

Abstract
S-nitrosoglutathione (GSNO) stabilizes the α-subunit of hypoxia inducible factor-1 (HIF-1) in normoxic cells, but not in the presence of PI3K inhibitors. In this report, the biochemical pathway by which GSNO alters PI3K/Akt activity to modify HIF-1 expression was characterized in Cos cells and primary pulmonary vascular endothelial cells. GSNO increased Akt kinase activity—and downstream HIF-1α protein accumulation and DNA-binding activity—in a dose- and time-dependent manner. The PI3K inhibitors, wortmannin and LY294002, blocked these responses. Neither glutathione nor 8-bromo-cyclic GMP mimicked the GSNO-induced increases in Akt kinase activity. GSNO-induced Akt kinase activity and downstream HIF-1α stabilization were blocked by acivicin, an inhibitor of γ−glutamyl transpeptidase (γGT), a transmembrane protein that can translate extracellular GSNO to intracellular S-nitrosocysteinylglycine. Dithiothreitol blocked GSNO-induced Akt kinase activity and HIF-1α stabilization. Moreover, the 3′-phosphatase of phosphoinositides, PTEN (phosphatase and tensin homolog deleted on chromosome ten) was S-nitrosylated by GSNO in pulmonary arterial endothelial cells, which was reversed by dithiothreitol and ultraviolet light. Interestingly, the abundance of S-nitrosylated PTEN also correlated inversely with PTEN activity. Taken together, these results suggest that GSNO induction of Akt appears to be mediated by S-nitrosylation chemistry rather than classic NO signaling through guanylate cyclase/cGMP. We speculate that γGT-dependent activation of Akt and subsequent activation of HIF-1 in vascular beds may be relevant to the regulation of HIF-1–dependent gene expression in conditions associated with oxyhemoglobin deoxygenation, as opposed to profoundly low Po2, in the pulmonary vasculature.
Keywords: phosphatidylinositol 3-kinase, hypoxia-inducible factor-1, S-nitrosothiol, γ-glutamyl transpeptidase, PTEN
CLINICAL RELEVANCE
S-nitrosoglutathione, formed as NO moves away from erythrocytes in response to hemoglobin desaturation, may signal hypoxia-inducible factor-1–mediated physiologic and gene regulatory events in pulmonary endothelial cells without profound hypoxia, through a thiol-based reaction.
Hypoxia-inducible factor (HIF)-1 promotes the expression of several genes that confer hypoxic tolerance through angiogenesis, erythropoeisis, vasodilation, and altered glucose metabolism. In hypoxia, the regulatory subunit of the HIF-1 heterodimer, HIF-1α, is stabilized through decreased activity of prolyl hydroxylases that target the protein for degradation (1). In normoxia, HIF-1 can also be activated by mitogen-activated protein kinase (MAPK)-dependent processes initiated by growth factors (2–4) or S-nitrosoglutathione (GSNO) (5). Indeed, exposure to GSNO in normoxia results in the accumulation of HIF-1α protein. Of note, inhibitors of the phosphatidylinositol 3′-kinase (PI3K)-initiated Akt activation prevent this effect, implicating this signaling pathway in the GSNO effect (6).
Redox-associated modification of cysteine thiols by nitric oxide (NO) in biology can regulate the function of proteins (7–9). S-nitrosylated proteins are involved in numerous signaling pathways. Transnitrosation, the transfer of NO in the form of nitrosonium (NO+) from one cysteine thiol to a second cysteine thiol, is believed to be responsible for many actions of GSNO. Intracellular transnitrosation reactions involving GSNO appear to require bioactivation by γ glutamyl transpeptidase (γGT). In this reaction, γGT cleaves GSNO to form cell-permeable S-nitrosocysteinyl glycine (CGSNO) and glutamate (10–13). CGSNO then transfers the NO+ to the recipient cysteine thiol as follows:
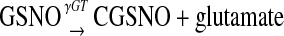 |
(1) |
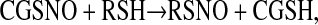 |
(2) |
where R = the target intracellular thiol (7, 13, 14).
In mammals, endovascular cells are rarely exposed to the Po2 values (< 10 mm Hg) normally required to achieve classical HIF-1 activation in vitro (15). GSNO formation in the presence of deoxygenated erythrocytes is proposed to reflect oxyhemoglobin desaturation, as opposed to low Po2 (10, 16, 17). As such, it is of interest that GSNO can induce Akt activation and subsequent downstream HIF-1 activity in relative normoxia. This may be relevant to the regulation of HIF-1–dependent genes in the pulmonary artery, as a result of changes in hemoglobin oxygen saturation rather than changes in Po2 (5, 7, 13). In this report, GSNO activation of Akt and the downstream effects on HIF-1 DNA binding and activity are shown to be relevant in primary pulmonary vascular endothelial cells, and to be both γGT dependent and cGMP independent. This may involve thiol modification of PI3K/Akt-regulatory enzymes, including its counter-regulatory phosphatase, PTEN (phosphatase and tensin homolog deleted on chromosome ten).
