

Abstract
The efficacy of a series of 2-aryl/alkyl selenazolidine-4(R)-carboxylic acids (SCAs) in reducing NNK [4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone]-induced lung adenomas in female AJ mice, a model for tobacco-related lung tumorigenesis, has been investigated. With selenazolidines in the diet for one month prior to carcinogen administration and during the subsequent four months of tumor development, 2-butylSCA, 2-cyclohexylSCA, 2-phenylSCA and 2-oxoSCA were chemopreventive, significantly reducing mean lung tumor numbers from the 10.9 of unsupplemented controls to 4.7, 5.3, 2.8 and 4.7 respectively. When selenazolidine supplementation began three days after carcinogen administration (i.e., post-initiation), 2-butylSCA, 2-cyclohexylSCA, and 2-oxoSCA were chemopreventive. In both regimens, selenocystine was also chemopreventive. In the post-initiation protocol, but with intervention at a precancerous stage (thirteen days), whole genome expression analysis of lung RNA identified six gene transcripts that weakly correlated with the efficacy of tumor reduction by the four selenocompounds at four months. None of these genes were among those identified as influenced by chemopreventive selenium compounds in human lung cancer cell lines. When supplementation was for one month-prior until 3 days-after carcinogen administration, 2-butylSCA, and 2-phenylSCA were chemopreventive but selenocystine was ineffective. Both 2-butylSCA and 2-phenylSCA retained their chemopreventive activity (44% and 40% tumor number reduction respectively), when the supplementation was shortened and restricted to a pre-initiation period (days -9 to -2). With supplementation spanning 2 days-prior until 3 days-after NNK, reductions in tumor numbers by 2-phenylSCA (26%) and 2-butylSCA (17%) did not achieve statistical significance. Thus, several 2-aryl/alkyl selenazolidines possess chemopreventive activity against NNK-induced lung tumors, and variously demonstrate pre-initiation and post-initiation efficacy.
Keywords: Lung Cancer, Cancer chemoprevention, Selenium, Selenazolidine, Tobacco derived nitrosamine
INTRODUCTION
Cancer is a continuing major health problem. The American Cancer Society’s estimate of new cancer cases in the U.S. in the current year exceeds 1.4 million, and on a tissue of origin basis, lung ranks ahead of colon but behind prostate and breast. For estimated deaths however (160,000), lung cancer exceeds prostate, breast, and colon combined. Nearly 90% of all lung cancers have been causally linked to cigarette smoking [1]. While cigarette smoke contains over 60 carcinogens [2,3], the tobacco-specific N-nitrosamines are believed to play a major role in the initiation and progression of the disease [4-6]. In particular, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is known to be a potent carcinogen in rodents and is strongly linked to the etiology of cancer in humans [5-11]. A mouse model of chemically induced lung cancer using NNK has been available and studied for over 30 years [12]. NNK treatment of female A/J mice results in the development of lung tumors that are similar in morphology, histogenesis, and molecular characteristics to human adenocarcinoma, the most common type of human lung cancer [13].
Selenium in many chemical forms has been investigated as a chemopreventive against, for the most part, chemically-induced tumors. The chemopreventive efficacy of different selenium compounds varied widely both within and between various chemically-induced tumor models. For example, while inorganic selenite was effective in most studies against 7, 12-dimethylbenz[a]anthracene (DMBA)-induced mammary tumors in rodents [14-18] although weak or an absence of chemopreventive activity has been reported in some rat studies, [19], it was ineffective in the murine NNK lung tumor model [20,21]. While selenocystine was not effective against DMBA-induced mammary tumors in mice [22], it was effective against NNK-induced lung tumors [20]. Another selenium-containing amino acid, selenomethionine, was either only weakly effective [20] or ineffective [23] in this model. The methylated cysteine congener of selenomethionine, Se-methylselenocysteine was also ineffective [20]. 2(R,S)-Methylselenazolidine-4(R)-carboxylic acid (methylSCA), a selenocysteine prodrug patterned after the sulfur-containing analog, thiazolidine, 2(R,S)-methylthiazolidine-4(R)-carboxylic acid [24], and designed to liberate selenocysteine after non-enzymatic ring opening and hydrolysis was also not an effective agent, while the 2-oxo congener, 2-oxoselenazolidine-4(R)-carboxylic acid (2-oxoSCA) had chemopreventive activity comparable to selenocystine [20]. 2-OxoSCA was designed to release selenocysteine via an enzymatic mechanism (by 5-oxoprolinase) rather than spontaneously.
In the present investigation, selenazolidines with other alkyl (butyl and cyclohexyl) and aryl (phenyl and 2′-hydroxyphenyl) substituents at the 2-position have been evaluated for chemopreventive activity (Figure 1). It is anticipated that these compounds will non-enzymatically release selenocysteine in much the same manner as 2-methylSCA, but at different rates, and by virtue of their differing lipid solubilities, likely possess differing distribution characteristics. In this comparative study, a single dietary level was used (15 ppm selenium) and chemopreventive efficacies were compared to selenocystine and 2-oxoSCA. In addition to tumor reduction, the effects of the selenocompounds in the diet on gene transcription changes in lung tissue shortly after NNK administration were evaluated. The rationale for this in vivo platform was that selenocompound reduction of NNK tumor numbers could result from a reduction or reversal of the NNK-induced gene transcription changes that sends lungs cell down a tumorigenic path. Previous comprehensive transcript analyses have only evaluated selenocompound-evoked gene transcription changes in professional lung cancer cell lines (25,20). With the RNA extracted from lung tissue ten days after NNK administration, we used Pavlidis Template Matching (PTM) or Significance Analysis for Microarrays (SAM) methodologies to identify the genes with the greatest significant differences between mice receiving NNK and selenocompounds compared to mice only receiving NNK, and the transcript changes that best corresponded to the efficacy of tumor reduction as determined at the four month end-point.
Figure 1.
