

Abstract
The presence of one P450 can influence the catalytic characteristics of a second enzyme through the formation of heteromeric P450 complexes. Such a complex has been reported for mixed reconstituted systems containing NADPH-cytochrome P450 reductase, CYP2B4 and CYP1A2, where a dramatic inhibition of 7-pentoxyresorufin-O-dealkylation (PROD) was observed when compared to simple reconstituted systems containing reductase and a single P450 enzyme. The goal of the present study was to characterize this interaction by examining the potential of the CYP1A2-CYP2B4 complex to be formed by charge-pair interactions. With ionic interactions being sensitive to the surrounding ionic environment, monooxygenase activities were measured in both simple systems and mixed reconstituted systems as a function of ionic strength. PROD was found to be decreased at high ionic strength in both simple and mixed reconstituted systems, due to disruption of reductase-P450 complexes. Additionally, the inhibition of PROD in mixed reconstituted systems was relieved at high ionic strength, consistent with disruption of the CYP2B4-CYP1A2 complex. When ionic strength was measured as a function of CYP1A2 concentration, a shift to the right in the inflection point of the biphasic curve occurred at high ionic strength, consistent with a loss in CYP1A2 affinity for CYP2B4. When this analysis was applied to the same systems using a different substrate, 7-EFC, evidence for a high affinity complex was not observed, demonstrating that the characteristics of the CYP1A2-CYP2B4 complex are influenced by the substrates present. These results support the role for a substrate specific electrostatic interaction between these P450 enzymes.
P450 catalyzes the oxidative metabolism of a wide variety of both exogenous and endogenous substrates. The enzyme system splits a dioxygen molecule, incorporating one atom of oxygen into bound substrate with the other atom of oxygen forming water. The P450 enzymes involved in foreign compound metabolism are generally found in the microsomal fraction, and receive electrons from NADPH via the flavoprotein NADPH cytochrome P450 reductase (reductase) (1;2). Electron transfer to this hemoprotein requires the formation of a 1:1 molar complex between the reductase and cytochrome P450 (3;4). The reductase sequentially transfers two electrons to P450, one before and one after the binding of molecular oxygen (5;6). In some cases cytochrome b5 can transfer the second electron to selective P450s (7). Since the identification of the components of the P450 system (8;9), several studies have been directed towards understanding the molecular association of P450 with its redox partners. Interactions between P450 and redox partners, cytochrome b5 and reductase, have been described as being predominately electrostatic in nature (10-18). An electron transfer complex is facilitated by the pairing of basic residues on the proximal face of P450 with acidic residues of P450 reductase (10;11;19) and cytochrome b5 (20;21). Although basic requirements for these interactions are understood, the organization of the P450 proteins and their redox partners remains unclear.
One of the major factors that confound our ability to understand the interactions of the P450 microsomal monooxygenase system is the multiplicity of cytochrome P450s. Over fifty functional P450s are expressed in human tissue (22). Additionally, when looking at the quantities of these proteins in their native membranes, P450s generally exist in a large excess over reductase. P450s must effectively interact with reductase or be metabolically silent. To further complicate our understanding of P450 architecture, these limiting concentrations of reductase are also required to transfer reducing equivalents to other microsomal proteins such as cytochrome b5, steroyl CoA desaturase, phospholipid desaturase and heme oxygenase (23;24). The presence of multiple P450 enzymes among limiting levels of reductase raises questions as to how the enzymes of the P450 system are organized in the microsomal membrane, and whether the presence of one P450 enzyme can affect the function of another P450.
Physical and functional P450-P450 interactions have previously been described by a number of laboratories (25-31). The potential for P450 enzymes to interact raises the possibility that both homomeric and heteromeric P450-P450 interactions can influence the function of these enzymes. The potential for one P450 enzyme to affect the catalytic behavior of another P450 enzyme using reconstituted systems and in rabbit liver microsomes has recently been examined (31-33). Functional interactions between CYP1A2 and CYP2B4 were demonstrated by comparing catalytic behavior in simple and mixed reconstituted systems. The results are consistent with CYP1A2 and CYP2B4 forming a heteromeric complex. Interestingly, the CYP1A2 moiety of this CYP1A2-CYP2B4 complex is capable of high affinity binding with reductase (31). The objective of the present study was to determine if complex formation between CYP1A2 and CYP2B4 is mediated by electrostatic interactions. Several groups have demonstrated the modulatory effects of electrostatic charge on various aspects of P450 metabolism, where multiple steps in this monooxygenase electron transport chain are affected (10;15;17;18;34-38). The effects of salt were shown to be dependent upon the P450s selected and the concentration of reductase (13;14;16). Many of these studies have focused on the effects of increasing ionic strength on the function of a particular P450 enzyme. The goal of the following experiments was to test the hypothesis that the heteromeric CYP1A2-CYP2B4 complex is governed by charge pairing and that the complex can be disrupted by changes in ionic strength.
MATERIALS AND METHODS
Chemicals
β-naphthoflavone (βNF), sodium phosphate, magnesium chloride, HEPES, EDTA, a protease inhibitor cocktail (containing: AEBSF, EDTA, Bestatin, Pepstatin A, and E-64) and glycerol were obtained from Sigma (St. Louis, MO). C41 cells were purchased through Avidis SA (Biopole Clermont-Limagne, France). Plasmid Mini, Midi, and Maxi kits were purchased from Qiagen Inc. (Valencia, CA). All restriction enzymes were purchased from New England Biolabs (Beverly, Ma). Protein extraction reagent BPER® and Alkaline Phosphatase immunoblot developing kit were obtained from Pierce Chemical Company (Rockford, IL). Polyclonal Goat anti-Rabbit CYP1A2 and CYP2B4 antibodies were purchased from Oxford Biomedical Research (Oxford, MI). The substrates, 7-pentoxyresorufin (7-PR) and reference standard resorufin were purchased from Sigma (St. Louis, MO); 7-ethoxy-4-trifluromethylcoumarin (7-EFC), and reference standard 7-hydroxy-4-trifluoromethylcoumarin (7-HFC) were purchased from Molecular Probes, Inc. (Eugene, Or).
Protein isolation
Recombinant rabbit NADPH cytochrome P450 reductase (plasmid: pSC-CPR, provided by Lucy Waskell (Univ. Michigan); constructed from plasmid pCWori-rabbit reductase and plasmid pOR263-rat reductase, utilizing a T7 promoter) was expressed in C41 E. coli, solubilized and purified according to a modification of previously described methods (39;40). Minor modifications were made for the expression and purification protocol of recombinant rabbit CYP2B4 (41) to improve efficiency of CYP2B4 protein purification. The CM Sepharose and Bio-beads steps of protein purification were eliminated. A protease inhibitor cocktail solution was used during solubilization. Alternating freeze-thaw with sonication was used to facilitate solubilization of CYP2B4. The final solubilized fraction from the (41) protocol was centrifuged at 105 g for 60 min rendering the supernatant clear of particulate matter prior to application to the DE52 column. CYP1A2 was purified from liver microsomes isolated from βNF treated rabbits as previously described (42). CYP2B4 had a specific content of 17.1 nmol/mg protein. Two preparations of CYP1A2 were used in this study, one of which had a specific content of 14.2 nmol/mg protein. Unfortunately, the supply of the other preparation was exhausted and we had not determined its specific content.
P450 levels were determined by measuring the carbon monoxy-ferrous complex using an extinction coefficient of 91 mM-1 cm-1 (43). The Lowry protein assay was used to determine total protein concentration of the purified protein preparations using a BSA standard curve (44). NADPH-cytochrome P450 reductase content was determined from the absolute spectrum at 456 nm, using an extinction coefficient of 21.4 mM-1 cm-1 (45). The purity of reductase, CYP2B4 and CYP1A2 were assessed by Coomassie Blue staining and immune blotting.
Reconstituted Systems
Catalytic activities of CYP1A2 and CYP2B4 were determined using binary reconstituted systems where (1) reductase and CYP2B4, or (2) reductase and CYP1A2, and mixed reconstituted systems (3) containing reductase, CYP2B4 and CYP1A2, were combined in DLPC. DLPC was prepared at a stock concentration of 8 mM in 50 mM potassium phosphate buffer, pH 7.25, containing 20% glycerol, 0.1 M NaCl, and 5 mM EDTA. The DLPC stock suspension was sonicated for approximately 30 min using a bath sonicator, leading to significant clarification of the solution. The sonicated DLPC was combined with reductase and P450 at the concentrations described in the results section and preincubated for 2 hr at room temperature. Unless otherwise stated, experiments used a DLPC:P450 molar ratio of 160:1, except when varying the CYP1A2 concentration, where 160:1 pertains to DLPC:CYP2B4. The optimal ratios of phospholipid to P450 were based on previous studies (46). After mixing the proteins with the liposomes, the reconstituted systems were preincubated at room temperature for 2 h before the addition of the other assay components. The reductase and P450 concentrations were approximately 4μM and 8μM, respectively during the preincubation step. These preincubation conditions permit the formation of stable interactions among the liposomal proteins (46). After preincubation, the reconstituted systems were diluted with buffer and other assay components, and assayed within 30 seconds at 37 °C. Unless otherwise stated, the final assay conditions for 7-pentoxyresorufin-O-deethlation (PROD), and 7-ethoxytriflurocoumarin (7-EFC) deethylation were the reconstituted system (containing 0.05 μM P450, 0.025 μM reductase with a single P450, and 0.05 μM for each P450 and 0.05 μM reductase for the mixed reconstituted systems), and substrate (2.5 μM for 7-PR; 10 μM for 7-EFC), in various buffers (pH = 7.4). The buffer and salt concentrations used are described in the figure legends. Reactions were initiated by the addition of NADPH to a final concentration of 0.5 mM. An Aminco Bowman Series 2 spectrofluorometer (Spectrum Unicam, Rochester, NY) was used to measure the rate of resorufin and 7-triflurocoumarin fluorescence using excitation and emission wavelengths of 559 and 585 for 7-PR, and 410 and 510 for 7-EFC. Rates of resorufin production were determined from the real time scans of fluorescence emission. Resorufin and 7-triflurocoumarin were used for generation of standard curves.
