

Abstract
Binding of Zn2+ has been shown previously to inhibit the ubiquinol cytochrome c oxidoreductase (cyt bc1 complex). X-ray diffraction data in Zn-treated crystals of the avian cyt bc1 complex identified two binding sites located close to the catalytic Qo site of the enzyme. One of them (Zn01) might interfere with the egress of protons from the Qo site to the aqueous phase. Using Zn K-edge x-ray absorption fine-structure spectroscopy, we report here on the local structure of Zn2+ bound stoichiometrically to noncrystallized cyt bc1 complexes. We performed a comparative x-ray absorption fine-structure spectroscopy study by examining avian, bovine, and bacterial enzymes. A large number of putative clusters, built by combining information from first-shell analysis and metalloprotein databases, were fitted to the experimental spectra by using ab initio simulations. This procedure led us to identify the binding clusters with high levels of confidence. In both the avian and bovine enzyme, a tetrahedral ligand cluster formed by two His, one Lys, and one carboxylic residue was found, and this ligand attribution fit the crystallographic Zn01 location of the avian enzyme. In the chicken enzyme, the ligands were the His121, His268, Lys270, and Asp253 residues, and in the homologous bovine enzyme they were the His121, His267, Lys269, and Asp254 residues. Zn2+ bound to the bacterial cyt bc1 complex exhibited quite different spectral features, consistent with a coordination number of 6. The best-fit octahedral cluster was formed by one His, two carboxylic acids, one Gln or Asn residue, and two water molecules. It was interesting that by aligning the crystallographic structures of the bacterial and avian enzymes, this group of residues was found located in the region homologous to that of the Zn01 site. This cluster included the His276, Asp278, Glu295, and Asn279 residues of the cyt b subunit. The conserved location of the Zn2+ binding sites at the entrance of the putative proton release pathways, and the presence of His residues point to a common mechanism of inhibition. As previously shown for the photosynthetic bacterial reaction center, zinc would compete with protons for binding to the His residues, thus impairing their function as proton donors/acceptors.
INTRODUCTION
The cytochrome (cyt) bc1 complexes are multisubunit integral membrane proteins that play a central role in the respiratory and photosynthetic electron-transfer chains of prokaryotic and eukaryotic organisms (see (1–3) for comprehensive reviews). They catalyze the transfer of electrons from a hydroquinone derivative (QH2) to a soluble electron carrier (a c-type cyt). This electron transfer is coupled to the pumping of protons across the energy-transducing membrane, and the resulting electrochemical potential of protons drives the synthesis of ATP via a chemiosmotic circuit. Several x-ray diffraction (XRD) structures of the mitochondrial cyt bc1 complex have been described during the last decade (4–9). These structures were supplemented recently by the XRD structure of the simpler bacterial cyt bc1 complex at 3.8-Å (10) and 3.2-Å resolution (11). The catalytic core of all cyt bc1 complexes comprises four redox-active metal centers: one heme c, bound to the cyt c1 subunit, one Fe2S2 center in the Rieske iron-sulfur protein (ISP), and two b-type hemes (bL and bH) bound to a single cyt b subunit. According to the modified Q-cycle mechanism (12), redox-coupled H+ translocation by the cyt bc1 complex involves two catalytic sites facing the two opposite sides of the energy-transducing membrane: the Qo site, at which QH2 oxidation is coupled to proton release, and the Qi site, where quinone (Q) reduction is coupled to H+ uptake. A key feature of the Q-cycle is the bifurcation of the electron-transfer chain at the Qo site: upon QH2 oxidation, one electron is delivered to the high-potential chain, reducing in sequence the Fe2S2 center and cyt c1; the second electron is transferred to a Q or a semiquinone (SQ) at the Qi site via the two low-potential hemes of cyt b. Electron transfer to cyt c1 involves a large movement of the ISP head domain (6,13). Reduction of the ISP by QH2 occurs when it is in a conformation docked onto cyt b, placing the Fe2S2 cluster in contact with the Qo site. A subsequent movement of the ISP head domain toward cyt c1 facilitates electron transfer to the latter redox partner. Although the availability of XRD structures has greatly contributed to the understanding and better definition of the catalytic mechanism, the molecular details of QH2 oxidation at the Qo site and the associated proton transfer events remain unclear (13–20).
Inhibitors of the cyt bc1 complex have proven to be powerful tools in elucidating several aspects of the catalytic mechanism of this enzyme. Most of these inhibitors are structural Q-analogs, and their interactions with the Qo or Qi sites have been extensively characterized at both functional and structural levels (4,6,21,22). Divalent metal ions are a distinct class of inhibitors of the cyt bc1 complexes (23–25). These ions also inhibit the catalytic cycle of other bioenergetically important proton-translocating proteins, like cytochrome c oxidase (26,27) and the bacterial photosynthetic reaction center (RC) (28,29). Recently, these inhibitors have received increased attention in view of their possible common mechanism of action in proton-translocating enzymes.
In the RC from Rhodobacter sphaeroides, stoichiometric binding of Zn2+ or Cd2+ inhibits proton transfer to the secondary photoreduced Q acceptor, QB (29). A high-affinity binding site for Zn2+ and Cd2+ has been located by XRD at the cytoplasmic surface of the RC (30). The Zn2+-ligand cluster is formed by the imidazole side chains of two histidines (HisH126 and HisH128) and by the side chain of an aspartic acid (AspH124) of the RC H-subunit. It has also been proposed that a water molecule interacts with Zn2+. This tetrahedral coordination geometry has been confirmed and refined by a subsequent extended x-ray-absorption fine-structure (EXAFS) study (31). Cd2+ binding also involves the same cluster of residues, and possibly three water molecules in an octahedral geometry (30). By examining the pH dependence of Cd2+ binding in native and mutated RCs, it has been shown clearly that inhibition of proton transfer by the metal ion is due to competition with protons for binding to HisH126 and HisH128 of the H-subunit, thus hampering the function of these residues as proton donors/acceptors along the proton pathway to the QB site (32). As a consequence, the structural definition of the Zn binding site has made it possible to determine the entry point of H+ and contributed to the complete definition of the proton pathway from the aqueous phase to the acceptor QB molecule (33).
This mechanism of Zn inhibition, i.e., competitive block of an entry or exit proton pathway, has been suggested to be responsible for Zn inhibitory effects observed in other redox enzymes, including the cyt bc1 complex (32). If this is correct, then location of Zn-binding sites and resolution of their local structure could become powerful tools to trace proton pathways in an entire class of membrane proteins.
Zn ions were first reported by Skulachev et al. (23) to inhibit mitochondrial respiration in micromolar concentrations. Subsequent studies identified the cyt bc1 complex as the primary target of Zn inhibition (34,35), and were aimed at localizing the binding site within the bovine-heart mitochondrial cyt bc1 complex (24). A radioactive binding assay revealed one high-affinity binding site (KD = 10−7 M at pH 7) and 3–4 additional low-affinity binding sites (KD > 2 × 10−6 M) (25). Inhibition by Zn2+ was found to be noncompetitive with respect to cyt c or QH2, as well as with respect to inhibitors of the Qo site. This indicated that the mechanism of Zn inhibition was different from that of other known cyt bc1 complex inhibitors (25). By studying the pH dependence of Zn inhibition, Link and von Jagow (25) concluded that Zn2+ binds close to a protonatable group of the enzyme, with pKa = 7.2, and proposed that zinc inhibits the enzyme activity by interfering with the pathway of protons released from the Qo site.
The effects of Zn2+ on the rates of specific electron-transfer steps of the cyt bc1 complex were investigated recently in chromatophores from the photosynthetic bacterium Rhodobacter capsulatus (36). Zn2+ was found to decelerate the flash-induced reduction of cyt c1, oxidation of cyt b, and generation of a transmembrane voltage. These observations have been tentatively explained by proposing that Zn2+ binds close to the Qo site, blocking the proton release channel(s).
Berry et al. (37) have located two Zn2+ binding sites in the chicken cyt bc1 complex by analysis of XRD data at 3.65 Å resolution from crystals treated with Zn2+. One binding site (Zn01) is located in the hydrophilic interface between cytochromes b and c1. Potential ligands include His121 of cyt c1, as well as the Asp253, Glu255, and His268 of cyt b. The other site (Zn02) is in a hydrophobic channel between the Qo site and the bulk lipid phase. Like the bovine cyt bc1 complex, the chicken enzyme is also inhibited by Zn2+, although the avian enzyme exhibits a lower affinity for this metal ion compared to its bovine counterpart (37). It has been tentatively proposed that Zn01 is homologous to the high-affinity site characterized by Link and von Jagow in the bovine enzyme (25), and that binding of Zn2+ at Zn01 interferes with the release of protons from the Qo site to the aqueous medium (37). From XRD data analysis, however, the coordination geometry of Zn01 cannot be assigned unambiguously. A considerable uncertainty exists, in particular, as to the effective involvement of His268 or Glu255. Coordination of Zn02 appears to be even more uncertain.
The identification of Zn01 with the high-affinity binding site characterized in the bovine cyt bc1 complex, and the determination of its local structure, would greatly help in elucidating the inhibition mechanism. X-ray absorption fine structure (XAFS) spectroscopy is an ideal tool for probing the coordination of a metal ion in a protein, particularly when a structural model based on XRD is available. XAFS can provide structural information at subatomic resolution (<0.2 Å) for metal clusters in a protein, in both crystalline and solution states (see, e.g., Hasnain and Hodgson (38)).
Guided by these considerations, we undertook XAFS spectroscopy studies of Zn2+-binding sites in noncrystallized purified cyt bc1 complexes treated with zinc ions in stoichiometric amounts. In this work, we have performed a comparative XAFS study by examining the avian cyt bc1 complex (for which XRD-Zn locations are available (37)), the bovine enzyme (in which Zn binding and inhibition have been extensively investigated (25)), and the bacterial cyt bc1 complex purified from Rb. capsulatus. In the latter system, Zn inhibition has been characterized in vivo using single-turnover experiments (36), and this system is readily amenable to future mutational studies. We therefore thought that comparison of XAFS data obtained using enzymes from different organisms (for which complementary information is available) could better reveal a possibly common structural basis of zinc inhibition.
We report here that for the bovine and avian cyt bc1 complexes, XAFS data define unambiguously the local structure of a tetrahedral site consistent with the XRD location of Zn01 in the avian complex. Moreover, we show that, although an octahedral coordination geometry is observed for the homologous bacterial, the corresponding binding cluster fit a site that is structurally similar to that seen with the avian and bovine enzymes, suggesting that the mechanism of proton egress from the Qo site of the cyt bc1 complexes is highly conserved among species.
MATERIALS AND METHODS
Sample preparation
The cyt bc1 complex was purified from chicken essentially as described in (6). Purification of the cyt bc1 complex from Rb. capsulatus was described in detail in (39). The cyt bc1 complex from bovine heart mitochondria was prepared as in (24). The concentration of the avian cyt bc1 complex was estimated spectrophotometrically using an extinction coefficient of 60 mM−1 cm−1 for the dithionite-reduced form at 558 nm versus 600 nm; analogous procedures were used for the bovine and bacterial enzymes, using extinction coefficients of 70 mM−1 cm−1 (at 562 nm versus 600 nm) and 59 mM−1 cm−1 (at 560 nm versus 600 nm), respectively (40, 41). To obtain Zn-cyt bc1 complexes, the avian and bovine enzymes were incubated at a concentration of 80 μM in 20 mM glycylglycine, pH 7.5, 0.01% dodecylmaltoside supplemented with ZnSO4 at a molar ratio of 0.9 Zn/bc1. Incubation was performed in a final volume of 1.2 ml for 15 h on ice. The same procedure was used for the bacterial enzyme, except that the final incubation volume was 3.4 ml and the protein concentration was 28 μM. A slightly substoichiometric Zn/bc1 ratio was chosen to maximize the occupancy of high-affinity binding site(s) while minimizing the possibility that additional lower-affinity sites are populated. Following incubation, samples were applied to a Sephadex G-25 column (PD10, Pharmacia, Peapack, NJ) and eluted with the same buffer without zinc. Zn stoichiometries were redetermined on the basis of the Zn and Fe content of the samples, measured by inductively coupled plasma-atomic emission spectroscopy (ICP-AES). The concentration of cyt bc1 complexes was estimated from the Fe content assuming five Fe atoms per complex (two for cyt b hemes, one for the cyt c1 heme, and two for the Fe2S2 center). ICP-AES analysis yielded the following Zn/bc1 stoichiometries: 0.79 ± 0.07, 0.80 ± 0.03 and 0.94 ± 0.03 for the bacterial, avian, and bovine enzymes, respectively. Stigmatellin was added to all samples from a 10-mM stock solution in ethanol, at a ratio of two molecules per cyt bc1 complex.