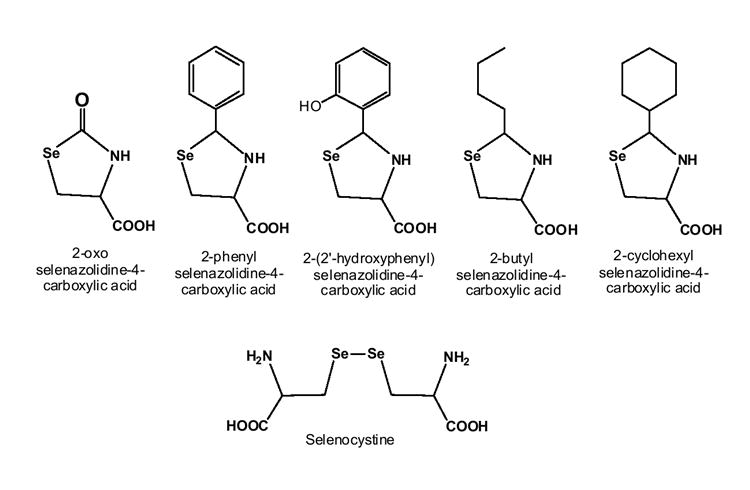
Structure of selenium compounds evaluated for chemopreventive activity.
MATERIALS AND METHODS
Chemicals and reagents
AIN-76A diet was obtained from Dyets, Inc., Bethlehem, PA . 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) was obtained from Toronto Research, Toronto, Canada and L-selenocystine, from Acros Organics (Morris Plains, NJ). 2-oxoSCA was synthesized as described by Xie et al.,[27] and the four other 2-substituted selenazolidines were synthesized in a similar manner to 2-methylSCA except the appropriate carbonyl compound was substituted for acetaldehyde in the synthesis [28].
Animals and treatments
Female A/J mice, 5 weeks of age, weighing 15-16g were obtained from The Jackson Laboratory, (Bar Harbor, ME) and housed on hardwood bedding at 22 ± 2 °C with 50 ± 10% humidity on a 12-hour light/dark cycle. They were randomized into groups of 14 - 15 mice each. Mice were stabilized on unsupplemented AIN-76A diet, ad libitum, for 1 week prior to supplementation with selenium compounds. The AIN-76A diet in the A/J mouse strain provides for more numerous and reproducible lung tumors in response to NNK than other rodent diets (29,30). The diet, which contains a basal level of 0.35 ppm selenium, was supplemented with the selenium compounds under investigation at 15 ppm (selenium). Previous studies have shown little or no toxicity of selenocystine and 2-oxoSCA at the 15 ppm level [20]. Since long-term selenazolidine stability when mixed into the diet has not been assessed, freshly prepared diet mixtures were provided to the animals every day. The various supplementation regimens employed, keyed to the results tables and figures, are shown in Figure 2. When required, mice received the NNK (10 μmol) by ip injection in 0.2 ml saline [21,29].
Figure 2. Selenium supplementation regimens.
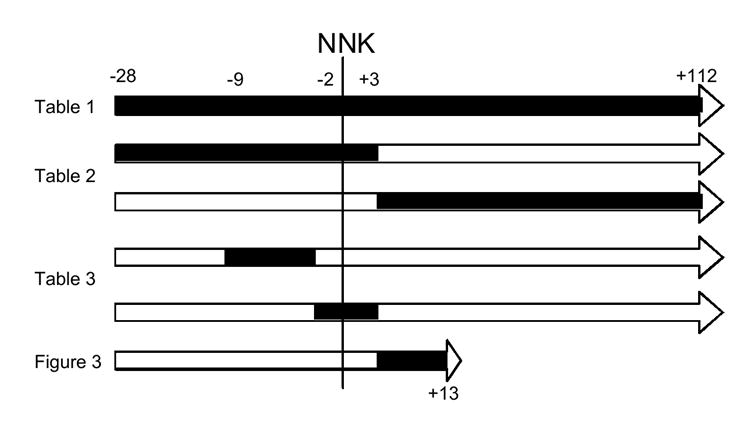
NNK administration is designated as day 0. Periods of dietary supplementation with selenocompounds are shown shaded. The regimens are keyed to the tables and figures in which the outcomes of supplementation can be found.
Lung tumor evaluation
Four months after NNK administration, animals were sacrificed and lungs were collected. Intact lungs were carefully removed and manually filled with Carnoy’s fixative through the trachea. After one week, visible surface adenomas were counted [31]. Both the percent of animals with lung tumors (i.e., incidence), as well as the mean number of tumors per mouse ± SEM (i.e., multiplicity) were evaluated by two independent observers and recorded. Statistical analysis of the results was performed by analysis of variance (ANOVA) followed by Fisher’s protected least significant difference multiple range test. Differences were considered significant when p < 0.05.
Microarray expression analysis
Thirteen days after receiving NNK, lungs were surgically removed, homogenized immediately in Trizol (Invitrogen), and frozen. RNA was later extracted from the homogenates following the Trizol protocol, resuspended in 100 μl DEPC H2O, quantified spectroscopically (A260), and 50 μl of the RNA sample was further purified using the Qiagen RNeasy Cleanup protocol. The purified RNA was again quantified spectroscopically (A260) and the quality determined using an Experion Automated Electrophoresis Station (BioRad) with standard sensitivity RNA chips. Agilent labeling kits were utilized to amplify and generate Cy-dye labeled cRNA for hybridization to Agilent 44K Whole Mouse Genome Oligonucleotide Microarrays. Transcript levels were assessed after competitive hybridization of four biological replicates (Cy5 channel) compared with a mixture of 4 mice that were untreated (Cy3 channel) and quantified by Agilent Feature Extraction software (v.8.1.1.1). This software preprocesses the data by subtracting local background, flags irregular spots and performs global linear regression (lowess) normalization, giving a log transformed ratio. This data is imported into TIGR MEV software for further analysis. A supervised strategy is used to identify the genes with the greatest significant differences between mice receiving NNK and selenocompounds compared to mice only receiving NNK using Pavlidis Template Matching (PTM) [32] or Significance Analysis for Microarrays (SAM) [33] methodologies.