Molecular Modeling
Experimental data were fit to two models using the DynaFit modeling program (47). The data were fit to a model where CYP1A2 and CYP2B4 could bind to reductase, but could not form kinetically observable CYP1A2-CYP2B4 complexes. This is referred to in the text as the simple competitive model. In the other model that was tested, CYP1A2 and CYP2B4 were able to form a CYP1A2-CYP2B4 complex. This complex had an altered ability to bind reductase; i.e. reductase bound to the CYP1A2 moiety of this complex with high affinity. According to this model, reductase would selectively bind to the CYP1A2 moiety of the P450-P450 complex and not be available for binding to CYP2B4. This model has been described in detail in previous reports (31).
RESULTS
Effect of Ionic Strength on P450-mediated Activities
As reported previously, the combination of CYP1A2 and CYP2B4 in a mixed reconstituted system leads to a significant inhibition of 7-PR dealkylation at subsaturating reductase. These results are consistent with the formation of a heteromeric complex between these P450s (31). The goal of this study is to determine if the interaction between these P450s is mediated by charge pairing. As reported with this and other enzyme systems, electrostatic interactions can be disrupted by increasing the ionic strength of the solvent (13-18). If CYP1A2 and CYP2B4 form charge pair complexes, disruption of the complex would be expected as the ionic strength is elevated. Because this P450-P450 complex inhibits 7-PR dealkylation, then reversal of the inhibitory effect would be expected with increasing ionic strength. However a complicating factor is that reductase and P450 have been shown to form charge-pair complexes. Disruption of the reductase P450 complex would be expected to lead to a continued decrease in monooxygenase activity. The effects of increasing concentrations of sodium phosphate (pH 7.4), potassium acetate (pH 7.4) and HEPES buffer (pH 7.4) on PROD in mixed and binary systems are shown in figure 1. As seen in figure 1a, PROD activity of the simple reconstituted system containing reductase and CYP2B4, reached a maximum at 50 mM sodium phosphate. In contrast, maximal PROD activity of the simple CYP1A2-containing reconstituted system was observed at 100 mM sodium phosphate. These results are consistent with disruption of the reductase P450 complexes at higher ionic strength (13-16).
Figure 1. Effect of CYP2B4 and CYP1A2 mediated PROD metabolism as a function of monovalent ion concentration.
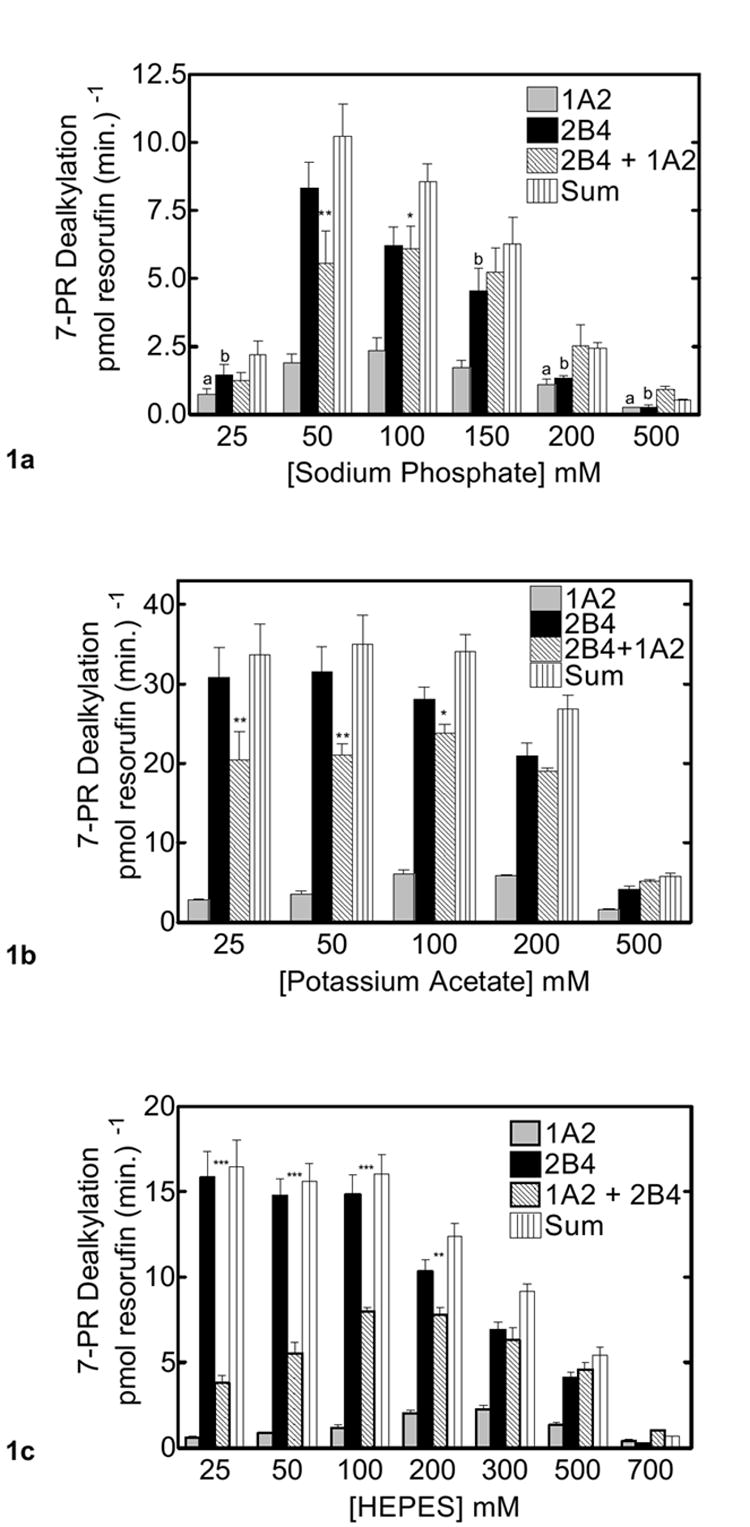
CYP2B4, CYP1A2 and reductase were reconstituted in binary (one P450 and reductase) and ternary (mixed P450s and reductase) reconstituted systems. The first and second bars in each group represent binary CYP1A2 and CYP2B4 reconstituted activities, respectively. The third bar represents the results from the mixed reconstituted system, and the fourth bar represents the sum of the rates of the binary systems (sum of the first and second bars). PROD activity was measured under subsaturating conditions (0.5:1.0 reductase:P450). Each binary reconstituted system contained 0.05 μM P450 (CYP1A2 or CYP2B4 alone), 0.025 μM reductase (CPR), and 8.0 μM DLPC. The mixed reconstituted system contained 0.05 μM of both CYP1A2 and CYP2B4, and 0.05 μM CPR. Groups represent the mean ± SEM for three determinations. Significant differences in activities between the sum of the two binary systems compared to the mixed reconstituted system are indicated (*, p < 0.05; **, p < 0.01; ***, p < 0.001). (a) Effect of varying sodium phosphate on PROD activity in simple and mixed reconstituted systems. The letter ‘a’ above the bars in this panel represents a significant difference from the CYP1A2 data measured at the highest activity (i.e. 100 mM). The letter ‘b’ above the bars represents a significant difference from the corresponding CYP2B4 data measured at its highest activity (i.e. 50 mM). (b) Effect of varying potassium acetate on PROD activity in simple and mixed reconstituted systems. (c) Effect of varying HEPES on PROD activity in simple and mixed reconstituted systems.
As previously reported (32), PROD activity in the mixed reconstituted system was lower than the sum of the rates of the binary systems. (The third bar in each group represents the results from the mixed reconstituted system, whereas the fourth bar represents the sum of the rates of the simple binary systems). This inhibition in PROD was observed at both 50 and 100 mM sodium phosphate. As the ionic strength of the buffer was increased above 100 mM, the inhibition observed at lower ionic strengths was diminished. In fact, at elevated buffer concentrations, the results in the mixed reconstituted system were similar to the sum of rates of the simple binary systems. These results are consistent with ionic strength disrupting the CYP1A2-CYP2B4 complex. Similar results were observed when using different ions to disrupt the functional interaction between CYP2B4 and CYP1A2. Figure 1b shows the effects of varying potassium acetate. Potassium acetate supports higher activities across ionic strengths compared to sodium phosphate, whereas the effects of HEPES (Fig 1c) are intermediate to those of sodium phosphate and potassium acetate. In each case, inhibition was observed in the mixed reconstituted system at low ionic strength. Consistent with the results in figures 1a and 1b, this inhibition in the mixed system was disrupted as ionic strength was elevated. These results are consistent with complex formation between CYP2B4 and CYP1A2 where this interaction is mediated at least in part by electrostatic attraction.
In order to determine if the valence of the cation affects the functional interaction between CYP1A2 and CYP2B4, we measured PROD activity when varying the divalent cation magnesium. HEPES buffer (50 mM, pH 7.4) containing varying concentrations of magnesium chloride were used to test the inhibition of PROD (Fig 2). Maximal activities for CYP2B4, CYP1A2, and the mixed system containing both P450s were observed at the lowest concentration tested, 25 mM magnesium chloride in 50 mM HEPES. In each of these systems, a sharp decline in activity occurred as the salt concentration was increased, consistent with disruption of ionic interactions between reductase and the P450s. Again, PROD was inhibited at lower ionic strength in the mixed reconstituted system. Consistent with the data in figure 1, as the magnesium chloride concentration was increased, the inhibition of PROD in the mixed reconstituted system was disrupted. Thus, the influence of a divalent cation on the mixed reconstituted system containing both CYP2B4 and CYP1A2 was similar to that of the other buffers tested. This phenomenon was observed regardless of the ions used, and is consistent with P450 charge pair associations not only of reductase and P450, but also of heteromeric P450 complexes.
