

Abstract
Uncoupling protein 2 (UCP-2) is an inner mitochondrial membrane proton carrier that modulates mitochondrial membrane potential (ΔΨm) and uncouples oxidative phosphorylation. We have shown that up-regulation of UCP-2 by Wy14,643, a selective peroxisome proliferator-activated receptor-α (PPARα) agonist, enhances cyanide cytotoxicity. The pathway by which Wy14,643 up-regulates UCP-2 was determined in a dopaminergic cell line (N27 cells). Since dopaminergic mesencephalic cells are a primary brain target of cyanide, the N27 immortalized mesencephalic cell was used in this study. Wy14,643 produced a concentration- and time-dependent up-regulation of UCP-2 that was linked to enhanced cyanide-induced cell death. MK886 (PPARα antagonist) or PPARα knock-down by RNA interference (RNAi) inhibited PPARα activity as shown by the peroxisome proliferator response element-luciferase reporter assay, but only partially decreased up-regulation of UCP-2. The role of oxidative stress as an alternative pathway to UCP-2 up-regulation was determined. Wy14,643 induced a rapid surge of ROS generation and loading cells with glutathione ethyl ester (GSH-EE) or pre-treatment with vitamin E attenuated up-regulation of UCP-2. On the other hand, RNAi knockdown of PPARα did not alter ROS generation, suggesting a PPARα-independent component to the response. Co-treatment with PPARα-RNAi and GSH-EE blocked both the up-regulation of UCP-2 by Wy14,643 and the cyanide-induced cell death. It was concluded that a PPARα-mediated pathway and an oxidative stress pathway independent of PPARα mediate the up-regulation of UCP-2 and subsequent increased vulnerability to cyanide-induced cytotoxicity.
Keywords: Wy14,643; PPAR alpha; Cyanide; UCP-2; Reactive oxygen species
Introduction
Uncoupling protein 2 (UCP-2) is an anion carrier expressed in the inner mitochondrial membrane. UCP-2 facilitates a proton leak across the inner membrane to modulate mitochondrial function by reducing the membrane potential (ΔΨm) and uncoupling oxidative phosphorylation to reduce ATP synthesis (Busquets et al., 2001; Rousset et al., 2004). Activation of UCP-2 stimulates the proton leak across the mitochondrial inner membrane to reduce ΔΨm. This action appears to protect cells against oxidative stress by reducing mitochondrial generation of reactive oxygen species (ROS) (Mattiasson et al., 2003). On the other hand, excess mitochondrial uncoupling due to UCP-2 over-expression sensitizes cells to cytotoxic agents, possibly by depleting cellular ATP as a result of reduced mitochondrial function (Li et al., 2005; Prabhakaran et al., 2005).
UCP-2 is regulated by gene transcription, mRNA translation and activation of the protein in mitochondria (Ledesma et al., 2002). Gene expression is stimulated by a diverse range of stimuli (Kim-Han and Dugan, 2005) and by factors that influence the promoter, including peroxisome proliferator-activated receptors (PPARs) (Murray et al., 2005; Nakatani et al., 2002). UCP-2 expression is up-regulated in several models of brain damage and neurodegenerative diseases (MacManus et al., 2004; Sullivan et al., 2003). In the mitochondrial inner membrane, the protein must undergo activation to catalyze the proton leak. UCP-2-mediated uncoupling is stimulated by superoxide and free fatty acids (Koshkin et al., 2003; Echtay et al., 2002; Medvedev et al., 2002), whereas UCP-2 mediated uncoupling is inhibited by purine nucleotides (GTP, ATP, GDP, ADP) through a high affinity binding site (Negre-Salvayre et al., 1997).
Recent studies have linked UCP-2 up-regulation to activation of PPARs (Nakatani et al., 2002; Murray et al., 2005). PPARs function as transcription factors to regulate expression of target genes involved in lipid and energy metabolism. PPARα is a PPAR subtype expressed in a variety of tissues, including brown adipose tissue, liver, and brain. In brain, PPARα regulates neuronal differentiation, anti-oxidative defense, and neuronal death (Cimini et al., 2005). For instance, activation of PPARα by a selective agonist, Wy14,643 [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid], enhanced low KCl media-induced death of cerebellar granule cells (Smith et al., 2001). Also, we have shown in primary cortical cells that Wy14,643 can enhance cyanide-induced mitochondrial dysfunction and switch the mode of death from apoptosis to necrosis. The enhancement of cyanide toxicity was linked to up-regulation of UCP-2 expression (Li et al., 2006).