MATERIALS AND METHODS
Materials
All chemicals used in this study were obtained from Sigma Chemical Co. (St Louis, MO). Lipofectamine and lipofectamine 2000 for transfections were obtained from Invitrogen (Carlsbad, CA). Ly294002 was obtained from Cell Signaling Technology, Inc. (Beverly, MA). HA antibodies (Y-11 or MMS-101P) were obtained from either Santa Cruz (Santa Cruz, CA) or Covance (Berkeley, CA), respectively. Anti-phospho(serine) Akt polyclonal antibody, anti-MAPK (p44/42) polyclonal antibody, and anti-PTEN polyclonal antibody were obtained from Cell Signaling Technology, Inc. Proteins A and G were obtained from Roche Diagnostics Corporation (Indianapolis, IN). Akt immunoprecipitation kinase assay was obtained from Upstate Biotechnology (Lake Placid, NY).
Cell Culture
Primary isolates of bovine pulmonary artery endothelial cells (BPAEC) from a local slaughterhouse were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum and 2.4 μg thymidine and were characterized as described previously (5). In these studies, BPAEC were used between passages 3 and 8. Primary murine lung vascular endothelial cells (MLVEC) were isolated from male C57Bl6/SvEv129 mice killed with Nembutol (5 mg/kg). The lungs were removed by thoracotomy after a wash with isopropanol and placed in heparinized Dulbecco's PBS. The lungs were incubated with collagenase at 37°C for 15 minutes, the tissue minced, and incubated for an additional 15 minutes. The tissue was spun at 500 × g for 5 minutes and the pellet washed with base media (DMEM D-valine substituted for L-valine). The minced lung was placed in two 6-well plates coated with 2% gelatin and incubated for 72 hours. Cells were sorted using uptake of acetylated LDL. LDL-positive cells were passaged. Cos 7 fibroblasts were grown in DMEM supplemented with 10% fetal bovine serum and 1% sodium pyruvate. Cells were maintained in a humidified 5% CO2 incubator at 37°C.
Transfections
Cos 7 cells were transiently co-transfected with plasmid DNA encoding (PEBB) HA-Akt (1 μg) and (PCMV) GFP-luciferase (35 ng) using lipofectamine in the absence of serum as described by the manufacturer. Luciferase activity was measured using a Lumat LB9507 luminometer (Berthhold Systems, Inc., Pittsburgh, PA). In experiments examining the effects of acivicin or LY294002, cells were treated for 30 minutes before incubation with S-nitrosoglutathione, 8-Bromo cGMP (8-Br cGMP), or glutathione (GSH). For the experiments using dithiothreitol (DTT), a thiol-reducing agent that reduces –SNO bonds to –SH, cells were exposed to DTT during the last 30 minutes of treatment after three washes with media to remove GSNO.
Aqueous Nitric Oxide Generation and Administration
Saturated aqueous NO· was prepared as previously described and stored in a −80°C freezer in a gas-tight vial (17). Before use, the vial containing aqueous NO· was thawed in a glove box deaerated with N2 gas. Aliquots of aqueous NO· needed to achieve the desired concentration used in the performed experiments were drawn into a 100-μl Hamilton syringe. The syringe was removed from the glove box and the aqueous NO· injected directly into the media overlaying the cells.
Akt Kinase Assay
Cells were lysed in buffer containing 50 mM Tris pH 7.6, 150 mM NaCl, 1% Tween-20, 10 mM sodium pyrophosphate, 10 mM NaF, 1 mM Na3VO4, 2 mM phenylmethanesulfonyl fluoride (PMSF), and 10 mg/ml each of aprotinin, leupeptin, pepstatin, and 1 μM microcystin. HA-Akt protein from the lysate was immunoprecipitated using anti-HA antibodies and protein G–conjugated beads for 2 to 24 hours at 4°C. Kinase activity of protein G–bound HA Akt protein was analyzed on a GSK-3–derived substrate peptide by 32P incorporation using the Akt immunoprecipitation kinase assay kit (Upstate USA, Inc., Charlottesville, VA) (18).
SNO-PTEN Assay
S-nitrosylated PTEN was detected by a modification of the biotin substitution method (19, 20). All experiments were fastidiously performed in a dark environment to prevent false-positive biotinylation due to ambient sunlight. Initially, all free thiols in whole cell lysate from treated or untreated Cos 7 or BPAEC were blocked by addition of 20 mM methyl methanethiosulfonate (MMTS) with 2% SDS (to ensure access of MMTS to buried cysteines) for 20 minutes at 50°C. Free MMTS was then removed by protein precipitation with addition of cold acetone at −20°C for 30 minutes. After centrifugation at 13,500 × g for 15 minutes at 4°C, the pellet was resuspended in 100 μl of HEN buffer (250 mM HEPES pH 7.7, 1 mM EDTA, 0.1 mM neocuproine) plus 1 μM sodium ascorbate for 1 hour at 25°C in the presence of the sulfhydryl-specific biotinylating reagent, N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propion-amide (biotin-HPDP, 1 mM). After 1 hour of incubation at room temperature, the free biotin-HPDP was removed by acetone precipitation. The pellet was resuspended in a neutralization buffer containing streptavidin-agarose beads (biotin binding capacity: 15–30 μg/ml) for 1 hour at 25°C to isolate the biotinylated proteins, representing those that were S-nitrosylated. After five washes with a high salt buffer (20 mM HEPES pH 7.7, 600 mM NaCl, 1 mM EDTA, 0.5% Triton X-100) to eliminate weak nonspecific binding, SDS PAGE sample buffer was added. Proteins were then resolved by SDS-PAGE. S-nitroso-PTEN protein bands were identified by Western blot using anti-PTEN antibody. To establish the overall proportion of PTEN that was S-nitrosylated, whole cell lysate from the same samples was simultaneously applied to SDS-PAGE and total PTEN protein quantified by Western blot with anti-PTEN antibody. Densitometry was performed to quantify the protein bands. Variability in protein loading was minimal as examined by MAPK protein abundance with anti-MAPK antibody on the same gels.
