCUSSION
From the close agreement between calculated and experimental dimerization propensities we conclude that the major features needed to understand the sequence dependence of GpA helix–helix interactions are contained within the parameters clsh, dsrot, and vdw, which describe steric clashes, rotamer freedom, and favorable van der Waals interactions. Because the success of the approach depends on the rules used to score the set of mutants, we must consider the underlying assumptions of the method as well as the parameters and coefficients when we attempt to infer properties of helix–helix interactions from these results.
In generating monomer and dimer “structures” for the sequences containing point mutants, we assume that each monomer will remain in essentially ideal α-helical geometry and that side chains will take conformations corresponding to rotamers compatible with a helical backbone. These assumptions reflect our expectations about the accessible conformations of a transmembrane polypeptide based on free-energy considerations. Monomeric helices within a bilayer are free to populate local conformational energy minima because they are not experiencing tight and specific interactions with other species; presumably these minima correspond to ideal helical geometry and favorable side-chain rotamer positions. The two-stage model argues for minimal perturbation of the ideal helical geometry of the monomer upon dimerization, because a large energy penalty would otherwise accompany changes in the backbone hydrogen bonds within a low dielectric environment. The predominance of low-energy rotamers for residues within mutant dimers can be rationalized in a similar fashion: altering a side chain from a preferred rotamer position in the monomer to a sterically unfavored side-chain dihedral angle upon dimerization would destabilize the dimer by several kilocalories per mole.
On the other hand, a sufficiently strong driving force for dimerization could overcome the energy costs of poor local geometry. Such a driving force is unlikely in this system, given the small interaction surface between GpA monomers (14), the lack of intermonomer hydrogen bonds or salt bridges, and the negligible role played by the hydrophobic effect in the thermodynamic cycle diagrammed in Fig. 2. Although these points do not apply to all membrane proteins, our assumptions provide a useful conceptual limit in which to consider transmembrane helix–helix interactions. We note that because our model is able to explain how changes as extensive as insertion of four residues affect GpA dimer stability, the structural assumptions are shown to be quite robust in this system.
Despite the fact that the range of the parameter ges exceeds the estimated range of the experimental dissociation free energies, hydrophobicity is without predictive value for the apolar substitutions of GpA. This contrasts with many studies of soluble proteins, where hydrophobicity has been shown to play an important role in folding and stability (26, 28, 29). However, this result is consistent with the two-stage model, which states that the energy from the hydrophobic effect is expended in the first stage of folding, upon insertion of a hydrophobic peptide into the bilayer as an α-helix. Because the lateral association of helices takes place within a nonpolar bilayer, the hydrophobic effect should have a negligible influence on the reaction. The regression analysis of the apolar subset of the data thus supports the central precept of the two-stage model: helical membrane folding is separated into two thermodynamically distinct steps, and so the hydrophobic effect does not modulate helix–helix association.
From a consideration of the molecular species in Fig. 2, we expect that the energy terms affecting helix–helix association will include helix–helix, helix–lipid, and lipid–lipid interaction enthalpies as well as peptide and lipid rotational, translational, and conformational entropies. Because we attempt to account for differences in dimer stabilities of sequence variants, we ignore terms (such as helix translational or rotational entropy) that should be independent of the peptide sequence. The absence of experimental information about lipid or detergent molecules precludes the design of structure-based parameters involving these species, and so our method parametrizes only two energy terms. Helix–helix interactions are represented by the empirical parameters clsh and vdw, which are defined to account for intermonomer steric clashes and favorable van der Waals contacts introduced between monomers by mutations. Peptide conformational entropy is represented by the term dsrot, which counts side-chain rotamer states; because the helix backbone should not be able to undergo changes in conformation, this term should suffice. We ignore vibrational entropy contributions as well as any intrinsic energetic differences between rotamers (21, 30).
Despite completely neglecting several energy terms, model III reproduces the experimental dimerization propensities remarkably well. We emphasize that the neglected energy terms may still contribute strongly to the net free energy of dimerization—this analysis merely shows that amino acid changes in the sequence of the interacting helices do not greatly modulate the total contribution from these terms. It should be stressed that our parameters were designed for simplicity: our goal was to determine whether these minimal concepts could account for the behavior of the GpA system. Rather than inferring that our simple empirical parameters manage to compensate for the neglected energy terms, we conclude that helix–helix interactions and side-chain conformational entropy are the dominant terms affecting the sequence dependence of GpA self-association.