Several studies have shown that Wy14,643 can modulate UCP-2 gene expression, but the pathway by which increased expression is mediated has not been established (Nakatani et al., 2002). It is possible that ligand-mediated activation of PPAR activates the ucp2 promoter to increase transcription (Medvedev et al., 2001). However, non-PPARα pathways may also mediate up-regulation since it has been shown that PPARα agonists can increase UCP-2 expression in PPARα deficient mice (Grav et al., 2003).
Cyanide is a potent neurotoxicant that inhibits cytochrome oxidase in mitochondrial complex IV to block oxidative metabolism, thus reducing ATP synthesis and enhancing ROS generation at complex I and III (Chen et al., 2003). In mice treated with cyanide, two distinct modes of cell death are produced through different signaling cascades, with apoptosis in the cortex and necrosis in the dopaminergic cells of the basal ganglia (Mills et al., 1999). We have shown that sensitivity of the either cortical or mesencephalic (dopaminergic) cells to cyanide is associated with the level of expression of UCP-2 (Prabhakaran et al., 2005; Li et al., 2006). In primary cortical cells, up-regulation of UCP-2 by Wy14,643 switches the cyanide-induced apoptosis to necrosis. It is apparent that a PPARα agonist can regulate the level of cell sensitivity to the mitochondrial toxin through modulation of UCP-2 expression.
The mesencephalic brain area is a primary target of in vivo cyanide toxicity (Mills et al., 1999). We have shown that overexpression of UCP-2 enhances cyanide-induced toxicity in primary mesencephalic cells by mediating a rapid reduction of mitochondrial function (Prabhakaran et al., 2005). Therefore, an immortalized mesencephalic cell line (N27 cell) that exhibits neurochemical properties of dopaminergic neurons, was used to study the pathways by which Wy14,643 up-regulates UCP-2 expression, leading to an enhancement of cyanide toxicity. It is demonstrated that Wy14,643 influences UCP-2 expression through two pathways, a PPARα dependent pathway and an oxidative stress mediated process independent of PPARα.
Materials and methods
Materials
4-Chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid (Wy14,643) and 3-[3-tert-butylsulfanyl-1-(4-chlorobenzyl)-5-isopropyl-1H-indol-2-yl]-2,2-dimethylpropionic acid (MK886) were purchased from BioMol (Plymouth Meeting, PA). 2′,7′-Dichlorofluorescein diacetate (DCF-DA) were purchased from Invitrogen (Carlsbad, CA). Pre-stained SDS-PAGE standards and Bio-Rad protein assay system were purchased from Bio-Rad (Hercules, CA). All other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO). Wy14,643, MK886 and vitamin E were dissolved in DMSO and the final concentration of DMSO did not exceed 0.1% (v/v). Other chemicals were dissolved in cell culture medium.
Cell culture
Rat immortalized mesencephalic IRB3AN27 neuronal cells (N27 cells), that display features of dopaminergic neurons (Adams et al., 1996), were plated at a density of 1 × 104 cells/cm2 on poly-L-lysine (5 μg/ml) coated 6 or 24-well plates. Cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, penicillin (100 units/ml) and streptomycin (100 μg/ml) at 37°C in an atmosphere of 5% CO2 and 95% air.
Cell viability assays
Cell viability was determined by quantitating succinate dehydrogenase catalyzed conversion of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) to formazan (Altman, 1976). The cultures were incubated with MTT (0.5 mg/ml) in medium for 3 h. The formazan salt was solubilized in DMSO and optical density determined at 570 nm. The values were expressed as percent viability relative to vehicle-treated control cultures.
Measurement of ROS generation
The DCF-DA assay was used to determine cellular ROS generation as previously described with minor modification (Zhang et al., 2006). In all experiments, pretreated cells were loaded with DCF-DA (30 μM) for 30 min at 37°C, then washed with PBS and fluorescence intensity monitored with a microtiter plate reader at excitation wavelength of 485 nm and emission wavelength of 535 nm. The data of the treatment groups were expressed as a percent of DCF fluorescence generated in control cells under identical incubation conditions.