PTEN Activity Assay
Fifteen micrograms of total protein from whole cell lysates were incubated with 300 μl of lysis buffer (see Akt Kinase Assay), containing no DTT, with 3μl of anti-PTEN antibody (Cell Signaling) overnight at 4°C. Protein G agarose beads were added to samples and incubated for 3 hours at 4°C. Beads were washed five times with 500 μl lysis buffer, then twice more with a low-stringency buffer containing 20 mM HEPES (pH 7.7), 50 mM NaCl, and 0.1 mM EDTA. Beads were resuspended in 50 μl assay buffer containing100 mM Tris-HCl, pH 7.5, and 100 μM phosphatidylinositol 3,4,5-trisphosphate diC8 (Echelon Biosciences Inc., St. Lake City, Utah). Samples were shaken at 37°C for 40 minutes in wells of a 96-well plate. Liberated inorganic phosphate was detected using a Malachite Green detection kit (R&D Systems, Inc., Minneapolis, MN) as described by the manufacturer.
Electromobility Shift Assay
Cells were gently scraped, quickly pelleted, and lysed at 4°C for 15 minutes with lysis buffer, containing 50 mmol/L Tris, pH 7.6, 150 mmol/L NaCl, 1% NP-40, 10 mmol/L Na pyrophosphate, 10 mmol/L NaF, 1 mmol/L Na3VO4, 2 mmol/L PMSF, and 10 μg/ml each of aprotinin, leupeptin, pepstatin, and PMSF. After centrifugation at 14,000 rpm for 10 minutes at 4°C, the whole cell lysate (5 μg) supernatant was then preincubated in binding buffer (10 mmol/L Tris, 50 mmol/L KCl, 50 mmol/L NaCl, 1 mmol/L MgCl2, 1 mmol/L EDTA, 5 mmol/L DTT, 0.1 μg calf thymus DNA, and 5% glycerol) for 5 minutes at 4°C. The radiolabeled oligonucleotide probe containing the HIF-1 DNA-binding sequence of the iNOS gene was added and incubated for 15 minutes. The mixture was loaded on a 5% nondenaturing polyacrylamide gel and electrophoresis was performed in 0.5 TBE (1× TBE is 89 mmol/L Tris-borate and 20 mmol/L EDTA [pH 8.0]) at 4°C. The gel was dried and autoradiography was performed.
Statistical Methods
Kinase activity was assessed as the average of triplicate samples, and each experiment was performed a minimum of three times.
Results were analyzed by ANOVA with calculation of standard error of the means. Gel experiments were also performed a minimum of three times.
RESULTS
GSNO Increases Akt Kinase Activity
Cos 7 cells were transiently transfected with a plasmid vector expressing a HA-tagged Akt construct. Akt kinase activity in the HA-Akt–transfected cells was significantly increased over the untransfected control. In addition, exposure to GSNO caused an increased Akt kinase activity over time with a 1.7-fold increase seen at 3 hours (Figure 1A). Moreover, Akt kinase activity was increased as a function of dose (Figure 1B). Western blot analysis of whole cell lysates confirmed expression of the HA-Akt protein (not shown). Taken together, these data show that GSNO increases Akt kinase activity in a dose- and time-dependent manner. Preincubation (30 min) with Ly294002, a potent inhibitor of PI3K, completely eliminated the effect of GSNO to increase Akt kinase activity; the level of Akt activity was reduced to the levels seen in the untransfected control (Figure 1C). Similar results were seen with wortmannin. The S-nitrosylation stability of GSNO was analyzed in the presence of LY294002, using the cupric chloride/cysteine chemiluminescence assay, and showed no change over a 5-hour period (data not shown) (21). Thus, GSNO-mediated activation of HA-Akt is blocked by inhibiting PI3K.
Figure 1.

GSNO increases Akt kinase activity in a time- and dose-dependent manner, an effect that is blocked by the phosphoinositol 3-kinase (PI3K) inhibitor, LY294002. Cos 7 cells were transiently co-transfected with plasmid DNA encoding HA-Akt and GFP-luciferase. Equal amounts of protein were immunoprecipitated with anti-HA antibody for each condition and Akt kinase activity assessed as described in Materials and Methods. Activity was corrected for transcription efficiency by luciferase activity. (A) Cells were treated with 100 μM GSNO for various times, or (B) were treated for 3 hours with varying concentrations of GSNO. (C) Additional Cos 7 cells were preincubated for 30 minutes with 100 μM Ly294002, a potent inhibitor of PI3K, then exposed to 100 μM GSNO for 3 hours. All cells were collected in duplicate or triplicate for each experiment and standard errors of the means were calculated (n = 7, *P = 0.002 for time course in A; n = 3, *P = 0.006 for dose response in B; n = 2, *P = 0.006 and #P = 0.006 for C).