Model III provides insight on the relative importance of the empirical parameters and suggests that stability “trade-offs” may be understood by using simple structure-based rules. Although the term clsh plays a dominant role in determining the predicted stability of many single-point mutations, second-site mutations can alleviate the clashes caused by a single mutation and thereby regain wild type calculated, and observed, dimerization propensities. This is entirely consistent with the large coefficient attributed to clsh by the regression, and reiterates a familiar point from other studies of molecular recognition: steric clashes provide excellent control over specific interactions (31). Our success in predicting the stabilities of multiple substitution mutations suggests that the concepts represented by the model may be useful in the rational redesign of helix–helix interactions.
Although this approach appears to yield considerable physical insight into the sequence specificity of GpA dimerization, it is important not to overinterpret the details of the model. Although the definitions of the empirical parameters are structure-based, they do not strictly correspond to physical energy terms. For example, the magnitudes of the model coefficients tell us that a mild clash should have the same energy cost as the loss of eight favorable van der Waals contacts, or about 1.5 kcal. By contrast, inspection of the Lennard–Jones potential reveals that a “mild” 0.3 Å clash has an astronomical energy penalty (18). These two different views of the energy associated with a steric clash can be reconciled with one another by viewing the regression coefficients in the context of the assumptions made by our method. Consider a substitution that results in a 0.3 Å clash and scores clsh = 1 under our method, assuming that no backbone atoms move at all. The energy of this artificially generated structure would indeed be very high; but in the real world, a slight separation of the helices might accommodate the clash. However, the nearly ideal packing of the wild-type GpA dimer interface means that such a separation of the helices would cause a decrease of the total intermonomer van der Waals interaction energy, resulting in an observed phenotype of perhaps 1 or 2. The experimentally observed reduced stability of the dimer would be “best-fit” by our regression analysis as if it resulted from the scored clash, because the method does not acknowledge any rearrangement of helices. Depending on the detailed geometry, some “mild” clashes will be more readily accommodated by rearrangements than others, so the clsh coefficient of our model simply corresponds to the single best-fit compromise over the existing data. This highlights the empirical nature of our approach and emphasizes the fact that physical insights obtained from these parameters should always be accompanied by a set of caveats. Indeed, a similar argument could be proposed for how slight deformations of the helix backbone, rather than translations, might avoid some clashes. Whatever the precise physical basis for the success of our model, the central point is that the properties of sequence variants can be inferred from the wild-type structure by using a few simple rules.
Our model provides a rational basis for the sequence specificity of GpA transmembrane helix dimerization. Three structure-based empirical parameters representing a set of simple concepts—predicted clashes, van der Waals contacts, and changes in side-chain rotamer freedom—are able to reproduce the qualitative dimerization propensities of more than 100 hydrophobic single-point mutants (and 30 insertion mutants) of the GpA transmembrane domain. The model demonstrates that the determinants of stability and specificity of transmembrane helix–helix interactions in detergent environments may be at least qualitatively understood by using extensions of the ideas outlined in the two-stage model for membrane protein folding (10). This finding suggests that arriving at an understanding of the structures and stabilities of α-helical integral membrane proteins may be feasible by using simple methods and ideas.
Acknowledgments
We thank P. B. Moore, J.-L. Popot, F. M. Richards, S. Kamtekar, J. H. Prestegard, C. E. Rogge, W. Russ, Y. Shamoo, and members of the Engelman lab for valuable discussions and critical reading of the manuscript. Funding for this work was provided by grants from the National Science Foundation, the National Institutes of Health, and the National Foundation for Cancer Research.
ABBREVIATION
- GpA
glycophorin A
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 (reference 1afo).