Figure 2. Effect of CYP2B4 and CYP1A2 mediated PROD metabolism as a function of divalent cation concentration.
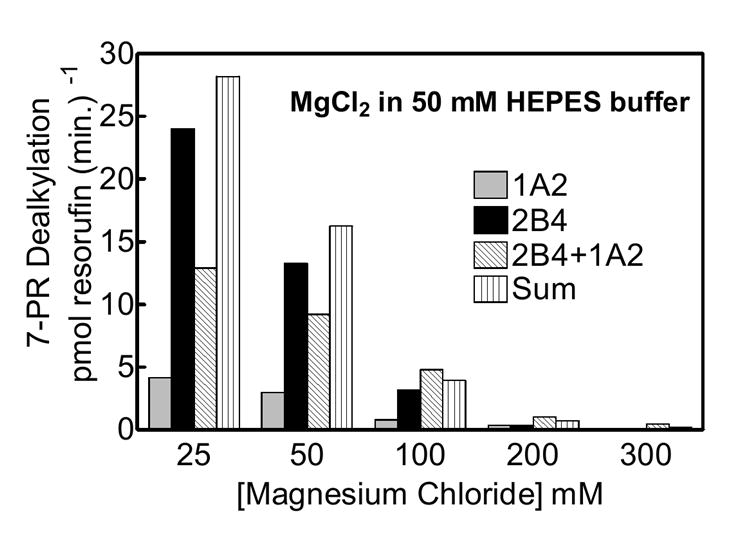
Experimental conditions are the same as figure 1 with the exception of buffer medium. Varying amounts of the divalent cation magnesium chloride in 50 mM HEPES buffer was used to test the effects of ionic strength on P450 metabolism in the binary and ternary systems shown.
The high affinity complex between CYP1A2 and CYP2B4 has been shown to be substrate-dependent (32). Therefore, we were interested in examining the effect of ionic strength on a substrate not exhibiting this pronounced inhibitory response. 7-ethoxytrifluromethylcoumarin (7-EFC) was selected, for like PROD, CYP2B4 more effectively catalyzes this substrate when compared to CYP1A2. The results are shown in figure 3. CYP2B4 activity reached a maximum at 50 mM HEPES, while CYP1A2 7-EFC activity achieved a maximum near 100 mM, consistent with the 7-PR results. A similar, but less dramatic inhibition was observed with 7-EFC in the mixed system as compared to PROD (Fig 1). At low ionic strength, the mixed reconstituted system exhibited a 40% inhibition (up to 100 mM HEPES) when compared to the sum of the rates of the binary systems (Fig 3). Although this effect was significant, it was not nearly as large as that observed with PROD under similar conditions (Fig. 1c). As the HEPES concentration was increased, inhibition of 7-EFC activity in the mixed RCS was diminished.
Figure 3. Effect of CYP2B4 and CYP1A2 mediated 7-EFC metabolism as a function of ionic strength.
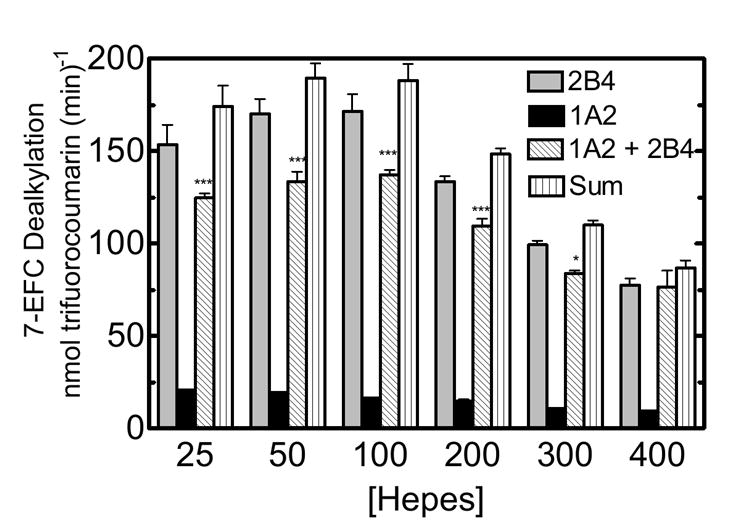
CYP2B4, CYP1A2 and reductase were reconstituted in binary (one P450) and ternary (mixed P450s) reconstituted systems. Experimental conditions are described in materials and methods and figure 1. Each group represents the mean ± SEM for three determinations. Significant differences in activities between the sum of the two binary systems compared to the mixed reconstituted system are indicated (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
CYP1A2 and CYP2B4 Interactions as a Function of Reductase Concentration
Previous studies in our lab have evaluated PROD activity in both simple and mixed reconstituted systems as a function of reductase concentration. The goal of the current study was to examine the effect of ionic strength on this response (Fig 4). PROD activity was examined as a function of reductase in both simple and mixed reconstituted systems in 50 mM HEPES buffer (Fig 4a). As mentioned previously, CYP2B4 (in the simple RCS) was shown to be a more effective enzyme for PROD when compared to CYP1A2. The mixed system containing both CYP1A2 and CYP2B4 at low ionic strength (50 mM Hepes) exhibited a substantial degree of inhibition at subsaturating reductase, rendering “S” shaped curves, rather than the hyperbolic shaped curves typically expected for simple Michaelis-Menten types of interactions. These results are similar to those published previously (31). CYP2B4 exhibited a typical hyperbolic curve. CYP1A2 alone did not exhibit a typical hyperbolic curve, but appeared to saturate as the reductase concentration reached 0.15 - 0.2 μM. This is consistent with our previous findings (31). When CYP2B4 and CYP1A2 were present together in the mixed reconstituted systems, a pronounced inhibition of PROD was observed at sub-saturating reductase. For example, at 0.05 μM reductase, mixed system activity was 80% inhibited when compared to CYP2B4 alone.
Figure 4. Effects of ionic strength on PROD as a function of reductase concentration.
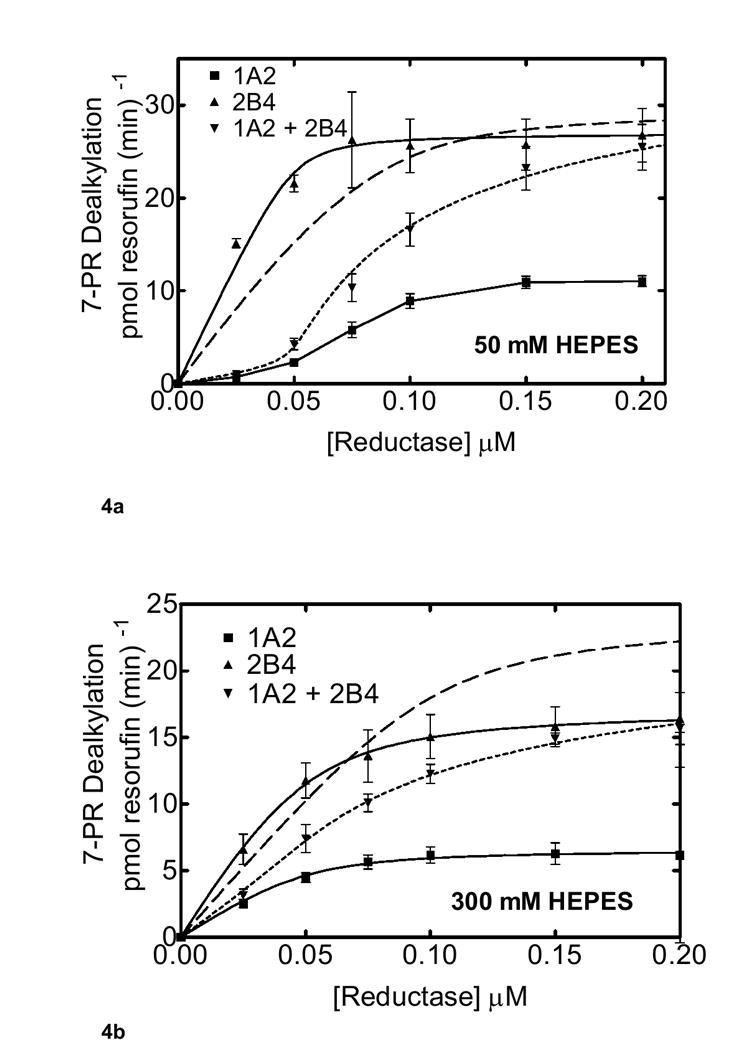
Pre-incubation of the binary systems containing 0.05 μM for both CYP2B4 and CYP1A2 in both the binary and ternary systems are described in Methods. The reductase concentration was varied as indicated. Groups represent the mean ± SEM for three determinations. The experimental data were fit to a model where CYP1A2 and CYP2B4 form a complex that influences reductase binding and are shown as a dotted line (Scheme 2). The dashed line shows the best fit based on model only allowing the formation of reductase-CYP1A2 and reductase-CYP2B4 complexes (Scheme 1). (a) Effect of mixed reconstitution of CYP1A2 and CYP2B4 on the reductase dependence of PROD at 50 mM HEPES. Concentrations of reductase were varied at the lower ionic strength (50 mM HEPES) buffer for CYP1A2, CYP2B4 and the mixed system containing both P450 enzymes. (b) Effect of mixed reconstitution of CYP1A2 and CYP2B4 on the reductase dependence of PROD (300 mM HEPES).