GSNO-Induced HIF-1 Expression Is Inhibited by PI3K/Akt Inhibitors
GSNO resulted in a transient, time-dependent increase in HIF-1α protein expression under conditions of normoxia in BPAEC and Cos 7 cells (Figure 2A). Maximum stabilization of HIF-1α occurred within 60 to 75 minutes of exposure to GSNO, but was transient, decreasing to basal levels within 3 hours. This was in contrast to GSNO induction of Akt, which showed a gradual increase in activity with time (Figure 1A). Consistent with previously published results, GSNO induced HIF-1 DNA-binding activity in normoxia (Figure 2B). Interestingly, the abundance of HIF-1α was substantially reduced in both BPAEC and Cos 7 in the presence of PI3K inhibitors, suggesting that activation of the PI3K/Akt pathway is necessary for GSNO-mediated HIF-1α protein accumulation and HIF-1 DNA binding activity (Figure 2C).
Figure 2.

GSNO causes an accumulation of HIF-1α protein and HIF-1α DNA binding in normoxia, but not in the presence of PI3K inhibitors. (A) BPAECs or Cos 7 cells were treated with 100 μM GSNO for various times. HIF-1α protein was detected in whole cell lysate in treated and untreated cells by Western blot analysis using anti–HIF-1α antibody. Blot is representative of several. (B) Cos 7 cells were treated with 100 μM GSNO or hypoxia for the times indicated. HIF-1 binding to DNA was determined by electromobility shift assay on whole cell lysate. (C) Cos 7 cells and BPAECs were treated with 100 μM GSNO in the presence or absence of the PI3K inhibitors 100nM wortmannin or 25 μM Ly 294002. HIF-1α protein was detected in whole cell lysate by Western blot analysis using anti–HIF-1α antibody.
GSNO-Mediated Activation of Akt Is cGMP Independent and Reversed by DTT
To test whether cGMP is required in the GSNO-mediated activation of Akt, according to the classical NO-guanylate cyclase pathway, a nonhydrolyzable cGMP analog, 8-Br cGMP, was tested. 8-Br cGMP did not increase Akt kinase activity in Cos cells compared with the increase seen with GSNO (Figure 3A). Similarly, GSNO increased Akt phosphorylation, which is required for Akt kinase activation, while 8-Br cGMP did not, in MLVEC (Figure 3A).
Figure 3.

Akt and HIF-1α activation by GSNO occurs through a thiol-dependent reaction. (A) Cos 7 cells were transfected as described in Figure 1, then treated with 100 μM GSNO or 100 μM 8-Br cGMP and Akt activity assessed. Results were corrected for transcription efficiency using luciferase activity (n = 3; *P = 0.007). Primary MLVEC were treated with 100 μM GSNO, ± Ly294002, or 8-Br cGMP for the times stated. Phosphorylated Akt protein was detected in whole cell lysate in treated and untreated cells by Western blot analysis using an anti-phospho(serine) Akt antibody. (B) Cos 7 cells were treated with GSNO, washed, then treated with 200 μM DTT. Akt kinase assays were preformed (n = 3; *P = 0.006), and HIF-1α protein was detected in whole cell lysate by Western blot analysis using anti–HIF-1α antibody.
To investigate whether the activation of Akt kinase by GSNO involves a thiol residue, the thiol-reducing agent DTT was used. Cos 7 cells transfected with the plasmid expressing HA-Akt were treated with DTT during the final 30 minutes of GSNO exposure. DTT significantly inhibited GSNO-mediated activation of Akt kinase, suggesting the mechanism of GSNO action involves a thiol residue. Similarly, DTT significantly decreased HIF-1α protein levels (Figure 3B).
GSNO-Mediated Activation of Akt and HIF-1α Requires γGT
GSNO often requires γGT, which degrades GSNO to membrane-permeable CGSNO, for bioactivity (5, 10, 11). To determine if γGT is required for GSNO-induced activation of Akt, Cos 7 cells transfected with the plasmid expressing HA-Akt were preincubated with acivicin, a potent inhibitor of γGT; Akt kinase activity was determined. Acivicin inhibited GSNO-induced Akt kinase activity (Figure 4A) and HIF-1α levels (Figure 4B).
Figure 4.

GSNO activation of Akt requires the GSNO membrane transporter, γGT, and is not duplicated by reduced glutathione. (A) Cos 7 cells were transfected as in Figure 1. Cells were preincubated with 100 μM acivicin in the absence or presence of GSNO. Akt kinase activity was assessed as described (n = 7, *P = 0.005). (B) BPAEC and Cos 7 cells were treated with GSNO, with or without acivicin, and HIF-1α protein was detected in whole cell lysate in treated and untreated cells by Western blot analysis using anti–HIF-1α antibody. (C) Akt-transfected Cos 7 cells were treated with 100 μM GSNO or 100 μM GSH and Akt kinase activity assessed (n = 5, *P = 0.003).
γGT is also involved in GSH turnover. To determine whether Akt kinase activity by GSNO is an effect of glutathione, experiments were performed in the presence of exogenous reduced GSH. Akt kinase activity was not significantly up-regulated by GSH, relative to GSNO (Figure 4C).