RESULTS
Pre- and post-initiation selenium supplementation
Four out of five selenazolidines investigated showed chemopreventive activity as characterized by a reduction in tumor number (Table 1). Prior studies have shown chemopreventive activity for both 2-oxoSCA and selenocystine using a -7 to +112 day supplementation (relative to NNK) and this was confirmed here with a -28 to +112 day regimen. 2-ButylSCA and 2-cyclohexylSCA reduced mean lung tumor numbers to a similar extent (57% and 51% respectively) as 2-oxoSCA (57%), while 2-phenylSCA caused an even greater reduction (74%). The reduction in tumor number by 2-phenylSCA was similar to that elicited by selenocystine (71%). The major effect of the selenium-containing compounds was a reduction in the number of tumors per animal rather than a decrease in the number of animals bearing tumors. 2-(2-Hydroxyphenyl)SCA not only failed to reduce the numbers of tumors, it increased the number by 60% although with large variation, the change did not achieve statistical significance.
Table 1.
NNK-induced lung tumors with selenium compounds included in the diet over both pre- and post-initiation periods
| Lung Tumors | ||
|---|---|---|
| Incidence (%) | Number (mean ± sem) | |
| Dietary supplementation: | ||
| None | 100 | 10.9 ± 0.9 |
| Selenocystine | 100 | 3.2 ± 0.7a |
| 2-oxoselenazolidine-4-(R)-carboxylic acid | 93 | 4.7 ± 0.9a |
| 2-butylselenazolidine-4-(R)-carboxylic acid | 100 | 4.7 ± 0.8a |
| 2-cyclohexylselenazolidine-(4R)-carboxylic acid | 100 | 5.3 ± 1.0a |
| 2-phenylselenazolidine-4-(R)-carboxylic acid | 93 | 2.8 ± 0.5a |
| 2-(2′-hydroxyphenyl)selenazolidine-4-(R)-carboxylic acid | 100 | 17.4 ± 2.0 |
|
| ||
= significantly different from no selenium supplementation
Selenium compounds were included in the diet at 15 ppm for the days −28 to +112 relative to NNK administration (10 μmol/animal, ip) on day 0. Lungs were removed at day +112 and after fixation, lung tumors counted.
Post initiation selenium supplementation
When the selenocompound supplementation began 3 days after carcinogen administration (i.e., post-initiation), 2-oxoSCA, 2-butylSCA, 2-cyclohexylSCA as well as selenocystine were chemopreventive (Table 2). The total lung tissue RNA of the post-initiation supplementation regimen, but with intervention at day 13 (i.e., before tumor development) was subjected to gene expression analysis. Using PTM to identify genes that best corresponded to the efficacy of tumor reduction, that is, the degree to which the four effective agents reduced tumor numbers at four months (40, 44, 59 and 63% for 2-oxoSCA, 2-cyclohexylSCA, 2-butylSCA and selenocystine respectively) only six genes of the 41,534 elements on the Agilent whole mouse genome array matched that efficacy profile at an R2 value greater than 0.6 (Table 3) and these genes demonstrate rather modest changes in expression, generally considerably less than a 2-fold change (Figure 3A). None of these genes had been identified as influenced by two other chemopreventive selenium compounds in human lung cancer cells in culture [25,26]. Examination of these six genes whose profiles best matched the tumor reduction efficacy, using ONCOMINE [34] identified stathmin 1 (Stmn1) and procollagen c-endopeptidase enhancer (Pcolce) as genes that have demonstrated increased expression in human lung cancers. Elevated Stmn1 protein has also been identified in poorly differentiated lung tumors [35]. When broadened to include other tumors, only Stmn1 (also known as oncoprotein 18) has been significantly associated with multiple human cancers. In most cancers, Stmn1 expression increases with cancer progression and our data demonstrate a decrease in Stmn1 expression in the lungs of mice with selenazolidines in their diets. The other genes identified, display more complex patterns of expression among tumor types, increasing expression in some and decreasing expression in others. The expression of these genes appears to be modulated by the selenazolidines since no differences was found in gene expression between supplemented animals that also received NNK and those that did not. Therefore, the gene expression response appears to be independent of carcinogen treatment.
Table 2.
NNK-induced lung tumors with selenium compounds included in the diet before and after day +3 relative to NNK administration
| Supplementation period | ||||
|---|---|---|---|---|
| days -28 to +3 | days +3 to +112
(post-initiation) |
|||
| Dietary supplementation: | Tumor number (mean ±sem) | |||
| Experiment 1: | ||||
| None | 10.9 ± 0.9 | |||
| Selenocystine | 11.9 ± 1.6 | 4.0 ± 0.8a (86%) | ||
| 2-oxoselenazolidine-4-(R)-carboxylic acid | 9.9 ± 1.1 | 6.5 ± 1.2a (93%) | ||
| 2-butylselenazolidine-4-R)-carboxylic acid | 7.7 ± 1.1a (93%) | 4.5 ± 0.9a (93%) | ||
| 2-phenylselenazolidine-4-(R)-carboxylic acid | 4.2 ± 0.8a (93%) | 13.0 ± 2.2 | ||
| Experiment 2: | ||||
| None | 15.3 ± 1.5 | |||
| 2-cyclohexylselenazolidine-4-(R)-carboxylic acid | 17.4 ± 2.6 (93%) | 8.6 ± 1.7a | ||
| 2-(2′-hydroxyphenyl)selenazolidine-4-(R)-carboxylic acid | 14.6 ± 2.0 | 10.2 ± 1.5 | ||
|
| ||||
= significantly different from no selenium supplementation.