For XAFS measurements, the Zn-cyt bc1 suspensions described above were supplemented with 2.5% w/v polyvinyl alcohol (PVA) (Fluka, Buchs, Switzerland, molecular weight ∼130,000) and dehydrated under N2 flow. Following this procedure, Zn-bc1 complexes are embedded at high concentration in dry PVA films, yielding samples that have the additional advantage of stability and ease of handling (31). After performing XAFS measurements, the portion of each PVA-Zn-cyt bc1 complex film exposed to x-rays was redissolved, and diluted for spectrophotometric measurements. Essentially, the same spectra were observed in the Zn-cyt bc1 complex suspensions before incorporation into PVA matrices and after XAFS measurements.
X-ray absorption fine structure
In the x-ray range, the smooth decrease of the absorption coefficient with energy is interrupted by sharp discontinuities (absorption edges). Above these edges, the absorption coefficient of molecular and condensed matter exhibits a series of relatively weak oscillations, named x-ray absorption fine structure (XAFS). These modulations are caused by an interference phenomenon between the primary photoelectron, generated by the absorption of the x-ray photon, and the electron waves back-scattered from the neighboring atoms (for a general review, see, e.g., Rehr and Albers (42)).
The EXAFS function 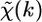 describes the oscillatory part of the signal, defined as the relative deviation of the measured absorption coefficient, μ, with respect to the atomic absorption coefficient, μ0, of the edge under consideration:
describes the oscillatory part of the signal, defined as the relative deviation of the measured absorption coefficient, μ, with respect to the atomic absorption coefficient, μ0, of the edge under consideration:
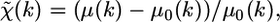 |
(1) |
where
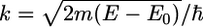 |
(2) |
is the wave number of the photoelectron, m its mass, and E0 the edge energy, i.e., the origin of its kinetic energy.
Adopting four main approximations (one electron approximation, a dipole approximation, a muffin-tin approximation for the scattering potentials, and a Gaussian radial distribution function), it has been demonstrated (43) that at sufficiently high energies above the edge (∼50 eV), 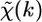 can be written as a sum of terms
can be written as a sum of terms 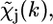 each relative to a particular scattering process (or path), of the form
each relative to a particular scattering process (or path), of the form
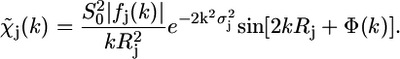 |
(3) |
Equation 3 contains physical quantities that can be calculated ab initio and structural parameters that can be extracted from the experimental spectrum. The physical quantities are the amplitude reduction factor, 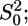 the effective scattering amplitude,
the effective scattering amplitude, 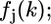 and the scattering phase shift,
and the scattering phase shift, 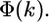 The structural parameters are the half path length, Rj, and the relative mean-square displacement (RMSD), or Debye-Waller (DW) factor of the path,
The structural parameters are the half path length, Rj, and the relative mean-square displacement (RMSD), or Debye-Waller (DW) factor of the path, 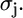 In the near-edge region, characterized by the so called x-ray absorption near-edge structure (XANES), it is not possible to express
In the near-edge region, characterized by the so called x-ray absorption near-edge structure (XANES), it is not possible to express 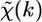 as a sum of terms given by Eq. 3. Instead, data analysis can be aided by simulations in the full multiple scattering regime (44).
as a sum of terms given by Eq. 3. Instead, data analysis can be aided by simulations in the full multiple scattering regime (44).
Data collection
Zn K-edge measurements were performed at the BM 8 GILDA beam-line of the European Synchrotron Radiation Facility in Grenoble, France. A Si(311) double-crystal monochromator employing dynamical sagittal focusing (45) was used; the photon flux was of the order of 1010 photons/s and the spot size was ∼1 × 1 mm2. Data were collected using a 13-element hyperpure Ge detector equipped with fast digital electronics with a peaking time equal to 1 μs (46). Samples were measured at room temperature in the region 9500–10,660 eV. For each sample we collected a minimum of three spectra to monitor possible modifications caused by the exposure to x-ray flux. Such modifications were not detected. The final spectra were obtained from the average of multiple scans, for a total integration time of 80 s/point for the avian, 60 s/point for the bovine, and 40 s/point for the bacterial cyt bc1 complexes.
Data analysis
EXAFS analysis was performed in three steps: background (i.e., atomic absorption cross section) subtraction, simulation of theoretical signals, and multiparameter fitting. The EXAFS oscillations were extracted from the raw data using the AUTOBK program (47) as implemented in the ATHENA package (48), using a linear function for the pre-edge region and a cubic spline to mimic the atomic background. Theoretical amplitude and phase-shift functions were calculated using the ab initio code FEFF 8.2. The 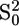 value was calculated by FEFF 8.2 from atomic overlap integrals of each different cluster taken into account, and was kept fixed during the analysis. The data were analyzed using the FEFFIT program (49) as implemented in the ARTEMIS package (version 0.8.006) (48), using as minimization algorithm a modified Levenberg-Marquardt method. The fits were performed directly in k space, with a k weight of 3 (43), minimizing the R-factor, defined as (50)
value was calculated by FEFF 8.2 from atomic overlap integrals of each different cluster taken into account, and was kept fixed during the analysis. The data were analyzed using the FEFFIT program (49) as implemented in the ARTEMIS package (version 0.8.006) (48), using as minimization algorithm a modified Levenberg-Marquardt method. The fits were performed directly in k space, with a k weight of 3 (43), minimizing the R-factor, defined as (50)
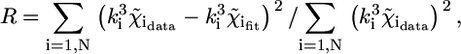 |
(4) |
where N is the number of experimental data points.
The reliability of the fitting procedure has been assured by a high determinacy of the system, which is described as the ratio between the number Nind of independent points in the XAFS data set and the number p of fitted parameters included in the model (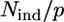 ) (51). The number of independent points has been calculated using (50)
) (51). The number of independent points has been calculated using (50)
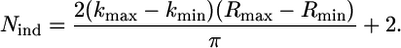 |
(5) |
In Eq. 5, kmax and kmin and Rmax and Rmin define the intervals in the reciprocal and real space in which the analysis was performed.
Confidence analysis was performed on the basis of the reduced χ-square, 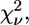 defined as (50)
defined as (50)
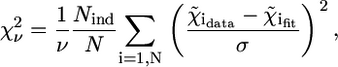 |
(6) |
where 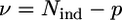 is the number of degrees of freedom in the fit. For each data set, a single value, σ2, for the variance of
is the number of degrees of freedom in the fit. For each data set, a single value, σ2, for the variance of 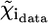 has been estimated over a range of high k values, where the XAFS oscillations are assumed to be indistinguishable from the random noise. This estimate was found to be consistent with Poisson statistics. Even in the presence of “good” fits, i.e., at relatively low values of the R-factor, the values calculated for
has been estimated over a range of high k values, where the XAFS oscillations are assumed to be indistinguishable from the random noise. This estimate was found to be consistent with Poisson statistics. Even in the presence of “good” fits, i.e., at relatively low values of the R-factor, the values calculated for 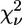 are usually much larger than 1. This situation is commonly encountered in XAFS analysis and attributed to small inadequacies of the model and/or to systematic experimental errors (see Kelly et al. (52) and references therein). In view of this, the standard fluctuation in
are usually much larger than 1. This situation is commonly encountered in XAFS analysis and attributed to small inadequacies of the model and/or to systematic experimental errors (see Kelly et al. (52) and references therein). In view of this, the standard fluctuation in 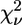 (
(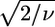 ) is rescaled to
) is rescaled to 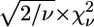 (50,52). The comparison between two different fits of the same data set (corresponding to two different clusters, a and b) was performed by means of
(50,52). The comparison between two different fits of the same data set (corresponding to two different clusters, a and b) was performed by means of 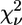 according to the following criterion (52). Fit to cluster b is considered significantly better than fit to cluster a when
according to the following criterion (52). Fit to cluster b is considered significantly better than fit to cluster a when
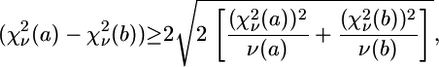 |
(7) |
which corresponds to 95% confidence level (2σ).
XANES simulations were performed using the ab initio code FEFF 8.2 in the framework of full multiple scattering theory (44). Scattering potentials were calculated by overlapping the free atom densities in the muffin-tin approximation and then using the partially nonlocal form for the exchange potentials (Dirac-Fock for core plus Hedin-Lundqvist for valence electrons plus a constant imaginary part) within a self-consistent field-iterative procedure.
We used MOLDRAW (53) to build the binding clusters and DeepView-Swiss-PdbViewer (GlaxoSmithKline R&D, Geneva, Switzerland) for alignment of different cyt bc1 complex structures and for testing allowed conformational motions of side chains.
RESULTS
Comparing Zn XAFS spectra of the different cyt bc1 complexes
Fig. 1 (solid lines) shows the near-edge region of the spectra recorded using the samples containing the bacterial, avian, and bovine cyt bc1 complexes treated with Zn2+, as described in Materials and Methods. Although the avian and bovine cyt bc1 complexes exhibit almost the same spectral features, this is not the case for the bacterial enzyme, for which we observe an enhancement of the white line (the intense absorption peak just above the edge) and some modifications in the region around 9670 eV and around 9685 eV. These differences in the XANES spectra indicate that the local structure around zinc must be significantly different in the bacterial cyt bc1 complex compared to avian and bovine enzymes.
FIGURE 1.

XANES spectra of the bacterial, avian, and bovine cyt bc1 complexes. Experimental spectra (normalized to μ0) are shown as solid lines; dotted lines represent simulations based on the structural parameters obtained by EXAFS analysis (see text and legend of Fig. 2 for details).
The experimental EXAFS functions and their Fourier transforms (FT) are shown in Figs. 2 and 3, respectively. Again, a strong similarity appears between the bovine and avian enzymes, whereas significant differences are present with the bacterial cyt bc1 complex. In the latter case, the first peak of the FT, due to the first ligands, is shifted to greater distances, and its amplitude is enhanced (Fig. 3). When considering more distant ligands, stronger contributions are observed over the 2–3 Å range in the FT amplitude of the bacterial cyt bc1 complex compared with the avian and bovine cases, although all three samples conspicuously show the triple peak feature in the range 2.9–4 Å, which is characteristic of histidine residues (54).
FIGURE 2.

Experimental k3 weighted EXAFS functions measured in the bacterial, avian, and bovine cyt bc1 complexes (solid lines). The dashed lines represent calculated best-fit EXAFS functions corresponding to the following clusters: two His, one Lys and one Asp or Glu residue for the avian and bovine enzymes (the corresponding structural parameters are given in Tables 2 and 3 (model m), respectively); one His, two Asp or Glu residues in monodentate coordination, one Gln or Asn residue, and two water molecules for the bacterial enzyme (see Table 4, model e, for the corresponding structural parameters).
FIGURE 3.

Amplitudes of the Fourier transforms (FT) of the k3 weighted EXAFS functions shown in Fig. 2, performed at Δk = 2.5–14.5 Å−1.
The strategy we adopted in data analysis was as follows. From a first-shell analysis we obtained information on the coordination number, the chemical nature, and distances of ligand atoms. Then, by investigating zinc binding sites in datasets of high-quality protein crystal structures (metal coordination sites in proteins, http://tanna.bch.ed.ac.uk/) and in the metalloprotein database (MDB) (http://metallo.scripps.edu/advanced/#advanced_form), we built a series of clusters made of all combinations of amino acid residues compatible with both EXAFS first-shell results and coordination chemistry information (55). In the last step of the analyses, we performed a series of fits to the experimental data looking for the cluster that minimized the R-factor (Eq. 4).