The data from the mixed reconstituted systems were analyzed using two models. In the first model, CYP2B4 and CYP1A2 could not form a heteromeric complex, and would compete for the available reductase (Scheme 1). Simulation of the expected data is shown as the dashed line in Fig. 4a. According to the second model (Scheme 2), CYP1A2 and CYP2B4 could form the heteromeric complex. Consequently, there are several potential complexes that could contribute to PROD, (a) reductase-CYP1A2, (b) reductase-CYP2B4, (c) reductase-CYP1A2-CYP2B4, (d) CYP1A2-CYP2B4-reductase, and (e) reductase-CYP1A2-CYP2B4-reductase. This model has been described in previous reports (31;48). A simulation of the expected data is shown as the dotted line in Fig. 4a, and fits the experimental data points quite well.
Scheme 1.
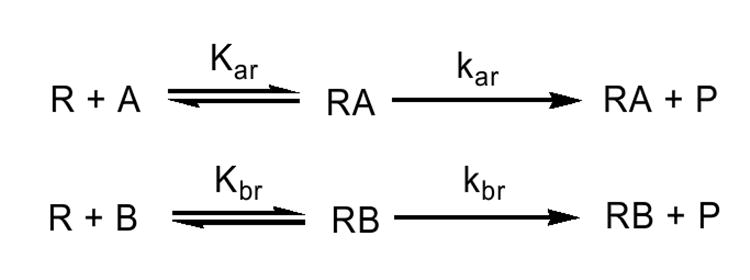
Model describing the interactions among reductase (R), CYP1A2 (A), and CYP2B4 (B) when only binary reductase-P450 complexes are formed.
Scheme 2.
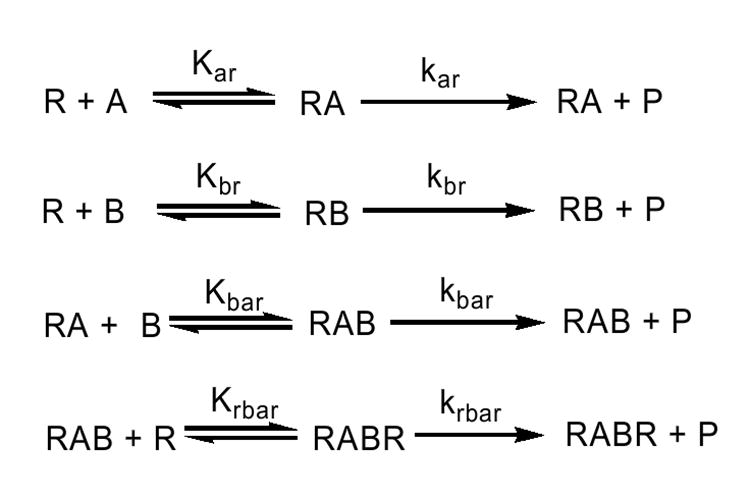
Simplified model describing the interactions among CYP1A2 (A), CYP2B4 (B) and reductase (R) in mixed reconstituted systems where CYP1A2-CYP2B4 complexes are possible.
As reported in figures 1 and 2, increasing ionic strength appeared to relieve the inhibition observed in the mixed system. To determine whether ionic strength can alter the effect of increasing reductase concentration, PROD was measured at high ionic strength (300 mM HEPES) in both the simple binary and the mixed reconstituted system (Fig 4b). CYP2B4 hyperbolic curves were found at both concentrations of HEPES (compare figure 4a and 4b). The shape of the CYP1A2-reductase curve appeared to convert to a hyperbolic function at the higher ionic strength (300 mM HEPES). When the mixed reconstituted systems were examined at 300 mM HEPES, the degree of inhibition at subsaturating reductase was attenuated in the mixed RCS (Fig 4b). For example, activity in the mixed RCS at 0.05 μM reductase was 50% that of CYP2B4 alone at 300 mM HEPES (Fig 4b), in contrast to the 80% inhibition observed at 50 mM HEPES (Fig 4a). Ionic strength appears to relieve the inhibition of PROD in the mixed reconstituted system. Again, these results are consistent with the ability of ionic strength to disrupt complex formation between CYP1A2 and CYP2B4.
In a manner similar to that shown in Fig. 4a, the experimental data in Fig. 4b was modeled based on (a) the model where both P450s compete for the reductase without being able to form CYP1A2-CYP2B4 complexes and (b) the model where CYP1A2-CYP2B4 complexes can form. Again, the experimental data fits the model allowing CYP1A2-CYP2B4 complexes (Fig. 4b, dotted line), whereas the simple competitive model (dashed line) exhibits significant deviations.
The kinetic constants for fitting of the experimental data of Figure 4 are shown in Table 1. As mentioned in our previous report, the experimental data for 7-pentoxyresorufin metabolism in the mixed reconstituted system is consistent with a model where CYP1A2 and CYP2B4 are capable of forming a heteromeric complex which binds reductase with high affinity (31). When comparing the effects of ionic strength on PROD, several features become evident. First, there is a generalized increase in each of the KD values for each of the complexes, which is consistent with ionic strength disrupting complex formation. Second, there was a decrease in the kcat values for the formation of the AR, BR and RBAR complexes. Interestingly, the kcat for product formation from the ternary BAR complex appears to be increased with increasing ionic strength. These data are consistent with the sigmoidal behavior of the mixed reconstituted system being a function of both the higher affinity of the complexes and the low kcat for the BAR complex.
Table 1. Kinetic constants for the simulated curves for 7-Pentoxyresorufin metabolism.
The experimental data for the reductase titrations of the mixed reconstituted systems (Fig 4) were simulated using (a) a model allowing only the formation of binary complexes, and (b) a model allowing the formation of CYP1A2-CYP2B4 complexes. The equilibrium and rate constants below are based on the model described in Scheme 1, which has been detailed in a previous report (31). Data fitting for the simpler model where both CYP1A2 and CYP2B4 compete for reductase (without P450-P450) complex formation utilize only the first 4 constants (Kar, Kbr, kar, and kbr)
| 50 mM HEPES | 300 mM HEPES | |
|---|---|---|
| Kar | 0.0017 | 0.0065 |
| kar | 36 | 18 |
| Kbr | 0.0015 | 0.0082 |
| kbr | 72 | 46 |
| Kbar | 0.00059 | 0.0024 |
| kbar | 5 | 18 |
| Krbar** | 0.001 | 0.005 |
| krbar* | 108 | 64 |
In these simulations, the rate constant for the RBAR complex was set as the sum of the AR and BR complexes. This was done to constrain the simulations under the assumption that the quaternary complex would have a rate constant similar to the sum of the binary complexes.
Under the conditions of this experiment, the value for Krbar could not be determined. Although the data were fit using these parameters, this value was not unique, and could have a range over 4 orders of magnitude without significantly affecting the values of the other rate constants.
In the next set of experiments, we tested the effect of increasing reductase concentrations with 7-EFC, a substrate for which CYP1A2 and CYP2B4 does not appear to exhibit such a high degree of inhibition (Fig 5). 7-EFC is predominately a CYP2B4 substrate, showing a much higher metabolic rate, when compared to the reductase-CYP1A2 binary system. As shown in Fig. 5a, CYP2B4 exhibited simple saturation kinetics. When examining the results from the mixed reconstituted system, 7-EFC showed a much smaller degree of inhibition when compared to the dramatic inhibition obtained with PROD (compare Fig 5a with Fig. 4a). In fact, the 7-EFC data could be described using the simple competitive model described in Scheme 1 (dashed line) where the P450s do not appear to form functionally detectable CYP1A2-CYP2B4 complexes. These data are consistent with the different P450 enzymes competing for the available reductase (Fig 5a). A similar effect was observed in both the simple and mixed systems at 300 mM HEPES buffer (Fig 5b) as compared to the results at 50 mM HEPES (Fig 5a). Again the results were consistent with the simple model where CYP2B4 and CYP1A2 compete for the available reductase. Taken together, these results are consistent with the previously reported formation of a high-affinity CYP1A2-CYP2B4 complex that is more effective at competing for reductase than is CYP2B4 alone when 7-PR is used as substrate (31). In contrast, the 7-EFC data can be described by a simple model where CYP1A2 or CYP2B4 can associate with reductase based on their relative affinities for the flavoprotein, but does not require the involvement of the high affinity CYP1A2-CYP2B4 complex.
Figure 5. Effect of ionic strength on CYP2B4 and CYP1A2 mediated 7-EFC metabolism as a function of reductase concentration.