Selenium compounds were included in the diet at 15 ppm for the days -28 to +3 and days +3 to +112 relative to NNK administration (10 μmol/animal, ip) on day 0. Lungs were removed at day +112 and after fixation, lung tumors counted. Tumor incidence is indicated in parentheses if less than 100%
Table 3.
Gene transcripts that correspond (R2 > 0.6) to the four-selenocompound NNK-tumor reduction profile
| Gene | R value | p Value |
|---|---|---|
| Mus musculus stathmin 1 (Stmn1), mRNA [NM_019641] | 0.831 | 5.59E-06 |
| Mus musculus solute carrier family 7 (cationic amino acid transporter, y+ system), member 10 (Slc7a10), mRNA [NM_017394] | 0.819 | 1.02E-05 |
| Mus musculus luteinizing hormone beta (Lhb), mRNA [NM_008497] | 0.796 | 2.75E-05 |
| Mus musculus nucleoporin 133 (Nup133), mRNA [NM_172288] | -0.796 | 2.73E-05 |
| Mus musculus procollagen C-proteinase enhancer protein (Pcolce), mRNA [NM_008788] | 0.792 | 3.12E-05 |
| Mus musculus transmembrane protein 46 (Tmem46), mRNA [NM_145463] | 0.785 | 4.23E-05 |
Figure 3. Heat maps depicting the gene expression profiles of mice injected with NNK and provided with selenium-supplemented diets.
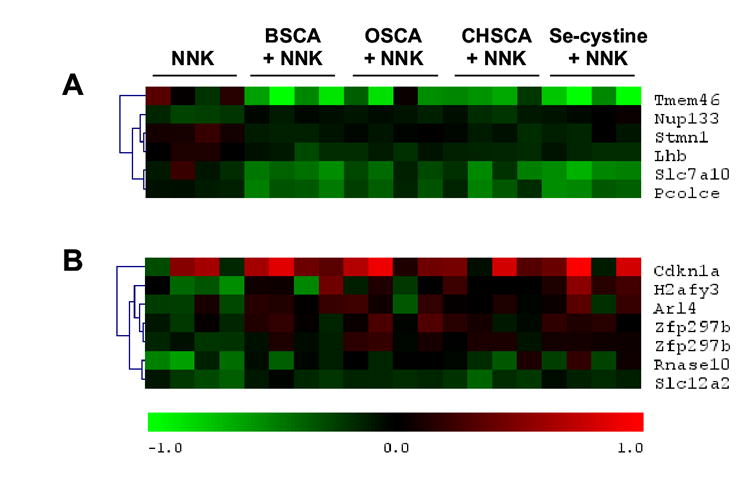
Treatment groups are indicated across the top; 2-butylSCA (BSCA), 2-oxoSCA (OSCA), 2-cyclohexylSCA (CHSCA), and selenocystine (Se-cystine). The accepted gene symbols are listed on the right and the genes are clustered such that genes with more similar profiles as measure by Euclidean distance in gene space are closer to each other. The scale for gene expression is in log2 and is shown at the bottom (the maximum and minimum display a 2-fold change). Panel A depicts the gene profiles that best matched the efficacy of tumor reduction of the selenocompounds. Panel B depicts those genes that best discriminate the selenocompounds treatments from the NNK treatment from among the selenocompound-responsive genes identified by others in lung tumor cell lines; methylseleninic acid responsive in H520 cells [25], 1,4-phenylenebis-(methylene)selenocyanate responsive in H460 cells [26].
When the gene expression changes of the present in vivo study were compared to the genes and related homologues identified in the previous cell culture studies (i.e., examination of 106 specific elements), the gene that best matched the efficacy profile was cyclin D2 (Ccnd2) with an R2 value of ~0.49. However, this gene displayed minimal expression variance among the samples (variance of ~0.013) and was in the lower 2/3 of expression variance among these 106 elements, and does not reliably distinguish selenium-supplemented from non-supplemented animals. To determine the correlation of any of the genes from the previous reports with the tumor reduction observed in this study, a two-class SAM was performed on these genes comparing animals injected with NNK alone to those injected with NNK and then placed on selenium enriched diets. A median false discover rate cutoff of ~10.8% found 6 genes that best discriminated the animals that received selenium supplementation compared to those that did not (Figure 3B), but most of these genes display only modest changes in expression and are poor predictors of selenium supplementation.
Pre-initiation selenium supplementation
In a third regimen, in which the supplementation period was dramatically shortened from the full duration regimen, (i.e., supplementation from day -28 to +3), selenocystine, 2-oxoSCA and 2-cycloSCA failed to significantly reduce tumor numbers (Table 2). However, 2-butylSCA and 2-phenylSCA were chemopreventive, reducing tumor numbers 29% and 61% respectively. In both of the selenium supplementation periods which were less than the full duration, again the major chemopreventive effect, where present, was a reduction in the number of tumors per animal, rather than a decrease in the number of animals bearing tumors.
Selenium supplementation effects on body weight increases
In the full duration, post- and pre-initiation supplementation regimens, reductions in body weight gains were observed for many of the selenium compounds (Table 4). 2-PhenylSCA caused the largest slowing of body weight gain, resulting in a 24-31% lower body weight (24% versus no NNK/no selenium supplementation, 31% versus +NNK/no selenium supplementation) with full duration (-28 to +112 day) supplementation. 2-(2′-hydroxyphenyl)SCA was the only selenium compound not showing slowed weight gain in the full duration regimen. For all compounds, the slowed weight gain was of lesser magnitude if the supplementation period covered only 109 days (+3 to +112) versus 140 (-28 to +112). For many compounds, supplementation for the period -28 to +3 days resulted in a reduced weight gain, even though there was no selenium supplementation during the subsequent 109 days.
Table 4.
