

Abstract
Adoptive transfer of Epstein Barr virus (EBV)–specific cytotoxic T-lymphocytes (EBV-CTLs) has shown that these cells persist in patients with EBV+ Hodgkin lymphoma (HD) to produce complete tumor responses. Treatment failure, however, occurs if a subpopulation of malignant cells in the tumor lacks or loses expression of EBV antigens. We have therefore determined whether we could prepare EBV-CTLs that retained the antitumor activity conferred by their native receptor while expressing a chimeric antigen receptor (CAR) specific for CD30, a molecule highly and consistently expressed on malignant Hodgkin Reed-Sternberg cells. We made a CD30CAR and were able to express it on 26% (± 11%) and 22% (± 5%) of EBV-CTLs generated from healthy donors and HD patients, respectively. These CD30CAR+ CTLs killed both autologous EBV+ cells through their native receptor and EBV−/CD30+ targets through their major histocompatibility complex (MHC)–unrestricted CAR. A subpopulation of activated T cells also express CD30, but the CD30CAR+ CTLs did not impair cellular immune responses, probably because normal T cells express lower levels of the target antigen. In a xenograft model, CD30CAR+ EBV-CTLs could be costimulated by EBV-infected cells and produce antitumor effects even against EBV−/CD30+ tumors. EBV-CTLs expressing both a native and a chimeric antigen receptor may therefore have added value for treatment of HD.
Introduction
Chemotherapy and radiotherapy cures more than 80% of patients with Hodgkin lymphoma (HD).1,2 However, a subset of patients have primary resistant disease or relapse even after high-dose chemotherapy and autologous stem-cell transplantation.3 Alternative therapeutic strategies are thus required to treat patients with resistant/relapsed disease, as well as to reduce the morbidity attributable to chemotherapy/radiotherapy.4
In almost 40% of HD patients, Hodgkin Reed-Sternberg (HRS) tumor cells express Epstein Barr virus (EBV)–associated antigens,5 and we have shown that the adoptive transfer of EBV-specific cytotoxic T cells (EBV-CTLs) is well tolerated and can induce disease responses including remission,6,7 so that in principle immunotherapy could be an alternative treatment for this disease. However, HD tumor cells lack expression of immunodominant EBV antigens, and only 3 weakly immunogenic antigens (EBNA1, LMP1, and LMP2) can be detected.5 In response to CTL therapy, subpopulations of EBV+ HD tumor cells may lack or lose expression of these weak antigens, allowing tumor escape and consequent treatment failure.
Almost all HRS cells overexpress the CD30 molecule, which is a member of the tumor necrosis factor family.8 Clinical trials of CD30 monoclonal antibodies (MAbs; conjugated with immunotoxins9,10 or radioisotopes11) produced modest clinical responses in patients with advanced/refractory HD. Treatment with monoclonal antibodies, however, is limited by the transience of their effects and by poor biodistribution in tumors.12 By contrast, T lymphocytes redirected to eliminate CD30+ tumor cells through the expression of a chimeric antigen receptor (CAR) specifically binding the CD30 molecule12,13 have the potential to generate a sustained antitumor effect.
Immunotherapy based on the transfer of redirected T cells is currently under investigation for a wide variety of tumors.14–18 Although preliminary clinical trials showed that redirected T cells can function in vivo, they do not expand or persist long term.19,20 Because incomplete activation of redirected T cells after engagement of the CAR contributes to the limited activity and persistence of redirected T cells, we and others have proposed grafting the CAR on antigen-specific T cells16,21 in order to provide them with costimulation from antigen-presenting cells (APCs) when their native T-cell receptor (TcR) is engaged. We chose EBV-specific CTLs as the vehicle for CARs, as most individuals are persistently infected with EBV and express viral antigens in epithelial cells and B lymphocytes.22 Hence, redirected T cells should receive appropriate costimulation for long-term persistence when they engage EBV-infected B cells with their native TcR, thereby increasing their antitumor activity mediated by engagement of their chimeric receptor. This strategy holds particular appeal for the treatment of EBV+ HD, because the administration of CAR+ CTLs with their native receptor specific for EBV antigens and their chimeric receptor specificity for CD30 should greatly reduce the risk of tumor escape due to EBV antigen or genome loss.
In the present study we explore the feasibility and value of redirecting EBV-CTLs to target CD30+ tumor cells, including HD tumor cells, ex vivo and in vivo. Because CD30 is also expressed on a subpopulation of activated T cells, we investigated whether these CD30-redirected EBV-CTLs would destroy T cells responding to unrelated immune stimuli and would thereby produce undesirable generalized immunosuppression.
Patients, materials, and methods
Tumor-cell lines
The HD-derived cell lines HDLM-2, L428, L540, and KM-H2 and the anaplastic large cell lymphoma (ALCL)–derived cell line Karpas-299 (all CD30+ and EBV−) were obtained from the German Collection of Cell Cultures (DMSZ, Braunschweig, Germany). Daudi, BJAB, Raji, HSB-2, and K562 were obtained from the American Type Culture Collection (ATCC; Rockville, MD). The SP-53 cell line was kindly provided by Dr Amin Hesham (M. D. Anderson Cancer Center). EBV-infected B-cell lines were generated as previously described.23,24 All cells were maintained in culture with RPMI-1640 medium (Hyclone, Logan, UT) containing 10% fetal bovine serum (FBS; Hyclone), 2 mM l-glutamine (GIBCO-BRL, Gaithersburg, MD). Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C.
Retroviral constructs
CD30ζ chimeric antigen receptor (CD30CAR).
As previously described,13 the CD30-specific single-chain Fv fragment was cloned in frame with the sequence encoding the human IgG1 CH2-CH3 domains and the transmembrane and cytoplasmic domain of the TcR receptor ζ chain. This construct was then subcloned into the SFG retroviral backbone.15 To produce the retroviral supernatant, 293T cells were cotransfected with Peg-Pam-e plasmid containing the sequence for MoMLV gag-pol and the DRF plasmid containing the sequence for the RD114 envelope,25 using the Fugene6 transfection reagent (Roche, Indianapolis, IN), according to the manufacturer's instructions.15,26 Supernatant containing the retrovirus was collected 48 and 72 hours later.
Luciferase vectors.
The generation of retrovirus vectors encoding Firefly Luciferase gene (FFLuc) or the fusion protein eGFP-Firefly Luciferase (eGFP-FFLuc) has previously been described.15 The FFLuc-specific vector, which also contains the puromycin resistance gene, was used for stable transduction of tumor-cell lines. To confirm transgene expression, 5 × 106 tumor cells were lysed and aliquots were diluted in 100 μL of D-luciferine according to the manufacturer's instructions (Promega, Madison, WI). Bioluminescence was measured in a luminometer (Monolight; BD Biosciences Pharmingen, San Diego, CA). The eGFP-FFLuc vector was used to transduce the EBV-CTLs. GFP expression by transduced cells was evaluated by fluorescence-activated cell sorter (FACS) analysis, whereas expression of FFLuc was detected using D-luciferine.
Preparation of CD30CAR EBV-CTLs
Generation and transduction of EBV-CTLs with CAR.
EBV-CTLs were generated from 8 EBV-seropositive healthy donors and from 4 patients with HD, as previously described.27 Established CTL lines, obtained after 3 stimulations with the autologous EBV-infected lymphoblastoid cell lines (LCLs), were transduced with retroviral supernatant. For transduction, 5 × 105 EBV-CTLs were plated per well in a retronectin-coated (FN CH-296; Takara, Otsu, Japan) 24-well plate in 0.5 mL complete media (RPMI1640 [Hyclone] 45%; Click's medium [Irvine Scientific, Santa Ana, CA] 45%; supplemented with 10% FBS and l-glutamine) containing recombinant human interleukin-2 (rhIL-2; 100 U/mL; Proleukine; Chiron, Emeryville, CA). Two milliliters of CD30CAR retroviral supernatant was added to each well. As negative controls, we used nontransduced (NT) EBV-CTLs and EBV-CTLs transduced with an irrelevant chimeric receptor (14g2aζCAR) directed against the “GD2” antigen expressed on neuroblastoma cells but absent on HD cells and EBV+ target cells.16 These 14g2aζCAR+ EBV-CTLs are referred to as control CTLs throughout the manuscript. Three days after transduction, cells were removed from the retronectin plates and expanded by weekly restimulation with LCLs in the presence of rhIL-2 (50-80 U/mL).
Phenotype.