First-shell analysis: the avian and bovine cyt bc1 complexes
It is well known that zinc can bind four, five, or six ligands in proteins, and that these ligands can be oxygen, nitrogen, or sulfur (55). A number of preliminary first-shell fits performed in r-space indicated that zinc binds four N or O atoms. Any attempt to insert sulfur atoms in the model failed to reproduce the experimental data. As far as the coordination number is concerned, we found that when this parameter was changed to 3 or 5, the R-factor increased appreciably. Moreover when we allowed the coordination number to vary, using Zn-N or Zn-O scattering paths, it converged to a value of 4. To properly distinguish between N and O atoms, we considered all structural models obtainable using their different combinations, keeping the coordination number fixed to 4 (Table 1). This yielded five different cluster types. All cluster types were built in a tetrahedral geometry, which is the geometry commonly found in tetracoordinated zinc clusters in proteins (55), using a starting distance of 2 Å. We considered as free parameters the Zn-N and Zn-O distances, the energy shift, and a single DW factor. Since, with the fitting range used, the number of independent points in the data was ∼10, the model was sufficiently overdetermined, having an 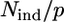 ratio of ∼2 (see Eq. 5).
ratio of ∼2 (see Eq. 5).
TABLE 1.
Results of first-shell analysis in the avian and bovine cyt bc1 complexes
| Avian cyt bc1 complex
| ||||
|---|---|---|---|---|
| Model | Zn-N (Å) | Zn-O (Å) | DW (N/O) (10−3 Å2) | R-factor (%) |
| 4N | 2.03 (1) | 7 (1) | 1.8 | |
| 3N, 1O | 2.02 (1) | 2.14 (3) | 3.0 (8) | 0.8 |
| 2N, 2O | 1.99 (87) | 2.04 (76) | 5 (70) | 1.9 |
| 1N, 3O | 2.22 (4) | 2.01 (1) | 4.7 ( 7) | 1.1 |
| 4O | 2.00 (1) | 8 (1) | 2.6 | |
| Bovine cyt bc1 complex
| ||||
| Model | Zn-N (Å) | Zn-O (Å) | DW (N/O) (10−3 Å2) | R-factor (%) |
| 4N | 2.01 (2) | 6 (2) | 3.6 | |
| 3N, 1O | 2.01 (1) | 2.13 (4) | 2.8 (7) | 1.2 |
| 2N, 2O | 1.97 (6) | 2.03 (8) | 5 (88) | 3.7 |
| 1N, 3O | 2.21 (5) | 2.00 (1) | 4.6 (9) | 1.9 |
| 4O | 1.99 (2) | 8 (1) | 5.1 | |
Δk = 2.5–14.6 Å−1 and ΔR = 1–2 Å. DW indicates the Debye-Waller factor. The 1σ-error on the least significant figure of the structural parameters is reported in parentheses.
As shown in Table 1 for both the avian and the bovine enzyme, the best fit (minimum R-factor) is obtained for three N and one O (3N, 1O) atoms as ligands. The cluster formed by one N and three O (1N, 3O) atoms also yielded a reasonable (although larger) R-factor. However, the latter combination can be excluded, because the distance found for the nitrogen is significantly larger (∼10%) than the value expected (2.05 Å) for Zn-N bond length in a tetracoordinated Zn cluster (http://tanna.bch.ed.ac.uk/). Conversely, the distances obtained for the cluster formed by three N and one O atoms matched those evaluated from databases of protein crystal structures (55).
First-shell analysis for the bacterial cyt bc1 complex
The zinc-binding site in the bacterial cyt bc1 complex was characterized by a high broadening in the first-neighbor distances (Fig. 3). In such cases, the first-shell analysis can provide only limited information with high precision (i.e., distance), whereas a considerable uncertainty is associated to other structural parameters, such as coordination number and atomic number (e.g., oxygen and nitrogen atoms can hardly be distinguished). A number of first-shell fits, performed in r-space with a k weight of 3, indicated that zinc binds five to six N or O atoms. Any attempt to insert sulfur atoms and decrease the coordination number led to unacceptable fits.
Discriminating between putative ligand clusters by multishell multiple-scattering analysis
In an attempt to define the clusters of amino acid residues forming the Zn2+ binding sites, multishell multiple-scattering analysis was subsequently performed. Within the restrictions of first-shell analysis results, we built a series of possible clusters as follows:
We associated the first ligands with one or more amino acid residues by examining a database of Zn binding sites in proteins whose crystallographic structures are known (http://tanna.bch.ed.ac.uk/cngroups.html). The Nδ1 and Nɛ2 atoms of the histidine imidazole ring, the Oδ1 and Oδ2 of the Asp and Glu residues are by far the most common nitrogen/oxygen ligands of zinc in proteins (55). In some cases, the Oδ1 atom of Asn and Gln can also bind to this metal. In a few cases, the Nz of Lys has also been observed to coordinate zinc. This coordination is rather uncommon, because it requires the deprotonation of the charged amino group of Lys, which is typically characterized by a high pKa value (∼10). The presence of a coordinating Lys residue in Zn2+ binding clusters is always associated with at least one coordinating carboxylic residue (see http://tanna.bch.ed.ac.uk/cngroups.html). Starting from these considerations, we could reasonably infer that if the first ligand was a nitrogen atom, it belonged to a His or a Lys, whereas if it was an oxygen atom, it belonged to Asp or Glu, or to Asn or Gln (since EXAFS is sensitive only to the atoms inside a sphere of ∼5-Å radius around the absorber, we cannot actually distinguish between Asp and Glu or between Asn and Gln). When considering the involvement of a Lys in coordination, we required the concomitant presence of at least one carboxylic group. The participation of a water molecule in zinc coordination has been observed in several cases (30,55) (see also http://tanna.bch.ed.ac.uk/). This consideration increased the number of possible oxygen donors that had to be taken into account.
-
Having selected the residues possibly present in the binding clusters, the coordinates of all atoms were set according to the following criteria. As first-neighbors distances, we used those found by first-shell analysis, and as intraligand distances and angles those reported by Engh and Huber (56) (Fig. 4). The 3-D geometries considered were those normally found in Zn2+-binding sites determined by high-resolution protein crystallography (55), i.e., tetrahedral for a coordination number of 4, trigonal bipyramidal for a coordination number of 5, and octahedral for a coordination number of 6. As far as carboxylate groups are concerned, we considered two different coordination modes, i.e., monodentate and bidentate binding (57). Also, in the case of His residues, two configurations were taken into account, depending on which nitrogen atom (Nδ1 or Nɛ2) binds the metal.
The orientation of the amino acid residues with respect to the Zn-first ligand direction was set according to the following angular values (with reference to Fig. 4): 128° and 127° for the Zn-Nɛ2-Cɛ1 and Zn-Nδ1-Cɛ1 angles (β) of the histidine residue; 110° for the Zn-Nz-Cɛ angle (δ) of lysine residue; 120° and 90° for the Zn-Oδ1-Cγ angles of carboxylic groups, in monodentate and bidentate binding modes, respectively (57); 120° for the Zn-Oδ1-Cγ and Zn-Oδ1-Cδ angles of asparagine and glutamine. Some of these angles (i.e., Zn-Oδ1-Cγ of carboxylic groups and Zn-Oδ1-Cγ angle of Asn or Zn-Oδ1-Cδ angle of Gln) were kept fixed during the fitting procedure, whereas angle δ of Lys and the β angles of the His residues were allowed to vary, because a large spread of these values has been encountered when examining crystallographic structures.
-
The models built as described above were fitted to the experimental data, and compared in terms of goodness of fit and consistency of the structural parameters obtained. All paths involving up to five scattering processes with an effective path length ≤5 Å were included (50,58). Fits were performed directly in k-space (with a k weight of 3) to avoid truncation and loss of information due to Fourier filtering (58). The fitting range was 2.5–14.6 Å−1 for avian and bovine cyt bc1 complex samples, and 2.5–11.6 Å−1 for the bacterial cyt bc1 complex sample. To avoid local minima, we fitted an extremely low number of parameters using the rigid-body refinement approach (54,59) and fixing the values of DW factors. With these prescriptions, the free parameters were a common shift in the energy origin E0, the variation of the first-ligand distance, and an angular parameter for each amino acid bound to the metal. The fitting range used and this number of free parameters led to a
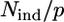 ratio between 3 and 9, i.e., the problem was considerably overdetermined.
ratio between 3 and 9, i.e., the problem was considerably overdetermined.As angular parameters we chose a bending angle for His residues (β in Fig. 4, accounting for rotation of the imidazole ring around an axis passing through the nitrogen bound to the metal and perpendicular to the imidazole plane), the Oδ1-Cγ-Oδ2 (γ) angle in carboxylic groups, the Oδ1-Cδ-Nɛ2 or Oδ1-Cγ-Nδ2 angle (η) in Gln and Asn residues, and the Zn-Nz-Cɛ angle in Lys residues (δ in Fig. 4). A simple model was chosen for the Debye-Waller (DW) factors, which were grouped and fixed to five values. The value obtained from first-shell analysis (3 × 10−3 Å2) was used for first neighbors, and the other four values were selected according to the results of density functional theory simulations performed on Zn-binding clusters in proteins (60). For the imidazole ring, the DW factor of the single scattering (SS) paths involving the two carbon atoms at a distance of ∼3 Å from zinc was set to 9 × 10−3 Å2; a value of 6 × 10−3 Å2 was chosen for the remaining SS paths involving more distant atoms and for all multiple scattering (MS) paths formed by three legs. We assigned a value of 4 × 10−3 Å2 to all SS and MS paths formed by three legs belonging to a carboxylic group, and a common value of 10−2 Å2 to the remaining paths.
FIGURE 4.

Sketch of the reference structural units used in multishell multiple scattering analysis. The values of distances and angles of amino acid residues were taken from (55). The Glu residue has the same structure as the Asp residue, provided Cγ and Cβ of Asp are replaced by Cδ and Cγ, respectively, of Glu. The structure of the Gln residue coincides with that of the Asn residue when Nδ2, Cγ, and Cβ of Asn are replaced by Nɛ2, Cδ, and Cγ, respectively, of Gln.
Avian and bovine cyt bc1 complexes
For the avian and bovine enzymes, the first-shell analysis indicated clearly Zn2+ coordination by three N and one O atoms. In attempting to associate each first ligand to an amino acid within the framework described above, we have considered four possible combinations of amino acids and water molecules: three His and one Asp or Glu; three His and one Gln or Asn; three His and one H2O; and two His, one Lys, and one Asp or Glu. The Asp or Glu residues were built in a monodentate configuration, since the bidentate configuration has never been observed in the case of a total coordination number of 4 (55). However, we considered two binding modes (Nδ1 or Nɛ2) for His residues (although the Nɛ2 binding mode is much more frequent than Nδ1 at 70% and 30%, respectively) (55), which increased the number of models to 15. For all clusters, the results of the fitting procedure are reported in Table 2 and Table 3 for the avian and the bovine enzymes, respectively. In both complexes, the minimum R-factors were obtained for clusters (models m and o) formed by two His, one Lys, and one carboxylic group. The Nɛ2 binding mode seems to be the most favored one in general, but close R-factor values are obtained for the cluster in which both His residues bind Zn2+ by Nɛ2 (model m) and the cluster in which one His coordinates in the Nɛ2 and the other in the Nδ1 configuration (model o).
TABLE 2.