Body weights in mice receiving NNK and selenium compounds in the diet
| Supplementation Period | |||
|---|---|---|---|
| -28 to +112 days | -28 to +3 days | +3 to +112 days | |
| Body weight (g) (mean ±sem) at +112 days | |||
| Dietary selenium component: | |||
| Selenocystine | 18.77 ± 0.77ab | 20.03 ± 0.61ab | 20.92 ± 0.34a |
| 2-oxoSCA | 20.33 ± 0.92ab | 22.02 ± 0.66a | 22.06 ± 0.79a |
| 2-butylSCA | 19.22 ± 0.72ab | 21.33 ± 0.46a | 22.05 ± 0.84a |
| 2-phenylSCA | 17.31 ± 1.20ab | 17.66 ± 0.95ab | 17.51 ± 1.20a |
| None | 24.95 ± 0.66b | 24.16 ± 0.61 | 24.16 ± 0.61 |
| 2-cyclohexylSCA | 21.74 ± 0.56a | 22.87 ± 0.69 | 21.28 ± 0.84a |
| 2-(2′-hydroxyphenyl)SCA | 22.83 ± 0.47 | 21.47 ± 0.56a | 23.95 ±0.46 |
|
| |||
= significantly different from no selenium supplementation (none).
b = significantly different from no selenium supplementation and without NNK administration (BW = 22.67 ± 1.50).
Selenium compounds were included in the diet at 15 ppm for the periods indicated and NNK was administered (10 μmol/animal, ip) on day 0. Animals were weighed on day +112 where NNK treatment = day 0.
Ultra-short selenium supplementation
Both the compounds identified as chemopreventive with -28 to +3 day supplementation, 2-butylSCA and 2-phenylSCA, were also effective when the supplementation period was reduced to a seven-day period terminating two days before NNK was administered, i.e., solely pre-initiation, (Table 5). When the ingestion period was adjusted to cover the time period immediately around NNK administration (-2 to +3 days), neither compound was effective in significantly reducing tumor number (Table 5).
Table 5.
NNK-induced lung tumors with 2-butyl- and 2-phenylselenazolidine-4-(R)-carboxylic acids included in the diet for abbreviated time periods prior to and during NNK administration
| Supplementation period | ||
|---|---|---|
| days -9 to -2
(pre-initiation) |
days -2 to +3 | |
| Dietary supplementation: | Tumor number (mean ±sem) | |
| None | 10.4 ± 1.2 | |
| 2-butylselenazolidine-4-(R)-carboxylic acid | 5.8 ± 1.0a | 8.6 ± 1.1 |
| 2-phenylselenazolidine-4-(R)-carboxylic acid | 6.2 ± 1.1a (93%) | 7.7 ± 1.1 |
|
| ||
= significantly different from no selenium supplementation.
Selenium compounds were included in the diet at 15 ppm for the days -9 to -2 and -2 to +3 relative to NNK administration (10 μmol/animal, ip) on day 0. Lungs were removed at day +112 and after fixation, lung tumors counted. Tumor incidence is indicated in parentheses if less than 100%.
DISCUSSION
Selenium in many chemical forms has been investigated as a chemopreventive against many chemically-induced tumors. Indications that selenium may provide a chemopreventive effect began to emerge 30-40 years ago [36]. A link between selenium and cancer also developed from ecological and mortality and disease data in the Linxian province of China, where a severe lack of micronutrients and a 100-fold excess of esophageal cancers and cancers of the gastric cardia compared to Caucasian Americans coexist, [37-39]. In the United States, selenium levels in forage crops inversely correlate with mortality from several types of cancer [40,41]. In a study in patients with histories of skin cancers, supplementation with selenized yeast showed benefits in overall cancer mortality and in prostate, colon, and lung cancer incidence [42]. Both animal [43] and human data [40] indicated that selenium doses needed for cancer chemoprevention were much higher than nutritional levels needed to maintain optimal antioxidant activity, (i.e., the levels of selenium necessary for discharging the effects of the element in supporting full and appropriate enzymatic activity of essential selenoenzymes [44]).
This study establishes that a range of 2-aryl/alkyl selenazolidine-4(R)-carboxylic acids at supranutritional dosing levels have chemopreventive activity against NNK-induced lung tumors in mice, that this property can manifest itself with much less than continuous administration in the diet, and that gene transcript changes seen with selenium compounds in lung tumor cell lines in continuous culture do not figure prominently with short-term selenocompound administration in intact lung tissue in vivo. With chemoprevention efficacy observable both with removal of selenazolidines from the diet 2 days prior to carcinogen (NNK) administration (Table 5), and with addition to the diet 3 days after carcinogen administration (Table 2), the selenazolidine chemopreventive properties appear related to both pre- and post-initiation events. For those supplementation regimens where selenazolidines are present when the carcinogen is administered, a direct effect on NNK metabolism could also be present, although from the absence of statistical significance to the reduction seen in a -2 to +3 day regimen for 2-butylSCA and 2-phenylSCA (Table 5), this contribution would likely to be only minor. The presence of chemoprevention activity in the -9 to -2 day pre-initiation regimen (Table 5) could also be an effect on NNK metabolism but if so it would be an indirect effect arising from an alteration in the transcription/translation of enzymes involved in NNK metabolism with a tipping of the balance of activation/inactivation enzyme activities towards inactivation. Alternatively, the pre-initiation efficacy could occur through a mechanism completely unrelated to NNK metabolism. The presence of chemoprevention efficacy when the selenazolidine administration is begun 3 days after the carcinogen (Table 2) indicates that for some selenazolidines, a mechanism that is unrelated to effects on NNK activation/inactivation enzyme activities is present in the post-initiation period. A similar NNK metabolism-unrelated mechanism has been observed for another chemopreventive organoselenium compound [23]. When 1,4-phenylenebis(methylene)selenocyanate (p-XSC) was added in the diet after NNK administration (15 ppm for the period +1 to +16 weeks) a 51% reduction in tumor numbers was observed. However, dietary p-XSC ingestion prior and during NNK administration (weeks -2 to +1 and -1 to +16) gave an even greater reduction in tumor numbers (78 and 76% respectively) [21,23], an observation which when linked with the ability of 7 days of prior p-XSC feeding to reduce the O6-methylguanine and 7-methylguanine residues present in lung tissue after NNK [43] suggests that p-XSC also elicits effects related to NNK metabolism. Whether it is due to a direct effect, an indirect effect, or a combination of both, on the enzymes responsible for the activation/inactivation of NNK has not been fully dissected. For the selenazolidines, the small, but statistically not significant tumor reduction seen with the -2 to +3 day supplementation regimen may represent a contribution of a direct effect on carcinogen metabolism, if 2 days is regarded as insufficient to establish any necessary enzyme activity changes. If a direct effect is present, the observation that the tumor reduction is much less than with the -28 to +3 regimen suggest that it only contributes partly to the chemopreventive activity, and enzyme activity changes established in a sustained pre-initiation period also play a role.