The following MAbs conjugated with phycoerythrin (PE), fluorescein isothiocyanate (FITC), and/or periodin chlorophyll protein (PerCP) were used: CD3, CD4, CD8, CD19, CD16, CD56, CD28, and CD30 (all from BD Biosciences). Expression of the CAR on EBV-CTLs was detected using Cy-5–conjugated goat anti–human IgG (H + L) Abs (Jackson ImmunoResearch Laboratories, West Grove, PA), which recognize the human IgG1-CH2CH3 component incorporated within the CAR. Cells were analyzed by a FACSCAlibur (BD Biosciences) equipped with the filter set for 4 fluorescence signals. The antigen specificity of the CTL native receptors was evaluated with EBV-specific tetramers, as previously described.27 We chose tetramers based on donor HLA type, which recognized the following: (a) EBV peptides: EBNA3A, HLA-B8: QAKWRLQTL, HLA-B7: RPPIFIRLL; LMP2, HLA-A2: CLGGLLTMV; BZLF1, HLA-B8: RAKFKQLL (listed in Khanna and Burrows28; Houssaint et al29); (b) cytomegalovirus (CMV) peptides: HLA-A2: NLVPMVATV and HLA-B7: TPRVTGGGAM30; (c) adenoviral peptides: HLA-A1: TDLGQNLLY; HLA-A24: TYFSLNNKF; HLA-B7: KPYSGTAYNSL.31 Tetramers were prepared by the Baylor College of Medicine core facility. For each sample, a minimum of 100 000 cells were analyzed using a FACSCalibur with the CellQuest software (BD Biosciences).
TcR Vβ usage by EBV-CTLs was studied using the TcR Vβ repertoire kit (IOTest Beta Mark kit; Immunotech, Marseille, France) according to the manufacturer's instructions.
Evaluation of cytotoxic activity.
To determine whether CD30CAR+ EBV-CTLs were able to maintain specificity for autologous LCLs and kill EBV−/CD30+ targets, we used a standard 51Cr-release assay.27 As EBV−/CD30+ tumor-cell lines we used the HD cell lines HDLM-2, L428, L540, and KM-H2 and the ALCL cell line Karpas-299 (Table S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). As CD30− target cells we used Raji, Daudi, BJAB, or SP-53 tumor cells (all < 0.1% CD30+). Autologous phytohemoagglutinin (PHA)–stimulated peripheral blood mononuclear cells (PBMCs) were used as target cells to exclude autoreactivity and were generated by adding 5 μg/mL of PHA (Sigma, St Louis, MO) to PBMCs on day 0 and then rhIL-2 (100 U/mL) on days 3 and 6 (Table S1). In some experiments, to evaluate susceptibility of activated T cells to CAR+ CTL–mediated killing, autologous T blasts were transduced with a retroviral vector to stably overexpress the CD30 molecule. In some experiments, before labeling, target LCLs were depleted or enriched for CD30 expression using immunomagnetic beads (MACS; Miltenyi, Auburn, CA). To determine whether killing was restricted by HLA class I or class II major histocompatibility complex (MHC) molecules or was dependent on CD30, target cells were preincubated for 30 minutes either with the MAb W6/32 (Dako, Carpinteria, CA), which recognizes a monomorphic HLA class I determinant, or the monoclonal antibody CR3/43 (Dako), recognizing HLA-DR, -DP, and -DQ, or with the MAb BerH2 (Dako), which recognizes the CD30 molecule. Killing by CAR+ CTLs was also evaluated in the presence of serum collected from patients with advanced HD and tested for the presence of soluble CD30 using a specific enzyme-linked immunosorbent assay (ELISA) kit (Alexis, Axxora, San Diego, CA). Serum samples with greater than 80 U/mL soluble CD30 were used.
Coculture experiments.
To evaluate the ability of CD30CAR+ CTLs to completely eliminate CD30+ tumors, we cocultured 106 EBV-CTLs in 24-well plates in the presence of nonirradiated CD30+ tumor cells (HDLM-2, L428, L540, or Karpas-299, at 4:1 effector-to–target cell [E/T] ratio), all in the presence of rhIL-2 (50 U/mL). Parallel cocultures were plated with control CTLs. Phenotypic analyses were performed on days 5 and 8. T cells were detected using CD3-PerCP, LCLs with CD19-PE, and tumor cells with CD30-FITC MAbs (BD Biosciences).
Elispot.
The following EBV peptides were used for analysis of EBV-specific T-cell populations according to the donors' HLA specificity: EBNA3A, HLA-B8: QAKWRLQTL, FLRGRAYGL; HLA-B7: RPPIFIRLL; LMP2, HLA-A2: CLGGLLTMV; BZLF1, HLA-B8: RAKFKQLL; BMLF1, HLA-A2: GLCTLVAML (listed in Khanna and Burrows28; Houssaint et al29). For some experiments, the CMV peptides A2-NLV and B7-TPR were used. Peptides were synthesized by Genemed Synthesis (South San Francisco, CA). In this paper, the peptides are referred to by the first 3 amino acids as underlined. To evaluate the functionality of CAR+ EBV-CTLs, we measured reactivity to identical EBV-derived peptide epitopes using the interferon-γ Elispot assay.27 Briefly, CTLs were plated in triplicate and serially diluted from 5 × 104 to 5 × 102 cells/well and then 100 μL of appropriately diluted peptides (5 μM) or autologous, irradiated LCLs (105 cells) were added to the wells as positive controls. Negative controls included CTLs alone and CTLs loaded with irrelevant peptides.
Proliferation of CAR+ CTLs.
Control and CAR+ CTLs were labeled with 1.5 μM of carboxyfluoroscein succinimidyl ester (CFSE; Invitrogen, Carlsbad, CA) following the manufacturer's instructions and then plated at 106 EBV-CTLs in 24-well plates, in the presence of irradiated autologous EBV+ or CD30+ cells (at 4:1 E/T ratio), and with rhIL-2 (50 U/mL). On day 5, cells were labeled with CD8 PerCP and EBV+ tetramers–PE. Samples were analyzed by FACS and cell division was assessed by CSFE dilution.
Generation of cytomegalovirus (CMV)– or adenovirus (Ad)–specific T cells
Ad-specific T cells were generated from 3 Ad-seropositive healthy donors, as previously reported.31 Briefly, PBMCs rested overnight in serum-free media were infected with Ad5f35 adenoviral vector for 2 hours at a multiplicity of infection (MOI) of 200 at 37°C and then plated at 2 × 106/well in a 24-well plate. Similarly, CMV-specific T cells were reactivated from 4 CMV-positive healthy donors by infecting PBMCs with Ad5f35-pp65 vector at an MOI of 50 for 2 hours at 37°C and then plating the cells at 2 × 106/well in a 24-well plate. Autologous, nontransduced, control CAR or CD30CAR+ EBV-CTLs were added to these cultures at a 5:1 (PBMCs/CTL) ratio. The percentage of tetramer+ T cells binding to the hexon-tetramer or to the pp65-tetramer was evaluated by FACS analysis after 8 to 9 days of coculture. Using this method, adeno-specific or CMV-specific T cells could be reactivated in all the tested donors. The presence of transduced CTLs was demonstrated in the cocultures using antihuman IgG (H + L) Ab. To confirm the functionality of the reactivated T cells, the frequency of interferon (IFN)-γ–producing cells was determined using the IFN-γ Elispot assay against CMV- or adenoviral-derived peptides.
In vivo experiments
To assess the homing, persistence, and antitumor effects of CD30CAR+ EBV-CTLs in vivo, we used a severe combined immunodeficiency (SCID) mouse model and the IVIS imaging system (Xenogen, Alameda, CA).
Transduction of tumor cells and EBV-CTLs with luciferase vectors.
The CD30+ tumor-cell line L428 was transduced with the FFLuc vector on a retronectin-coated plate and then selected in puromycin (Sigma). CAR+ EBV-CTLs were first enriched to ensure CAR expression on greater than 90% of EBV-CTLs and then transduced on a retronectin-coated plate with the eGFP-FFluc vector. Phenotypic analysis confirmed that the GFP was expressed on CAR+ CTLs.
Mice.
Six- to 8-week-old CB17/SCID mice were purchased from Harlan-Sprague (Indianapolis, IN). All mouse experiments were performed in accordance with Baylor College of Medicine Animal Husbandry and Institutional Animal Care and Use Committee (IACUC) guidelines.
Efficacy of CD30CAR+ CTLs against EBV+ tumors.
