

Abstract
Due to their pharmacological importance in the oxidation of amine neurotransmitters, the membrane-bound flavoenzymes monoamine oxidase A and monoamine oxidase B have attracted numerous investigations and, as a result, two different mechanisms; the single electron transfer and the polar nucleophilic mechanisms, have been proposed to describe their catalytic mechanisms. This review compiles the recently available structural data on both enzymes with available mechanistic data as well as current NMR data on flavin systems to provide an integration of the approaches. These conclusions support the proposal that a polar nucleophilic mechanism for amine oxidation is the most consistent mechanistic scheme as compared with the single electron transfer mechanism mechanism.
Keywords: Monoamine oxidases, MAO A, MAO B, flavin coenzymes, amine neurotransmitter, QSAR, proton abstraction, expression in Pichia pastoris, x-ray structures
One of the pathways for control of amine neurotransmitter levels in the cell is through their oxidation by the mitochondrial outer membrane enzymes monoamine oxidases A and B (MAO A and MAO B). In addition to oxidation of amines such as dopamine and serotonin, these enzymes also function to oxidize ingested amines such as phenethylamine and tyramine to prevent their functioning as false neurotransmitters. Their structures, functions, and mechanisms have been the focus of extensive investigations with approximately 20, 000 papers in the literature since the 1950s. This intensive research effort has its origins by the observation that MAO inhibitors were originally found to function as antidepressants. Observed clinical side effects have reduced the clinical interest in covalent MAO inhibitors as antidepressants in favor of the serotonin reuptake inhibitors. A renewed interest in the inhibition of MAO B has resulted from the observed age-related increase of MAO B levels in humans [1] and possible connection to neurodegenerative diseases of the elderly such as Parkinson’s Disease. Thus, newer MAO B specific inhibitors that are non-covalently bound may serve to useful neuroprotectants.
Mechanistic studies of MAO catalyzed amine oxidation are of intrinsic biological interest in that a number of amine oxidizing flavoenzymes are known and have not been investigated to the level that MAO has been. Other flavin-dependent amine oxidizing enzymes include a bacterial trimethylamine dehydrogenase, plant and mammalian polyamine oxidase, and more recently intensive interest has been focused on LSD-1; a lysyl demethylase that specifically demethylates histone -N-methyl lysyl residues that is involved with the regulation of gene expression [2]. The advantage of MAO A or B as a model system to study flavin dependent amine oxidation is the simplicity of the substrates used (benzylamine or phenethylamine) thus facilitating the synthesis of analogs or isotopically-labeled forms which are not readily done with the substrates for the other known amine oxidases. A disadvantage for using MAO A or B in such studies is that they are both bound to the outer mitochondrial membrane and the preparation of purified preparations in the past was difficult since most mammalian tissues contained both forms which were not readily separable on solubilization and purification.
Sources of Purified Preparations of MAO A and MAO B
In studies requiring purified preparations of MAO A and/or MAO B through the mid 1990s, bovine liver mitochondria served as the most convenient source of MAO B [3] while human placental mitochondria served as the most ready source of MAO A [4]. Preparations from either source required extensive facilities, access to source tissue, and resulted in preparations that were of variable activities. In addition, the spectral properties of preparations from either of these sources exhibited variable levels of inactive flavin anionic semiquinone [5] which probably originated from reaction of the either enzyme with reactive oxygen radicals liberated during the bulk disruption of mitochondrial preparations. In addition to these difficulties, the reliance on natural sources of MAO A or B also precluded more detailed studies of the enzyme using site-directed mutagenesis approaches.
The first successful heterologous expression of human MAO activity was reported in 1990 [6] using Saccharomyces cerevisiae as the host expression system. The expressed enzymes were found to be bound to the outer mitochondrial membrane and exhibited sensitivities to classic MAO inhibitors. The levels of MAO expression were found to be too small for this system to be viable as a source for purified enzymes in reagent quantities. Further work [7] demonstrated that S. cerevisiae could function (with a different promoter) as a reasonable source of human MAO A with levels of expression that would permit the isolation of tens of mg of purified enzyme. Unfortunately, despite numerous attempts in the author’s laboratory, this system was not capable of expressing large quantities of human MAO B.