S-Nitroso-PTEN Accumulates in the Presence of GSNO in a Time- and Dose-Dependent Manner
Akt must be translocated to the plasma membrane for activation (18). This translocation occurs when Akt binds to a plasma membrane phospholipid that has been phosphorylated by PI3K at the inositol 3′-OH group. Conversely, activation of Akt is inhibited by the phosphatase, PTEN, which dephosphorylates the same 3′-OH group. GSNO could augment the membrane association and activation of Akt by modifying a PTEN cysteine residue, rendering the enzyme inactive. To test this possibility, BPAEC, Cos 7 cells, and MLVEC were treated with GSNO, S-nitroso-cysteinylglycine (CGSNO), S-nitroso-N'-acetylcysteine (SNOAc), aqueous nitric oxide (AqNO), or L-S-nitrosocysteine (L-SNOcys). The ability of PTEN to be S-nitrosylated (SNO-PTEN) was measured using the biotin-for-SNO exchange method (19, 20). Western blot band densities of SNO-PTEN protein were normalized to each other within the experiment to assess the relative level of modification. Total PTEN bands from the same lysate were also similarly normalized to each other. Therefore, the “ratio” of SNO-PTEN to total PTEN represents a relative level of modification rather than the absolute number of PTEN molecules that are S-nitrosylated.
In all three cell lines examined, GSNO treatment resulted in an increase in the relative proportion of SNO-PTEN to total PTEN (Figures 5A), which was attenuated with the thiol-reducing agent, DTT, or ultraviolet light exposure, implicating an S-nitrosylation event. Incubation with GSNO showed an increasing SNO-PTEN: PTEN ratio over time (Figure 5A, first panel), attenuation by acivicin indicating a requirement for the γGT receptor (second panel), and an effect of GSNO dose (third panel). Treatment of MLVEC with nitrosothiols other than GSNO were less efficient in S-nitrosylating PTEN (Figure 5B). Reduced glutathione (GSH) did not mimic the GSNO effect.
Figure 5.

S-nitroso-PTEN accumulates in the presence of GSNO and is inactivated in a time- and dose-dependent manner. (A) BPAEC, Cos 7, or MLVEC were treated with GSNO, GSH, S-nitroso-cysteinylglycine (CGSNO), S-nitroso-N'-acetylcysteine (SNOAc), or aqueous nitric oxide (AqNO) for the times and doses stated. Some cells were treated with DTT during the final 30 minutes of GSNO treatment or were preincubated with 100 μM acivicin. Lysate from some GSNO-treated cells was treated with ultraviolet light before analysis. Total PTEN protein was then detected in the lysate samples by Western blot analysis using anti-PTEN antibody. MAPK protein levels were assessed using anti-MAPK antibody to control for protein loading. S-nitrosylated PTEN (SNO-PTEN) was isolated from whole cell lysate using the biotin switch method. SNO-PTEN protein was detected by Western blot analysis using anti-PTEN antibody. Relative protein densities of the SNO-PTEN bands were compared with that of total PTEN. The data shown are single experiments. Similar patterns of results were seen in at least two trials for each cell line. (B) MLVEC cells were treated as stated. Data shown are Western blots representing total PTEN from whole cell lysate, and SNO-PTEN isolated from the same lysate using the biotin switch method. The graph represents the ratio of SNO-PTEN: total PTEN and is representive of several experiments illustrating the same pattern of SNO-PTEN: total PTEN ratios. (C) BPAEC and MLVEC cells were treated as described above. Western blots show bands representing total PTEN and SNO-PTEN isolated from the same lysate. The graph below represents the relative PTEN phosphatase activity on phosphatidylinositol 3,4,5-trisphosphate (PIP3). Arrows associate enzyme activity with the correlating SNO-PTEN and total PTEN bands detected by Western blot.
In both BPAEC and MLVEC, S-nitrosylation of PTEN corresponded to a decrease in its phosphatase activity, and the largest reduction in enzyme activity followed treatment with GSNO (Figure 5C). In MLVEC, the decrease in SNO-PTEN phosphatase activity by GSNO was a function of the dose; not mimicked by reduced glutathione; and dissipated by treatment with acivicin, DTT, and strong ultraviolet light. More importantly, the down-regulation of PTEN enzyme activity after treatment with GSNO was inversely related to the accumulation of SNO-PTEN and to Akt kinase activity noted in previous figures.
DISCUSSION
In this work, the cellular pathway through which GSNO activates Akt leading to the stabilization and activation of HIF-1 in normoxia was examined. The data demonstrate that: (1) GSNO activates the PI3K/Akt pathway; (2) the mechanism involves signaling based on NO-thiol chemistry, as opposed to cGMP-based signaling; and (3) the activity of the surface enzyme, γGT, is required. They also confirm previous observations that Akt activation by GSNO results in HIF-1 accumulation and activation under nonhypoxic conditions, and extend these observations to primary pulmonary endothelial cells.
The concentration of GSNO used to study Akt activation was chosen to maximize the response seen in statically grown culture cells. This dose is higher than that normally measured in the plasma (22). However, lower concentrations of GSNO (10μM), similar to those found in the airway (23) and the brainstem (24), were found to stimulate Akt activity, increase SNO-PTEN levels, and decrease PTEN activity, albeit to a lesser degree than a larger dose GSNO. In addition, local concentrations of GSNO may be higher than that measured in serum, especially in disease states where endogenous S-nitrosothiol levels may increase due to increased blood flow and shear stress (increased SNO delivery), inflammation (increase SNO production), or increased SNO transfer (hypoxia).