To evaluate whether CAR+ EBV-CTLs retained their antitumor activity against the native EBV+ tumor (LCLs), we used SCID mice that were sublethally irradiated (230 cGy) and injected subcutaneously the following day with 107 autologous LCLs resuspended in matrigel.32 A third group of mice received a subcutaneous implant of HLA-mismatched LCLs. Ten to 15 days later, when a tumor was measurable (0.5-0.8 cm in maximum diameter), mice received 107 eGFP-FFluc–labeled CD30CAR+ EBV-CTLs or control CTLs intravenously. After CTL transfer, mice received 500 to 1000 U rhIL-2 intraperitoneally twice weekly. CTLs trafficking toward EBV+ tumors and their expansion at the tumor site were measured using the bioluminescence system. Thirty days after CTL transfer, mice were killed and evaluated for presence of EBV+ tumor.
To confirm that increased bioluminescence signals corresponded to increased T-cell numbers rather than exclusively to increased transgene expression in a fixed number of cells, we killed mice that showed increasing levels of bioluminescence. Cells from the tumor site were isolated and stained with antihuman CD45 and CD3 antibodies.
Antitumor activity against CD30+ HD tumors.
We used an intraperitoneal tumor xenograft model to test the efficacy of CAR+ CTLs against EBV−/CD30+ HD21 and to evaluate the effects of concomitant stimulation of the native TcR receptor on these CD30CAR+ EBV-CTLs using EBV-infected cells (LCLs). To assess the antitumor activity of CAR+ CTLs, CD30+ tumor cells labeled with FFLuc gene were injected intraperitoneally (5 × 106 cells) in sublethally irradiated (230 cGy) SCID mice. Five to 7 days later, the mice received 2 injections 4 days apart of either control or CD30CAR+ EBV-CTLs (107). rhIL-2 (500-1000 U) was administered intraperitoneally every other day. To test the effects of native TcR stimulation, a third group of mice received intraperitoneal injections of irradiated (40 Gy) autologous LCLs (4 × 106) twice/week for 2 weeks. Tumor growth was monitored twice a week by injecting mice intraperitoneally with D-luciferin (150 mg/kg). Photon emission was analyzed using the Xenogen-IVIS Imaging System. Briefly, a constant region-of-interest (ROI) was drawn over the tumor region and the intensity of the signal measured as total photon/s/cm2/steradian (p/s/cm2/sr) as previously validated.15
Statistical analysis
All in vitro data are presented as mean plus or minus the SD. Student t test was used to determine the statistical significance of differences between samples, and P value less than .05 was accepted as indicating a significant difference. For the bioluminescent experiments, intensity signals were log transformed and summarized using mean plus or minus standard deviation at baseline and multiple subsequent time points for each group of mice. Changes in intensity of signal from baseline at each time point were calculated and compared using paired t tests or Wilcoxon signed-ranks test.
Results
EBV-CTLs can express the CD30CAR while retaining their phenotype and native receptor function
The transduction efficiency of CD30CAR was 26% (± 11%; Figure 1A) in 8 different EBV-CTL lines from healthy donors. After transduction, EBV-CTLs were maintained in culture by weekly restimulation with autologous LCLs in the presence of rhIL-2. To ensure that CD30CAR-transduced CTLs retained the same phenotypic and functional characteristics as the control EBV-CTLs, we monitored their growth kinetics and immunophenotype for a minimum of 4 weeks. Expansion of CD30CAR+ CTLs was comparable to that of control CTLs (Figure 1B). In addition, expression of the CD30CAR was retained for the entire culture period (Figure 1C). The CTLs ceased to proliferate and progressively died when restimulation with LCLs and rhIL-2 was stopped, confirming that the presence of the CD30CAR did not alter the requirement for stimulation with antigen and exogenous cytokine and demonstrating that autonomous growth had not developed (data not shown).
Figure 1.

Growth kinetics, immunophenotype, and functionality of EBV-CTL lines are retained after transduction with CD30CAR. EBV-CTL lines were expanded from PBMCs obtained from 8 healthy EBV-seropositive donors by weekly stimulation with irradiated autologous LCLs and biweekly feeding with rhIL-2. EBV-CTLs were transduced with the CD30CAR or an irrelevant CAR (control CTLs) after the third stimulation. (A) CD30CAR expression was evaluated by flow cytometry using a goat anti–human IgG (H + L) Ab (—) or the isotype control (- - -). (B) The growth of EBV-CTLs was transduced with the CD30CAR (■) or an irrelevant CAR (□). The arrow indicates time of retroviral transduction. (C) The expression of CAR on transgenic CTLs over time is shown. The number of stimulations after transduction is indicated. Bars represent average (± SD) for the 8 donors. (D) The immunophenotype of the EBV-CTLs was transduced with the CD30CAR (■) compared with EBV-CTLs transduced with an irrelevant CAR (□). ▩ show the phenotype of the CD30CAR+ CTLs stimulated with CD30+ tumor cells. Means (± SD) are shown for the 8 donors. (E) CD30CAR can be detected on both CD8+-transduced (left plot) and CD4+-transduced (right plot) EBV-CTLs.
Transduction with CD30CAR did not significantly change the immunophenotype of the EBV-CTLs (CD3+/CD8+, 84% ± 11%; CD3+/CD4+, 11% ± 8%). The majority of control EBV-CTLs were CD3+/CD8+ T cells (84% ± 12%), with 12% (± 11%) of CD3+/CD4+ T cells. Less than 2% of the CTLs were CD3−/CD56+/CD16+ (Figure 1D). The costimulatory molecule CD28 was expressed on 86% (± 15%) of control CTLs and 78% (± 23%) of CD30CAR+ CTLs. The immunophenotype of transgenic CTLs as well as the expression of CD30CAR remained unchanged after stimulation with CD30+ HD tumor cells (Table 1; Figure 1D). Expression of CD30CAR was detected on both CD8 and CD4 CTLs (Figure 1E).
Table 1.
Evaluation of CAR receptor expression on EBV-CTL after stimulation with EBV+ and CD30+ tumor cells
| CD30CAR+ cells | Stimulation |
||
|---|---|---|---|
| Second | Third | Fourth | |
| After stimulation with EBV+ tumor cells | 19 ± 10 | 23.8 ± 5 | 16.8 ± 8 |
| After stimulation with CD30+ tumor cells | 18.2 ± 14 | 26.4 ± 10 | 21 ± 11 |
Data are percentages of CD30CAR+ cells. Evaluation was of CD30CAR expression on transduced CTLs after the indicated stimulation with autologous LCLs or CD30+ tumor cells. Analysis was performed by FACS after staining with antihuman IgG (H+L) Ab.
EBV-CTLs expressing the CD30CAR kill CD30+ target cells in an MHC-unrestricted manner
To discover whether transgenic expression of CD30CAR on EBV-CTLs would render them capable of killing CD30+ tumor cells, we used both a standard 5-hour 51Cr-release assay and a long-term coculture assay.
Cytotoxicity assay.
CTLs expressing CD30CAR lysed the MHC-mismatched CD30+ HD cell line HDLM-2 at a significantly higher rate (50% ± 20% at 20:1 E/T ratio) compared with control CTLs (12% ± 11%; P < .05; Figure 2A). The specificity of the CD30-mediated killing was demonstrated with antibody blocking experiments (Figure 2B right panel). When CD30+ targets were incubated with the CD30 MAb, lysis was significantly inhibited (13% ± 10% at 20:1 E/T ratio vs 46% ± 20% in the presence of the isotype control; P < .05). Monoclonal antibodies to HLA molecules had no effect. Hence tumor killing is MHC unrestricted and occurs through the CD30 receptor. The cytotoxic activity of CD30CAR+ CTLs was confirmed using additional CD30+ HD cell lines, including L428, L540, and KM-H2 (56% ± 28%, 57% ± 19%, and 58% ± 21% 51Cr release, respectively, at 20:1 E/T ratio) and the CD30+ Karpas-299 cell line (56% ± 23%; Figure 2A). In contrast, killing of CD30− targets (BJAB, Raji, SP-53, or Daudi) was low and comparable between CD30CAR+ and control CTLs (13% ± 9% and 10% ± 6%, respectively; Figure 2A). These data further confirm that killing of MHC-mismatched CD30+ target cells only occurs through engagement of the chimeric and not the native receptor (see also Figure S1A,B).
Figure 2.