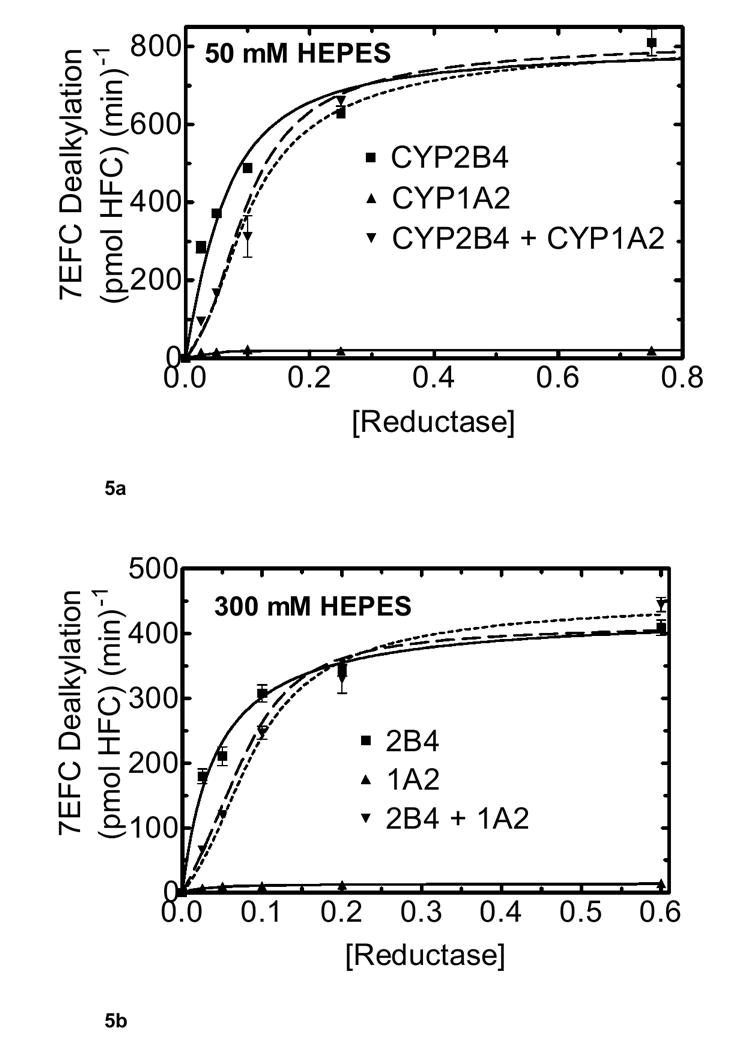
Pre-incubation conditions are the same as in figure 4 described in Materials and Methods. The final concentration of CYP2B4 and CYP1A2 each were 0.05 μM, and DLPC was 8.0 μM. The experimental data were effectively simulated using a model only allowing the formation of reductase-CYP1A2 and reductase-CYP2B4 complexes (dashed line). The dotted line shows a simulation using the model described in Scheme 2 allowing the formation of CYP2B4-CYP1A2 complexes. (a) Effect of mixed reconstitution of CYP1A2 and CYP2B4 on the reductase dependence of 7-EFC dealkylation at 50 mM HEPES. Concentrations of reductase were varied at the lower ionic strength (50 mM HEPES) buffer for CYP1A2, CYP2B4 and the mixed system. (b) Effect of mixed reconstitution of CYP1A2 and CYP2B4 on the reductase dependence of 7-EFC dealkylation (300 mM HEPES).
The kinetic constants for 7EFC metabolism in the mixed reconstituted systems are shown in Table 2. Although, the data can be fit to the more complex model allowing functionally detectable CYP1A2-CYP2B4 complexes, the data also reasonably fit the simpler model where both CYP1A2 and CYP2B4 both compete for NADPH-cytochrome P450 reductase. This model is illustrated in Scheme 1 and can be described by the kinetic constants for the AR and BR complexes in Table 2 (Kar, Kbr, kar, and kbr). The equilibrium constants for the formation of the AR and BR complexes are not affected by the increase in ionic strength, exhibiting less than a 2 fold change. This is in contrast to the results with 7-PR.
Table 2. Kinetic constants for the simulated curves for 7-Ethoxyfluorocoumarin metabolism.
The experimental data for the reductase titrations of the mixed reconstituted systems (Fig 5) were simulated using (a) a model allowing only the formation of binary complexes, and (b) a model allowing the formation of CYP1A2-CYP2B4 complexes. The equilibrium and rate constants below are based on the model described in Scheme 1, which has been detailed in a previous report (31). Data fitting for the simpler model where both CYP1A2 and CYP2B4 compete for reductase (without P450-P450) complex formation utilize only the first 4 constants (Kar, Kbr, kar, and kbr)
| 50 mM HEPES | 300 mM HEPES | |
|---|---|---|
| Kar | 0.004 | 0.0073 |
| kar | 56 | 35 |
| Kbr | 0.036 | 0.0177 |
| Kbr | 2144 | 1080 |
| Kbar | 0.093 | 0.00255 |
| Kbar | 434 | 35 |
| Krbar** | 0.01 | 0.01 |
| krbar* | 2197 | 1115 |
In these simulations, the rate constant for the RBAR complex was set as the sum of the AR and BR complexes. This was done to constrain the simulations under the assumption that the quaternary complex would have a rate constant similar to the sum of the binary complexes.
Under the conditions of this experiment, the value for Krbar could not be determined. Although the data were fit using these parameters, this value was not unique, and could have a range over 4 orders of magnitude without significantly affecting the values of the other rate constants.
To further examine the effects of ionic strength on complex formation between these two P450s, PROD activity was measured as a function of CYP1A2 concentration. PROD was measured in mixed RCS in both low (50 mM) and high (300 mM) ionic strength HEPES buffer as a function of CYP1A2 (Fig 6). Varying CYP1A2 concentrations in low ionic strength HEPES produced a biphasic CYP2B4-mediated PROD response. PROD was dramatically inhibited by the addition of even small amounts of CYP1A2 to the RCS leading to 90% inhibition at 50 nM CYP1A2 consistent with the formation of a high affinity complex with the reductase (32). When extrapolating the extremes of this curve (Fig. 6 at 50 mM HEPES), a breakpoint was observed at about 25 mM CYP1A2. These results point to an extremely high affinity complex between reductase and CYP1A2, similar to previous results (31). Interestingly, varying CYP1A2 at a higher HEPES concentration (300 mM) resulted in a shallower curve with the intersection of the extremes shifted to a higher concentration of CYP1A2. The tendency for more curvature and the shift in the point of intersection at higher ionic strength (indicated where the hashed lines cross), are consistent with a decrease in the ability of CYP1A2 to compete with CYP2B4 for reductase. Greater amounts of CYP1A2 are required to produce the same amount of inhibition. In other words, the high affinity reductase-CYP1A2-CYP2B4 complex formed at 50 mM HEPES is disrupted at 300 mM HEPES.
Figure 6. Effect of ionic strength on PROD activity was measured as a function of CYP1A2 concentration.
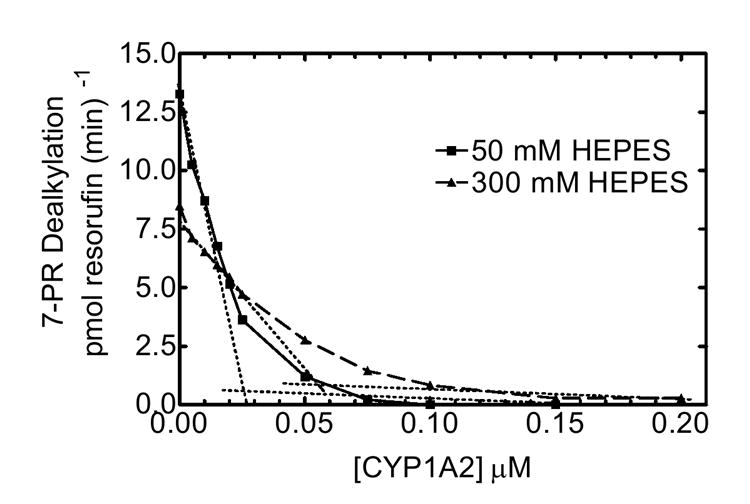
PROD activity was measured in mixed reconstituted systems in both low (50 mM) and high (300 mM) ionic strength HEPES buffer, using a fixed CYP2B4 (0.05 μM) and reductase (0.025 μM) concentration, while varying CYP1A2.
To test whether a change in ionic strength can affect the curvature in mixed reconstituted systems using a substrate that does not appear to form this high affinity CYP2B4-CYP1A2 complex, 7-EFC activity was measured as a function of CYP1A2, using a fixed CYP2B4 and reductase concentration (Fig 7). 7- EFC dealkylation was examined in simple and mixed reconstituted systems using low (50 mM) and high (300 mM) ionic strength HEPES buffer. Varying CYP1A2 concentrations in both low and high ionic strength HEPES produced a less dramatic modulation of CYP2B4-mediated 7-EFC when compared to results with 7-PR (Fig 6). Overall 7-EFC reaction rates were lower at higher ionic strength HEPES. Again, this is consistent with electrostatic reductase-P450 interactions being disrupted by increasing ionic strength solutions. Addition of CYP1A2 inhibited 7EFC activity at both low and high ionic strength HEPES. The breakpoint of the curve, examined at 50 mM HEPES was near 0.05 μM CYP1A2 (Fig 7). A repeat of this experiment at a higher HEPES concentration (300 mM) did not appear to shift the inflection point of 7-EFC activity, consistent with ionic strength not affecting the ability of CYP1A2 to inhibit metabolism of this CYP2B4-selective substrate. Collectively, these results are consistent with a substrate-specific functional interaction between CYP2B4 and CYP1A2, where 7-PR facilitates the formation of CYP1A2-CYP2B4 complexes. Complex formation between these proteins appears to be disrupted by an increase in ionic strength as evidenced by the shift in the breakpoint in Fig. 6. In contrast, 7-EFC does not appear to facilitate the formation of functionally detectable CYP1A2-CYP2B4 complexes, and although the overall activities are sensitive to changes in ionic strength, there is no ionic strength-dependent shift in the breakpoint (Fig. 7).
Figure 7. Effect of ionic strength on 7-EFC dealkylation as a function of CYP1A2 concentration.
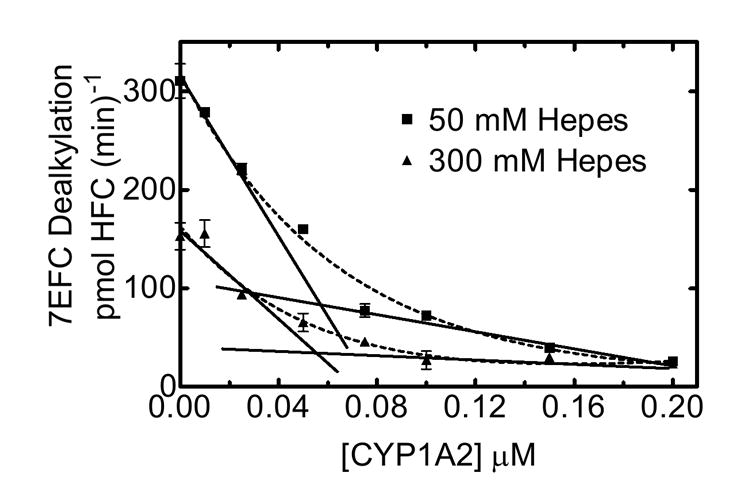
Reaction rates for PROD using binary and mixed reconstituted systems were carried out at low (50 mM) and high (300 mM) ionic strength HEPES buffer, using a fixed CYP2B4 (0.05 μM) and reductase (0.025 μM) concentrations, while varying CYP1A2 as described in Methods.