Glutathione analysis
After the treatments, cells were lysed by freezing and thawing in HCl (10 mM) and centrifuged for 15 min at 4°C and 2000 x g. The supernant was removed and neutralized with buffer (143 mM NaH2PO4, 6.3 mM EDTA, pH 7.4). The reaction mixture, containing 5,5′-dithiobis-2-nitrobenzoic acid (1 mM) and NADPH (0.34 mM), was added to samples and the reaction was started by adding 8.5 IU/ml glutathione reductase (Vandeputte et al., 1994). Total glutathione (GSH+GSSG) levels were determined by measuring the increase in absorbance at 415 nm and compared to a standard curve. The reduced glutathione (GSH) was determined as the difference between the total glutathione values and samples pretreated with 2-vinylpyridine to selectively remove GSH.
Measurement of mitochondrial uncoupling
Cells were washed with ice-cold PBS, gently scraped off the culture plates and then centrifuged at 500 x g for 5 min. The cells were re-suspended in medium (250 mM sucrose, 1 mM EDTA, 50 mM KCl, 2 mM KH2PO4, 25 mM Tris-HCl, pH 7.4) and then digitonin (40 μg/ml) was used to permeabilize the cells. Oxygen consumption was measured polarographically at 37°C with a Clark oxygen electrode (Rank Brothers, Ltd., Cambridge, UK) that was interfaced with a microcomputer to provide a real time display of oxygen concentration. Basal oxygen consumption was measured, and then the state 4 respiratory rate was determined in permeablized cells in the presence of succinate (5 mM), ADP (1 mM) and the F1F0-ATP synthase inhibitor, oligomycin (10 μg/ml). After addition of ADP and oligomycin, UCP-2 mediated proton conductance was estimated as an increased palmitic acid-induced respiration compared with state 4 respiration induced by oligomycin (Echtay et al., 2002).
Transient transfection and PPAR reporter assay
The ability of Wy14,643 to activate PPARα was assessed using a reporter assay system (peroxisome response element-luciferase) as described by Kehrer et al. (2001). The luciferase reporter construct PPRE3-TK-LUC was a kind gift from Dr R. M. Evans (Salk Institute for Biological Studies, San Diego, CA). pRL-CMV-RLUC (Promega, Madison, WI), a reporter vector containing Renilla luciferase (RLUC), was used as an internal control for normalizing transfection efficiency. PPRE3-TK-LUC and pRL-CMV-RLUC were transfected into N27 cells by using the Lipofectamine 2000™ (Invitrogen, Carlsbad, CA) for 24 h followed by the different treatments for an additional 6 h, then the cells were lysed and analyzed by using a dual-luciferase reporter gene assay system (Promega, Madison, WI). The luciferase activity was normalized to the internal control (RLUC activity).
Western blot analysis
After the various treatments or transient transfection, cells were washed with ice-cold PBS and harvested by centrifugation at 500 x g for 5 min. Cell pellets were lysed in a buffer containing 220 mM mannitol, 68 mM sucrose, 20 mM HEPES, pH 7.4, 50 mM KCl, 5 mM EGTA, 1 mM EDTA, 2 mM MgCl2, 1 mM dithiothreitol, 0.1% Triton X-100, and protease inhibitors on ice for 15 min. After centrifugation, supernatants were taken as whole-cell protein extraction. The protein content in the extractions was determined by the Bradford assay (Bio-Rad, Hercules, CA). Samples containing 30 μg of protein were boiled in Laemmli buffer for 5 min and then subjected to electrophoresis in 12% SDS-polyacrylamide gel, followed by transfer to a polyvinylidene difluoride membrane. After blocking with Tris-buffered saline containing 5% nonfat dry milk and 0.1% Tween 20, the membrane was exposed to primary UCP-2 antibody or β-actin antibody for 3 h at room temperature on a shaker. The UCP-2 antibody was a rabbit anti-mouse polyclonal antibody (1:2000) directed toward the C-terminal domain of UCP-2 (Alpha Diagnostic International, Inc., San Antonio, TX). Antibody specificity was determined by using a 14-amino acid UCP-2 blocking peptide according to the manufacturer’s protocol. The UCP-2 antibody was detected with a fluorescein-linked anti-rabbit IgG (second antibody). The signal was then amplified by detection of the secondary antibody with an anti-fluorescein alkaline phosphatase conjugate followed by fluorescent ECF substrate according to the ECF Western Blotting Kit™ (Amersham, Piscataway, NJ). Densitometric analysis was performed using Scion Image software (Scion Corporation, Frederick, MD). Data were normalized to the internal control (β-actin) and then expressed as relative density of each band compared with the respective vehicle control band. For each study, Western blot analysis was conducted two to three times and representative blots are shown.