CD30CAR-transduced EBV-CTLs specifically lyse CD30+ targets. (A) The results of a standard 51Cr-release assay of several CD30+ tumor-cell lines, at a CTL/tumor-cell ratio of 20:1, are shown. Bars represent the mean (± SD) of the EBV-CTLs generated from 8 donors and transduced with the CD30CAR (■) or an irrelevant CAR (□; P < .05). (B) Killing (shown is the percentage of lysis at 20:1 E/T ratio) of the CD30+ targets (■) by CD30CAR is inhibited by incubation with CD30 MAb (▒) but not by isotype control MAb (□) or by class I MHC MAb (▩), indicating that killing of CD30CAR is not MHC restricted. (*P < .05). Bars represent SD. (C) EBV-CTLs expressing the CD30CAR can eliminate CD30+ tumor cells in a long-term culture assay. EBV-CTLs obtained from healthy donors and transduced either with irrelevant CAR (left panels) or CD30CAR (right panels) were cocultured with the indicated CD30+ tumor-cell lines (ratio 5:1). After 5 to 7 days of culture, cells were collected and stained with CD3-PerCP and CD30-FITC to evaluate the growth of CD30+ tumor cells. No CD30+ cells were detectable after coculture with the CD30CAR+ EBV-CTLs, whereas CD30+ cells were detectable when tumor cells were cocultured with control CTLs. The phenotypes shown are representative of 4 performed experiments.
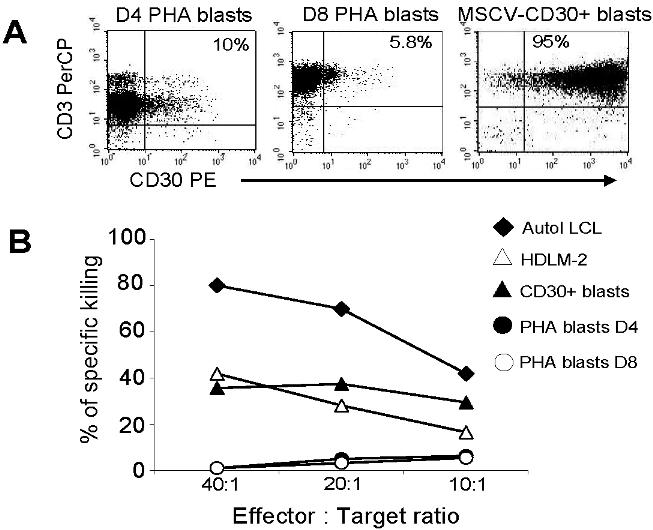
To exclude acquisition of autoreactivity by transduced CTLs, we tested their cytotoxicity against autologous PHA-activated blasts. These cells express negligible amounts of CD30 (4.5% ± 2%) by day 8 when they were used as target cells (Table S2). As expected, no significant lysis of autologous PHA blasts (< 10%, at 20:1 E/T ratio) was produced by nontransduced or CAR+ EBV-CTLs (Figure 2A; Figure S2). Both HSB-2 and K562 express the CD30 molecule, so a contribution from natural killer (NK) activity to the killing of these cells could not be ruled out, although NK cells represented less than 2% of the population, as assessed by immunophenotype (CD3−CD56/16+ were < 2%).
To determine whether killing mediated by CD30CAR was impaired by the presence of soluble CD30, we repeated the cytotoxicity assay in the presence of serum obtained from patients with advanced HD, in which we measured high levels of soluble CD30 molecule (> 80 U/mL). Using a 1:1 serum-medium ratio, killing of CD30+ targets by redirected CTLs was not significantly affected (lysis of HDLM-2 at 20:1 E/T ratio 74% ± 9% without serum vs 61% ± 8% with serum; data not shown). This result was expected, as the single chain for this CD30 molecule was previously reported not to be inhibited by the specific soluble molecule.13
Coculture experiments.
To evaluate the long-term ability of EBV-CTLs expressing the CD30CAR to eliminate CD30+ tumor cells, we cocultured 106 EBV-CTLs with nonirradiated CD30+ tumor cells (HDLM-2 or L428 or Karpas-299) in the presence of rhIL-2 (50 U/mL). Parallel cocultures were plated with control CTLs. Cells were collected after 5 to 8 days of culture and tumor cells were enumerated by CD30 staining and FACS analysis. In the presence of CD30CAR+ CTLs, we found complete elimination of CD30+ tumor (CD30+ cells < 1%), whereas tumor cells overgrew (CD30+ cells, 30%-45%) in cultures with control CTLs (Figure 2C).
EBV-CTLs continue to kill EBV-expressing targets through their native receptor
We also used the standard 5-hour 51Cr-release assay to confirm that transgenic expression of CD30CAR on EBV-CTLs did not reduce their ability to recognize the targets of their native receptors (ie, autologous EBV+ LCLs). As shown in Figure 3A, CTLs expressing CD30CAR, while able to kill CD30+ allogeneic tumor cells, retained their cytotoxic activity against autologous LCLs (65% ± 14%, at 20:1 E/T ratio). As LCLs express the CD30 molecule, these CD30CAR+ CTLs were able to kill MHC class I–mismatched LCLs (36% ± 20% at 20:1 E/T ratio; Figure 3B; Figure S1A,B). Such killing was not inhibited by MAbs to MHC class I molecules (51% ± 16% at 20:1 E/T ratio; Figure 3C). Hence, blocking interaction between the native TcR and its cognate antigen still allows MHC-independent killing by engagement of the CAR with the CD30 molecule on LCLs (Figure 3B left panel). Conversely, when target HLA-matched or autologous LCLs were incubated with MAbs to CD30, killing through the native TcR continued unabated.
Figure 3.

EBV-CTLs expressing the CD30CAR retain their ability to kill EBV+ tumor cells. (A)The mean (± SD) of 51Cr release from target cells exposed to EBV-CTLs from 8 donors transduced with an irrelevant CAR (left) and CD30CAR (right) is shown. CD30CAR+ CTLs lysed autologous LCLs (□) and the EBV−/CD30+ HD-derived cell line HDLM-2 (●; P < .05), whereas control CTLs showed significant lysis only of autologous LCLs. Autoreactivity was excluded by the absence of lysis of autologous PHA blasts (*). (B) CD30CAR+ CTLs (■) retain their killing activity against autologous LCLs and acquire the ability to kill allogeneic LCLs (* P < .05). As LCLs express CD30, this suggests that the observed lysis of allogeneic LCLs is mediated by the CAR. Bars indicate SD. Panel C shows that killing (at 20:1 E/T ratio) by CD30CAR+ CTLs of autologous LCLs (■) is not inhibited by incubation with class I MHC MAb (▩). This suggests that that killing of LCLs can still be mediated by the engagement of the CAR with the CD30 molecule expressed on LCLs. Bars indicate SD. (D) The expression of CD30 antigen on LCLs after depletion or enrichment for the CD30 molecule using immunomagnetic beads (MACS system) in 1 representative donor is shown. The percentage of specific 51Cr release at 40:1 E/T ratio of CTLs against CD30-depleted and CD30-selected autologous LCLs in 3 donors is shown in the bottom panel. Bars indicate SD.
To determine whether CD30CAR+ CTLs retained full activity mediated through their native receptor, we quantified EBV-mediated killing by labeling LCLs after depletion of CD30+ cells, using the MACS system. LCLs depleted for CD30 were then used as target cells in these 51Cr-release assays (Figure 3D). We observed that lysis of CD30− LCLs by CAR+ CTLs was comparable with that of control EBV-CTLs, suggesting that recognition and lysis of EBV-infected cells was retained after transduction (Figure 3D).
The generation of antigen-specific T cells is not impaired by the presence of EBV-CTLs expressing the CD30CAR
As CD30 is expressed by a subpopulation of activated T cells after exposure to viral antigens and mitogens,33 it is possible that CD30CAR+ CTLs would delete viral or other antigen-specific T cells during exposure to an infectious agent, with deleterious consequences for immunity. We therefore determined if cultured CD30CAR+ CTLs would display a reduced EBV-antigenic repertoire (self-destruction) or if they could inhibit the ability of cocultured primary T cells to respond to other antigenic stimuli, such as CMV and adenovirus (bystander destruction). We first measured CD30 up-regulation on T cells during our culture conditions. We collected PBMCs from 8 healthy EBV-seropositive donors and activated them with irradiated autologous LCLs. Expression of CD30 on CD3+ T cells was then evaluated daily by flow cytometry. CD30 was not detectable on resting T cells (< 1%) but was transiently up-regulated on a subpopulation of CD3+ T cells (14% ± 8%) between days 5 and 7 after the first stimulation with LCLs (Table 2). After the second and subsequent stimulations, CD30 continued to be expressed by only a minor fraction of CD3+ T cells in most of the donors (Table 2). In addition, we monitored the expression of CD30 on CD30CAR+ CTLs after antigenic stimulation and observed that expression remained low (Table 2). To confirm that lack of killing of activated T cells was due to their low expression of CD30 compared with HRS cells rather than to an inherent lack of sensitivity to CTLs, we used primary T cells expressing transgenic CD30 at levels comparable with HD tumor cell lines. As shown in Figure S3, these CD30+ T cells were lysed at a comparable level as HD lines. No killing of CD30+ blasts occurred with control CTLs.