Thus, as with p-XSC, there may be mutliple mechanisms responsible for the chemoprevention activity of selenazolidines. It is also evident that all the selenazolidines do not possess the same combination of properties since in contrast to 2-butylSCA and 2-phenylSCA, 2-oxoSCA and 2-cyclohexylSCA were not effective in the -28 to +3 day regimen, and in contrast to 2-oxoSCA, 2-cyclohexylSCA and 2-butylSCA, 2-phenylSCA was not effective in the +3 to +112 day. Thus 2-butylSCA may be the only selenazolidine possessing both pre-initiation and post-initiation mechanisms of action. 2-PhenylSCA chemopreventive action appears to reside in the pre-inititation period while 2-oxoSCA and 2-cyclohexylSCA action appears to reside in the post-initiation period. Whether the differences among the selenazolidines are inherent to the parent compounds, or a consequence of the rate at which they are converted to selenocysteine in vivo remains for elucidation. Selenocystine chemoprevention activity appears to reside with a post-initiation mechanism of action (Table 2). From other studies, it is evident, albeit at lower doses, that selenomethionine, a selenoamino acid that is present in selenized yeast, possesses neither mechanism of action [45,20]. It should be noted that the contributions of pre-initiation and post-initiation effects cannot be discerned from regimens that provide continuous supplementation through both pre-carcinogen and post-carcinogen periods (as in Table 1), a frequent experimental design for chemopreventive compound evaluation.
All the chemoprevention efficacy observed above was with the selenium supplementation at a single and high level (15 ppm). It will remain for future investigations to determine to what extent this level can be titrated down while still retaining efficacy. The 15 ppm level did result in some retardation of body weight increases but this was not a necessary accompaniment for chemoprevention. This is apparent with both 2-oxoSCA and 2-phenylSCA where body weight gains were diminished similarly in the -28 to +3 and +3 to +112 day regimens with each compound, yet chemoprevention was not seen with the -28 to +3 regimen with 2-oxoSCA, or the +3 to +112 regimen with 2-phenylSCA.
The regimen used for the gene array analysis (+3 to harvest at +13) was designed to provide insight into the post-initiation mechanism of SCA and selenocystine chemoprevention. It was based on the hypothesis that the selenocompound reduction of NNK tumor numbers resulted from a counteracting of any NNK-induced gene transcription changes that result ultimately in the progression of initiated cells into tumors, and the extent to which this counteraction occurs would be proportional to the degree to which tumor numbers are ultimately reduced. This approach yields information not possible with in vitro studies that examine selenium compound gene transcription effects in professional lung cancer cell lines. It does, however suffer from a number of limitations not the least of which are the mixed population of cell types from which the RNA is extracted, not all of which may have been subject to NNK modification, and even within a single cell type, the percentage of cells affected may be very low. From the gene array results it was also evident that there was considerable inter-animal variation. Despite these limitations, six genes have been identified, five of which correlated with tumor reduction in a positive manner, that were not evident in studies with lung tumor cell lines. The corollary of this is that of the gene transcripts that had been subject to selenium compound modulation in tumor cells, only one of over a hundred matched the efficacy profile. In further array analysis (not shown), these gene changes were not confined to animals co-treated with NNK; they were also present in animals receiving the selenocompound in the absence of any NNK administration. In a reverse consideration from the foregoing, an examination of the gene transcripts that had been subject to selenium compound modulation in tumor cells, revealed that only one of over a hundred matched the efficacy profile.
Although this study has not identified the mechanism, it firmly establishes that 2-substituted selenazolidine-4(R)-carboxylic acids possess chemopreventive activity against NNK-induced lung tumors in a murine model. Dependent on the nature of the 2-substituent, the chemopreventive activity can arise from changes elicited in the post-initiation period, similar to selenocystine, or in the pre-initiation period. One selenazolidine, 2-butylSCA, demonstrated activity through both pre- and post-initiation events. Extension of these studies to other chemically induced tumors and in other organs will be needed to establish the utility of these compounds as generalized chemopreventive agents.