A breakthrough in MAO expression came with the successful demonstration that the heterologous expression of human MAO B [8] and later human MAO A [9] in large quantities (∼200 mg of purified enzyme from 2 L of culture was possible using the methylotrophic yeast; Pichia pastoris. The expressed enzymes are found in the outer membrane of the yeast mitochondria and constitute approximately 30% of the protein in those membrane preparations. The purified proteins contain all of the post-translational modifications (eg. stoichiometric covalent FAD incorporation) found in preparations from natural sources and comparable activity levels. Not only does this expression system serve as a convenient and reliable source of human MAO A and MAO B but it also allows the production of site-directed mutants important as structural and mechanistic probes. Recent unpublished work in this laboratory has shown it also possible to express rat MAO A and rat MAO B in Pichia pastoris. Previous work has shown rat MAO A can be expressed well in S. cerevisiae [10]. The successful expression of human MAO A and MAO B in the baculovirus insect cell system also results in the formation of active enzymes located in the mitochondrial outer membrane [11]. This system has been commercialized as a convenient source for membrane bound human MAO A and human MAO B (“Supersomes from BD Gentest, Woburn, MA, USA) although the reported specific activities (based on units of enzyme activity/mg mitochondrial protein) are lower than observed in the Pichia expression system.
Structures of Human MAO B and of MAO A
With purified preparations of human MAO A and MAO B in hand, a collaboration was initiated between our laboratories to crystallize and determine the structures of the full length recombinant enzymes. The formation of diffracting crystals of both enzymes were challenging projects with the structural elucidation of human MAO A more difficult than that of MAO B. The initial report of the MAO B structure [12] and subsequent structures of this enzyme [13] have resulted in new insights into the active site. Subsequent successes in the crystallization and structural elucidation of human MAO A [14] resulted in a detailed comparison of the active sites of both enzymes which are important for mechanistic comparisons. During this period, a structure of rat MAO A was also reported [15] that provides some useful comparative insights.
Human MAO B crystallizes as a dimer and exhibits hydrodynamic properties of a dimeric form in two different detergent solutions. The structure of a single monomer of the dimeric form is shown in Fig.1. For a substrate molecule to reach the flavin center, it must first negotiate a protein loop at the entrance to one of two cavities before reaching the flavin coenzyme. The first cavity has been termed the “entrance cavity” is very hydrophobic in nature and exhibits a volume of 290 Å3. Separating the “entrance cavity” from the similarly hydrophobic” substrate cavity (volume=390 Å3) is an isoleucine199 side chain which serves as a “gate” between the two cavities. Depending on the substrate or bound inhibitor, it can exist in either an open or a closed form which has been shown to be important in defining the inhibitor specificity of hMAO B [16]. At the end of the substrate cavity is the FAD coenzyme which is covalently bound in an 8α-thioether linkage [17] to cysteine397. Analysis of the active site cavities using the computer program “GRID” (www.moldiscovery.com) show them both to be very hydrophobic with sites for favorable amine binding about the flavin involving two nearly parallel tyrosyl (398 and 435) residues (Fig.2) that form what has been termed an “aromatic cage” which, as will be described below, has catalytic significance.
Fig.1.
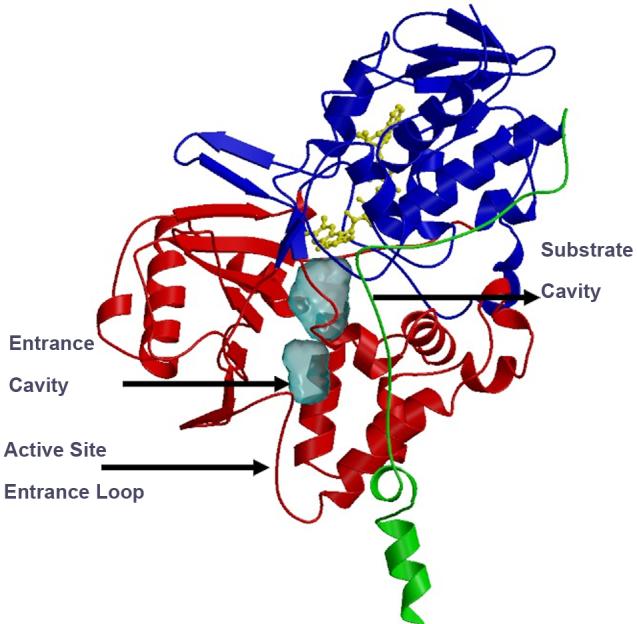
Ribbon diagram of monomeric unit of human MAO B structure. The covalent flavin moiety is shown in a ball and stick model in yellow. The flavin binding domain is in blue, the substrate domain in red and the membrane binding domain in green.
Fig.2.