Structural results from multishell multiple-scattering analysis in the avian cyt bc1 complex
| Model | Ligand cluster | Zn-N (Å) | Zn-O (Å) | β (°) His | γ (°) Asp/Glu | δ (°) Lys | R (%) | 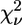 |
|---|---|---|---|---|---|---|---|---|
| a | 3 His (3Nɛ2) | 2.014 (9) | 130 (2) | 13.0 | 31.0 (7.6) | |||
| 1 Asp/Glu | 2.12 (2) | 119 (1) | ||||||
| b | 3 His (3Nδ1) | 2.012 (9) | 130 (2) | 14.9 | 107.4 (26.2) | |||
| 1 Asp/Glu | 2.12 (2) | 119 (1) | ||||||
| c | 3 His (2Nɛ2, 1Nδ1) | 2.012 (9) | 130 (2) | 13.4 | 61.8 (15.1) | |||
| 1 Asp/Glu | 2.12 (2) | 119 (1) | ||||||
| d | 3 His (1Nɛ2, 2Nδ1) | 2.012 (9) | 130 (2) | 14.0 | 83.9 (20.5) | |||
| 1 Asp/Glu | 2.11 (2) | 119 (1) | ||||||
| e | 3 His (3Nɛ2) | 2.014 (9) | 130 (2) | 12.7 | 44.4 (10.8) | |||
| 1 Gln/Asn | 2.12 (2) | 125 (2) | ||||||
| f | 3 His (3Nδ1) | 2.012 (9) | 130 (2) | 14.6 | 114.5 (27.9) | |||
| 1 Gln/Asn | 2.12 (2) | 125 (2) | ||||||
| g | 3 His (2Nɛ2, 1Nδ1) | 2.013 (9) | 130 (2) | 13.7 | 49.6 (12.1) | |||
| 1 Gln/Asn | 2.12 (2) | 125 (2) | ||||||
| h | 3 His (1Nɛ2, 2Nδ1) | 2.014 (9) | 130 (2) | 13.2 | 87.7 (21.4) | |||
| 1 Gln/Asn | 2.12 (2) | 125 (2) | ||||||
| i | 3 His (3Nɛ2) | 2.011 (9) | 133 (2) | 13.4 | 81.2 (19.5) | |||
| 1 H2O | 2.11 (2) | |||||||
| j | 3 His (3Nδ1) | 2.010 (9) | 133 (2) | 15.0 | 90.8 (21.8) | |||
| 1 H2O | 2.11 (2) | |||||||
| k | 3 His (2Nɛ2, 1Nδ1) | 2.012 (9) | 133 (2) | 13.7 | 71.1 (17.1) | |||
| 1 H2O | 2.12 (2) | |||||||
| l | 3 His (1Nɛ2, 2Nδ1) | 2.011 (9) | 133 (2) | 14.3 | 80.2 (19.3) | |||
| 1 H2O | 2.11 (2) | |||||||
| m | 2 His (2Nɛ2) | 2.03 (1) | 130 (5) | 12.2 | 37.8 (9.4) | |||
| 1 Asp/Glu | 2.11 (2) | 121 (2) | ||||||
| 1 Lys | 1.98 (2) | 127 (12) | ||||||
| n | 2 His (2Nδ1) | 2.03 (1) | 128 (2) | 13.1 | 99.0 (24.5) | |||
| 1 Asp/Glu | 2.10 (2) | 121 (1) | ||||||
| 1 Lys | 1.97 (3) | 130 (16) | ||||||
| o | 2 His (1Nɛ2, 1Nδ1) | 2.03 (1) | 129 (4) | 12.6 | 61.6 (15.3) | |||
| 1 Asp/Glu | 2.10 (2) | 120 (1) | ||||||
| 1 Lys | 1.97 (2) | 128 (14) |
The bending angle of the His imidazole group is indicated by β. Angles γ and δ refer to the Oδ1-Cγ-Oδ2 angle of Asp or Glu and the Zn-Nz-Cɛ angle of Lys, respectively (Fig. 4). The 1σ-error on the least significant figure of the structural parameters is reported in parentheses, as is the 1σ-uncertainty in 
TABLE 3.
Structural results obtained from multishell multiple scattering analysis in the bovine cyt bc1 complex
| Model | Ligand cluster | Zn-N (Å) | Zn-O (Å) | β (°) His | γ (°) Asp/Glu | δ (°) Lys | R (%) |  |
|---|---|---|---|---|---|---|---|---|
| a | 3 His (3Nɛ2) | 2.00 (1) | 130 (2) | 15.1 | 43.6 (10.6) | |||
| 1 Asp/Glu | 2.11 (2) | 117 (1) | ||||||
| b | 3 His (3Nδ1) | 1.99 (1) | 132 (1) | 16.2 | 108.7 (26.5) | |||
| 1 Asp/Glu | 2.10 (2) | 113 (1) | ||||||
| c | 3 His (2Nɛ2, 1Nδ1) | 2.00 (1) | 130 (2) | 15.3 | 71.9 (17.5) | |||
| 1 Asp/Glu | 2.11 (2) | 117 (1) | ||||||
| d | 3 His (1Nɛ2, 2Nδ1) | 1.99 (1) | 131 (2) | 15.8 | 91.7 (22.3) | |||
| 1 Asp/Glu | 2.10 (2) | 114 (1) | ||||||
| e | 3 His (3Nɛ2) | 2.00 (1) | 131 (1) | 14.6 | 52.4 (12.8) | |||
| 1 Gln/Asn | 2.11 (2) | 120 (2) | ||||||
| f | 3 His (3Nδ1) | 2.00 (1) | 131 (2) | 16.0 | 102.8 (25.1) | |||
| 1 Gln/Asn | 2.11 (2) | 120 (2) | ||||||
| g | 3 His (2Nɛ2, 1Nδ1) | 2.00 (1) | 131 (2) | 15.2 | 69.6 (17.0) | |||
| 1 Gln/Asn | 2.11 (2) | 120 (2) | ||||||
| h | 3 His (1Nɛ2, 2Nδ1) | 2.00 (1) | 131 (2) | 15.5 | 91.3 (22.3) | |||
| 1 Gln/Asn | 2.11 (2) | 120 (2) | ||||||
| i | 3 His (3Nɛ2) | 2.00 (1) | 132 (2) | 15.0 | 81.2 (19.5) | |||
| 1 H2O | 2.11 (2) | |||||||
| j | 3 His (3Nδ1) | 2.00 (1) | 132 (2) | 15.7 | 139.7 (33.6) | |||
| 1 H2O | 2.11 (2) | |||||||
| k | 3 His (2Nɛ2, 1Nδ1) | 2.00 (1) | 132 (2) | 15.1 | 115.8 (27.8) | |||
| 1 H2O | 2.11 (2) | |||||||
| l | 3 His (1Nɛ2, 2Nδ1) | 2.00 (1) | 132 (2) | 15.3 | 128.1 (30.4) | |||
| 1 H2O | 2.11 (2) | |||||||
| m | 2 His (2Nɛ2) | 2.01 (1) | 134 (5) | 13.2 | 44.4 (11.0) | |||
| 1 Asp/Glu | 2.11 (2) | 125 (2) | ||||||
| 1 Lys | 1.98 (3) | 119 (11) | ||||||
| n | 2 His (2Nδ1) | 2.01 (1) | 132 (2) | 15.2 | 67.0 (16.6) | |||
| 1 Asp/Glu | 2.11 (3) | 113 (1) | ||||||
| 1 Lys | 1.98 (2) | 120 (6) | ||||||
| o | 2 His (1Nɛ2, 1Nδ1) | 2.01 (1) | 133 (3) | 13.2 | 56.7 (14.0) | |||
| 1 Asp/Glu | 2.11 (2) | 125 (2) | ||||||
| 1 Lys | 1.98 (3) | 118 (9) |
Angles β, γ, and δ, as well as 1σ-uncertainties, are defined as in Table 2.
In the case of these two models (m and o) that minimized the R-factor, we examined the possibility of a structural heterogeneity of the two His residues involved in the binding cluster. Best fitting performed by considering independent structural parameters for the two His residues led to a difference of 0.05 Å between the first-ligand distances (Zn-N) and to a difference of 8° between the ring-bending angle (β) of the two His residues. However, the 1 σ-error of these parameters increased considerably (∼5 times and 2 times for the first-ligand distances and bending angle, respectively). The goodness of fit was comparable, since the slight decrease (0.2) found in the R-factor was accompanied by a slight increase (0.4) in the reduced χ2, presumably caused by the two additional free parameters of the model. Interestingly, introducing a possible heterogeneity in the two His residues did not change the best-fit structural parameters of the other two residues that form the binding cluster.
The small difference between the R-factors of the different models tested (see Tables 2 and 3) is reasonable in view of the high structural similarity between the clusters. However, when the reduced χ2 statistics are considered and the crystallographic information available is taken into account (see Discussion), the binding clusters of the avian and bovine cyt bc1 complexes can be identified with high levels of confidence.
Bacterial cyt bc1 complex
In the case of the bacterial cyt bc1 complex, the most probable configuration indicated by first-shell analysis included a group of five or six N or O atoms. This first-shell information was clearly not exhaustive, leaving, in principle, a very large, practically unmanageable, number of clusters to scrutinize. However, a large number of ab initio simulations (not shown) strongly suggested that the presence of a prominent contribution in the FT amplitude over the 2–3 Å range (Fig. 3) can only be reproduced when residues such as carboxylic acids or Gln or Asn are included in the binding cluster. Moreover, as already noted, the triple peak observable in the FT amplitude between 2.9 and 4 Å is diagnostic of the presence of one or more His residues (54). Based on this evidence, we examined all the clusters deposited in the metalloprotein database MDB (http://metallo.scripps.edu/advanced/#advanced_form) that contained one or two His and no cysteine or methionine, and with a total coordination number, n, of 5 or 6. This yielded the following possibilities: 1), two His, two carboxylic acids in a monodentate binding mode, and one H2O (PDB codes 1AH7, 1FOJ, 1QMD, and 1QH5) (n = 5); 2), one His, two carboxylic acids in a monodentate binding mode, one Gln or Asn, and one H2O (PDB codes 1BH5, 1FRO, and 2USH) (n = 5); 3), one His, one carboxylic acid in a bidentate binding mode, one Gln or Asn, and one H2O (PDB code 1F83) (n = 5); 4), one His, three carboxylic acids in a monodentate binding mode, and one Gln or Asn (PDB code 1USH) (n = 5); and 5), one His, two carboxylic acids in a monodentate binding mode, one Gln or Asn, and two H2O (PDB codes 1BH5 and 1QIP) (n = 6). Fitting to these clusters yielded the results summarized in Table 4.
TABLE 4.
Structural parameters determined by fitting the experimental data of the bacterial cyt bc1 complex to the model clusters extracted from the metalloprotein database MDB
| Model | Ligand cluster n | Zn-N (Å) | Zn-O (Å) | β (°) His | γ (°) Asp/Glu | η (°) Gln/Asn | R (%) |  |
|---|---|---|---|---|---|---|---|---|
| a | 2 His (2 Nɛ2) | 2.05 (2) | 120 (5) | 11.0 | 8.05 (2.3) | |||
| 2 Asp/Glu m | 2.14 (3) | 123 (13) | ||||||
| 1 H2O | 2.09 (5) | |||||||
| n = 5 | ||||||||
| b | 1 His (1Nɛ2) | 2.01 (4) | 120 (11) | 9.3 | 5.4 (1.6) | |||
| 2 Asp/Glu m | 2.08 (6) | 124 (3) | ||||||
| 1 Gln/Asn | 2.18 (9) | 115 (6) | ||||||
| 1 H2O | 2.10 (17) | |||||||
| n = 5 | ||||||||
| c | 1 His (1 Nɛ2) | 2.01 (7) | 128 (40) | 14.5 | 24.3 (7.4) | |||
| 1 Asp/Glu b | 2.11 (7) | 122 (2) | ||||||
| 1 Gln/Asn | 2.10 (8) | 115 (7) | ||||||
| 1 H2O | 2.10 (17) | |||||||
| n = 5 | ||||||||
| d | 1 His (1 Nɛ2) | 2.14 (4) | 146 (14) | 10.3 | 5.7 (1.7) | |||
| 3 Asp/Glu m | 2.06 (2) | 124 (1) | ||||||
| 1 Gln/Asn | 2.22 (5) | 115 (4) | ||||||
| n = 5 | ||||||||
| e | 1 His (1Nɛ2) | 1.99 (3) | 128 (17) | 8.6 | 4.1 (1.2) | |||
| 2 Asp/Glu m | 2.10 (7) | 123 (3) | ||||||
| 1 Gln/Asn | 2.28 (4) | 111 (6) | ||||||
| 2 H2O | 2.10 (7) | |||||||
| n = 6 |
For details, go to (http://metallo.scripps.edu/advanced/#advanced_form). Monodentate and bidentate binding configurations are indicated by m and b, and n is the coordination number. Other symbols and values in parentheses are as in Table 2. See text for explanation.
We observed that, among the clusters described above, only one contained two His residues (model a in Table 4). Two coordinating His residues have been found in the high-affinity Zn2+-inhibitory binding sites of photosynthetic RC from Rb. sphaeroides (30,31) and bovine cytochrome c oxidase (61). Two His residues also seem to be present in one of the two Zn2+ sites found by XRD in the avian cyt bc1 complex (37). In view of this feature of Zn2+ inhibitory binding sites characterized so far in redox-active membrane proteins, we tested additional clusters containing two His residues and compatible with first-shell results. These structures were built, starting from cluster a in Table 4, by considering the different coordination modes of carboxylates and by exchanging water molecules with Gln or Asn residues. Water molecules were added or eliminated to maintain a coordination number of 5 or 6. The fitting results obtained are given in Table 5. An analogous procedure was used to generate other putative clusters, starting with one that minimizes the R-factor (Table 4, model e). Testing these new possibilities yielded the results shown in Table 6. For all the clusters reported in Tables 4–6, only the results obtained for the Nɛ2 binding mode of His are shown. When the corresponding clusters including Nδ1 as His ligands were tested, the respective R-factors did not change appreciably. None of the additional tested clusters (Tables 5 and 6) improved the R-factor obtained for model e in Table 4.