Acknowledgments
We gratefully acknowledge the technical assistance of Tenley Schofield, Jonathan Constance and Matthew Honeggar. This project was supported by a USPHS Grants GM 058913 (MRF) and CA 115616 (PJM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wynder EL. The past, present, and future of the prevention of lung cancer. Cancer Epidemiol Biomark Prev. 1998;7:735–748. [PubMed] [Google Scholar]
- 2.Hoffmann D, Hoffmann I, El-Bayoumy K. The less harmful cigarette: A controversial issue. A tribute to Ernst L. Wynder. Chem Res Toxicol. 2001;14:767–790. doi: 10.1021/tx000260u. [DOI] [PubMed] [Google Scholar]
- 3.Hecht SS. Cigarette smoking and lung cancer: Chemical mechanisms and approaches to prevention. Lancet Oncol. 2002;3:461–469. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 4.Hoffmann D, Djordjevic MV, Fan J, Zang E, Glynn T, Connolly GN. Five leading U.S. commercial brands of moist snuff in 1994; Assessment of carcinogenic N-nitrosamines. J Natl Cancer Inst. 1995;87:1862–1869. doi: 10.1093/jnci/87.24.1862. [DOI] [PubMed] [Google Scholar]
- 5.Murrah VA, Gilchrist EP, Moyer MP. Morphologic and growth effects of tobacco-associated chemical carcinogens and smokeless tobacco extracts on human oral epithelial cells in culture. Oral Surg Oral Med Oral Pathol. 1993;75:323–332. doi: 10.1016/0030-4220(93)90145-t. [DOI] [PubMed] [Google Scholar]
- 6.Brunnemann KD, Prokopczyk B, Kjordjevic MV, Hoffmann D. Formation and analysis of tobacco-specific N-nitrosamines. Crit Rev Toxicol. 1996;26:121–137. doi: 10.3109/10408449609017926. [DOI] [PubMed] [Google Scholar]
- 7.Adams JD, Lee SJ, Vinchkoski N, Castonguay A, Hoffmann D. On the formation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone during smoking. Cancer Lett. 1983;17:339–346. doi: 10.1016/0304-3835(83)90173-8. [DOI] [PubMed] [Google Scholar]
- 8.Chhabra SK, Souliotis VL, Kyrtopoulos SA, Anderson LM. Nitrosamines, alcohol, and gastrointestinal tract cancer: recent epidemiology and experimentation. In Vivo. 1996;10:265–284. [PubMed] [Google Scholar]
- 9.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 10.Peterson LA, Thomson NM, Crankshaw DL, Donaldson EE, Kenney PJ. Interactions between methylating and pyridyloxobutylating agents in A/J mouse lungs: implications for 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-Induced lung tumorigenesis. Cancer Res. 2001;61:5757–5763. [PubMed] [Google Scholar]
- 11.Fischer S, Spiegelhalder B, Eisenbarth J, Preussmann R. Investigations on the origin of tobacco-specific nitrosamines in mainstream smoke of cigarettes. Carcinogenesis. 1990;11:723–730. doi: 10.1093/carcin/11.5.723. [DOI] [PubMed] [Google Scholar]
- 12.Shimkin MB, Stoner GD. Lung tumors in mice: application to carcinogenesis bioassay. Adv Cancer Res. 1975;21:1–58. doi: 10.1016/s0065-230x(08)60970-7. [DOI] [PubMed] [Google Scholar]
- 13.Malkinson AM. Primary lung tumors in mice: an experimentally manipulable model of human adenocarcinoma. Cancer Res. 1992;52:2670s–2676s. [PubMed] [Google Scholar]
- 14.Ip C. Prophylaxis of mammary neoplasia by selenium supplementation in the initiation and promotion phases of chemical carcinogenesis. Cancer Res. 1981;41:4386–4390. [PubMed] [Google Scholar]
- 15.Ip C, Sinha D. Anticarcinogenic effect of selenium in rats treated with dimethylbenz[a]anthracene and fed different levels and types of fat. Carcinogenesis. 1981;2:435–438. doi: 10.1093/carcin/2.5.435. [DOI] [PubMed] [Google Scholar]
- 16.Medina D, Lane HW, Tracey CM. Selenium and mouse mammary tumorigenesis: an investigation of possible mechanisms. Cancer Res. 1983;43:2460s–2464s. [PubMed] [Google Scholar]
- 17.Ip C, White G. Mammary cancer chemoprevention by inorganic and organic selenium: single agent treatment or in combination with vitamin E and their effects on in vitro immune functions. Carcinogenesis. 1987;8:1763–1766. doi: 10.1093/carcin/8.12.1763. [DOI] [PubMed] [Google Scholar]
- 18.Ip C, Hayes C, Budnick RM, Ganther HE. Chemical form of selenium, critical metabolites, and cancer prevention. Cancer Res. 1991;51:595–600. [PubMed] [Google Scholar]
- 19.Nayini J, El-Bayoumy K, Sugie S, Cohen LA, Reddy BS. Chemoprevention of experimental mammary carcinogenesis by the synthetic organoselenium compound, benzylselenocyanate, in rats. Carcinogenesis. 1989;10:509–512. doi: 10.1093/carcin/10.3.509. [DOI] [PubMed] [Google Scholar]
- 20.Li L, Xie Y, El-Sayed WM, Szakacs JG, Franklin MR, Roberts JC. Chemopreventive activity of selenocysteine prodrugs against tobacco-derived nitrosamine (NNK) induced lung tumors in the A/J mouse. J Biochem Mol Toxicol. 2005;19:396–405. doi: 10.1002/jbt.20105. [DOI] [PubMed] [Google Scholar]
- 21.El-Bayoumy K, Upadhyaya P, Desai DH, Amin S, Hecht SS. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenicity in mouse lung by the synthetic organoselenium compound, 1,4phenylenebis(methylene)selenocyanate. Carcinogenesis. 1993;14:1111–1113. doi: 10.1093/carcin/14.6.1111. [DOI] [PubMed] [Google Scholar]
- 22.Lane HW, Teer P, Dukes J, Johnson J, White MT. The effect of four chemical forms of selenium on mammary tumor incidence in BALB/c female mice treated with 7-12-dimethylbenz[a]anthracene. Cancer Lett. 1990;50:39–44. doi: 10.1016/0304-3835(90)90176-x. [DOI] [PubMed] [Google Scholar]