RNA Interference
Small interfering RNA corresponding to the UCP-2 gene was designed as recommended (Elbashir et al., 2001) and synthesized by Ambion, Inc. (Austin, TX) with 5′ phosphate, 3′ hydroxyl, and two base overhangs on each strand. The gene-specific sequences were used for UCP-2 interference: sense 5′-GAACGGGACACCUUUAGAGtt-3′ and antisense 5′-CUCUAAAGGUGUCCCGUUCtt-3′; annealing for duplex siRNA formation was performed as described by the manufacturer. Pre-designed siRNA for PPARα (Ambion Inc, Austin, TX) was used to knock-down PPARα expression. The Silencer Negative Control siRNA 1, which does not target rat, mouse, or humans genes, was used as a negative control (Ambion, Austin, TX). Transient transfections of the UCP-2 and PPARα siRNA were performed with Lipofectamine 2000™ (Invitrogen, Carlsbad, CA).
Statistics
Data were expressed as mean ± S.E.M., and statistical significance was assessed by one-way ANOVA or two-way ANOVA. The Tukey-Kramer multiple range test and Fisher’s LSD test were used for post hoc comparisons. Differences were considered significance at P<0.05.
Results
Wy14,643 potentiates cyanide-induced toxicity by up-regulating UCP-2
Examination of N27 cells by phase contrast microscopy showed that incubation with cyanide (400 μM) for 24 h did not produce a significant level of cell death. Wy14,643 (100 μM) produced a low-level cytotoxicity as compared to control cells, whereas pretreatment with Wy14,643 potentiated the toxicity of cyanide (Fig. 1A, B). Co-treatment with Wy14,643 and cyanide decreased cell density and altered the morphology of surviving cells. The mode of death was predominantly necrotic as determined by PI staining (data not shown). Quantitation of cell death showed that Wy14,643 sensitized cells to cyanide (Fig 1B). Western blots of UCP-2 showed that Wy14,643 induced a concentration- and time-dependent up-regulation of UCP-2 (Fig. 2A, B). Up-regulation of UCP-2 expression was detected as early as 6 h and reached a maximum within 12 h. To determine whether UCP-2 up-regulation would increase mitochondrial uncoupling, state 4 respiration was measured. After 12 h of Wy14,643 treatment, a marked increase of palmitic acid-induced state 4 respiration was observed (150% of control cells) (Fig. 2C), reflecting that an increased mitochondrial uncoupling accompanied UCP-2 up-regulation.
Fig. 1.
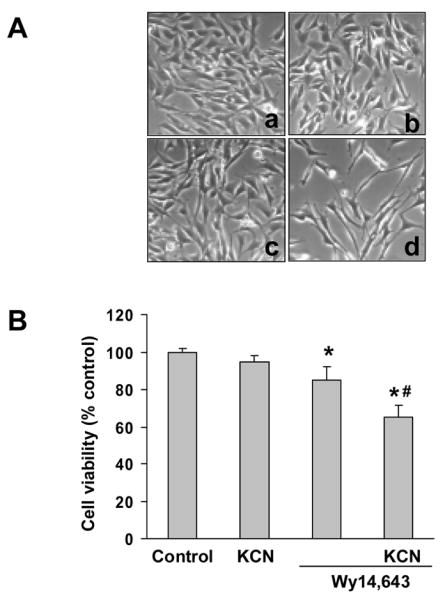
Wy14,643 enhancement of cyanide-induced cell death. (A) N27 cells were incubated with Wy14,643 (100 μM) or DMSO (control) for 5 h followed by cyanide (400 μM) for 24 h. Magnification is 200X for phase contrast micrographs of cells (a. Solvent control; b. KCN; c. Wy14,643; d. Wy14,643 + KCN). (B) Cells were pretreated with Wy14,643 (100 μM) or DMSO (control) 5 h before cyanide and cell viability was assessed 24 h later. Values were expressed as percent viability relative to vehicle-treated control cultures. *Significantly different from control group. #Significantly different from Wy14,643 group. P < 0.05.
Fig. 2.