TABLE 5.
Structural parameters obtained by fitting the spectrum of the bacterial cyt bc1 complex to additional clusters containing two His residues
| Model | Ligand cluster n | Zn-N (Å) | Zn-O (Å) | β (°) His | γ (°) Asp/Glu | η (°) Gln/Asn | R (%) |  |
|---|---|---|---|---|---|---|---|---|
| a | 2 His (2 Nɛ2) | 2.05 (3) | 115 (5) | 10.6 | 7.4 (2.2) | |||
| 2 Asp/Glu m | 2.11 (2) | 124 (2) | ||||||
| 1 Gln/Asn | 2.18 (5) | 116 (4) | ||||||
| n = 5 | ||||||||
| b | 2 His (2 Nɛ2) | 2.08 (3) | 137 (7) | 15.4 | 18.4 (5.5) | |||
| 1 Asp/Glu m | 2.18 (8) | 122 (4) | ||||||
| 1 Asp/Glu b | 2.11 (7) | 121 (4) | ||||||
| n = 5 | ||||||||
| c | 2 His (2 Nɛ2) | 2.02 (3) | 118 (4) | 11.5 | 13.1 (3.8) | |||
| 2 Asp/Glu m | 2.15 (3) | 123 (1) | ||||||
| 2 H2O | 2.09 (1) | |||||||
| n = 6 | ||||||||
| d | 2 His (2 Nɛ2) | 2.05 (5) | 134 (7) | 11.8 | 12.8 (3.9) | |||
| 1 Asp/Glu m | 2.19 (6) | 123 (4) | ||||||
| 1 Asp/Glu b | 2.16 (9) | 126 (2) | ||||||
| 1 H2O | 1.93 (7) | |||||||
| n = 6 | ||||||||
| e | 2 His (2 Nɛ2) | 2.02 (3) | 135 (6) | 10.2 | 10.6 (3.3) | |||
| 1 Asp/Glu m | 2.13 (10) | 123 (11) | ||||||
| 1 Asp/Glu b | 2.13 (9) | 124 (2) | ||||||
| 1 Gln/Asn | 2.13 (10) | 116 (15) | ||||||
| n = 6 | ||||||||
| f | 2 His (2 Nɛ2) | 2.07 (3) | 135 (5) | 23.6 | 37.9 (10.8) | |||
| 2 Asp/Glu b | 2.14 (3) | 122 (2) | ||||||
| n = 6 | ||||||||
| g | 2 His (2 Nɛ2) | 2.09 (4) | 135 (5) | 17.8 | 34.2 (10.2) | |||
| 1 Asp/Glu b | 2.25 (7) | 124 (3) | ||||||
| 2H2O | 2.11 (3) | |||||||
| n = 6 | ||||||||
| h | 2 His (2 Nɛ2) | 2.06 (2) | 137 (5) | 11.7 | 13.2 (3.8) | |||
| 1 Asp/Glu m | 2.28 (5) | 118 (3) | ||||||
| 3 H2O | 2.12 (2) | |||||||
| n = 6 |
Symbols are as in Table 2. See text for details.
TABLE 6.
Structural parameters derived by fitting the data obtained in the bacterial cyt bc1 complex to clusters built from a model that minimizes the R-factor
| Model | Ligand cluster n | Zn-N (Å) | Zn-O (Å) | β (°) His | γ (°) Asp/Glu | η (°) Gln/Asn | R (%) | 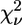 |
|---|---|---|---|---|---|---|---|---|
| a | 1 His (1 Nɛ2) | 2.01 (8) | 130 (32) | l9.6 | 13.4 (4.3) | |||
| 1 Asp/Glu m | 2.12 (10) | 123 (13) | ||||||
| 1 Asp/Glu b | 2.09 (10) | 125 (3) | ||||||
| 1 Gln/Asn | 2.13 (10) | 115 (15) | ||||||
| 1 H2O | 2.09 (10) | |||||||
| n = 6 | ||||||||
| b | 1 His (1 Nɛ2) | 2.00 (4) | 132 (20) | 11.1 | 12.2 (3.8) | |||
| 1 Asp/Glu m | 2.12 (10) | 123 (14) | ||||||
| 1 Asp/Glu b | 2.08 (8) | 122 (4) | ||||||
| 1 Gln/Asn | 2.12 (12) | 115 (20) | ||||||
| n = 5 | ||||||||
| c | 1 His (1 Nɛ2) | 2.01 (7) | 128 (4) | 16.8 | 26.9 (8.0) | |||
| 2 Asp/Glu b | 2.11 (7) | 122 (2) | ||||||
| 1 Gln/Asn | 2.10 (8) | 115 (8) | ||||||
| n = 6 |
Simulations of the XANES spectra
To test the reliability of EXAFS results, we performed simulations of the XANES spectra based on the atomic structure of the clusters that minimize the R-factor of the extended spectra. The results obtained are shown in Fig. 1 as dashed lines. For the bacterial cyt bc1 complex, the simulation based on the most probable sixfold-coordinated cluster with first-shell ligands in the octahedral geometry (Table 4, model e) is in very good agreement with the experimental data, both in the white-line intensity and in the overall spectral features. We performed a number of tests with alternative structures, all of which gave poorer agreement. In particular, it is important to note that any simulation in which one of the six ligands was removed, i.e., the coordination number changed from 6 to 5, led to a significant decrease in the intensity of the white line. A similar strong correlation between the coordination number and the white-line intensity has been observed for other metalloproteins, for example, for the two His and one carboxylate motif of tyrosine hydroxylase (62). This observation provided further proof of the reliability of the EXAFS analysis for the bacterial enzyme.
Analogous simulations performed for the avian and bovine complex, starting from the tetrahedral cluster determined from EXAFS analysis, yielded a poorer description of the experimental spectrum compared to the bacterial complex. In the EXAFS analysis, the 3-D cluster was built according to an exact tetrahedral coordination without taking into account the relative orientation of the residues. Actually, the EXAFS spectrum is substantially insensitive to these structural parameters. By comparison, the simulations performed for the avian and bovine enzyme showed that the XANES region was influenced by the relative orientation of the cluster residues. We found particularly effective rotations of the His residues around the Zn-N axis and of the Asp/Glu around the Zn-Cγ/Cδ axis. The best description obtained by sampling these two parameters is shown in Fig. 2 as a dashed line. The decrease of the experimental white-line intensity in the XANES spectra of the avian and bovine complexes, as compared to the bacterial enzyme, is well simulated. However, as a whole, the simulated XANES spectra do not provide as good a quantitative agreement for those complexes as for the bacterial cyt bc1 complex. We believe that this is due to a limitation of the simulation procedure rather than to an erroneous structural determination. A possible source of this pronounced disagreement is the approximation of the scattering potential in the muffin-tin scheme (62).
DISCUSSION
The high-affinity Zn-binding site of the avian and bovine cyt bc1 complexes
First-shell analysis of the EXAFS data of the avian cyt bc1 complex identifies unequivocally three N and one O atoms as Zn2+ ligands. By subsequent multishell multiple-scattering analysis we found that the cluster that minimizes the R-factor, among the many that were tested, is formed by two His, one Lys, and one carboxylic acid (Asp or Glu) (Table 2, model m). When comparing models m, n, and o on the basis of reduced-χ2 statistics by means of Eq. 7, it appears that the fit to the model m (both His in the Nɛ2 configuration) is significantly better than model n (both His in Nδ1), whereas the difference with model o (one His in the Nɛ2 and the other in the Nδ1 configuration) is not significant. Cluster m is characterized by 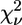 = 37.8, a value lower than all the other models considered, except for cluster a, formed by three His and one carboxylic acid, for which
= 37.8, a value lower than all the other models considered, except for cluster a, formed by three His and one carboxylic acid, for which 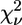 = 31.0 (Table 2). However, this difference in the reduced χ2 values is not statistically significant, since the condition of Eq. 7 is not satisfied. When comparing the fit to model a with the other fits summarized in Table 2 by means of Eq. 7, it appears that the fits that are significantly worse correspond to models b, d, f, h, i, j, k, l, and n. In summary, we can discard the clusters formed by three His and one H2O molecule (for all configurations of His coordination), that is, models i–l. Of the remaining three clusters, on a purely statistical basis, we can exclude only those configurations that include more than one His bound at Nδ1, that is, models b, d, f, h, and n.
= 31.0 (Table 2). However, this difference in the reduced χ2 values is not statistically significant, since the condition of Eq. 7 is not satisfied. When comparing the fit to model a with the other fits summarized in Table 2 by means of Eq. 7, it appears that the fits that are significantly worse correspond to models b, d, f, h, i, j, k, l, and n. In summary, we can discard the clusters formed by three His and one H2O molecule (for all configurations of His coordination), that is, models i–l. Of the remaining three clusters, on a purely statistical basis, we can exclude only those configurations that include more than one His bound at Nδ1, that is, models b, d, f, h, and n.
Further progress in the identification of the Zn2+-binding site can be made by comparison with the XRD investigations. Two Zn2+-binding sites have been located by XRD close to the stigmatellin-binding site (37). This study, performed in Zn-treated crystals of the chicken cyt bc1 complex in the absence of stigmatellin, located a first site (Zn01) in which Zn2+ appears to be coordinated by His121 of cyt c1 and Asp253 of cyt b. His268 and Glu255, although not refining into a coordinating position, were tentatively proposed as possible additional ligands. XRD studies have located a second site (Zn02) in the hydrophobic channel between the Qo site and the bulk lipid phase, but here, only one potential ligand (Met125 of cyt b) could be identified.
Among the three putative Zn2+-binding clusters defined by our EXAFS analysis (models m or o, a or c, and e or g in Table 2), only models m and o (two His, one Lys, and one carboxylic acid) fit well the crystallographic Zn01 site. In the crystallographic structure, only two His residues are found within a radius of 10 Å from the Zn2+ ion, so the other two clusters, which both include three His residues, can be eliminated. Thus, by combining our EXAFS analysis with XRD data (37), we identify a ligand cluster characterized by a tetrahedral geometry, and formed by the His121 of cyt c1, and His268, Lys270, and Asp253 of cyt b. This binding site is depicted in Fig. 5 A, based on the Zn-crystal structure of chicken cyt bc1 complex (37). Stigmatellin from the superimposed 2BCC structure is also shown below the Zn2+-binding site.
FIGURE 5.

View of the proposed Zn2+-binding sites in the avian (A) and bovine (B) cyt bc1 complex. The site of Zn2+ (orange sphere) is in the interface between cyt b (yellow) and cyt c (blue). (A) Structure obtained from the XRD data of the Zn crystal of chicken cyt bc1 complex (37). Coordinates are from bczn3ref.pdb file, available at http://sb20.lbl.gov/cytbc1/PDB/. Stigmatellin, below the Zn2+ site, is from the superimposed avian cyt bc1 structure 2BCC. (B) Coordinates are from the structure of the bovine cyt bc1 complex obtained in the presence of stigmatellin (1PP9). Zn2+ ion was superimposed after alignment of this structure with that of the Zn crystal of the avian complex (A).
EXAFS analysis provides high-resolution structural information on this site. We found a considerable contraction of the Zn-N distance obtained for His residues (2.03 ± 0.01 Å) compared to the XRD Zn-N bond length for His121 (2.9 Å). EXAFS analysis does not reveal a significant structural heterogeneity in the His residues of the binding cluster. In fact, when considering independent structural parameters for the two His residues, only slight, not significant, differences are found in the Zn-N distances and bending angle β, without any improvement of the fit. Moreover, the structural parameters of the other two ligand residues are not affected when a possible His heterogeneity is considered (see, in Results, Avian and bovine cyt bc1 complex). The carboxylic acid predicted by the EXAFS analysis was identified to be the Asp253 rather than the Glu255, as Asp253 appears to be located closer to the Zn ion in the crystallographic structure (Zn-Oδ2(Asp253) = 2.7 Å compared to Zn-Oɛ1(Glu255) = 3.7 Å). The Zn-O distance resulting from EXAFS analysis (2.11 ± 0.02 Å) is considerably shorter than the corresponding XRD length. Similar, although less pronounced, systematic differences have been reported when comparing local structures in metalloproteins determined by XAFS and XRD at resolutions between 3.0 and 2.0 Å (63).