The findings that GSNO activation of PI3K/AKT is cGMP independent and is reversed by the thiol-modifying agent, DTT, are in apparent contrast to those of Kawasaki and coworkers, who demonstrated a cGMP-dependent mechanism (25). This apparent contradiction may be explained by the following. First, cGMP-independent and cGMP-dependent pathways may complement one another to produce cell-specific regulation (26). Indeed, activation of the PI3K pathway by GSNO appears to be cell type–specific (25). Second, pathways of cellular GSNO bioactivity may be regulated by certain cell membrane proteins (11, 13, 27). For instance, GSNO bioactivation depends on the abundance of cell surface γGT, which is known to be steadily lost over time in cells grown in culture (28). Since it is not known whether the cells used by Kawasaki and colleagues have intact γGT, it is possible to speculate that the differences reflect, in part, a change from S-nitrosylation to a cGMP-based mechanism as surface γGT is lost.
One known downstream consequence of Akt activation by GSNO is the stabilization and activation of HIF-1 (6, 18). GSNO is believed to activate HIF-1 by both PI3K/Akt and non-PI3K/Akt mechanisms. PI3K/Akt may modify HIF-1 abundance by inhibiting its non-pVHL–mediated degradation through altered heat shock protein (hsp90) synthesis (29), by interfering with regulators of proteosome degradation such as the forkhead transcription factor, FOX04 (30), and/or by increasing general cap-dependent mRNA translation of HIF-1α (31). Non-PI3K/Akt mechanisms include S-nitrosylation events that either impair the activity of the prolyl hydroxylases or directly S-nitrosylate HIF-1α at Cys-800 (32, 33). It is not known whether some or all of these events take place in a particular cell upon GSNO treatment or if they are cell type specific. Of note in this regard, we have recently published our finding that GSNO SNO-modifies Hsc70 in airway epithelial cells (34), but a causal relationship between the modification and HIF1α stabilization remains to be shown.
The activity of PI3K and Akt is opposed by the phosphatase PTEN, which specifically dephosphorylates membrane inositol phospholipids at the 3′-OH group of the inositol ring, preventing Akt translocation to the membrane for activation (35). PTEN thereby inhibits PI3K-dependent activation of Akt. Deletion or inactivation of PTEN results in constitutive Akt activation (39). It is known that PTEN contains an essential cysteine within its catalytic domain that, when mutated, renders the phosphatase impotent (36). Recent studies suggest that PTEN is redox regulated (36–38), and that this cysteine has particular redox vulnerability due to its signature motif [C(X5)R] and a local pKa < 5 (40–43). Thus, one could hypothesize that other redox alterations of the PTEN cysteine residue, in this case S-nitrosylation by GSNO, could likewise inactivate the phosphatase, and account for the positive effect of GSNO on the PI3K/Akt pathway. In fact, PTEN has been shown to be oxidized by L-S-nitrosocysteine in epidermoid carcinoma cells resulting in decreased activity (44). Here, using direct methods, we demonstrate that PTEN can be SNO-modified in pulmonary vascular endothelial cells and that this modification is increased by GSNO, markedly weakening the phosphatase. Although glutathionylation (S-S formation) by GSNO cannot be completely excluded, (1) the effects of reduced glutathione on Akt activation were significantly less than SNO-glutathione; (2) the effects of the biotin switch were performed in the presence of ascorbate, which is highly selective for S-nitrosylated versus S-oxidized proteins (45), and strong ultraviolet light eliminated the signal; and (3) a dose of DTT strong enough to break S-S bonds would denature proteins and result in cell death, which was not observed. Taken together, these data favor the concept that nitrosative stress induced by GSNO on PTEN preferentially results in the S-nitroso- form of PTEN, as opposed to PTEN disulfide.
There appears to be some increase in SNO-PTEN with a corresponding reduction in activity by reduced glutathione and other nitrosothiols, albeit to a lesser degree. The effect of reduced glutathione could be explained by its ability to be S-nitrosylated, thereby participating in transnitrosation events within the cell. The reduced efficacy of the other S-nitrosylating agents, on the other hand, may be due to S-nitrosothiol–specific regulatory mechanisms influencing intracellular bioavailability. Nonetheless, these data suggest that they do not S-nitrosylate, nor inactivate PTEN as readily as GSNO, an abundant endogenously produced S-nitrosylating agent.
Inhibition of PTEN by GSNO-produced S-nitrosylation would be predicted to increase Akt activity, increasing downstream HIF-1 activity. Data presented here support that proposition. These observations may help explain HIF-1 activation in pulmonary endothelial cells in the absence of profound hypoxia. Typically, the oxygen level seen in mammalian vascular endothelial cells does not compare with the oxygen levels required in vitro to induce HIF-1 (15). Activation of HIF-1 by GSNO in normoxia may be physiologically important during hemoglobin deoxygenation, where glutathione accelerates the movement of NO away from intact erythrocytes producing an increase of GSNO (10, 17). GSNO, therefore, may signal physiologic (10, 16) and gene regulatory (5, 22) events in response to oxyhemoglobin desaturation, as opposed to low pO2. One such signaling target of GSNO, defined here, is the activation of Akt through PI3K.