- 23.Prokopczyk B, Amin S, Desai DH, Kurtzke C, Upadhyaya P, El-Bayoumy K. Effects of 1,4-phenylenebis(methylene)selenocyanate and selenomethionine on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced tumorigenesis in A/J mouse lung. Carcinogenesis. 1997;18:1855–1857. doi: 10.1093/carcin/18.9.1855. [DOI] [PubMed] [Google Scholar]
- 24.Nagasawa HT, Goon DJW, Zera RT, Yuzon DL. Prodrugs of L-cysteine as liver-protective agents. 2(RS)-methylthiazolidine-4(R)-carboxylic acid, a latent cysteine. J Med Chem. 1982;25:489–491. doi: 10.1021/jm00347a001. [DOI] [PubMed] [Google Scholar]
- 25.Swede H, Dong Y, Reid M, Marshall J, Ip C. Cell cycle arrest biomarkers in human lung cancer cells after treatment with selenium in culture. Cancer Epidemiol Biomark Prev. 2003;12:1248–1252. [PubMed] [Google Scholar]
- 26.El-Bayoumy K, Das A, Narayanan B, Narayanan N, Fiala ES, Desai D, Rao CV, Amin S, Sinha R. Molecular targets of the chemopreventive agent 1,4-phenylenebis(methylene) selenocyanate in human non small cell lung cancer. Carcinogenesis. 2006;27:1369–1376. doi: 10.1093/carcin/bgi328. [DOI] [PubMed] [Google Scholar]
- 27.Xie Y, Short MD, Cassidy PB, Roberts JC. Selenazolidines as novel organoselenium delivery agents. Bioorg Med Chem Lett. 2001;11:2911–2915. doi: 10.1016/s0960-894x(01)00590-x. [DOI] [PubMed] [Google Scholar]
- 28.Short MD, Xie Y, Li L, Cassidy PB, Roberts JC. Characteristics of selenazolidine prodrugs of selenocysteine: toxicity and glutathione peroxidase induction in V79 cells. J Med Chem. 2003;46:3308–3313. doi: 10.1021/jm020496q. [DOI] [PubMed] [Google Scholar]
- 29.Hecht SS, Morse MA, Amin S, Stoner GD, Jordan KG, Choi C-I, Chung F-L. Rapid single-dose model for lung tumor induction in A/J Mice by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and the effect of diet. Carcinogenesis. 1989;10:1901–1904. doi: 10.1093/carcin/10.10.1901. [DOI] [PubMed] [Google Scholar]
- 30.Hecht SS, Morse MA, Eklind KI, Chung FL. A/J mouse lung tumorigenesis by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and its inhibition by arylalkyl isothiocyanates. Exp Lung Res. 1991;17:501–511. doi: 10.3109/01902149109064435. [DOI] [PubMed] [Google Scholar]
- 31.Malkinson AM, Deer DS. Major effect on susceptibility to urethane-induced pulmonary adenoma by a single gene in BALB/cBy mice. J Natl Cancer Inst. 1983;70:931–936. [PubMed] [Google Scholar]
- 32.Pavlidis P, Noble WS. Analysis of strain and regional variation in gene expression in mouse brain. Genome Biol. 2001;2:RESEARCH0042. doi: 10.1186/gb-2001-2-10-research0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen G, Wang H, Gharib TG, Huang CC, Thomas DG, Shedden KA, Kuick R, Taylor JM, Taylor JM, Kardia SL, Misek DE, Giordano TJ, Iannettoni MD, Orringer MB, Hanash SM, Beer DG. Overexpression of oncoprotein 18 correlates with poor differentiation in lung adenocarcinomas. Mol Cell Proteomics. 2003;2:107–116. doi: 10.1074/mcp.M200055-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Shamberger RJ. Relationship of selenium to cancer. I. Inhibitory effect of selenium on carcinogenesis. J Natl Cancer Inst. 1970;44:931–936. [PubMed] [Google Scholar]
- 37.Blot WJ, Li JY, Taylor PR, Guo W, Dawsey S, Wang GQ, Yang CS, Zheng SF, Gail M, Li GY. Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J Natl Cancer Inst. 1993;85:1483–1492. doi: 10.1093/jnci/85.18.1483. [DOI] [PubMed] [Google Scholar]
- 38.Taylor EW, Nadimpalli RG, Ramanathan CS. Genomic structures of viral agents in relation to the biosynthesis of selenoproteins. Biol Trace Elem Res. 1997;56:63–91. doi: 10.1007/BF02778984. [DOI] [PubMed] [Google Scholar]
- 39.Mark SD, Qiao YL, Dawsey SM, Wu YP, Katki H, Gunter EW, Fraumeni JF, Jr, Blot WJ, Dong ZW, Taylor PR. Prospective study of serum selenium levels and incident esophageal and gastric cancers. J Natl Cancer Inst. 2000;92:1753–1763. doi: 10.1093/jnci/92.21.1753. [DOI] [PubMed] [Google Scholar]
- 40.Shamberger RJ, Willis CE. Selenium distribution and human cancer mortality. CRC Crit Rev Clin Lab Sci. 1971;2:211–221. doi: 10.3109/10408367109151308. [DOI] [PubMed] [Google Scholar]
- 41.Clark LC, Cantor KP, Allaway WH. Selenium in forage crops and cancer mortality in U.S. counties. Arch Environ Health. 1991;46:37–42. doi: 10.1080/00039896.1991.9937427. [DOI] [PubMed] [Google Scholar]
- 43.Clark LC, Combs GF, Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, Krongrad A, Lesher JL, Jr, Park HK, Sanders BB, Jr, Smith CL, Taylor JR. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276:1957–1963. [PubMed] [Google Scholar]
- 43.Combs GF, Jr, Gray WP. Chemopreventive agents: Selenium. Pharmacol Ther. 1998;79:179–192. doi: 10.1016/s0163-7258(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 44.Combs GF., Jr Current evidence and research needs to support a health claim for selenium and cancer prevention. J Nutr. 2005;135:343–347. doi: 10.1093/jn/135.2.343. [DOI] [PubMed] [Google Scholar]
- 45.Prokopczyk B, Cox JE, Upadhyaya P, Amin S, Desai D, Hoffmann D, El-Bayoumy K. Effects of dietary 1,4 phenylenebis(methylene)selenocyanate on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced DNA adduct formation in lung and liver of A/J mice and F344 rats. Carcinogenesis. 1996;17:749–753. doi: 10.1093/carcin/17.4.749. [DOI] [PubMed] [Google Scholar]