Table 2.
Evaluation of CD30 expression on T cells during the generation of EBV-CTLs and on CD30CAR+ EBV-CTLs
| Stimulation |
||||
|---|---|---|---|---|
| First | Second | Third | Fourth or more | |
| CD3+ T cells/EBV-CTLs | 14 ± 8 | 5 ± 4 | 6 ± 6 | 3 ± 5 |
| CD30CAR+ EBV-CTLs | 3.7 ± 1.8 | 7.2 ± 5.5 | 7.3 ± 4.5 | 1.8 ± 0.4 |
Data are percentages of T cells. Evaluation of CD30 expression on CD3+ T cells/CTLs and on CD30CAR+ CTLs 5 to 7 days after the indicated stimulation with autologous LCLs. Analysis was performed by FACS after staining with CD30 PE-labeled MAb. CD30 expression on circulating T cells is less than 1%.
Antigenic repertoire of EBV-CTLs.
Because weekly restimulation with LCLs is used to expand transduced CTLs, and because this antigen-specific stimulation can induce up-regulation of CD30, we investigated the effects of CD30CAR expression by EBV-CTLs on their own EBV-specific TcR repertoire. We evaluated the frequency of CTLs responding to available HLA class I–restricted EBV peptides using specific tetramers before and after expression of CD30CAR. The antigenic repertoire of the EBV-CTLs did not change after transduction in any of the 5 donors tested. Instead, the percentage of tetramer+ CTLs recognizing lytic, immunodominant, and less immunogenic latent EBV antigens was comparable in CTLs expressing CD30CAR and CTLs expressing an irrelevant CAR (Figure 4A). CD30CAR+ EBV-CTLs were consistently detectable within the tetramer+ population (Figure 4B), further indicating that the reactivation of these cells was not impaired. To show that these tetramer+ CAR+ cells retained function, we monitored CFSE partitioning (and thus proliferation) after culture with EBV+ or CD30+ cells. As shown in Figure 4C, CFSE partitioning occurred in the CAR+ tetramer+ CTL population. Finally, we used interferon-γ Elispot assays to directly demonstrate that the EBV-CTLs transduced with the CD30CAR retained equal functional reactivity to EBV-derived peptide epitopes compared with control CTLs (Figure 4D).
Figure 4.

EBV-CTLs redirected with CD30CAR retain their polyclonal EBV specificity. (A)The frequencies of tetramers recognizing lytic (BZLF1-RAK) or latent (EBNA3C-RPP and LMP2-CLG) EBV-associated antigens in control and transgenic EBV-CTLs generated from 3 different donors are shown. The bottom panels show that the same frequency of EBV-specific tetramers is maintained after transduction with CD30CAR. (B) The CD30CAR is also detectable on tetramer+ CTLs. (C) CFSE-labeled control (left panels) and CD30CAR+ CTLs (right panels) nonstimulated (top panels), stimulated with EBV+ (middle panels), or stimulated with CD30+ cells (bottom panels) are shown. After LCL stimulation (middle panels), both control and CD30CAR+ EBV-tetramer+ CTLs proliferate, as shown by decrease of CFSE+ cells. After stimulation with CD30+ cells (bottom panels), only CAR+ CTLs proliferate. (D) The frequency (mean ±SD) of T cells responding to EBV-specific peptides in control and CD30CAR+ EBV-CTLs from a representative donor, assessed by IFN-γ Elispot assay, is shown. Tetramer and Elispot analyses are representative of a total of 5 donors.
To determine whether there were changes in CTL clonality after transduction, we analyzed the TcR-combinatorial diversity of control and EBV-CTLs expressing CD30CAR by monoclonal antibodies recognizing TcR-Vβ–specific regions. Redirected CTLs had a TcR-Vβ repertoire superimposable on that of nontransduced EBV-CTLs, with maintained heterogeneity and no specific overrepresentation or underrepresentation of any Vβ family (data not shown).
Reactivation of CMV- or Ad-specific T-cell responses is not affected.
We also determined if CD30CAR-grafted CTLs could affect the reactivation of bystander antigen-specific T-cell responses. We added CD30CAR+ EBV-CTLs to PBMCs, which were then stimulated to reactivate CMV- or Ad-specific CTLs. Virus-specific CTLs were successfully reactivated in all donors (Figure 5A; Table 3), even though CD30CAR+ CTLs persisted to the end of the coculture experiments (Figure 5B). These surviving antigen-specific T cells were also functional, because there was no change in the frequency of IFN-γ–responding T cells in cocultures containing CD30CAR+ CTLs (Figure 5C). In only 1 of the 7 donors (Figure 5D), the generation of Ad-specific T cells was impaired when CD30CAR+ EBV-CTLs were added to the culture. However, even in this donor, the generation of Ad-specific T cells was not completely abolished.
Figure 5.

Reactivation of viral-specific T cells is not impaired in the presence of EBV-CTLs expressing the CD30CAR. Autologous EBV-CTLs engineered to express the CD30CAR were added to cultures of PBMCs stimulated to reactivate CMV- or adenovirus-specific CTLs. (A) The percentage of pp65-tetramer+ T cells generated in a representative donor by day 9 of culture in the presence of nontransduced EBV-CTLs (top plot), EBV-CTLs transduced with an irrelevant CAR (middle plot), or the CD30 CAR+ (bottom plot) are shown. (B) Cells from the coculture were stained with the goat anti–human IgG (H + L) Ab to demonstrate the continued presence of the CD30CAR+ CTLs throughout the culture. As expected, no CAR+ CTLs were detectable in cocultures where NT CTLs were added. In contrast, 21% and 25% CAR+ CTLs were detectable at the end of the cocultures where irrelevant CAR or CD30CAR+ CTLs were added, respectively. (C) The IFN-γ–specific Elispot assay of coculture from 2 representative donors is shown. Mean frequency (± SD) of IFN-γ–producing T cells in response to the CMV-specific peptides NLV and TRP is shown. (D) The percentage of adeno-tetramer+ T cells (shown is the analysis with the 2 available tetramers) is shown in the only donor whose viral-specific response was reduced when CD30CAR+ EBV-CTLs were added to the culture (see also Table 3).
Table 3.
Effects of CD30CAR+ EBV-CTLs on reactivation of CMV- or Ad-specific T-cell responses
| Donor | Tetramers | Tetramer+ T cells in coculture |
||
|---|---|---|---|---|
| NT EBV-CTLs | Irrelevant CAR+ EBV-CTLs | CD30CAR+ EBV-CTLs | ||
| 1 | pp65 A2 NVL | 25.7 ± 5.2 | 31.9 ± 18.1 | 27.3 ± 8.7 |
| 2 | pp65 B7 TPR | 4.0 ± 1.9 | 3.3 ± 0.6 | 5.1 ± 2.1 |
| 3 | pp65 A2 NVL | 27.3 ± 19.9 | 28.9 ± 9.6 | 33.6 ± 8.5 |
| 4 | pp65 A2 NVL | 15.6 | 22.2 | 10.9 |
| 5 | Hexon A1 TDL | 8.5 | 3.4 ± 0.6 | 3.1 ± 0.9 |
| 5 | Hexon B7 KPY | 0.2 | 0.4 ± 0.5 | 0.2 ± 0.1 |
| 6 | Hexon A24 TYF | 9.6 ± 13.2 | 1.0 ± 0.5 | 0.8 ± 0.6 |
| 7 | Hexon A1 TDL | 15.1 ± 5 | 7.9 ± 5.9 | 2.7 ± 2.8 |
| 7 | Hexon A24 TYF | 4.6 ± 2 | 3.3 ± 3.4 | 1.0 ± 0.7 |
Data are percentages of tetramer+ T cells. Coculture experiments (tetramer+ T cells in coculture with NT EBV-CTLs, irrelevant CAR+ EBV-CTLs, or CD30CAR+ EBV-CTLs) repeated 3 times for each donor, except for donor no. 4 (done only once). The tetramer used is as follows: (a) CMV peptides: HLA-A2, NLVPMVATV and HLA-B7, TPRVTGGGAM; (b) adenoviral peptides: HLA-A1, TDLGQNLLY; HLA-A24, TYFSLNNKF; HLA-B7, KPYSGTAYNSL.31
NT EBV-CTL indicates nontransduced EBV-CTLs.