References
- 1.Haltia T, Freire E. Biochim Biophys Acta. 1995;1241:295–322. doi: 10.1016/0304-4157(94)00161-6. [DOI] [PubMed] [Google Scholar]
- 2.Popot J, Saraste M. Curr Opin Biotechnol. 1995;6:394–402. doi: 10.1016/0958-1669(95)80068-9. [DOI] [PubMed] [Google Scholar]
- 3.Tribet C, Audebert R, Popot J. Proc Natl Acad Sci USA. 1996;93:15047–15050. doi: 10.1073/pnas.93.26.15047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riley M, Wallace B, Flitsch S, Booth P. Biochemistry. 1997;36:192–196. doi: 10.1021/bi962199r. [DOI] [PubMed] [Google Scholar]
- 5.von Heijne G, Manoil C. Protein Eng. 1990;4:109–112. doi: 10.1093/protein/4.2.109. [DOI] [PubMed] [Google Scholar]
- 6.Engelman D, Steitz T, Goldman A. Annu Rev Biophys Biophys Chem. 1986;15:321–353. doi: 10.1146/annurev.bb.15.060186.001541. [DOI] [PubMed] [Google Scholar]
- 7.O’Neil K T, DeGrado W F. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 8.Munoz V, Serrano L. Proteins. 1994;20:301–311. doi: 10.1002/prot.340200403. [DOI] [PubMed] [Google Scholar]
- 9.Minor, D. L. & Kim, P. S. (1994) 371, 264–267. [DOI] [PubMed]
- 10.Popot J, Engelman D. Biochemistry. 1990;29:4031–4037. doi: 10.1021/bi00469a001. [DOI] [PubMed] [Google Scholar]
- 11.von Heijne G. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 12.Lemmon M, Flanagan J, Treutlein H, Zhang J, Engelman D. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 13.Lemmon M, Treutlein H, Adams P, Brunger A, Engelman D. Nat Struct Biol. 1994;1:157–163. doi: 10.1038/nsb0394-157. [DOI] [PubMed] [Google Scholar]
- 14.MacKenzie K, Prestegard J, Engelman D. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 15.Ponder J W, Richards F M. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 16.Schrauber H, Eisenhaber F, Argos P. J Mol Biol. 1993;230:592–612. doi: 10.1006/jmbi.1993.1172. [DOI] [PubMed] [Google Scholar]
- 17.Dunbrack R L, Karplus M. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 18.Levitt M. J Mol Biol. 1974;82:393–420. doi: 10.1016/0022-2836(74)90599-3. [DOI] [PubMed] [Google Scholar]
- 19.Freund, J. E. & Williams, F. J. (1991) Dictionary/Outline of Basic Statistics [Dover, New York; originally published by McGraw–Hill, New York (1966)].
- 20.Fleming K, Ackerman A, Engelman D. J Mol Biol. 1997;272:266–275. doi: 10.1006/jmbi.1997.1236. [DOI] [PubMed] [Google Scholar]
- 21.Pickett S D, Sternberg M J. J Mol Biol. 1993;231:825–839. doi: 10.1006/jmbi.1993.1329. [DOI] [PubMed] [Google Scholar]
- 22.Mingarro I, Whitley P, Lemmon M, von Heijne G. Protein Sci. 1996;5:1339–1341. doi: 10.1002/pro.5560050712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mingarro I, Elofsson A, von Heijne G. J Mol Biol. 1997;272:633–641. doi: 10.1006/jmbi.1997.1276. [DOI] [PubMed] [Google Scholar]
- 24.Sandberg W S, Terwilliger T C. Science. 1989;245:54–57. doi: 10.1126/science.2787053. [DOI] [PubMed] [Google Scholar]
- 25.Lim W A, Sauer R T. J Mol Biol. 1991;219:359–376. doi: 10.1016/0022-2836(91)90570-v. [DOI] [PubMed] [Google Scholar]
- 26.Eriksson A E, Baase W A, Zhang X J, Heinz D W, Blaber M, Baldwin E P, Matthews B W. Science. 1992;255:178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 27.Li S, Deber C. Nat Struct Biol. 1994;1:368–373. doi: 10.1038/nsb0694-368. [DOI] [PubMed] [Google Scholar]
- 28.Murphy K P, Gill S J. J Mol Biol. 1991;222:699–709. doi: 10.1016/0022-2836(91)90506-2. [DOI] [PubMed] [Google Scholar]
- 29.Bryson J W, Betz S F, Lu H S, Suich D J, Zhou H X, O’Neil K T, DeGrado W F. Science. 1995;270:935–941. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 30.Stapley B, Doig A. J Mol Biol. 1997;272:456–464. doi: 10.1006/jmbi.1997.1250. [DOI] [PubMed] [Google Scholar]
- 31.Lesser D, Kurpiewski M, Jen-Jacobson L. Science. 1990;250:776–786. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]