In conclusion, the endogenous S-nitrosothiol, S-nitrosoglutathione (GSNO), activates PI3K/Akt, resulting in the stabilization of HIF-1α in normoxia in Cos 7 cells and in primary pulmonary vascular endothelial cells. In our experiments, this occurred not through classic NO· diffusion with activation of guanylyl cyclase and cGMP, but through a thiol-based reaction requiring the membrane protein, γGT. One target of this modification was the Akt inhibitory enzyme, PTEN. We propose that this complex pathway (Figure 6) may be exploited to alter hypoxia-mimetic states in diseases that are characterized by limited oxygen availability.
Figure 6.

Proposed pathway by which GSNO can activate HIF 1 through PI3K/Akt in normoxia. This schematic illustrates the proposed regulation of HIF-1 in normoxia by the endogenous S-nitrosothiol, GSNO, through PI3K/Akt activation. In pulmonary vascular endothelial cells and Cos 7 cells, this appears to occur not through classic NO· diffusion with activation of guanylyl cyclase, but through a thiol-based reaction requiring the membrane protein, γGT. One target of this –SNO modification is activation of PI3K. Another target is PTEN, the counter-regulatory phosphatase of the PI3K/Akt pathway.
Acknowledgments
The authors thank Dr. Kodi S. Ravichandran for the HA-Akt plasmid, Dr. James Garrison for assistance with PTEN activity assays, and Katie Brown-Steinke for expert assistance with cell transfections and mouse cell isolation.
This work was supported by a Child Health Research Career Development grant from the National Institutes of Health (HD01421–01, D.J.C.), K08 GM074880–01 (D.J.C.), and RO1 HL068173 (L.A.P.).
Originally Published in Press as DOI: 10.1165/rcmb.2006-0289SM on May 31, 2007
Conflict of Interest Statement: B.G. has been a consultant to and a minority shareholder of Nitrox LLC, a company with an interest in S-nitrosoglutathione signaling in pulmonary hypertension. None of the other authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Semenza GL. O2-regulated gene expression: transcriptional control of cardiorespiratory physiology by HIF-1. J Appl Physiol 2004;96:1173–1177. [DOI] [PubMed] [Google Scholar]
- 2.Zhou J, Callapina M, Goodall GJ, Brune B. Functional integrity of nuclear factor kappa B, phosphatidylinositol 3′-kinase, and mitogen-activated protein kinase signaling allows tumor necrosis factor alpha-evoked Bcl-2 expression to provoke internal ribosome entry site-dependent translation of hypoxia-inducible factor 1alpha. Cancer Res 2004;64:9041–9048. [DOI] [PubMed] [Google Scholar]
- 3.Qian D, Lin HY, Wang HM, Zhang X, Liu DL, Li QL, Zhu C. Normoxic induction of the hypoxic-inducible factor-1 alpha by interleukin-1 beta involves the extracellular signal-regulated kinase 1/2 pathway in normal human cytotrophoblast cells. Biol Reprod 2004;70:1822–1827. [DOI] [PubMed] [Google Scholar]
- 4.Kasuno K, Takabuchi S, Fukuda K, Kizaka-Kondoh S, Yodoi J, Adachi T, Semenza GL, Hirota K. Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J Biol Chem 2004;279:2550–2558. [DOI] [PubMed] [Google Scholar]
- 5.Palmer LA, Gaston B, Johns RA. Normoxic stabilization of hypoxia-inducible factor-1 expression and activity: redox-dependent effect of nitrogen oxides. Mol Pharmacol 2000;58:1197–1203. [DOI] [PubMed] [Google Scholar]
- 6.Sandau KB, Faus HG, Brune B. Induction of hypoxia-inducible-factor 1 by nitric oxide is mediated via the PI 3K pathway. Biochem Biophys Res Commun 2000;278:263–267. [DOI] [PubMed] [Google Scholar]
- 7.Gaston B, Doctor A, Singel D, Stamler JS. S-Nitrosothiol signaling in respiratory biology. Am J Respir Crit Care Med 2006;173:1186–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 2005;6:150–166. [DOI] [PubMed] [Google Scholar]
- 9.Forrester MT, Stamler JS. A classification scheme for redox-based modifications of proteins. Am J Respir Cell Mol Biol 2007;36:135–137. [DOI] [PubMed] [Google Scholar]
- 10.Lipton A, Johnson M, Macdonald T, Lieberman M, Gozal D, Gaston B. S-nitrosothiols signal the ventilatory response to hypoxia. Nature 2001;413:171–174. [DOI] [PubMed] [Google Scholar]
- 11.Hogg N, Singh RJ, Konorev E, Joseph J, Kalyanaraman B. S-Nitrosoglutathione as a substrate for gamma-glutamyl transpeptidase. Biochem J 1997;323:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaman K, Palmer LA, Doctor A, Hunt JF, Gaston B. Concentration-dependent effects of endogenous S-nitrosoglutathione on gene regulation by specificity proteins Sp3 and Sp1. Biochem J 2004;380:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaston B, Carver J, Doctor A, Palmer LA. S-nitrosylation signaling in cell biology. Mol Interv 2003;3:253–263. [DOI] [PubMed] [Google Scholar]
- 14.Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron 1997;18:691–696. [DOI] [PubMed] [Google Scholar]
- 15.Jiang BH, Semenza GL, Bauer C, Marti H. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol 1996;271:1172–1180. [DOI] [PubMed] [Google Scholar]
- 16.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 1996;380:221–226. [DOI] [PubMed] [Google Scholar]