Our study also confirmed the involvement of His268 in Zn2+ coordination, a feature that was uncertain based on crystallographic data, due to the large XRD Zn-Nδ1 distance (4.2 Å). As for the fourth ligand, EXAFS analysis indicates clearly and with high confidence a Lys nitrogen atom, and indeed, Lys270 is located in the vicinity of Zn01 (Fig. 5 A). This residue was not considered as a possible ligand in the earlier XRD study (37), in view of the large Zn-Nz distance (6.5 Å). However, by exploring the allowed conformational motions of Lys270 and His268, we found that their N atoms can be brought at coordination distances of ∼2 Å. The large Zn-N distances obtained from XRD data for both Lys270 and His268 might be due to a number of factors, including 1), the relatively low resolution of the crystallographic structure (3.85 Å); 2), the large B-factors obtained for the potential ligands in the crystallographic structure when refined assuming full occupancy of the binding site (∼110 for His268), indicating a high degree of disorder (37); 3), possible disruption of coordination bonds caused by x-ray irradiation, which usually involves exposure to a higher dose in XRD as compared to XAFS measurements (for an extensive discussion, see Yano et al. (64)); 4), the amorphous state of the sample in XAFS measurements, which were performed by incorporating the protein in a PVA film. Concerning this latter aspect, we have recently shown that the local structure and dynamics of the heme iron in cyt c are essentially coincident when the protein is in solution or embedded in a PVA film (65).
At variance with the crystallographic study of Zn-binding sites (37), XAFS data reported in this article were obtained in the presence of stigmatellin. We have chosen to do so to fix the ISP domain in a well defined (i.e., proximal) position. It has been reported that in the absence of stigmatellin, the conformation of the ISP domain is affected by the redox state of its Fe2S2 cluster, moving away from the proximal position when the cluster is oxidized (66). In contrast, in the presence of stigmatellin, the ISP domain is located in the proximal position (6,66). In fact, Lys270 is in the ef loop of cyt b, in a region that has been reported to move in the presence of stigmatellin (67,68). However, it is unlikely that the ligand field of the Zn2+-binding cluster changes upon binding stigmatellin, because XAFS measurements performed on the bovine cyt bc1 complex in the presence and absence of stigmatellin yielded quite similar spectra that could be described by essentially the same structural parameters (not shown). The only difference was a slightly more pronounced damping of oscillations in the absence of the inhibitor, which suggests a larger dynamical and/or static disorder in its absence (not shown).
We performed the XAFS measurements on samples incubated at substoichiometric Zn/cyt bc1 ratios. The consistency of the binding cluster determined by EXAFS analysis with the crystallographic Zn01 site indicates that Zn01 is a high-affinity site. The lower-affinity Zn02 site seen in XRD studies is likely to be the result of prolonged (one-week) incubation of the cyt bc1 complex crystal in ZnCl2.
XAFS spectra of the avian and bovine cyt bc1 complexes exhibit a striking similarity, both in the XANES and in the extended region. This is confirmed by multishell multiple-scattering analysis, which indicates that in both cases, the clusters that minimize the R-factor are formed by two His, one Lys, and one carboxylic acid (Asp or Glu) (models m and o in Table 3). Confidence analysis based on reduced χ2 statistics carried out with the bovine enzyme leads to conclusions similar to those drawn in the case of the avian complex. In particular, a cluster formed by three His and one water molecules could be excluded.
Examination of the crystallographic structure of the bovine complex (1PP9) and its superposition with that of the Zn-crystal of the avian cyt bc1 complex revealed that a site that is highly homologous to Zn01 is also present in the bovine enzyme. In addition, since in this case only two His residues are found within a radius of 10 Å from the Zn2+ position, we could exclude clusters liganded with three His side chains. Therefore, we proposed a Zn2+-binding cluster formed by His121, His267, Lys269, and Asp254. The location of these residues is shown in Fig. 5 B, obtained from the crystallographic coordinates of the bovine cyt bc1 complex in the presence of stigmatellin (1PP9). Although, as expected, the resulting distances between Zn2+ and ligand atoms greatly exceeded the ligand bond length determined by EXAFS analysis, in this case, too, the conformational mobility of the amino acid chains of the proposed cluster allowed ligand atoms to be brought to coordinating distances. In particular, His267 and Lys 269 could be adjusted so that the Zn-N distance becomes <2 Å in both cases.
The observations by Lorusso et al. (24) on the effect of Zn2+ on the enzymatic properties of bovine heart cyt bc1 complex are consistent with the proposed location of the Zn2+-binding site in the vicinity of the catalytic Qo site. These authors observed that addition of Zn2+ to the reduced cyt bc1 complex caused a red shift in the absorption spectrum of cyt bL and a decrease in the signal intensity of the EPR spectrum of the Fe2S2 center.
The coincidence of the bovine Zn2+-binding site with the Zn01 of the avian cyt bc1 complex has remarkable functional implications. It strongly suggests that these structurally homologous sites can be identified with the high-affinity site characterized by Link and von Jagow (25) as the inhibitory site interfering with a proton extrusion pathway. Notably, this site shares with the Zn inhibitory binding site of the photosynthetic reaction center from Rb. sphaeroides the presence of two His residues and one carboxylic acid (30,31). These structural features suggest that in the case of the cyt bc1 complex, too, Zn inhibits proton transfer, because it binds to the His residues involved in proton-transfer steps, thereby impairing their function as proton donors/acceptors along a proton transfer pathway.
The Zn2+ binding site of Rb. capsulatus cyt bc1 complex
The existence of a high-affinity Zn2+-binding site in the cyt bc1 complex of the photosynthetic bacterium Rb. capsulatus was proven by the observation of a well structured XAFS signal in samples characterized by a Zn/cyt bc1 stoichiometry of ∼1. Interestingly, the bacterial cyt bc1 complex exhibited quite distinct spectral features compared with the avian and bovine enzymes, revealing a different local structure for the Zn-binding site. First-shell analysis indicated a coordination number of 5 or 6, but provided limited information on the number of N and O atoms involved in binding. As a consequence, a large number of putative binding clusters were tested when performing multishell multiple-scattering fitting of the EXAFS signal. Among the considered possibilities, the cluster corresponding to the minimum R-factor (R = 8.6) was characterized by an octahedral geometry, and formed by one His, two carboxylic acids in monodentate binding mode, one Gln or Asn, and two water molecules (Table 4, model e). This cluster yielded the smallest 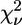 among all tested models. A reasonably good, although higher, R-factor (R = 9.6) was obtained for a similar cluster, equally characterized by a coordination number of 6, and formed by the same residues, in which one of the carboxylic acids binds in the bidentate mode and a single water molecule is involved (Table 6, model a). When the reduced χ2 values corresponding to these two clusters are compared using Eq. 7, it appears that the former (Table 4, model e) is significantly better. A systematic comparison of model e of Table 4 with all the other tested models by means of eqn.7 indicated that the difference in
among all tested models. A reasonably good, although higher, R-factor (R = 9.6) was obtained for a similar cluster, equally characterized by a coordination number of 6, and formed by the same residues, in which one of the carboxylic acids binds in the bidentate mode and a single water molecule is involved (Table 6, model a). When the reduced χ2 values corresponding to these two clusters are compared using Eq. 7, it appears that the former (Table 4, model e) is significantly better. A systematic comparison of model e of Table 4 with all the other tested models by means of eqn.7 indicated that the difference in 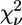 values is significant in all cases except for the model e in Table 5, characterized by a coordination number of six, and four clusters (Table 4, models a, b, and d, and Table 5, model a), all with a coordination number of 5. The hexacoordinated cluster could be ruled out on the basis of XRD structural information (see below). All the clusters with a coordination number of 5, besides any statistical evaluation of confidence levels, were highly unlikely based on XANES simulations. Any attempt to simulate the near-edge spectral region with clusters characterized by a coordination number of 5 yielded a white-line amplitude significantly lower than the experimental one. Conversely, the simulation based on the sixfold-coordinated octahedral cluster minimizing the R-factor (Table 4, model e) yielded the experimentally observed amplitude of the white line, and reproduced with a remarkable accuracy the other XANES features (Fig. 1, dotted line in upper trace).
values is significant in all cases except for the model e in Table 5, characterized by a coordination number of six, and four clusters (Table 4, models a, b, and d, and Table 5, model a), all with a coordination number of 5. The hexacoordinated cluster could be ruled out on the basis of XRD structural information (see below). All the clusters with a coordination number of 5, besides any statistical evaluation of confidence levels, were highly unlikely based on XANES simulations. Any attempt to simulate the near-edge spectral region with clusters characterized by a coordination number of 5 yielded a white-line amplitude significantly lower than the experimental one. Conversely, the simulation based on the sixfold-coordinated octahedral cluster minimizing the R-factor (Table 4, model e) yielded the experimentally observed amplitude of the white line, and reproduced with a remarkable accuracy the other XANES features (Fig. 1, dotted line in upper trace).
By aligning the crystallographic structure of the cyt bc1 complex of Rb. capsulatus (1ZRT) with that of Zn crystals of the avian complex, in the region homologous to that of the Zn01 site, a group of residues that fit the Zn-binding cluster (Table 4, model e) indicated by EXAFS analysis as the most probable one, could be identified in the bacterial enzyme. This cluster included the His276, Asp278, Glu295, and Asn279 residues of the cyt b subunit (Fig. 6). After exploring the allowed motion of these residues, it appears that the cluster can rearrange to accommodate a coordinated Zn ion in a pseudooctahedral geometry, which includes, additionally, two water molecules characterized by the first-ligand distances reported in Table 4, model e. Thus, we proposed this cluster as the inhibitory Zn2+-binding site responsible for the effects previously observed in chromatophores from Rb. capsulatus (36,69).
FIGURE 6.

View of the proposed Zn2+-binding cluster in the bacterial cyt bc1 complex. Coordinates are from 1ZRT. Zn2+ ion was superimposed after alignment of this structure with that of the Zn crystal of the avian complex (Fig. 5 A, legend).
As shown in Fig. 6, alignment of the crystallographic structure of the cyt bc1 complex from Rb. capsulatus with that of Zn crystals of the avian counterpart located the proposed binding cluster somewhat displaced with respect to the position of the Zn2+ bound to the chicken complex, and closer to the Qo site. In addition, structural alignment could also suggest a contiguous, different Zn2+-binding site, which involves the His276, Asp278 (i.e., two residues belonging to the cluster we propose), and, additionally, His291 (see Fig. 6). Indeed, in an earlier study these residues were proposed as ligands of Zn2+ in the Rb. capsulatus complex (70). As the coordination number of 6 emerged clearly from the XANES analysis, depending on the bidentate or monodentate binding mode of the carboxylic acid, this cluster could be completed by two or three water molecules. Although there is no example of such Zn-binding sites in crystallographic data bases, a cluster formed by two His, one Asp, and three water molecules has been found by XRD analysis to bind Cd2+ in the RC of Rb. sphaeroides (30). Moreover the location of this Cd2+-binding site coincided with that of the high-affinity Zn2+-binding site (30). In the case of Zn2+, the coordination with two water molecules was lost, and the tetrahedral geometry was obtained. These features therefore make the cluster formed by His291, His276, Asp278, and three water molecules a possible alternative to model e of Table 4. However, as shown in Table 5, such a cluster (model h) provided a worse fit to the EXAFS spectrum measured in the bacterial complex, and it is noteworthy that the difference in goodness of fit was significant. The same conclusions hold for a putative binding cluster formed by His291, His276, Asp278 in bidentate configuration, and two water molecules (Table 5, model g).
As mentioned above, also the hexacoordinated cluster formed by two His, two Asp or Glu (one in monodentate and the other in bidentate configuration), and one Gln or Asn (Table 5, model e) deserves attention. Although it yields a worse fit than model e of Table 4, the difference in the reduced χ2 is not highly significant. However, inspection of the crystallographic structure of Rb. capsulatus shows that no cluster formed by these residues can be located in the region identified by the alignment of this structure with that of the Zn crystal of the avian enzyme. In addition, when considering His291, His276, and Asp278 as participating residues, a second carboxylic residue and a Gln or Asn are not found at coordinating distances, even considering large structural rearrangements. When moving to a possible contiguous cluster involving His276, Asp278, Glu295, and Asn279, a second His residue at coordinating distances is lacking. By integrating EXAFS and XANES analyses with the crystallographic information available for the avian and bacterial cyt bc1 complexes, we conclude therefore that the most probable binding site is formed by the His276, Asp278, Glu295, and Asn279 residues and two water molecules.