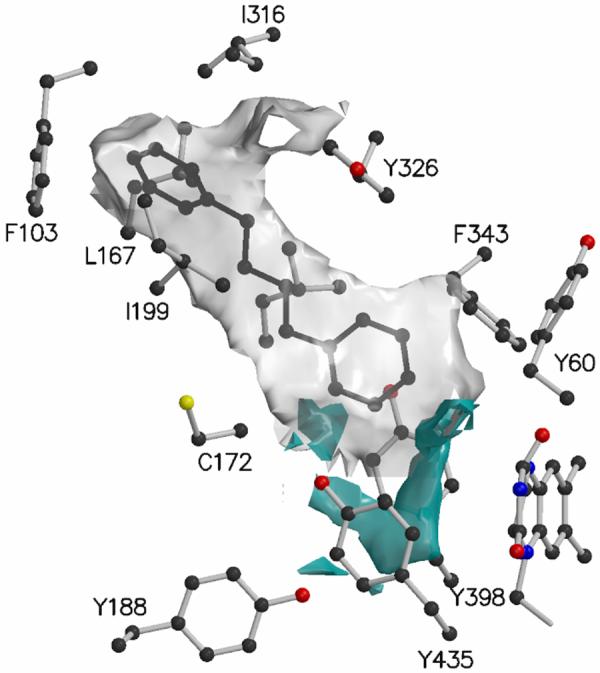
Isocontour of GRID molecular interaction fields computed with an aromatic sp2 carbon probe (gray) and a neutral NH2 probe (cyan) of the coordinates representation by the fused entrance and substrate cavities in the human MAO B complexed with the inhibitor 1,4-diphenyl-2-butene.
In contrast to human MAO B, human MAO A crystallizes as a monomer (Fig.3) [14] in agreement with the prediction by Andres et al. [18] based on genetic and modeling studies. The monomeric form of human MAO A also contrasts to the structural properties of rat MAO A [15] which crystallizes as a dimer. Both human and rat MAO A’s differ from human MAO B in that they have only single substrate binding cavities with protein loops at the entrances of either cavities. Human MAO A has a cavity size of 550 Å3 whereas the rat enzyme has a somewhat smaller active site cavity (450 Å3). As in MAO B, the ri face of the covalent FAD is situated as one of the faces of the substrate binding site in either the human or rat enzyme opposite to the entrance and also has the two nearly parallel tyrosyl residues (407 and 444) forming an “aromatic cage” in front of the flavin. The substrate binding sites in MAO A is also quite hydrophobic as found for MAO B.
Fig.3.
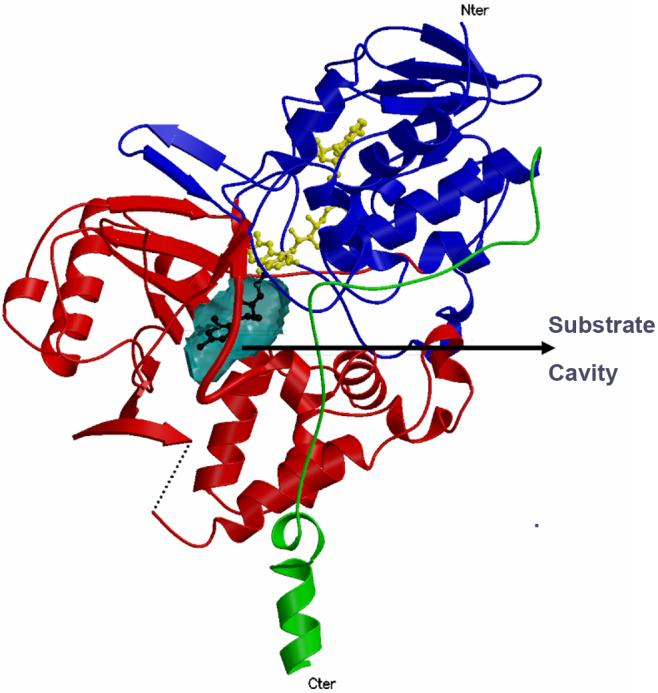
Ribbon diagram of the human MAO A structure. The coloring of the domains follow that shown in Fig.1.
A comparison of the active sites for human MAO B and MAO A are shown in Fig. 4. MAO B has a bipartite elongated cavity that occupies a combined volume close to 700 Å3 when the side chain of Ileu199 is in the “open” conformation. MAO A has a single cavity that exhibits a “rounder” shape and is larger in volume than the “substrate” cavity of MAO B. Analysis of residue side chains in either active site shows the substrate to have less freedom for rotation in the MAO B site than in MAO A. This difference in substrate conformational freedom was predicted on comparison of QSAR effects on catalytic properties of the two enzymes (see below). The structural basis for differences between the two cavities can be partially attributed to conformational differences of a six residue segment (residues 200-215) that constitutes what is termed a “cavity shaping loop” in MAO A and MAO B. This loop is in a more extended conformation in MAO A and is in a more compact conformation in MAO B. Aside from this difference in structure, the Cα chain trace comparison between the two enzymes (∼70% amino acid sequence identity) is 0.7Å [14].
Fig.4.

Comparison of the active site cavities of human MAO A (left) and human MAO B (right). Clorgyline is in the active site of MAO A and Deprenyl is in the active site of MAO B. Both of these inhibitors form covalent N(5) flavocyanine adducts with the respective flavin coenzymes. The active site “shaping loop” structure is denoted in red for MAO A and green for MAO B.