DISCUSSION
Early studies using microsomal preparations showed that steps of the P450 reaction cycle were sensitive to the concentration of monovalent cations (34). Using a reconstituted system containing phospholipid, P450, and reductase, Coon et al. (38) demonstrated that high ionic strength led to a decrease in benzphetamine demethylation from phenobarbital-treated rabbit liver. The authors suggested a chaotropic role for salt during complex formation between the flavoprotein and P450. Shortly thereafter, cytochrome b5 was shown to be reduced by flavoprotein under high concentrations of potassium chloride (35). It became clear from these early studies, that electron transfer among the various microsomal cytochromes and their subsequent catalysis was modulated by ionic strength.
Complementary charge pairing between P450 and reductase, and the effects of salt on this interaction has been reported (10-18;30;36;37;49). Both monovalent ions and divalent cations were shown to affect overall P450 reaction rates. Electrostatic interactions between reductase and P450 were shown to involve the pairing of positively charged residues on the P450 with negatively charged residues on the reductase (10;11). The degree of stimulation at lower ionic strength and inhibition at higher ionic strength depends on the P450 enzyme and ion species selected.
The effect of ionic strength on mixed reconstituted systems containing multiple P450s has not previously been shown. In the present study, we measured the effects of salt on a previously characterized functional interaction between CYP1A2 and CYP2B4. Two processes in P450 metabolism appear to be affected by ionic strength: reductase-P450 and P450-P450 interactions. Although optimal reconstituted activity varies for each P450 system tested, overall reaction rates decline in higher ionic strength buffers. The decline in overall reaction rates is consistent with a disruption in the 1:1 charge pair complex between reductase and P450 required for activity (10). In addition to reductase-P450 separation, ionic strength was shown to disrupt complex formation between CYP1A2 and CYP2B4. Interestingly, the pronounced inhibition in PROD activity observed when these two P450s are present together in the ternary reconstituted system at sub-saturating reductase, is relieved with increasing concentrations of salt (Fig 1). These results are consistent with charge pairing in the formation of the CYP1A2-CYP2B4 complex.
Tamura et al. (50), reported that the anion in various salts is the effective inhibitor of cytochrome b5 reduction by NADH-cytochrome b5 reductase. Of the carboxylates and halides tested in their study, acetate was one of the least effective ions in disrupting this reaction. Cytochrome P450s appear to behave similarly, for higher overall PROD activity was found using acetate buffer both in the single and mixed reconstituted systems in the present study suggesting that this buffer does not appear to disrupt reductase-P450 complexes as effectively as other ions; however, elevated acetate concentration did appear to be capable of disrupting CYP1A2-CYP2B4 complexes. Several groups have investigated the effects of divalent cations on various cytochromes and their redox partners (13;17;34;50-52). Although more pronounced, the effects of elevated concentrations of divalent cations show the same trend as monovalent ions on relieving the inhibition of PROD activity in this mixed reconstituted system. Tamura et al. (52) showed that small increases in the concentration of divalent cations inhibited cytochrome b5 reduction by NADH:cytochrome b5 reductase more effectively than alkali halides or phosphate buffer. However, divalent cations were shown to activate cytochrome b5 reduction by NADPH-cytochrome P450 reductase (52). The behavior of cytochromes within various electron transfer complexes, are clearly affected by ionic strength. Our results support the hypothesis that CYP1A2-CYP2B4 interactions (as shown by PROD inhibition) are also disrupted by ionic strength, regardless of the ion used.
Recently, Yun et al. (14) showed that reductase-supported CYP1A2 mediated EROD activity increased dramatically up to 50 mM potassium phosphate, and declined at higher buffer strengths. In contrast, when using cumene hydroperoxide as an electron provider, increases in buffer concentrations led to continually increased EROD metabolic activity. Since cumene hydroperoxide – mediated reactions do not require a charge-paired reductase-P450 complex, CYP1A2 peroxygenation is not inhibited at higher ionic strengths, as indicated by the roughly linear increases in activity while varying phosphate (14). However, reductase supported P450 activity behaves quite differently. Consistent with our results, P450 activity reported by Yun et al. (14) reached a maximum at 50-100 mM buffer, followed by a decline in activity. These results are consistent with a disruption in the charge complex between P450 and reductase at higher ionic strengths (13-16).
There are several lines of evidence supporting the potential for one P450 to affect the function of another P450 enzyme. Tan et al. (49) described substrate-induced competition between human CYP2A6 and CYP2E1 for reductase, where metabolism was not significantly altered by ionic strength. Kaminsky and Guengerich (27) observed generalized inhibition with mixtures of different P450s during warfarin metabolism that was attributed to P450 aggregation. Previous studies by Yamazaki et al. (30) reported testosterone 6β-hydroxylation by CYP3A4 was enhanced by the presence of either CYP1A1 or CYP1A2, whereas other P450s did not stimulate this reaction. Interestingly, CYP1A1 was shown to physically interact with other P450s specifically binding to CYP3A2 (29). In this study, CYP1A1 (P450 c) specifically bound to CYP3A2 (P450 2a) by chemical crosslinking methodologies. Results from our laboratory (31;32) are consistent with CYP1A2 and CYP2B4 being capable of forming a heteromeric complex (in the presence of certain substrates such as 7-PR). The CYP1A2 moiety of this complex can bind reductase with high affinity, effectively drawing reductase away from CYP2B4. These results support a functional role for these interactions. Taken together, the data support the idea that enzymes from the CYP1A subfamily are capable of forming ternary complexes with another P450 and reductase, and that such an interaction can have a significant influence on P450 function.
One explanation that is currently being evaluated, is that CYP1A2 forms homomeric complexes, where the presence of reductase disrupts self-association as demonstrated in previous studies by Kawato’s group (53;54). This could explain the non hyperbolic kinetics reported for CYP1A2 with 7-PR as substrate (Fig. 4a; (31)). Although higher ionic strength solutions are predicted to disrupt CYP1A2 reductase complex formation, CYP1A2 self-association resulting from electrostatic interactions could also be disrupted by high ionic strength leading to the hyperbolic dependence of reductase observed in Figure 4b. As predicted for CYP1A2, the potential for functional homomeric P450 complexes raises the question as to how these proteins are organized around reductase in the presence of multiple P450s. A disrupted CYP1A2 homo-oligomer would free more CYP1A2 active sites to pair with complementary charged reductase residues. Both increased concentrations of ions and reductase could facilitate dispersion of these putative CYP1A2 oligomers.
Although the working model that we are using suggests that substrates influence the interactions among the P450s, which in turn affects reductase binding, we are not certain of the actual characteristics of these complexes. There several possible explanations for the responses reported in this manuscript. (1) CYP1A2-CYP2B4 complexes that have a high affinity for reductase may not exist in the absence of substrate. In this case, different substrates could alter the ability of the proteins to form these complexes, probably through a conformational change, which could alter the relative affinity for reductase binding or catalysis. According to this model, CYP1A2 and CYP2B4 would exist as monomers in the absence of substrate with some, but not all, substrates promoting the formation of CYP1A2-CYP2B4 complexes. (2) A second possibility is that the P450s form complexes in the absence of substrate. In this case, the binding of some substrates to one of the P450s would lead to disruption of this complex. (3) A third possibility is that the P450s exist as a mixture of both homomeric and heteromeric P450-P450 complexes. Substrate binding may not necessarily disrupt these complexes, but may alter the relative ability of reductase to bind to one of the contributing P450 ‘subunits’. With this model, 7EFC could bind to both moieties of the CYP2B4-CYP1A2 complex, but the substrate would neither disrupt the P450-P450 complex, nor alter the relative affinity of either P450 for the reductase. In contrast, the 7PR data could be explained by the substrate eliciting a conformational change in one of the P450s of the CYP1A2-CYP2B4 complex (e.g. CYP1A2) that would lead to a decrease in both the Km and the Vmax for reductase binding to the CYP1A2 moiety of the ternary reductase-CYP1A2-CYP2B4 complex (based on the kinetic parameters shown in Table 1).
With the multiplicity of P450 proteins in the microsomal monooxygenase system, differences in their quaternary structure, overall charge, and hydrodynamic characteristics, individual P450s may behave differently. It is well known that CYP1A2 and CYP2B4 exhibit different physicochemical properties. CYP1A2 has been reported to be more hydrophobic (53), has a lower solubility (55), and has a tendency to form larger aggregates (56;57) than CYP2B4. Although it is apparent that P450 aggregates when in solution, the physical state of the enzyme incorporated into membranes is less clear. The physical characteristics of P450 enzymes are also dependent on the solution into which these proteins are surrounded. Several investigators have studied the effects of concentration and ionic strength on CYP1A2 solubility (14;58). Yun et al. (14) reported higher aggregation, as measured by increased light scattering, at higher CYP1A2 concentration, as well as in lower ionic strength potassium phosphate buffer. The authors showed that conformational changes (increased helical content) induced by ionic strength, were accompanied by increases in activity (14).