EBV-CTLs generated from patients with HD can be grafted with a functional CD30CAR
To ensure that the approach we describe would also be feasible using T cells from patients with active HD, we generated EBV-CTLs from 4 patients with relapsed HD and transduced them with the CD30CAR. As observed for healthy donors, EBV-CTLs were efficiently grafted with the CD30CAR, as 22% (± 5%) of EBV-CTLs stained with the antihuman IgG (H + L) Ab (Figure 6A). Transduction with CD30CAR did not result in significant modification of their immunophenotype, growth pattern, or EBV-antigen specificity compared with control EBV-CTLs (Figure 6B,C). As observed for CTLs from healthy donors, CD30CAR+ EBV-CTLs from HD patients became able to lyse CD30+ cell lines (killing of HDLM-2 was 43% ± 18% at 20:1 E/T ratio vs 2% ± 3% of control EBV-CTLs; P < .05) while retaining their ability to kill autologous LCLs (59% ± 17% vs 57% ± 9% of control EBV-CTLs; Figure 4D; Figure S1C,D).
Figure 6.

EBV-CTLs generated from patients with HD can be grafted with a functional CD30CAR. EBV-CTL lines were expanded from PBMCs of 4 patients with HD. (A) The expression of CD30CAR on 2 representative CTL lines by flow cytometry using a goat anti–human IgG (H + L) Ab (solid line) is shown. The dotted line shows the isotype control. (B) The immunophenotype of EBV-CTLs generated from these 4 HD patients and transduced with the CD30CAR (■) compared with EBV-CTLs transduced with an irrelevant CAR (□) is shown. Mean and SD are shown. (C) The frequency of tetramers recognizing the lytic (BZLF1-RAK) EBV-associated antigen in EBV-CTLs generated from 1 of these patients is shown. The bottom panels show that the same frequency of EBV-specific tetramers is maintained after transduction with CD30CAR and that the CD30CAR is also detectable on tetramer+ T cells. (D) The killing of LCLs and CD30+ tumor cell lines in a standard 51Cr-release assay at a CTL/tumor cell ratio of 20:1 is shown. Bars represent the mean plus or minus the SD of the EBV-CTLs transduced with the CD30CAR (■) or an irrelevant CAR (□). CD30CAR+ CTLs lysed both autologous LCLs and CD30+ target cells, whereas control CTLs showed significant lysis only of autologous LCLs.
Redirected EBV-CTLs home to native EBV+ tumors in vivo and subsequently expand
To demonstrate that CD30CAR+ EBV-CTLs maintain their functional activity in vivo, we used a mouse xenograft model.32 First, CTLs expressing the CD30CAR were FACS sorted to obtain a population of greater than 90% CAR+ cells (Figure 7A). These cells were then transduced with the eGFP-FFluc vector. As a control, we transduced NT EBV-CTLs with the eGFP-FFLuc vector (Figure 7A). Both GFP-FFluc–labeled NT and CAR+ EBV-CTLs were then injected intravenously in sublethally irradiated mice bearing subcutaneous autologous LCL tumor and their homing and expansion were monitored by in vivo imaging. Mice received 500 to 1000 U of rhIL-2 intraperitoneally twice weekly. As shown in Figure 7B, by day 7 after CTL transfer, both NT and CD30CAR+ EBV-CTLs localized at the tumor site. In addition, the bioluminescence signal from mice that received transduced or control CTLs increased over the following 2 weeks, confirming their ability to expand in response to the native antigen (Figure 7B,C). As expected, the expansion of EBV-CTLs at the site of EBV+ HLA-mismatched LCL tumors was significantly reduced (P = .05; Figure 7C), presumptively due to lack of stimulation through the (MHC restricted) native antigen receptor.34 We confirmed that the increase in bioluminescence correlated with an increase in T-cell numbers rather than simply an increase in transgene expression by a fixed number of cells using FACS analysis quantification of CD3+ cells at tumor sites in mice with different levels of bioluminescence (Figure S4). The expansion of both control and CD30CAR+ CTLs at the tumor site was associated with improved antitumor activity. At 30 days after CTL transfer, mice were evaluated for presence of EBV+ tumor, and 2 (29%) of 7 mice that received control CTLs and 3 (30%) of 9 mice that received CD30CAR+ CTLs were tumor free compared with 0 (0%) of 6 control mice. These findings are in line with previous observations by Lacerda et al,32 who used a similar model to evaluate the efficacy of EBV-CTL adoptive transfer in a SCID mouse model. These results imply that CD30CAR+ EBV-CTLs can receive adequate costimulation from autologous EBV+ B cells not just in vitro but in vivo as well.
Figure 7.

CD30CAR+ EBV-CTLs can control tumor growth in vivo while retaining their ability to migrate to EBV+ tumor and expand. To evaluate in vivo homing, NT EBV-CTLs or CTLs transduced with CD30CAR and sorted for transgene expression were injected intravenously in SCID mice implanted subcutaneously with autologous LCLs.32 Both NT and CAR+ EBV-CTLs were labeled with the eGFP-FFLuc gene to monitor their trafficking and expansion using an in vivo imaging system (Xenogen-IVIS Imaging System). (A) Circa 50% of CD30CAR+ EBV-CTLs, as assessed by the goat anti–human IgG (H + L) Ab, are expressing the FFLuc transgene as GFP+. (B) The signal of EBV-CTLs is localized to the EBV+ tumors and is elevated in mice receiving either control (top panels) or CD30CAR+ EBV-CTLs (bottom panels). (C) The bioluminescence fold expansion of CTLs at the tumor site is shown. To evaluate the contribution of costimulation21 by EBV antigen, EBV-CTLs transduced with irrelevant CAR or CD30CAR were injected intraperitoneally in SCID mice bearing EBV−/CD30+ L428 tumor that was transgenic for FFLuc. EBV-CTLs were transferred 7 days after tumor implant. Tumor growth was monitored using the in vivo imaging system. (D) By 7 days after CTL infusion, tumor growth measured as maximum photon/s/cm2/sr (p/s/cm2/sr) was significantly greater in mice receiving control CTLs (top panels) compared with mice receiving CD30CAR+ EBV-CTLs (middle panels). Persistence of tumor control can be observed in mice receiving CD30CAR+ EBV-CTLs and intraperitoneal injection of irradiated EBV-infected cells, which thus provide the appropriate costimulation (bottom panels). Panel E illustrates the results of 6 mice per group implanted with the CD30+ L428 cell line. Bars represent average of light emission (± SD) (P < .05).
CD30CAR-expressing EBV-CTLs have antitumor activity in vivo against EBV−/CD30+ tumors
To assess whether CD30CAR+ EBV-CTLs can modify the growth of an established EBV−/CD30+ tumor and to evaluate the costimulatory effects of EBV-infected cells on the antitumor activity of CAR+ CTLs, we used an intraperitoneal SCID xenograft model.21 Sublethally irradiated SCID mice were implanted with FFLuc-labeled CD30+ tumor cells (L428) in the peritoneum. Light emission was monitored as an indication of tumor growth. Once progressive increase of bioluminescence occurred (usually 7 days after tumor injection), mice received control EBV-CTLs or CD30CAR+ EBV-CTLs intraperitoneally followed by intraperitoneal rhIL-2 on alternate days. To evaluate costimulation from EBV-infected cells, a group of mice also received irradiated autologous EBV+ LCLs intraperitoneally twice weekly for 2 weeks. After adoptive transfer, we observed a reduction of light emission in mice treated with CD30CAR+ EBV-CTLs (Figure 7D,E), indicating control of tumor growth for more than 2 weeks. In addition, the control of tumor growth was sustained for up to 4 weeks in mice that also received costimulation from autologous EBV-infected cells (Figure 7D,E). In contrast, photon emissions, and thus tumor size, increased in mice receiving control EBV-CTLs, regardless of costimulation from autologous EBV-infected cells (Figure 7D,E).
Discussion
Adoptive transfer of viral antigen–specific CTLs has had clinical value in patients with EBV+ HD.6,7 To increase the activity of these cells and reduce the potential for tumor escape by target antigen down-regulation, we have modified these viral antigen–specific cells so that they express a transgenic artificial receptor specific for the HD-associated antigen CD30.8 We have shown that EBV-CTLs expressing this CD30-specific receptor retain their ability to kill EBV+ tumors and acquire the ability to recognize and kill CD30+ HD tumor cells in vitro and in a SCID mouse model in vivo. The redirected EBV-CTLs also retained their phenotype and polyclonal specificity for EBV-derived antigens. Moreover, T cells from HD patients, which may have a low endogenous level of the ζ chain,6 were also able to produce EBV-CTLs that were able to express functional levels of CD30CAR, confirming the feasibility of our approach for patients with this disease. When injected in vivo, the redirected EBV-CTLs retained the ability to migrate to EBV+ tumors, to expand locally in response to EBV antigens through their native TcR, and to kill CD30+ tumor cells through their chimeric receptor, suggesting that they will receive sufficient and appropriate costimulation from persistently infected normal and malignant host cells to be able to expand and survive long term in humans with HD.