Glu295 is likely involved in H-bonding with stigmatellin (11). The fact that an almost stoichiometric binding of Zn2+ was obtained in our sample supplemented with this inhibitor could be thought to contraindicate Glu295 as a Zn2+ ligand. However, if this residue binds more tightly to the metal ion, it may well participate in the coordination cluster without feeling the presence of stigmatellin. Glu295 is also an attractive candidate for Zn2+ ligation because it is probably a ligand for ubiquinol at the Qo site and, in the absence of the metal ion, could carry the proton released by ubiquinol oxidation to one of the other Zn2+ ligands, participating in a proton pathway toward the surface of the complex (see below).
The crystallographic structure of the bacterial bc1 complex has been recently determined at higher resolution (3.2 Å) for the enzyme isolated from the related species Rb. sphaeroides (11). We aligned the structures of Rb. capsulatus and sphaeroides (2FYN) with that of the avian Zn-crystal complex, and examined the region of the Zn-binding site identified by our XAFS analysis. This procedure shows a close structural homology that includes all the residues in the vicinity of the proposed Zn-binding cluster. In particular, His276, Asp278, Glu295, and Asn279 are located at positions compatible with Zn coordination in the Rb. sphaeroides structure as well. This structural homology is consistent with the idea that in both species the proposed residues play a role in Zn binding at a site that interferes with the proton pathway from the Qo site to the aqueous phase (see below).
In Rb. capsulatus chromatophores the inhibitory action of Zn2+ depends on pH with an apparent pKa of ∼7.0 in the acidic range (69). This agrees with the value pKa = 7.2 found for Zn2+ inhibition in the mitochondrial bovine cyt bc1 complex (25). Studies on Cd2+ inhibition in Rb. sphaeroides reaction centers showed a similar pH dependence, with an apparent pKa of ∼6.8, which has been attributed to the His residues involved in divalent metal-ion binding and proton uptake (32). The presence of His residues in the Zn2+-binding sites identified by our analysis (one His residue in the cluster of the Rb. capsulatus cyt bc1 complex and two His residues in the site of the avian and bovine cyt bc1 complexes) points to a common mechanism of zinc inhibition. Accordingly, zinc would compete with protons for binding to the His residues, thus hampering their functions as proton donor/acceptors along the proton pathway. Tentative proton pathways connecting the Qo site with the water phase have been discussed in relation to the catalytic mechanism (7,17,71–73). A first pathway connects Glu272, which has been proposed as a proton acceptor from the Qo site (20), to the external phase through an H-bonded water network involving residues in the vicinity of the Zn01 site (7,71,72). Recently (17), another putative proton pathway has been considered, going directly through the His-residue-rich region of the Zn01 site. Zinc binding to the clusters shown in Fig. 5, A and B, could well interfere with proton egress along this latter route. As suggested in (70), an additional effect of Zn binding could consist of stiffening the ef loop of cyt b, the dynamics of which is considered to be associated to the ISP domain movement (74,75). Binding of Zn2+ to the proposed clusters could in fact hamper the flexibility of the ef loop by bridging Asp253 and His268 (Asp252 and His267) in the avian (bovine) enzymes, and His121 of cyt c1 could further reduce the mobility of the ef loop. The Zn2+-binding cluster proposed for the bacterial enzyme includes Glu295, which corresponds to Glu272 of the avian structure. Both structural data (6,72,76) and mutational studies (19,20,72) suggest that this residue is closely associated, possibly via a proton-coupled electron-transfer mechanism with QH2 oxidation at the Qo site. If this is also the case for the bacterial cyt bc1 complex, then binding of Zn2+ would indeed interfere with the entrance of the proton release pathway.
Acknowledgments
Measurements at ESRF were performed within the public user program. We are grateful to the staff of the GILDA beamline of ESRF for excellent support. The authors thank A. Y. Mulkidjanian, S. S. Klishin, and B. A. Melandri for stimulating discussions.
This work was supported by the Ministero dell'Università e della Ricerca of Italy (grant PRIN 2005, “Molecular mechanisms, physiology and pathology of membrane bioenergetics systems”, No. 2005052128), by a National Institutes of Health grant (GM 38237) to F.D., and by a National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grant (DK44842) to E.A.B.
Editor: Jill Trewhella.
References
- 1.Berry, E. A., M. Guergova-Kuras, L. S. Huang, and A. R. Crofts. 2000. Structure and function of cytochrome bc complexes. Annu. Rev. Biochem. 69:1005–1075. [DOI] [PubMed] [Google Scholar]
- 2.Darrouzet, E., J. W. Cooley, and F. Daldal. 2004. The cytochrome bc1 complex and its homologue the b6f complex: similarities and differences. Photosynth. Res. 79:25–44. [DOI] [PubMed] [Google Scholar]
- 3.Cooley, J. W., E. Darrouzet, and F. Daldal. 2004. Bacterial hydroquinone: cytochrome c oxidoreductases: physiology, structure, and function. In Respiration in Archaea and Bacteria, Vol. 1. D. Zannoni, editor. Kluwer Academic Publishers, Norwell, MA. 41–55.
- 4.Xia, D., C.-A. Yu, H. Kim, J.-Z. Xia, A. M. Kachurin, L. Zhang, L. Yu, and J. Deisenhofer. 1997. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science. 277:60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwata, S., J. W. Lee, K. Okada, J. K. Lee, M. Iwata, B. Rasmussen, T. A. Link, S. Ramaswamy, and B. K. Jap. 1998. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 281:64–71. [DOI] [PubMed] [Google Scholar]
- 6.Zhang, Z., L. Huang, V. M. Shulmeister, Y. I. Chi, K. K. Kim, L. W. Hung, A. R. Crofts, E. A. Berry, and S. H. Kim. 1998. Electron transfer by domain movement in cytochrome bc1. Nature. 392:677–684. [DOI] [PubMed] [Google Scholar]
- 7.Hunte, C., J. Koepke, C. Lange, T. Robmanith, and H. Michel. 2000. Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure. 8:669–684. [DOI] [PubMed] [Google Scholar]
- 8.Gao, X., X. Wen, L. Esser, B. Quinn, L. Yu, C. A. Yu, and D. Xia. 2003. Structural basis for the quinone reduction in the bc1 complex: a comparative analysis of crystal structures of mitochondrial cytochrome bc1 with bound substrate and inhibitors at the Qi site. Biochemistry. 42:9067–9080. [DOI] [PubMed] [Google Scholar]
- 9.Palsdottir, H., C. G. Lojero, B. L. Trumpower, and C. Hunte. 2003. Structure of the yeast cytochrome bc1 complex with a hydroxyquinone anion Qo site inhibitor bound. J. Biol. Chem. 278:31303–31311. [DOI] [PubMed] [Google Scholar]
- 10.Berry, E. A., L.-S. Huang, L. K. Saechao, N. G. Pon, M. Valkova-Valchanova, and F. Daldal. 2004. X-ray structure of Rhodobacter capsulatus bc1: comparison with its mitochondrial and chloroplast counterparts. Photosynth. Res. 81:251–275. [DOI] [PubMed] [Google Scholar]
- 11.Esser, L., X. Gong, S. Yang, L. Yu, C.-A. Yu, and D. Xia. 2006. Surface-modulated motion switch: capture and release of iron-sulfur protein in the cytochrome bc1 complex. Proc. Natl. Acad. Sci. USA. 103:13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crofts, A. R., S. W. Meinhardt, K. R. Jones, and M. Snozzi. 1983. The role of the quinone pool in the cyclic electron-transfer chain of Rhodopseudomonas sphaeroides: a modified Q-cycle mechanism. Biochim. Biophys. Acta. 723:202–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crofts, A. R. 2004. The cytochrome bc1 complex: function in the context of structure. Annu. Rev. Physiol. 66:689–733. [DOI] [PubMed] [Google Scholar]
- 14.Berry, E. A., and L.-S. Huang. 2003. Observations concerning the quinol oxidation site of the cytochrome bc1 complex. FEBS Lett. 555:13–20. [DOI] [PubMed] [Google Scholar]
- 15.Osyczka, A., C. C. Moser, F. Daldal, and P. L. Dutton. 2004. Reversible redox energy coupling in electron-transfer chains. Nature. 427:607–612. [DOI] [PubMed] [Google Scholar]
- 16.Rich, P. R. 2004. The quinone chemistry of bc complexes. Biochim. Biophys. Acta. 1658:165–171. [DOI] [PubMed] [Google Scholar]
- 17.Mulkidjanian, A. Y. 2005. Ubiquinol oxidation in the cytochrome bc1 complex: reaction mechanism and prevention of short-circuiting. Biochim. Biophys. Acta. 1709:5–34. [DOI] [PubMed] [Google Scholar]
- 18.Osyczka, A., C. C. Moser, and P. L. Dutton. 2005. Fixing the Q cycle. Trends Biochem. Sci. 30:176–182. [DOI] [PubMed] [Google Scholar]
- 19.Osyczka, A., H. Zhang, C. Mathé, P. R. Rich, C. C. Moser, and P. L. Dutton. 2006. Role of the PEWY glutamate in hydroquinone-quinone oxidation-reduction catalysis in the Qo site of cytochrome bc1. Biochemistry. 45:10492–10503. [DOI] [PubMed] [Google Scholar]
- 20.Wenz, T., P. Hellwig, F. MacMillan, B. Meunier, and C. Hunte. 2006. Probing the role of E272 in quinol oxidation of mitochondrial complex III. Biochemistry. 45:9042–9052. [DOI] [PubMed] [Google Scholar]
- 21.von Jagow, G., and T. A. Link. 1986. Use of specific inhibitors on the mitochondrial bc1 complex. Methods Enzymol. 126:253–271. [DOI] [PubMed] [Google Scholar]
- 22.Crofts, A. R. 1985. The mechanism of the ubiquinol: cytochrome c oxidoreductases of mitochondria and of Rhodopseudomonas sphaeroides. In The Enzymes of Biological Membranes. A. N. Martonosi, editor. Plenum Publishing, New York. 347–382.