Our studies and others demonstrate that P450 activity is sensitive to reconstitution conditions. Changes in reconstitution conditions such as: pre-incubation time and temperature (5;46) protein, detergent and lipid-P450 ratios (5;46;53;59;60) as well as the buffers used (14;16;61) can lead to dramatic changes in monooxygenase activities. Optimal ionic strength relative to P450 monooxygenase activity, depends on the reaction system under which these proteins are examined (35;37). Because of this variability, it is often difficult to compare results in the literature, derived from different laboratories and their systems used. Although great strides have been made to identify, isolate and characterize individual P450s, there is no consensus as to how multiple P450s are organized in the lipid membrane, and how they interact with their redox partners. Collectively, the studies presented in this paper support the potential for P450-P450 interactions, and that these interactions are sensitive to changes in ionic strength. Heteromeric assembly of P450s has been suggested to help in the sequential processing of a parent drug and its metabolites (29). The efficiency of drug biotransformation may be influenced by such P450 complexes.
Supplementary Material
Acknowledgments
We would like to thank George F. Cawley and Travis Broome for the preparation of CYP1A2 and NADPH-cytochrome P450 reductase.
These studies were supported by a US Public Health Service Research Grant from the National Institute of Environmental Health Sciences ES004344 (WLB), and a predoctoral fellowship from the Stanley S. Scott Cancer Center (RWK)
Abbreviations
- CYP; P450
cytochrome P450
- PROD
7-pentoxyresorufin-O-deethlation
- 7-PR
7-pentoxyresorufin
- 7-EFC
7-ethoxy-4-trifluromethylcoumarin
- 7-HFC
7-hydroxy-4-trifluoromethylcoumarin
- ER
endoplasmic reticulum
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- DLPC
L-α-dilauroyl-sn-glycero-3-phosphochiline
Footnotes
SUPPORTING INFORMATION AVAILABLE Sample DynaFit files for fits of the experimental kinetic data for both (1) simple competition between different P450s for reductase and (2) the more complex model allowing for the formation of P450-P450 complexes are available free of charge via the Internet at http://pubs.acs.org. These programs were used for the fits shown in figures 4 and 5 and the resulting kinetic parameters shown in Tables 1 and 2.
References
- 1.Estabrook RW, Franklin MR, Cohen B, Shigamatzu A, Hildebrandt AG. Biochemical and genetic factors influencing drug metabolism. Influence of hepatic microsomal mixed function oxidation reactions on cellular metabolic control. Metabolism. 1971;20:187–199. doi: 10.1016/0026-0495(71)90091-6. [DOI] [PubMed] [Google Scholar]
- 2.Gigon PL, Gram TE, Gillette JR. Studies on the rate of reduction of hepatic microsomal cytochrome P-450 by reduced nicotinamide adenine dinucleotide phosphate: effect of drug substrates. Mol Pharmacol. 1969;5:109–122. [PubMed] [Google Scholar]
- 3.Miwa GT, West SB, Huang MT, Lu AYH. Studies on the association of cytochrome P-450 and NADPH-cytochrome c reductase during catalysis in a reconstituted hydroxylating system. J Biol Chem. 1979;254:5695–5700. [PubMed] [Google Scholar]
- 4.Miwa GT, Lu AYH. The association of cytochrome P-450 and NADPH-cytochrome P-450 reductase in phospholipid membranes. Arch Biochem Biophys. 1984;234:161–166. doi: 10.1016/0003-9861(84)90337-0. [DOI] [PubMed] [Google Scholar]
- 5.Peterson JA, Ebel RE, O’Keeffe DH, Matsubara T, Estabrook RW. Temperature dependence of cytochrome P-450 reduction. A model for NADPH-cytochrome P-450 reductase:cytochrome P-450 interaction. J Biol Chem. 1976;251:4010–4016. [PubMed] [Google Scholar]
- 6.Tamburini PP, Gibson GG, Backes WL, Sligar SG, Schenkman JB. Reduction kinetics of purified rat liver cytochrome P-450. Evidence for a sequential reaction mechanism dependent on the hemoprotein spin state. Biochem. 1984;23:4526–4533. doi: 10.1021/bi00315a004. [DOI] [PubMed] [Google Scholar]
- 7.Schenkman JB, Jansson I. The many roles of cytochrome b5. Pharmacol Ther. 2003;97:139–152. doi: 10.1016/s0163-7258(02)00327-3. [DOI] [PubMed] [Google Scholar]
- 8.Lu AY, Strobel HW, Coon MJ. Hydroxylation of benzphetamine and other drugs by a solubilized form of cytochrome P-450 from liver microsomes: lipid requirement for drug demethylation. Biochem Biophys Res Commun. 1969;36:545–551. doi: 10.1016/0006-291x(69)90339-8. [DOI] [PubMed] [Google Scholar]
- 9.Lu AYH, Junk KW, Coon MJ. Resolution of the cytochrome P-450-containing omega- hydroxylation system of liver microsomes into three components. J Biol Chem. 1969;244:3714–3721. [PubMed] [Google Scholar]
- 10.Bernhardt R, Makower A, Janig GR, Ruckpaul K. Selective chemical modification of a functionally linked lysine in cytochrome P-450 LM2. Biochim Biophys Acta. 1984;785:186–190. doi: 10.1016/0167-4838(84)90143-2. [DOI] [PubMed] [Google Scholar]
- 11.Nadler SG, Strobel HW. Role of electrostatic interactions in the reaction of NADPH- cytochrome P-450 reductase with cytochromes P-450. Arch Biochem Biophys. 1988;261:418–429. doi: 10.1016/0003-9861(88)90358-x. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu T, Tateishi T, Hatano M, Fujii-Kuriyama Y. Probing the role of lysines and arginines in the catalytic function of cytochrome P450d by site-directed mutagenesis. Interaction with NADPH-cytochrome P450 reductase. J Biol Chem. 1991;266:3372–3375. [PubMed] [Google Scholar]
- 13.Yamazaki H, Ueng YF, Shimada T, Guengerich FP. Roles of divalent metal ions in oxidations catalyzed by recombinant cytochrome P450 3A4 and replacement of NADPH-- cytochrome P450 reductase with other flavoproteins, ferredoxin, and oxygen surrogates. Biochem. 1995;34:8380–8389. doi: 10.1021/bi00026a020. [DOI] [PubMed] [Google Scholar]
- 14.Yun CH, Song M, Ahn T, Kim H. Conformational change of cytochrome p450 1A2 induced by sodium chloride. J Biol Chem. 1996;271:31312–31316. doi: 10.1074/jbc.271.49.31312. [DOI] [PubMed] [Google Scholar]
- 15.Davydov DR, Kariakin AA, Petushkova NA, Peterson JA. Association of cytochromes P450 with their reductases: opposite sign of the electrostatic interactions in P450BM-3 as compared with the microsomal 2B4 system. Biochem. 2000;39:6489–6497. doi: 10.1021/bi992936u. [DOI] [PubMed] [Google Scholar]
- 16.Yun CH, Ahn T, Guengerich FP. Conformational change and activation of cytochrome P450 2B1 induced by salt and phospholipid. Arch Biochem Biophys. 1998;356:229–238. doi: 10.1006/abbi.1998.0759. [DOI] [PubMed] [Google Scholar]
- 17.Tamura M, Yoshida S, Tamura T, Saitoh T, Takeshita M. Effect of divalent cations on NADH-dependent and NADPH-dependent cytochrome b5 reduction by hepatic microsomes. Arch Biochem Biophys. 1990;280:313–319. doi: 10.1016/0003-9861(90)90335-v. [DOI] [PubMed] [Google Scholar]
- 18.Nisimoto Y, Edmondson DE. Effect of KCl on the interactions between NADPH:cytochrome P-450 reductase and either cytochrome c, cytochrome b5 or cytochrome P-450 in octyl glucoside micelles. Eur J Biochem. 1992;204:1075–1082. doi: 10.1111/j.1432-1033.1992.tb16731.x. [DOI] [PubMed] [Google Scholar]
- 19.Bernhardt R, Kraft R, Otto A, Ruckpaul K. Electrostatic interactions between cytochrome P-450 LM2 and NADPH-cytochrome P-450 reductase. Biomed Biochim Acta. 1988;47:581–592. [PubMed] [Google Scholar]
- 20.Bridges A, Gruenke L, Chang YT, Vakser IA, Loew G, Waskell L. Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem. 1998;273:17036–17049. doi: 10.1074/jbc.273.27.17036. [DOI] [PubMed] [Google Scholar]
- 21.Omata Y, Sakamoto H, Robinson RC, Pincus MR, Friedman FK. Interaction between cytochrome P450 2B1 and cytochrome bs: inhibition by synthetic peptides indicates a role for P450 residues Lys-122 and Arg-125. Biochem Biophys Res Commun. 1994;201:1090–1095. doi: 10.1006/bbrc.1994.1817. [DOI] [PubMed] [Google Scholar]
- 22.Nelson DR. Comparison of P450s from human and fugu: 420 million years of vertebrate P450 evolution. Arch Biochem Biophys. 2003;409:18–24. doi: 10.1016/s0003-9861(02)00553-2. [DOI] [PubMed] [Google Scholar]
- 23.Schacter BA, Nelson EB, Marver HS, Masters BS. Immunochemical evidence for an association of heme oxygenase with the microsomal electron transport system. J Biol Chem. 1972;247:3601–3607. [PubMed] [Google Scholar]
- 24.Backes WL. NADPH-Cytochrome P450 Reductase: Function. In: Schenkman JB, Greim H, editors. Cytochrome P450. Springer-Verlag; Berlin: 1993. pp. 15–34. [Google Scholar]
- 25.Zhang S, Cawley GF, Eyer CS, Backes WL. Altered ethylbenzene-mediated hepatic CYP2E1 expression in growth hormone-deficient dwarf rats. Toxicol Appl Pharmacol. 2002;179:74–82. doi: 10.1006/taap.2002.9349. [DOI] [PubMed] [Google Scholar]