Mechanistic Proposals for MAO Catalyzed Amine Oxidation
The catalytic mechanism of MAO catalysis consists of a reductive half reaction where the Cα-H bond of the amine is cleaved with the transfer of two reducing equivalents to the flavin to form, respectively, the imine and flavin hydroquinone as shown in the reaction pathway in Fig. 5. All evidence to date supports the deprotonated form of the amine substrate as the species that enters the substrate binding site for interaction with the flavin. This view is based on analysis of para-substituted benzylamine binding to MAO A [19] and from analysis of the binding of para-substituted dimethylbenzylamine or para-substituted α-methylbenzylamine analogs to MAO B [20]. Further support that the amine substrate is in its neutral, deprotonated form on entering the substrate cavity stems from consideration of the energetics involved in placing a charged moiety in the hydrophobic cavity environment of either enzyme. In addition, previous work with p-dimethylaminobenzylamine oxidation by bovine MAO B [21] demonstrates the protonated form of the imine product is formed at a rate identical with the spectral changes observed for reduction of the flavin. This property is also observed with human MAO A and with active mutant forms of either enzyme. If imine formation from the imine involves only α-CH bond cleavage and subsequent electron rearrangement without any change in the number of H atoms bound to the amine N, this behavior is expected. To date, no data are available on the process involved for deprotonation of the amine substrates which, at physiological pH values, would be predominantly in its protonated forms in solution. The entrances to the active sites of either MAO A or MAO B are proposed to be at the surface of the negatively charged outer membrane which could function to increase the effective substrate concentration by electrostatic attraction which may explain why the observed Km values for the substrates are lower in membrane-bound forms of MAO than in detergent solutions.
Fig.5.

A schematic of the catalytic reaction pathway followed by MAO A and by MAO B. With most substrates, both enzymes follow the lower branch. MAO B follows the upper branch with the substrate phenethylamine.
Proposed Mechanism of C-H Bond Cleavage in MAO Catalysis
Investigations of the stereochemistry of MAO catalysis demonstrated that the pro-R H of several differing primary amine substrates are abstracted [22] using mitochondrial membrane preparations probably containing both MAO A and MAO B. Purified recombinant human MAO A and MAO B have been shown in this laboratory to exhibit the same stereochemistry in C-H bond cleavage as shown earlier for the membrane bound rat enzymes [M. Li, Unpublished observations]. These experiments demonstrate that there is a rigid stereo-chemical relation between the pro-R hydrogen of the substrate and the acceptor group on the enzyme during catalysis.
The cleavage of C-H bonds can occur by three different pathways. In one case, the H is abstracted as a hydride ion in which both bonding electrons follow the H to its acceptor. This hydrogen transfer mechanism is favored for the flavin-dependent amino acid oxidases [23]. A second case is the homolytic cleavage of C-H to form a carbon-based substrate radical and a hydrogen atom. Such reactions are usually catalyzed by radical generating systems as exemplified by several B12 enzymes or by ribonucleotide reductase as examples [24]. The third possibility is a proton abstraction reaction with a proton and a carbanionic substrate intermediate as products. To distinguish among these possibilities, a classical physical organic approach is to use para-substituted aromatic substrate analogs to determine the effect of electron withdrawing or donating substituents on reaction rates. For enzyme catalyzed reactions, the influence of substituents on reaction rates is complex and requires a considerable amount of work to delineate electronic, steric, and hydrophobic contributions. Fortunately, in the case of MAO catalysis, a series of benzylamine analogs can be readily synthesized in both their α,α-1H or α,α-2H forms. Large deuterium kinetic isotope effects (Dk values of 5-10) are observed in both steady state and in reductive half reaction stopped flow experiments demonstrating that C-H bond cleavage is rate limiting in catalysis [19, 25]. In the case of human MAO A, the rate of catalysis or the limiting rate of flavin coenzyme reduction increases with increasing electron withdrawing power of the para substituent which is characteristic of a proton abstraction reaction step [19].
(σ is a measure of electron donating or withdrawing power of the para substiuent)_ If the reaction were to be a hydride reaction, one would expect to observe an increase of rate with decreasing electron withdrawing power. For a hydrogen atom transfer reaction, one would expect to see little influence of electron withdrawing substituents on the rate. This latter behavior is what is observed with MAO B [25]. On the surface, this difference in behavior would indicate MAO A
(Es is the Taft steric constant and π is the hydrophobicity constant for the para substituents) and MAO B function by different mechanisms in catalysis. A comparison of catalytic site structures of both enzymes show that it is not necessary to propose separate mechanistic pathways. In the case of MAO A, the catalytic site is quite open which allows the aromatic ring of the substituted benzylamine to access different conformations as required for transmission of aromatic ring electronic effects to the benzyl carbon where C-H cleavage occurs. Structural studies show the MAO B active site is very constrained resulting in a reduction of the conformational sampling of the bound p-substituted benzylamine analogs to adopt conformations allowing the transmission of aromatic ring electronic effects to the benzyl carbon. In the absence of other data, our view is that both enzymes function by a proton abstraction mechanism and predict that a similar substituent electronic behavior would be observed in MAO B if the active site were to be altered in structure through application of multiple site-directed mutations to alter the limitations of substrate rotation in the substrate binding site. This type of analysis which involves both steady state and pre-steady state kinetic approaches coupled with deuterium kinetic isotope data on two related enzyme systems constitutes the most exhaustive QSAR approach published.