- 23.Skulachev, V. P., V. V. Christyakov, A. A. Jasaitis, and E. G. Smirnova. 1967. Inhibition of the respiratory chain by zinc ions. Biochem. Biophys. Res. Commun. 26:1–6. [DOI] [PubMed] [Google Scholar]
- 24.Lorusso, M., T. Cocco, A. M. Sardanelli, M. Minuto, F. Bonomi, and S. Papa. 1991. Interaction of Zn2+ with the bovine-heart mitochondrial bc1 complex. Eur. J. Biochem. 197:555–561. [DOI] [PubMed] [Google Scholar]
- 25.Link, T. A., and G. von Jagow. 1995. Zinc ions inhibit the QP center of bovine heart mitochondrial bc1 complex by blocking a protonatable group. J. Biol. Chem. 270:25001–25006. [DOI] [PubMed] [Google Scholar]
- 26.Kannt, A., T. Ostemann, H. Muller, and M. Ruitenberg. 2001. Zn2+ binding to the cytoplasmic side of Paracoccus denitrificans cytochrome c oxidase selectively uncouples electron transfer and proton translocation. FEBS Lett. 503:142–146. [DOI] [PubMed] [Google Scholar]
- 27.Faxen, K., L. Salomonsson, P. Ädelroth, and P. Brzezinski. 2006. Inhibition of proton pumping by zinc ions during specific steps in cytochrome c oxidase. Biochim. Biophys. Acta. 1757:388–394. [DOI] [PubMed] [Google Scholar]
- 28.Utschig, L. M., Y. Ohigashi, M. C. Thurnauer, and D. M. Tiede. 1998. A new metal-binding site in photosynthetic bacterial reaction centers that modulates QA to QB electron transfer. Biochemistry. 37:8278–8281. [DOI] [PubMed] [Google Scholar]
- 29.Paddock, M. L., M. S. Graige, G. Feher, and M. Y. Okamura. 1999. Identification of the proton pathway in bacterial reaction centers: inhibition of proton transfer by binding of Zn2+ or Cd2+. Proc. Natl. Acad. Sci. USA. 96:6183–6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Axelrod, H. L., E. C. Abresch, M. L. Paddock, M. Y. Okamura, and G. Feher. 2000. Determination of the binding site of the proton trasfer inhibitors Cd2+ and Zn2+ in bacterial reaction centers. Proc. Natl. Acad. Sci. USA. 97:1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giachini, L., F. Francia, A. Mallardi, G. Palazzo, E. Carpenè, F. Boscherini, and G. Venturoli. 2005. Multiple scattering x-ray absorption studies of Zn2+ binding sites in bacterial photosynthetic reaction centers. Biophys. J. 88:2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paddock, M. L., L. Sagle, A. Tehrani, J. T. Beatty, G. Feher, and M. Y. Okamura. 2003. Mechanism of proton transfer inhibition by Cd2+ binding to bacterial reaction centers: determination of the pKA of functionally important histidine residues. Biochemistry. 42:9626–9632. [DOI] [PubMed] [Google Scholar]
- 33.Ädelroth, P., M. L. Paddock, L. B. Sagle, G. Feher, and M. Y. Okamura. 2000. Identification of the proton pathway in bacterial reaction centers: both protons associated with reduction of QB to QBH2 share a common entry point. Proc. Natl. Acad. Sci. USA. 97:13086–13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleiner, D., and G. von Jagow. 1972. On the inhibition of mitochondrial electron transport by Zn2+ ions. FEBS Lett. 20:229–232. [DOI] [PubMed] [Google Scholar]
- 35.Kleiner, D. 1974. The effect of Zn2+ ions on mitochondrial electron transport. Arch. Biochem. Biophys. 165:121–125. [DOI] [PubMed] [Google Scholar]
- 36.Klishin, S. S., W. Junge, and A. Y. Mulkidjanian. 2002. Flash-induced turnover of the cytochrome bc1 complex in chromatophores of Rhodobacter capsulatus: binding of Zn2+ decelerates likewise the oxidation of cytochrome b, the reduction of cytochrome c1 and the voltage generation. Biochim. Biophys. Acta. 1553:177–182. [DOI] [PubMed] [Google Scholar]
- 37.Berry, E. A., Z. Zhang, H. D. Bellamy, and L. Huang. 2000. Crystallographic location of two Zn2+-binding sites in the avian cytochrome bc1 complex. Biochim. Biophys. Acta. 1459:440–448. [DOI] [PubMed] [Google Scholar]
- 38.Hasnain, S. S., and K. O. Hodgson. 1999. Structure of metal centres in proteins at subatomic resolution. J. Synchrotron Rad. 6:852–864. [Google Scholar]
- 39.Valchova-Valchanova, M. B., A. S. Saribas, B. R. Gibney, P. L. Dutton, and F. Daldal. 1998. Isolation and characterization of a two-subunit cytochrome b-c1 subcomplex from Rhodobacter capsulatus and reconstitution of its ubihydroquinone oxidation (Qo) site with purified Fe-S protein subunit. Biochemistry. 37:16242–16251. [DOI] [PubMed] [Google Scholar]
- 40.Berry, E. A., L.-S. Huang, and V. J. DeRose. 1991. Ubiquinol-cytochrome c oxidoreductase of higher plants. J. Biol. Chem. 266:9064–9077. [PubMed] [Google Scholar]
- 41.Robertson, D. E., H. Ding, P. R. Chelmiski, C. Slaughter, J. Hsu, C. Moomaw, M. Tokito, F. Daldal, and P. L. Dutton. 1993. Hydroubiquinone- cytochrome c2 oxidoreductase from Rhodobacter capsulatus: Definition of a minimal, functional isolated preparation. Biochemistry. 32:1310–1317. [DOI] [PubMed] [Google Scholar]
- 42.Rehr, J. J., and R. C. Albers. 2000. Theoretical approaches to X-ray absorption fine structure. Rev. Mod. Phys. 72:621–654. [Google Scholar]
- 43.Lee, P. A., P. H. Citrin, P. Eisenberger, and B. M. Kincaid. 1981. Extended x-ray absorption fine structure: its strengths and limitations as a structural tool. Rev. Mod. Phys. 53:769–806. [Google Scholar]
- 44.Ankudinov, A. L., B. Ravel, J. J. Rehr, and S. D. Conradson. 1998. Real space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Phys. Rev. B. 58:7565–7576. [Google Scholar]
- 45.Pascarelli, S., F. Boscherini, F. D'Acapito, J. Hardy, C. Meneghini, and S. Mobilio. 1996. X-ray optics of a dynamical sagittal focussing monochromator on the GILDA beamline at ESRF. J. Synchrotron Rad. 3:147–155. [DOI] [PubMed] [Google Scholar]
- 46.Ciatto, G., F. D'Acapito, F. Boscherini, and S. Mobilio. 2004. Treatment of EXAFS data taken in the fluorescence mode in non-linear conditions. J. Synchrotron Rad. 11:278–283. [DOI] [PubMed] [Google Scholar]
- 47.Newville, M., P. Livins, Y. Yacoby, E. A. Stern, and J. J. Rehr. 1993. Near-edge x-ray-absorption fine structure of Pb: a comparison of theory and experiment. Phys. Rev. B. 47:14126–14131. [DOI] [PubMed] [Google Scholar]
- 48.Ravel, B., and M. Newville. 2005. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12:537–541. [DOI] [PubMed] [Google Scholar]
- 49.Newville, M., B. Ravel, D. Haskel, J. J. Rehr, E. A. Stern, and Y. Yacoby. 1995. Analysis of multiple scattering XAFS data using theoretical standards. Physica B (Amsterdam). 208/209:154–155. [Google Scholar]
- 50.Stern, E. A., M. Newville, B. Ravel, Y. Yacoby, and D. Haskel. 1995. The UWXAFS analysis package: philosophy and details. Physica B. 208/209:117–120. [Google Scholar]
- 51.Levina, A., R. S. Armstrong, and P. A. Lay. 2005. Three-dimensional structure determination using multiple-scattering analysis of XAFS: applications to metalloproteins and coordination chemistry. Coord. Chem. Rev. 249:141–160. [Google Scholar]
- 52.Kelly, S. D., K. M. Kenner, G. E. Fryxell, J. Liu, S. V. Mattigod, and K. F. Ferris. 2001. X-ray-absorption fine-structure spectroscopy study of the interactions between contaminant tetrahedral anions and self-assembled monolayers on mesoporous supports. J. Phys. Chem. B. 105:6337–6346. [Google Scholar]
- 53.Ugliengo, P., D. Viterbo, and G. Chiari. 1993. MOLDRAW: molecular graphics on a personal computer. Z. Kristallogr. 207:9–23. [Google Scholar]
- 54.Cheung, K., R. W. Strange, and S. Hasnain. 2000. 3D EXAFS refinement of the Cu site of azurin sheds light on the nature of structural change at the metal centre in an oxidation-reduction process: an integrated approach combining EXAFS and crystallography. Acta Crystallogr. D. 56:697–704. [DOI] [PubMed] [Google Scholar]
- 55.Alberts, I. L., K. Nadassy, and S. J. Wodak. 1998. Analysis of zinc binding sites in protein crystal structures. Protein Sci. 7:1700–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Engh, R., and R. Huber. 1991. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A. 47:392–400. [Google Scholar]
- 57.Ryde, U. 1999. Carboxylate binding modes in zinc proteins: a theoretical study. Biophys. J. 77:2777–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scherk, C. G., A. Ostermann, K. Achterhold, O. Iakovleva, C. Nazikoll, B. Krebs, E. V. Knapp, W. Meyer-Klaucke, and F. G. Parak. 2001. The X-ray absorption spectroscopy Debye-Waller factors of an iron compounds and met-myoglobin as a function of temperature. Eur. Biophys. J. 30:393–403. [DOI] [PubMed] [Google Scholar]
- 59.Binsted, N., R. W. Strange, and S. S. Hasnain. 1992. Constrained and restrained refinement in EXAFS data analysis with curved wave theory. Biochemistry. 31:12117–12125. [DOI] [PubMed] [Google Scholar]
- 60.Dimakis, N., and G. Bunker. 2004. XAFS Debye-Waller factors for Zn metalloproteins. Phys. Rev. B. 70:195114. [Google Scholar]
- 61.Francia, F., L. Giachini, F. Boscherini, G. Venturoli, G. Capitanio, P. L. Martino, and S. Papa. 2007. The inhibitory binding site(s) of Zn2+ in cytochrome c oxidase. FEBS Lett. 581:611–616. [DOI] [PubMed] [Google Scholar]
- 62.Mijovilovich, A., and W. Meyer-Klauke. 2003. Simulating the XANES of metalloenzymes: a case study. J. Synchrotron Rad. 10:64–68. [DOI] [PubMed] [Google Scholar]
- 63.Samar Hasnain, S., and R. W. Strange. 2003. Marriage of XAFS and crystallography for structure-function studies of metalloproteins. J. Synchrotron Rad. 10:9–15. [DOI] [PubMed] [Google Scholar]
- 64.Yano, J., J. Kern, K.-D. Irgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni, and V. K. Yachandra. 2005. X-ray damage of the Mn4Ca complex in single crystals of photosystem II: a case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. USA. 102:12047–12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giachini, L., F. Francia, L. Cordone, F. Boscherini, and G. Venturoli. 2007. Cytochrome c in a dry trehalose matrix: structural and dynamical effects probed by x-ray absorption spectroscopy. Biophys. J. 92:1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brugna, M., S. Rodgers, A. Schricker, G. Montoya, M. Kazmeier, W. Nitschke, and I. Sinning. 2000. A spectroscopic method for observing the domain movement of the Rieske iron-sulfur protein. Proc. Natl. Acad. Sci. USA. 97:2069–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berry, E. A., L. S. Huang, Z. Zhang, and S. H. Kim. 1999. Structure of the avian mitochondrial cytochrome bc1 complex. J. Bioenerg. Biomembr. 31:177–190. [DOI] [PubMed] [Google Scholar]
- 68.Esser, L., B. Quinn, Y. F. Li, M. Zhang, E. Berry, L. Yu, C. A. Yu, and D. Xia. 2004. Crystallographic studies of quinol oxidation site inhibitors: a modified classification of inhibitors for the cytochrome bc1 complex. J. Mol. Biol. 341:281–302. [DOI] [PubMed] [Google Scholar]
- 69.Klishin, S. S. 2005. Resolution of partial steps in the catalytic cycle of Rhodobacter capsulatus cytochrome bc1 complex with help of zinc ions. PhD thesis, Lomonosov University, Moscow.
- 70.Mulkidjanian, A. Y. 2007. Proton translocation by the cytochrome bc1 complex of phototrophic bacteria: introducing the activated Q-cycle. Photochem. Photobiol. Sci. 6:19–34. [DOI] [PubMed] [Google Scholar]
- 71.Izrailev, S., A. R. Crofts, E. A. Berry, and K. Shulten. 1999. Steered molecular dynamics simulations of the Rieske subunit motion in the cytochrome bc1 complex. Biophys. J. 77:1753–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crofts, A. R., S. Hong, N. Ugulava, B. Barquera, R. Gennis, M. Guergova-Kuras, and E. A. Berry. 1999. Pathways for proton release during ubihydroquinone oxidation by the bc1 complex. Proc. Natl. Acad. Sci. USA. 96:10021–10026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hunte, C., H. Palsdottir, and B. L. Trumpower. 2003. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett. 545:39–46. [DOI] [PubMed] [Google Scholar]
- 74.Darrouzet, E., and F. Daldal. 2002. Movement of the iron-sulfur subunit beyond the ef loop of cytochrome b is required for multiple turnovers of the bc1 complex but not for single turnover Qo site catalysis. J. Biol. Chem. 277:3471–3476. [DOI] [PubMed] [Google Scholar]
- 75.Darrouzet, E., and F. Daldal. 2003. Protein-protein interactions between cytochrome b and the Fe-S protein subunits during QH2 oxidation and large-scale domain movement in the bc1 complex. Biochemistry. 42:1499–1507. [DOI] [PubMed] [Google Scholar]
- 76.Berry, E. A., and L. Huang. 2003. Observations concerning the quinol oxidation site of the cytochrome bc1 complex. FEBS Lett. 555:13–20. [DOI] [PubMed] [Google Scholar]