- 17.Doctor A, Platt R, Sheram ML, Eischeid A, McMahon T, Maxey T, Doherty J, Axelrod M, Kline J, Gurka M, et al. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc Natl Acad Sci USA 2005;102:5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carver DJ, Aman MJ, Ravichandran KS. SHIP inhibits Akt activation in B cells through regulation of Akt membrane localization. Blood 2000;96:1449–1456. [PubMed] [Google Scholar]
- 19.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 2001;3:193–197. [DOI] [PubMed] [Google Scholar]
- 20.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001;86:PL1. [DOI] [PubMed] [Google Scholar]
- 21.Fang K, Ragsdale N, Carey R, Macdonald T, Gaston B. Reductive assays for S-nitrosothiols: implications for measurements in biological systems. Biochem Biophys Res Commun 1998;252:535–540. [DOI] [PubMed] [Google Scholar]
- 22.Foster MW, Pawloski JR, Singel DJ, Stamler JS. Role of circulating S-nitrosothiols in control of blood pressure. Hypertension 2005;45:15–17. [DOI] [PubMed] [Google Scholar]
- 23.Gaston B, Reilly J, Brazen JM, Gackler J, Ramdev P, Arnelle D, Mulllins ME, Sugarbaker DJ, Chee C, Singel DJ, et al. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci USA 1993;90:10957–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kluge I, Gutteck-Amsler U, Zollinger M, Don KQ. S-nitrosoglutathione in rat cerebellum: identification and quantification by liquid chromatography-mass spectrometry. J Neurochem 1997;69:2599–2607. [DOI] [PubMed] [Google Scholar]
- 25.Kawasaki K, Smith RS Jr, Hsieh CM, Sun J, Chao J, Liao JK. Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis. Mol Cell Biol 2003;23:5726–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen L, Patel RP, Teng X, Bosworth CA, Lancaster JR Jr, Matalon S. Mechanisms of cystic fibrosis transmembrane conductance regulator activation by S-nitrosoglutathione. J Biol Chem 2006;281:9190–9199. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Hogg N. The mechanism of transmembrane S-nitrosothiol transport. Proc Natl Acad Sci USA 2004;101:7891–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mischeck U, Meyer J, Galla HJ. Characterization of gamma-glutamyl transpeptidase activity of cultured endothelial cells from porcine brain capillaries. Cell Tissue Res 1989;256:221–226. [DOI] [PubMed] [Google Scholar]
- 29.Zhou J, Schmid T, Frank R, Brune G. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1 alpha from pVHL-independent degradation. J Biol Chem 2004;279:13506–13513. [DOI] [PubMed] [Google Scholar]
- 30.Tang TT, Lasky LA. The forkhead transcription factor FOXO4 induces the down-regulation of hypoxia-inducible factor 1 alpha by a von Hippel-Lindau protein-independent mechanism. J Biol Chem 2003;278:30125–30135. [DOI] [PubMed] [Google Scholar]
- 31.Karni R, Dor Y, Keshet E, Meyuhas O, Levitzki A. Activated pp60c-Src leads to elevated hypoxia-inducible factor (HIF)-1alpha expression under normoxia. J Biol Chem 2002;277:42919–42925. [DOI] [PubMed] [Google Scholar]
- 32.Yasinska IM, Sumbayev VV. S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett 2003;549:105–109. [DOI] [PubMed] [Google Scholar]
- 33.Sumbayev VV, Budde A, Zhou J, Brune B. HIF-1 alpha protein as a target for S-nitrosation. FEBS Lett 2003;535:106–112. [DOI] [PubMed] [Google Scholar]
- 34.Zaman K, Carraro S, Doherty J, Henderson EM, Lendermon E, Liu L, Verghese G, Zigler M, Ross M, Park E, et al. S-nitrosylating agents: a novel class of compounds that increase CFTR expression and maturation in epithelial cells. Mol Pharmacol 2006;70:1435–1442. [DOI] [PubMed] [Google Scholar]
- 35.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 1998;273:13375–13378. [DOI] [PubMed] [Google Scholar]
- 36.Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 2002;277:20336–20342. [DOI] [PubMed] [Google Scholar]
- 37.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, And Downes CP. Redox regulation of PI 3-kinase signaling via inactivation of PTEN. EMBO J 2003;22:5501–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Reversible oxidation and activation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc Natl Acad Sci USA 2005;101:16419–16424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA 1999;96:4240–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denu JM, Dixon JE. Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Curr Opin Chem Biol 1998;2:633–641. [DOI] [PubMed] [Google Scholar]
- 41.Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol 1999;9:125–128. [DOI] [PubMed] [Google Scholar]
- 42.Maehama T, Taylor GSM, Dixon JE. PTEN and myotubularin: novel phospho-inositide phosphatases. Annu Rev Biochem 2001;70:247–279. [DOI] [PubMed] [Google Scholar]
- 43.Leslie NR, Downes CR. PTEN function: how normal cells control it and tumour cells lose it. Biochem J 2004;382:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu CX, Li S, Whorton AR. Redox regulation of PTEN by S-nitrosothiols. Mol Pharmacol 2005;68:847–854. [DOI] [PubMed] [Google Scholar]
- 45.Forrester MT, Foster MW, Stamler, JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–13983. [DOI] [PubMed] [Google Scholar]