With strong evidence for a proton abstraction from a benzyl carbon, the question arose as what base on the enzyme would be strong enough since the pKa of a benzyl proton is expected to be ∼25 [26]? Structural analysis of the active sites in either MAO A or in MAO B show there are no basic amino acid residues that could possibly function as a proton acceptor [13,14]. The structure of the bound flavin ring in either MAO A or in MAO B offers some clues as to how such an proton transfer step could occur. The isoalloxazine ring in either enzyme is “bent” conformation which is approximately 30° from planarity about the N(5), N(10) axis whereas the ring geometry is in an energetically favorable flat conformation either free in solution or in most other flavoenzymes [13] (Fig. 6). The functional consequences of this bent conformation is that the N atom at the 5 position of the flavin ring is in a configuration closer to sp3 rather than to sp2 as shown by previous 15N and 13C NMR data on other flavoenzymes and on model flavin systems [27]. This results in a higher electron density at N(5) and lowered electron density at the C4a position of the flavin ring. The significance of this flavin conformation is that it facilitates the nucleophilic attack of the basic substrate amine lone pair on the C (4a) position of the coenzyme resulting in a flavin-substrate adduct that would be isoelectronic with the reduced flavin ring. For a flavin ring to have such a conformation, it must acquire a proton at the N(5) position. Recent 15N NMR studies [28] have estimated the pK of the N(5) proton of the reduced flavin ring to be ∼ 24 demonstrating that the N(5) position of the flavin ring is indeed very basic in its reduced form. Therefore, the formation of a flavin C(4a) substrate adduct would require proton addition to the N(5) position of the flavin coenzyme and therefore could constitute the strong base required to abstract the proton from the benzyl carbon of benzylamine analog substrates to account for the positive ρ value observed in QSAR plots of MAO A kinetic data [19]. A mechanistic scheme for the reductive phase of the catalytic mechanism of MAO that is consistent with both data and expectations based on structural information and model behaviors is shown in Fig. 7 and is derived from the original flavin nucleophilic mechanism proposed by Hamilton [29]. It is shown as a likely mechanism for MAO catalysis and may be applicable to the reaction mechanisms of other amine oxidizing flavoenzymes.
Fig.6.
Crystallographic data of the flavin conformation in MAO B. The flavin ring is oriented perpendicular to the plane of the drawing to show the aromatic flavin ring deviates from planarity.
Fig.7.
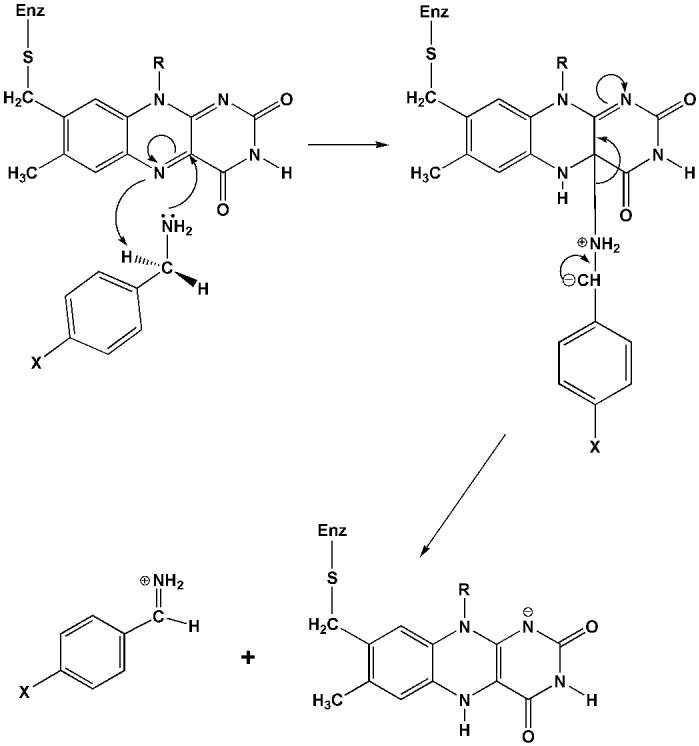
Suggested polar nucleophilic mechanistic scheme for the reductive half reaction in MAO A or in MAO B catalysis where the products of amine oxidation are the protonated imine and the flavin hydroquinone.