Although CAR-targeting CD30 and other antigens can readily be expressed in primary T cells, which will then kill the relevant tumor targets in vitro,13 it has become evident from clinical trials that CAR-expressing primary T cells have limited persistence in vivo after adoptive transfer.19,20 This lack of persistence has been attributed to incomplete stimulation after engagement of the CAR,19,20 a phenomenon particularly problematic when tumors that lack expression of costimulatory molecules are targeted. To date, the primary strategy to overcome the limitations of these first-generation CARs has been to incorporate additional costimulatory endodomains in the chimeric receptor.15,35–38 In a range of studies, both in vitro and in vivo, the addition of such endodomains improves CAR+ T-cell function, increasing proliferation, cytokine secretion, and cytotoxic effector activity.15,21,35 While promising, this approach has the limitation that it can never precisely mimic the physiologic sequence of costimulatory events that follow engagement of the native antigen receptor. It is now clear that optimum recruitment of an immune response requires several discrete families of costimulatory molecules to be used in sequence. Moreover, recruitment of each class of effector cells (for example, CD4 helper versus CD8 cytotoxic) has a preferential requirement for a different costimulatory receptor-ligand interaction.
Because of these inherent limitations to chimeric receptor manipulation, a second strategy for improving efficacy has been investigated. Instead of expressing the CAR in an undefined population of primary T cells, it is possible to transduce cultured cytotoxic T-lymphocyte lines with specificity for antigens known to be expressed in vivo on professional APCs. Through an appropriate sequence of costimulation, these APCs produce effective and sustained immune responses. In this way, chimeric T cells receive appropriate costimulatory signals in the correct sequence when they engage their native receptors and are thus better able to persist and kill their tumor targets when they engage their chimeric receptor. A number of preclinical studies have suggested the benefit of this overall concept.16,21 We chose to investigate the value of expressing CD30CAR in EBV-CTLs because the lifelong expression of EBV antigens in infected individuals ensures the long-term persistence of infused CTLs and because EBV-CTLs have been shown to traffic to and destroy EBV-expressing malignancies.23,39 We reasoned that EBV-CTLs expressing the chimeric receptor would retain these desirable characteristics. Moreover, because the tumor cells from almost half of the patients with HD also express EBV antigens themselves, tumor cells could be targeted by both native and chimeric receptors on a single T cell, potentially enhancing effectiveness and reducing the risk of a tumor escape by antigen modulation/mutation.40 By using redirected EBV-CTLs, costimulation will be provided locally by EBV-infected HRS cells in EBV+ HD tumors and systemically by the EBV-infected memory B cells that persist lifelong in lymphoid tissues.
We chose to target CD30 because this antigen is constitutively expressed on virtually all HRS cells both at diagnosis and at relapse.8 We used a derived antibody rather than an alpha-beta TcR as the basis for the synthetic receptor to minimize issues of competition for receptor formation41 and to allow binding even to malignant target cells in which the antigen processing machinery had become impaired.40 The CD30 antigen is not detected on cells of the peripheral blood or on resting lymphocytes, but it is present on a subpopulation of physiologically activated T cells and on thymic medulla.33 Expansion of an anti-CD30–specific CTL population could therefore be self-defeating, with autodestruction as the CTLs became activated or the destruction of T cells responding to other infectious antigens. Our experiments demonstrated that these hypothetical concerns were not realized in practice, because CD30CAR+ EBV-CTLs do not lose their own target antigen specificities nor do they impede reactivation of CMV- and adenovirus-specific CTLs. Preservation of virus-specific CTL reactivation may relate to the weak expression of CD30 on activated T cells, which is probably insufficient for killing by CD30CAR effector cells. Alternatively, CD30 may be present on only a minor subset of activated T cells, which is nonessential to an effective immune response. This latter explanation is favored by studies in CD30 knockout mice, in which the number of circulating B and T lymphocytes, and the function of their immune system, is largely unimpaired.42 Although clinical trials will be required to confirm the safety of this strategy, we believe that discrimination between levels of CD30 expression on physiologically activated T cells and on HRS tumor cells will occur in vivo and that activated T cells will be spared by the CD30CAR+ EBV-CTLs that can kill tumor targets. As far as expression of CD30 on thymic medulla is concerned, impaired negative selection of T cells has been described in CD30 knockout mice.42 However, this process is minimal in adults. Expression of CD30 on other nonhematopoietic cells is limited to malignancies such as embryonal carcinomas and some mesenchymal tumors.42
Our clinical trials of adoptive transfer of EBV-CTLs for therapy of posttransplantation lymphoproliferative disease (PTLD) and EBV+ HD demonstrated that infused EBV-CTLs localized to the tumor and expanded.7,23,39 Moreover, although these cells lacked (central) memory markers CCR7 and CD62L (data not shown), they persisted for longer than 8 years and could be expanded by appropriate antigenic stimuli, thereby ensuring both early and long-term control of disease.6,7,23,39 While the pathway of differentiation of human T cells into the memory compartment remains ill defined, it seems likely that even terminally differentiated effector cells can nonetheless revert to effector memory.43 This observation appears to hold both for EBV-specific7,23,39 and melanoma antigen (MART-1)–specific effector cells.43 Our SCID mouse xenograft model indicated that we can anticipate that the CAR+ CTLs will behave in a similar manner, because the phenotype of these cells and their homing ability to EBV+ cells was identical to that of nontransduced EBV-CTLs. Prolonged T-cell culture may reduce the efficacy of T-cell therapy,44 but our approach uses a polyclonal CTL population that has been validated for its ability to expand, persist, and function in vivo.7,23,39
CD30 is also expressed by EBV-negative HRS cells, so that in principle our approach could be extended to all HD, regardless of the presence of EBV antigens. However, EBV-negative HD, unlike EBV+ disease, does not produce chemokines for which CTLs express receptors.45,46 Hence optimal effects on EBV-negative HD will probably require the CD30CAR+ CTLs to be further modified with transgenic chemokine receptors to ensure adequate accumulation at tumor sites.47
In conclusion, the adoptive transfer of EBV-CTLs grafted with a CAR targeting the CD30 molecule may enhance immunotherapy of patients with EBV+ HD. Acquisition of antitumor effect against CD30+ tumor cells should be benefited by local and systemic stimulation of the native receptor by EBV-infected B cells, without harm to the function of the immune system as a whole.
Supplementary Material
Acknowledgments
We are grateful for the persistent and reliable support of Tatiana Gotsolva and Fernando Jimenez of the Flow Cytometry Core Facility (Texas Children's Hospital, Houston, TX) for FACS analyses. We thank Martin Pule, MD (University College London, London, United Kingdom) and Elio Vanin, PhD (Children's Memorial Research Center, Chicago, IL) for assistance in vector production.
This work was supported by National Institute of Allergy and Infectious Diseases (NIAID) R21 (B.S.), a Doris Duke Distinguished Clinical Scientist Award (H.E.H.), National Institutes of Health (NIH) CA61384 (C.M.R.), American Italian Cancer Foundation (A.D.), Leukemia Lymphoma Translational Research (G.D.) and Doris Duke Clinical Scientist Development Award (G.D.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: B.S. designed and performed in vitro and in vivo experiments, analyzed the data, and wrote the manuscript. C.M.R., H.E.H., and M.K.B. designed experiments and edited the manuscript. A.D. and L.Z. performed many of the in vitro experiments. H.A. and A.H. provided vital reagents for the research. A.E.F. performed many of the in vivo the experiments. G.D. designed experiments, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Barbara Savoldo, MD, Center for Cell and Gene Therapy, Baylor College of Medicine, 6621 Fannin St, MC 3-3320, Houston, TX 77030; e-mail: bsavoldo@bcm.tmc.edu.