- 26.West SB, Lu AYH. Reconstituted liver microsomal enzyme system that hydroxylates drugs, other foreign compounds and endogenous substrates. V. Competition between cytochromes P-450 and P-448 for reductase in 3,4-benzpyrene hydroxylation. Arch Biochem Biophys. 1972;153:298–303. doi: 10.1016/0003-9861(72)90448-1. [DOI] [PubMed] [Google Scholar]
- 27.Kaminsky LS, Guengerich FP. Cytochrome P-450 isozyme/isozyme functional interactions and NADPH-cytochrome P-450 reductase concentrations as factors in microsomal metabolism of warfarin. Eur J Biochem. 1985;149:479–489. doi: 10.1111/j.1432-1033.1985.tb08950.x. [DOI] [PubMed] [Google Scholar]
- 28.Dutton DR, McMillen SK, Sonderfan AJ, Thomas PE, Parkinson A. Studies on the rate-determining factor in testosterone hydroxylation by rat liver microsomal cytochrome P-450: evidence against cytochrome P-450 isozyme:isozyme interactions. Arch Biochem Biophys. 1987;255:316–328. doi: 10.1016/0003-9861(87)90399-7. [DOI] [PubMed] [Google Scholar]
- 29.Alston K, Robinson RC, Park SS, Gelboin HV, Friedman FK. Interactions among cytochromes P-450 in the endoplasmic reticulum. Detection of chemically cross-linked complexes with monoclonal antibodies. J Biol Chem. 1991;266:735–739. [PubMed] [Google Scholar]
- 30.Yamazaki H, Inoue K, Shaw PM, Checovich WJ, Guengerich FP, Shimada T. Different contributions of cytochrome P450 2C19 and 3A4 in the oxidation of omeprazole by human liver microsomes: effects of contents of these two forms in individual human samples. J Pharmacol Exp Ther. 1997;283:434–442. [PubMed] [Google Scholar]
- 31.Backes WL, Batie CJ, Cawley GF. Interactions among P450 enzymes when combined in reconstituted systems: formation of a 2B4-1A2 complex with a high affinity for NADPH-cytochrome P450 reductase. Biochem. 1998;37:12852–12859. doi: 10.1021/bi980674a. [DOI] [PubMed] [Google Scholar]
- 32.Cawley GF, Batie CJ, Backes WL. Substrate-dependent competition of different P450 isozymes for limiting NADPH-cytochrome P450 reductase. Biochem. 1995;34:1244–1247. doi: 10.1021/bi00004a018. [DOI] [PubMed] [Google Scholar]
- 33.Cawley GF, Zhang S, Kelley RW, Backes WL. Evidence supporting the interaction of CYP2B4 and CYP1A2 in microsomal preparations. Drug Metab Dispos. 2001;29:1529–1534. [PubMed] [Google Scholar]
- 34.Hildebrandt A, Estabrook RW. Evidence for the participation of cytochrome b 5 in hepatic microsomal mixed-function oxidation reactions. Arch Biochem Biophys. 1971;143:66–79. doi: 10.1016/0003-9861(71)90186-x. [DOI] [PubMed] [Google Scholar]
- 35.Prough RA, Masters BS. The mechanism of cytochrome b5 reduction by NADPH-cytochrome c reductase. Arch Biochem Biophys. 1974;165:263–267. doi: 10.1016/0003-9861(74)90163-5. [DOI] [PubMed] [Google Scholar]
- 36.Voznesensky AI, Schenkman JB. The cytochrome P450 2B4-NADPH cytochrome P450 reductase electron transfer complex is not formed by charge-pairing. J Biol Chem. 1992;267:14669–14676. [PubMed] [Google Scholar]
- 37.Voznesensky AI, Schenkman JB. Quantitative analyses of electrostatic interactions between NADPH-cytochrome P450 reductase and cytochrome P450 enzymes. J Biol Chem. 1994;269:15724–15731. [PubMed] [Google Scholar]
- 38.Coon MJ, Hoeven TA, Kaschnitz RM, Strobel HW. Biochemical studies on cytochrome P-450 solubilized from liver microsomes: partial purification and mechanism of catalysis. Ann N Y Acad Sci. 1973;212:449–457. doi: 10.1111/j.1749-6632.1973.tb47613.x. [DOI] [PubMed] [Google Scholar]
- 39.Yasukochi Y, Masters BS. Some properties of a detergent-solubilized NADPH-cytochrome c(cytochrome P-450) reductase purified by biospecific affinity chromatography. J Biol Chem. 1976;251:5337–5344. [PubMed] [Google Scholar]
- 40.Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 41.Saribas AS, Gruenke L, Waskell L. Overexpression and Purification of the Membrane-Bound Cytochrome P450 2B4. Protein Expression and Purification. 2001;21:303–309. doi: 10.1006/prep.2000.1377. [DOI] [PubMed] [Google Scholar]
- 42.Coon MJ, Van Der Hoeven TA, Dahl SB, Haugen DA. Methods in Enzymology. Academic Press; 1984. Two Forms of Liver Microsomal Cytochrome P-450, P-450LM2 and P-450LM4 (Rabbit Liver) pp. 109–123. [DOI] [PubMed] [Google Scholar]
- 43.Omura T, Sato R. The Carbon Monoxide-binding Pigment of Liver Microsomes: II. Solubilization, Purification, and Properties. J Biol Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- 44.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein Measurement with the Folin Phenol Reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 45.Vermilion JL, Coon MJ. Purified liver microsomal NADPH-cytochrome P-450 reductase. Spectral characterization of oxidation-reduction states. J Biol Chem. 1978;253:2694–2704. [PubMed] [Google Scholar]
- 46.Causey KM, Eyer CS, Backes WL. Dual role of phospholipid in the reconstitution of cytochrome P- 450 LM2-dependent activities. Mol Pharmacol. 1990;38:134–142. [PubMed] [Google Scholar]
- 47.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 48.Backes WL, Kelley RW. Organization of multiple cytochrome P450s with NADPH-cytochrome P450 reductase in membranes. Pharmacology & Therapeutics. 2003;98:221–233. doi: 10.1016/s0163-7258(03)00031-7. [DOI] [PubMed] [Google Scholar]
- 49.Tan YZ, Patten CJ, Smith P, Yang CS. Competitive interactions between cytochromes P450 2A6 and 2E1 for NADPH-cytochrome P450 oxidoreductase in the microsomal membranes produced by a baculovirus expression system. Arch Biochem Biophys. 1997;342:82–91. doi: 10.1006/abbi.1997.9995. [DOI] [PubMed] [Google Scholar]
- 50.Tamura M, Yubisui T, Takeshita M. The inhibitory effect of halides and carboxylates on hepatic NADH: cytochrome b5 oxidoreductase. Biochem J. 1985;230:273–276. doi: 10.1042/bj2300273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strittmatter P, Velick SF. The purification and properties of microsomal cytochrome reductase. J Biol Chem. 1957;228:785–799. [PubMed] [Google Scholar]
- 52.Tamura M, Yubisui T, Takeshita M. The opposite effect of bivalent cations on cytochrome b5 reduction by NADH:cytochrome b5 reductase and NADPH:cytochrome c reductase. Biochem J. 1988;251:711–715. doi: 10.1042/bj2510711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamada M, Ohta Y, Bachmanova GI, Nishimoto Y, Archakov AI, Kawato S. Dynamic interactions of rabbit liver cytochromes P450IA2 and P450IIB4 with cytochrome b5 and NADPH-cytochrome P450 reductase in proteoliposomes. Biochem. 1995;34:10113–10119. doi: 10.1021/bi00032a003. [DOI] [PubMed] [Google Scholar]
- 54.Gut J, Richter C, Cherry RJ, Winterhalter KH, Kawato S. Rotation of cytochrome P-450. II. Specific interactions of cytochrome P-450 with NADPH-cytochrome P-450 reductase in phospholipid vesicles. J Biol Chem. 1982;257:7030–7036. [PubMed] [Google Scholar]
- 55.Guengerich FP, Martin MV, Guo Z, Chun YJ. Purification of functional recombinant P450s from bacteria. Methods Enzymol. 1996;272:35–44. doi: 10.1016/s0076-6879(96)72006-2. [DOI] [PubMed] [Google Scholar]
- 56.Dean WL, Gray RD. Hydrodynamic properties of monomeric cytochromes P-450LM2 and P-450LM4 in n-octylglucoside solution. Biochem Biophys Res Commun. 1982;107:265–271. doi: 10.1016/0006-291x(82)91699-0. [DOI] [PubMed] [Google Scholar]
- 57.Sevrukova IF, Kanaeva IP, Koen YM, Samenkova NF, Bachmanova GI, Archakov AI. Catalytic activity of cytochrome P4501A2 in reconstituted system with Emulgen 913. Arch Biochem Biophys. 1994;311:133–143. doi: 10.1006/abbi.1994.1218. [DOI] [PubMed] [Google Scholar]
- 58.Haugen DA, Coon MJ. Properties of electrophoretically homogeneous phenobarbital-inducible and beta-naphthoflavone-inducible forms of liver microsomal cytochrome P-450. J Biol Chem. 1976;251:7929–7939. [PubMed] [Google Scholar]
- 59.Kanaeva IP, Nikityuk OV, Davydov DR, Dedinskii IR, Koen YM, Kuznetsova GP, Skotselyas ED, Bachmanova GI, Archakov AI. Comparative study of monomeric reconstituted and membrane microsomal monooxygenase systems of the rabbit liver. II. Kinetic parameters of reductase and monooxygenase reactions. Arch Biochem Biophys. 1992;298:403–412. doi: 10.1016/0003-9861(92)90428-y. [DOI] [PubMed] [Google Scholar]
- 60.Yun CH, Song M, Kim H. Conformational Change of Cytochrome P450 1A2 Induced by Phospholipids and Detergents. J Biol Chem. 1997;272:19725–19730. doi: 10.1074/jbc.272.32.19725. [DOI] [PubMed] [Google Scholar]
- 61.Church GM, Gilbert W. Genomic Sequencing (DNA Methylation/UV Crosslinking/Filter Hybridization/Immunoglobulin Genes) Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.