Supporting evidence for the proposed polar nucleophilic mechanism shown in Fig. 7 is provided by structural experiments to probe the binding of the MAO B specific inhibitor farnesol [16]. This isoprenoid structure binds to MAO B with the -OH group situated directly in front (3.4 Å) of the C(4a) position of the flavin ring and the 1-CH2 directly in front (also 3.4 Å) of the flavin N(5) site (Fig.8). These data are supportive of farnesol being a substrate analog that binds in a catalytic relevant conformation with turnover not being observed due to the decreased nucleophilicity of the -OH moiety relative to that of an amine group. Replacement of the OH with an amine does result in catalytic turnover with kcat = 30 min-1 and Km= 10 μM values for farnesylamine oxidation by MAO B (M. Li, Unpublished observations). Anaerobic incubation of either MAO A or MAO B with farnesylamine results in the rapid reduction of their respective flavin coenzymes.
Fig.8.

Ball and stick representation of the structure of the -CH2-OH moiety of bound trans-trans farnesol and the flavin ring in the structure of human MAO B. The indicated distances are between the oxygen of farnesol and the C(4a) of the flavin ring and the 1′ carbon of farnesol and the N(5) position of the flavin ring.
A survey of the literature reveals that the single electron transfer (SET) mechanism (Fig.9) for MAO catalysis originally proposed by Silverman and co-workers [30] is also favored by a number of investigators. This mechanism is based on the rationale that one-electron oxidation of the substrate amine nitrogen results in labilization of the α-CH bond to allow abstraction by a basic residue normally associated with proteins. An SET mechanism would also rationalize the observed ring opening observed with cyclopropylamine substrate analogs [30]. In spite of considerable experimental effort including stopped flow detection of flavin radicals and the influence of magnetic field on MAO B reaction rates as a probe for intermediate radical pair formation [31], no direct evidence has been found for any radical intermediate during substrate oxidation. Studies suggesting the formation of a catalytically significant tyrosyl radical in MAO A on one electron reduction of the flavin [32] have been shown to be incorrect using both published EPR experiments [32] and unpublished pulsed EPR experiments. Perhaps the most definitive evidence against the SET mechanism is the finding that the covalent flavin of MAO B exhibits one electron oxidation-reduction potentials of +0.043 V. for the Ox/Sq couple and + 0.037 V for the Sq/Hq couple [34]. Since the potential required for one-electron oxidation of primary or secondary amines is in the range of +1.0 to +1.5 V ([35], the possibility of 1 electron amine oxidation by a ground state flavin is quite remote. Recent theoretical calculations on MAO catalysis have demonstrated the plausibility of the polar nucleophilic mechanism for MAO catalysis shown in Fig. 7 [36] and have also shown that the QSAR data on MAO A can be represented by this mechanism. It would be of interest to also test the SET mechanism from a computational perspective, however, that type of mechanism is a formidable challenge to the computational chemists and has, to our knowledge, not been done.
Fig.9.

Proposed Single Electron Transfer Mechanism for the reductive half reaction in MAO catalysis [30].
Functional Role of the Aromatic Cage in MAO Catalysis
Structural studies show that two tyrosyl residues (398 and 435 in human MAO B) are situated directly in front of the re faces of the covalent flavins in both MAO A and in MAO B. Aromatic residues are found in front of the flavin ring in structural studies of the amine oxidizing enzymes polyamine oxidase and trimethylamine dehydrogenase [37]. To investigate possible functional roles of these tyrosyl side chains in the active sites of MAO A and MAO B, a number of site directed mutants of Tyr435 were formed and expressed in human MAO B and their functional and structural properties compared [38]. Mutagenesis of Tyr398 has not been done at this time since structural data show it is in a required cis peptide linkage with Cys398 which forms a covalent linkage with the flavin and it is not apparent how mutations of this residue would influence the protein folding and/or covalent flavin linkage in MAO B. Similar mutations with MAO A resulted in a decreased stability of these mutants on extraction and solubization from the membrane which therefore precluded their purification and characterization. A comparison of the catalytic properties of the MAO B mutants showed that a correlation exists between kcat/Km and the interaction energy of the dipole moment of the substrate amine moiety and the dipole moment of the side chain amino acid substituent at position 435. One of the conclusions from this study is that one of the functional roles of the “aromatic cage” is to polarize the amine moiety of the substrate to make it a more effective nucleophile in accord with the mechanism shown in Fig. 10. Another role appears to be provide a path for guiding the substrate amine towards the reactive positions on the flavin ring. Recent theoretical calculations [39] provide additional support for this interpretation of the experimental data.
Fig.10.

Schematic of the proposed effect of the dipole moments of the tyrosyl residues (398 and 435 in MAO B) on the electron lone pair of the substrate amine moiety before its attack on the flavin ring as depicted in the scheme in Fig.7.