References
- 1.Yung L, Linch D. Hodgkin's lymphoma. Lancet. 2003;361:943–951. doi: 10.1016/S0140-6736(03)12777-8. [DOI] [PubMed] [Google Scholar]
- 2.Aleman BM, Raemaekers JM, Tirelli U, et al. Involved-field radiotherapy for advanced Hodgkin's lymphoma. N Engl J Med. 2003;348:2396–2406. doi: 10.1056/NEJMoa022628. [DOI] [PubMed] [Google Scholar]
- 3.Diehl V, Franklin J, Pfreundschuh M, et al. Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin's disease. N Engl J Med. 2003;348:2386–2395. doi: 10.1056/NEJMoa022473. [DOI] [PubMed] [Google Scholar]
- 4.Ng AK, Mauch PM. Late complications of therapy of Hodgkin's disease: prevention and management. Curr Hematol Rep. 2004;3:27–33. [PubMed] [Google Scholar]
- 5.Wu TC, Mann RB, Charache P, et al. Detection of EBV gene expression in Reed-Sternberg cells of Hodgkin's disease. Int J Cancer. 1990;46:801–804. doi: 10.1002/ijc.2910460509. [DOI] [PubMed] [Google Scholar]
- 6.Roskrow MA, Suzuki N, Gan Y, et al. Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the treatment of patients with EBV-positive relapsed Hodgkin's disease. Blood. 1998;91:2925–2934. [PubMed] [Google Scholar]
- 7.Bollard CM, Aguilar L, Straathof KC, et al. Cytotoxic T lymphocyte therapy for Epstein-Barr virus+ Hodgkin's disease. J Exp Med. 2004;200:1623–1633. doi: 10.1084/jem.20040890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falini B, Pileri S, Pizzolo G, et al. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 1995;85:1–14. [PubMed] [Google Scholar]
- 9.Falini B, Bolognesi A, Flenghi L, et al. Response of refractory Hodgkin's disease to monoclonal anti-CD30 immunotoxin. Lancet. 1992;339:1195–1196. doi: 10.1016/0140-6736(92)91135-u. [DOI] [PubMed] [Google Scholar]
- 10.Schnell R, Staak O, Borchmann P, et al. A phase I study with an anti-CD30 ricin A-chain immunotoxin (Ki-4.dgA) in patients with refractory CD30+ Hodgkin's and non-Hodgkin's lymphoma. Clin Cancer Res. 2002;8:1779–1786. [PubMed] [Google Scholar]
- 11.Schnell R, Dietlein M, Staak JO, et al. Treatment of refractory Hodgkin's lymphoma patients with an iodine-131-labeled murine anti-CD30 monoclonal antibody. J Clin Oncol. 2005;23:4669–4678. doi: 10.1200/JCO.2005.09.098. [DOI] [PubMed] [Google Scholar]
- 12.Abken H, Hombach A, Reinhold U, Ferrone S. Can combined T-cell- and antibody-based immunotherapy outsmart tumor cells? Immunol Today. 1998;19:2–5. doi: 10.1016/s0167-5699(97)01191-2. [DOI] [PubMed] [Google Scholar]
- 13.Hombach A, Heuser C, Sircar R, et al. An anti-CD30 chimeric receptor that mediates CD3-zeta-independent T-cell activation against Hodgkin's lymphoma cells in the presence of soluble CD30. Cancer Res. 1998;58:1116–1119. [PubMed] [Google Scholar]
- 14.Dotti G, Heslop HE. Current status of genetic modification of T cells for cancer treatment. Cytotherapy. 2005;7:262–272. doi: 10.1080/14653240510027217. [DOI] [PubMed] [Google Scholar]
- 15.Vera J, Savoldo B, Vigouroux S, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B-lymphocyte derived malignant cells. Blood. 2006;108:3890–3897. doi: 10.1182/blood-2006-04-017061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. 2002;99:2009–2016. doi: 10.1182/blood.v99.6.2009. [DOI] [PubMed] [Google Scholar]
- 17.Lamers CH, Sleijfer S, Vulto AG, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 18.Cooper LJ, Topp MS, Serrano LM, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–1644. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 19.Walker RE, Bechtel CM, Natarajan V, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96:467–474. [PubMed] [Google Scholar]
- 20.Mitsuyasu RT, Anton PA, Deeks SG, et al. Prolonged survival and tissue trafficking after adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96:785–793. [PubMed] [Google Scholar]
- 21.Cooper LJ, Al Kadhimi Z, Serrano LM, et al. Enhanced antilymphoma efficacy of CD19-redirected influenza MP1-specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood. 2005;105:1622–1631. doi: 10.1182/blood-2004-03-1208. [DOI] [PubMed] [Google Scholar]
- 22.Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol. 2001;1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- 23.Rooney CM, Smith CA, Ng CY, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92:1549–1555. [PubMed] [Google Scholar]
- 24.Savoldo B, Goss J, Liu Z, et al. Generation of autologous Epstein-Barr virus-specific cytotoxic T cells for adoptive immunotherapy in solid organ transplant recipients. Transplantation. 2001;72:1078–1086. doi: 10.1097/00007890-200109270-00017. [DOI] [PubMed] [Google Scholar]
- 25.Kelly PF, Carrington J, Nathwani A, Vanin EF. RD114-pseudotyped oncoretroviral vectors. Biological and physical properties. Ann N Y Acad Sci. 2001;938:262–276. [PubMed] [Google Scholar]
- 26.Dotti G, Savoldo B, Pule M, et al. Human cytotoxic T lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood. 2005;105:4677–4684. doi: 10.1182/blood-2004-08-3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savoldo B, Goss JA, Hammer MM, et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs). Blood. 2006;108:2942–2949. doi: 10.1182/blood-2006-05-021782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khanna R, Burrows SR. Role of cytotoxic T lymphocytes in Epstein-Barr virus-associated diseases. Annu Rev Microbiol. 2000;54:19–48. doi: 10.1146/annurev.micro.54.1.19. [DOI] [PubMed] [Google Scholar]
- 29.Houssaint E, Saulquin X, Scotet E, Bonneville M. Immunodominant CD8 T cell response to Epstein-Barr virus. Biomed Pharmacother. 2001;55:373–380. doi: 10.1016/s0753-3322(01)00082-8. [DOI] [PubMed] [Google Scholar]
- 30.Sili U, Huls MH, Davis AR, et al. Large-scale expansion of dendritic cell-primed polyclonal human cytotoxic T-lymphocyte lines using lymphoblastoid cell lines for adoptive immunotherapy. J Immunother. 2003;26:241–256. doi: 10.1097/00002371-200305000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Leen AM, Sili U, Savoldo B, et al. Fiber-modified adenoviruses generate subgroup cross-reactive, adenovirus-specific cytotoxic T lymphocytes for therapeutic applications. Blood. 2004;103:1011–1019. doi: 10.1182/blood-2003-07-2449. [DOI] [PubMed] [Google Scholar]
- 32.Lacerda JF, Ladanyi M, Louie DC, et al. Human Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes home preferentially to and induce selective regressions of autologous EBV-induced B cell lymphoproliferations in xenografted C.B-17 scid/scid mice. J Exp Med. 1996;183:1215–1228. doi: 10.1084/jem.183.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Croft M. Costimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 34.Koehne G, Doubrovin M, Doubrovina E, et al. Serial in vivo imaging of the targeted migration of human HSV-TK-transduced antigen-specific lymphocytes. Nat Biotechnol. 2003;21:405–413. doi: 10.1038/nbt805. [DOI] [PubMed] [Google Scholar]
- 35.Pulè MA, Straathof KC, Dotti G, et al. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–941. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 36.Brentjens RJ, Latouche JB, Santos E, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9:279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 37.Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161:2791–2797. [PubMed] [Google Scholar]
- 38.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 39.Heslop HE, Ng CY, Li C, et al. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2:551–555. doi: 10.1038/nm0596-551. [DOI] [PubMed] [Google Scholar]
- 40.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 41.Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol. 2002;2:512–519. doi: 10.1038/nri841. [DOI] [PubMed] [Google Scholar]
- 42.Amakawa R, Hakem A, Kundig TM, et al. Impaired negative selection of T cells in Hodgkin's disease antigen CD30-deficient mice. Cell. 1996;84:551–562. doi: 10.1016/s0092-8674(00)81031-4. [DOI] [PubMed] [Google Scholar]
- 43.Powell DJ, Jr, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105:241–250. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maggio EM, van den BA, Visser L, et al. Common and differential chemokine expression patterns in rs cells of NLP, EBV positive and negative classical Hodgkin lymphomas. Int J Cancer. 2002;99:665–672. doi: 10.1002/ijc.10399. [DOI] [PubMed] [Google Scholar]
- 46.van den Berg A, Visser L, Poppema S. High expression of the CC chemokine TARC in Reed-Sternberg cells: a possible explanation for the characteristic T-cell infiltratein Hodgkin's lymphoma. Am J Pathol. 1999;154:1685–1691. doi: 10.1016/S0002-9440(10)65424-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Stasi A, Dotti G, Foster A, et al. Improved homing of antigen-specific T cells to Hodgkins Disease (HD) tumor cells by forced expression of CCR4 receptor [abstract]. Blood. 2006:143e. Abstract 472. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}