Concluding Remarks
The focus of this review is to illustrate the important role of integrating structural information as a guide for the interpretation of mechanistic studies on enzymes such at MAO which has important consequences for our knowledge of amine oxidizing flavoenzymes in general. Since the focus of this review is on monoamine oxidases, comparisons could not be included with the well studied amino acid oxidase flavoenzyme class which, in our opinion, constitute a differing class of C-N bond dehydrogenating enzymes. In one aspect, the acquisition of three-dimensional structural data is akin to “turning on a light in a darkened room” in that it not only aids in the interpretation of existing kinetic and mechanistic data but also provides insights into the design of new experiments that would not have been feasible with that structural information. A key component that made possible all of the results presented in this review is the development of excellent expression systems for both human MAO B and human MAO A and improvements made to purification procedures such that the production of these recombinant enzymes and their mutant forms is now routine. For those advances, the authors are grateful to past and present graduate and post-doctoral students for their contributions.
Acknowledgements
The work described in this review was supported by NIH grant GM-29433, by Ministry of Science and Education Grants (Italy) FIRB and COFIN04, and by Fondazione MINTAS. C.B. is supported by an Investigator Fellowship from Collegio Ghislieri, Pavia, Italy. The following former and present graduate and postdoctoral students are acknowledged for their contributions to the work described her include: Paige Newton Vinson, Richard Miller, Min Li, Franta Hubalek, Luigi DeColibus, Milagros Aldeco, and Jin Wang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kumar MJ, Nicholls DG, Andersen JK. J. Biol. Chem. 2003;278:46432–46439. doi: 10.1074/jbc.M306378200. [DOI] [PubMed] [Google Scholar]
- 2.Forneris F, Binda C, Dall,Aglio A, Fraaije MW, Battaglioli E, Mattevi A. J. Biol. Chem. 2006;281:35289–35295. doi: 10.1074/jbc.M607411200. [DOI] [PubMed] [Google Scholar]
- 3.Salach JI. Arch. Biochem. Biophys. 1979;192:128–137. doi: 10.1016/0003-9861(79)90078-x. [DOI] [PubMed] [Google Scholar]
- 4.Weyler W, Salach JI. J. Biol. Chem. 1985;260:13199–13207. [PubMed] [Google Scholar]
- 5.Yue KT, Bhattacharyya AK, Zhelyaskov VR, Edmondson DE. Arch. Biochem. Biophys. 1993;300:179–185. doi: 10.1006/abbi.1993.1025. [DOI] [PubMed] [Google Scholar]
- 6.Urban P, Andersen JK, Hsu HPP, Pompom D. FEBS Lett. 1991;286:142–146. doi: 10.1016/0014-5793(91)80960-b. [DOI] [PubMed] [Google Scholar]
- 7.Weyler W, Titlow CC, Salach JI. Biochim. Biophys. Res. Commun. 1990;173:1205–1211. doi: 10.1016/s0006-291x(05)80914-3. [DOI] [PubMed] [Google Scholar]
- 8.Newton-Vinson AP, Hubalek F, Edmondson DE. Protein Expr. & Purif. 2000;20:334–345. doi: 10.1006/prep.2000.1309. [DOI] [PubMed] [Google Scholar]
- 9.Li M, Hubalek F, Newton-Vinson P, Edmondson DE. Prot. Exp. & Purif. 2002;24:152–162. doi: 10.1006/prep.2001.1546. [DOI] [PubMed] [Google Scholar]
- 10.Hiro I, Tsugeno Y, Hirashiiki I, Ogata F, Ito A. J. Biochem. (Toko) 1996;120:759–765. doi: 10.1093/oxfordjournals.jbchem.a021476. [DOI] [PubMed] [Google Scholar]
- 11.Rebrin I, Geha RM, Chen K, Shih JC. J. Biol. Chem. 2001;276:29499–29506. doi: 10.1074/jbc.M100431200. [DOI] [PubMed] [Google Scholar]
- 12.Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Nature, Structural Biology. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- 13.Binda C, Li M, Hubalek F, Restelli N, Edmondson DE, Mattevi A. Proc. Natl. Acad. Sci. USA. 2003;100:9750–9755. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeColibus L, Li M, Binda C, Edmondson DE, Mattevi A. Proc. Natl. Acad Sci. USA. 2005;102:12684–12689. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma J, Yoshimura M, Yamashita E, Nakagawa A, Ito A, Tsukihara T. J. Mol. Biol. 2004;338:103–114. doi: 10.1016/j.jmb.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 16.Hubalek F, Binda C, Khalil A, Li M, Mattevi A, Castagnoli N, Edmondson DE. J. Biol. Chem. 2005;280:15761–15766. doi: 10.1074/jbc.M500949200. [DOI] [PubMed] [Google Scholar]
- 17.Kearney EB, Salach JI, Walker WH, Seng RL, Kenney W, Zeszotek E, Singer TP. Eur. J. Biochem. 1971;24:321–327. doi: 10.1111/j.1432-1033.1971.tb19689.x. [DOI] [PubMed] [Google Scholar]
- 18.Andres AM, Soldevila M, Navarro A, Kidd KK, Oliva B, Bertranpetit J. Hum. Genet. 2004;115:377–386. doi: 10.1007/s00439-004-1179-6. [DOI] [PubMed] [Google Scholar]
- 19.Miller JR, Edmondson DE. Biochemistry. 1999;38:13670–13683. doi: 10.1021/bi990920y. [DOI] [PubMed] [Google Scholar]
- 20.Edmondson DE, Bhattacharrya AK, Xu J. Biochim. Biophys. Acta. 2000;1479:52–58. doi: 10.1016/s0167-4838(00)00055-8. [DOI] [PubMed] [Google Scholar]
- 21.Edmondson DE, Bhattacharyya AK, Walker MC. Biochemistry. 1993;32:5196–5202. doi: 10.1021/bi00070a031. [DOI] [PubMed] [Google Scholar]
- 22.Yu P, Bailey BA, Durden DA, Boulton AA. Biochem. Pharm. 1986;35:1027–1036. doi: 10.1016/0006-2952(86)90094-8. [DOI] [PubMed] [Google Scholar]
- 23.Umhau S, Pollegioni L, Molla G, Diederichs K, Welte W, Pilone M;, Ghisla S. Proc. Natl. Acad. Sci. USA. 2000;97:12463–12468. doi: 10.1073/pnas.97.23.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stubbe J, van Der Donk WA. Chem. Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 25.Walker MC, Edmondson DE. Biochemistry. 1994;33:7088–7098. doi: 10.1021/bi00189a011. [DOI] [PubMed] [Google Scholar]
- 26.Bordwell FG, Cheng J-P, Satish AV, Twyman CL. J. Org. Chem. 1992;57:6542–6546. [Google Scholar]
- 27.Rüterjans H, Fleischmann G, Knauf M, Löhr F, Blümel M, Lederer F, Mayhew SG, Müller F. Biochem. Soc. Trans. 1996:116–121. doi: 10.1042/bst0240116. [DOI] [PubMed] [Google Scholar]
- 28.Macheroux P, Ghisla S, Sanner C, Rüterjans H, Müller F. BMC Biochem. 2005;6:26. doi: 10.1186/1471-2091-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamilton GA. In: Progress in Bioorganic Chemistry. Kaiser ET, Kezdy FJ, editors. Vol. 1. J. Wiley & Sons; NY: 1971. pp. 83–157. [Google Scholar]
- 30.Lu X, Ji H, Silverman RB. In: Flavins and Flavoproteins 2002. Chapman S, Perham R, Scrutton N, editors. Agency for Sci. Publ.; Berlin: 2002. pp. 817–830. [Google Scholar]
- 31.Miller JR, Edmondson DE, Grissom CB. J. Am. Chem. Soc. 1995;117:7830–783. [Google Scholar]
- 32.Rigby SEJ, Hynson RMG, Ramsay RR, Munro AW, Scrutton NS. J. Biol. Chem. 2005;280:4627–4631. doi: 10.1074/jbc.M410596200. [DOI] [PubMed] [Google Scholar]
- 33.Ramsay RR, Upadhyay AK, Li M, Edmondson DE. In: Flavins & Flavoproteins. Nishino T, Miura R, Fukui K, Tanokura M, editors. ArchiTect, Inc.; 2005. pp. 137–142. [Google Scholar]
- 34.Newton-Vinson P, Edmondson DE. In: Flavins and Flavoproteins. Ghisla S, Kroneck P, Macheroux P, Sund H, editors. Agency for Scientific Publications; Berlin: 1999. pp. 431–434. [Google Scholar]
- 35.Portis LC, Klug JT, Mann CK. J. Org. Chem. 1974;39:3488–3494. [Google Scholar]
- 36.Erdem S, Karahan Ö, Yildiz I, Yelekci K. Org & Biomolec. Chem. 2006;4:646–658. doi: 10.1039/b511350d. [DOI] [PubMed] [Google Scholar]
- 37.Binda C, Mattevi A, Edmondson DE. J. Biol. Chem. 2002;277:23973–23976. doi: 10.1074/jbc.R200005200. [DOI] [PubMed] [Google Scholar]
- 38.Li M, Binda C, Mattevi A, Edmondson DE. Biochemistry. 2006;45:4775–4784. doi: 10.1021/bi051847g. [DOI] [PubMed] [Google Scholar]
- 39.Akyüz MA, Erdem SS, Edmondson DE. J. Neur. Trans. 2007 doi: 10.1007/s00702-007-0670-3. in press.