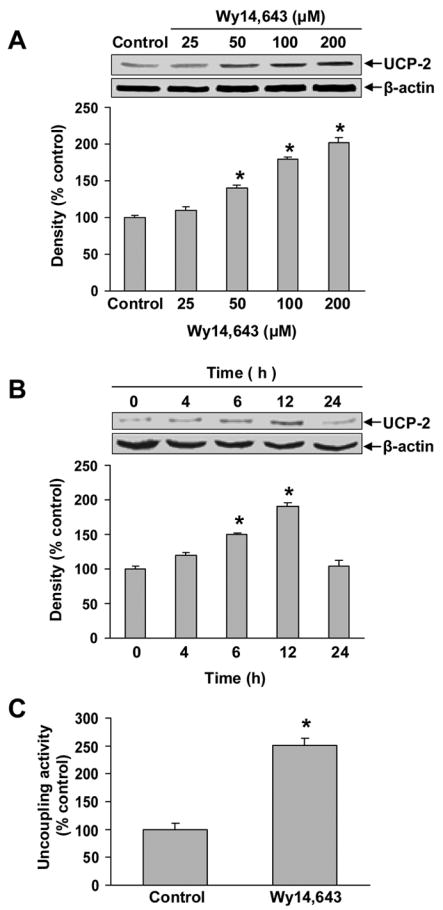
Wy14,643-induced up-regulation of UCP-2 and enhanced mitochondrial uncoupling. (A–B) Western blot of UCP-2 after treatment with varying concentrations of Wy14,643 for 12 h (panel A) or with Wy14,643 (100 μM) for 0–24 h (panel B). β-actin was used as an equal loading control. Data represent mean ± SEM for three separate experiments. (C) Cells were pretreated with Wy14,643 (100 μM) for 12 h or DMSO (control) and mitochondrial respiratory function was then measured. Mitochondrial uncoupling was determined by the increase of state 4 respiration produced by addition of a UCP-2 activator (palmitic acid, 300 μM). Data are expressed as percent increase above oligomycin-induced state 4 respiration (mean ± SEM). Values are relative O2 consumption (mean ± SEM). *Significantly different from control group, P < 0.05.
Western blot analysis showed that the Wy14,643-induced up-regulation of UCP-2 was significantly enhanced when cells were also treated cyanide (Fig. 3A). Knock-down of UCP-2 expression by RNAi markedly reduced the UCP-2 up-regulation, and importantly reversed the Wy14,643 enhancement of cyanide toxicity (Fig. 3B), thus linking UCP-2 up-regulation to cell death. It should be noted that induction of UCP-2 expression by Wy14,643 was not totally blocked in cells transfected with UCP-2 RNAi. This may be attributed in part to a 60–70% transfection efficiency of the UCP-2 RNAi in these cells (data not shown). These results are consistent with our previous work with primary cortical cells that showed constitutively expressed UCP-2 was not totally knocked-down after transfection with RNAi (Li, et al., 2005).
Fig. 3.
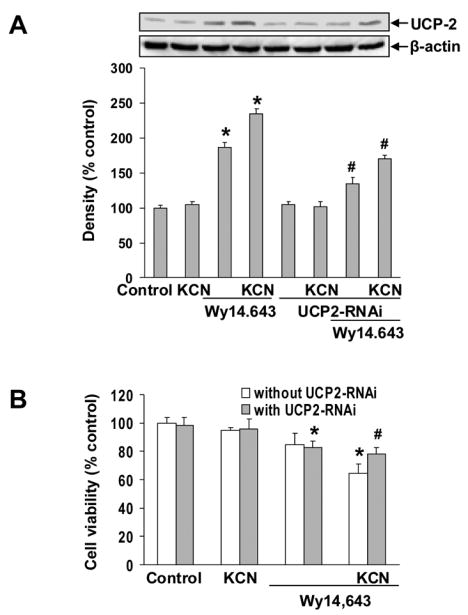
Knock-down of UCP-2 blocked Wy14,643-induced expression of UCP-2 and enhancement of cyanide toxicity. (A) Cells were transfected with UCP-2 RNAi and 24 h later treated with Wy14,643 (100 μM) for 12 h. UCP-2 was detected by Western blot analysis. Data represent mean ± SEM for three separate experiments. (B) Effect of UCP-2 RNAi on cyanide toxicity. Cell viability was assessed by MTT assay. Data represent mean ± SEM. *Significantly different from control group. #Significantly different from Wy14,643+KCN group. P < 0.05.
PPARα antagonism and RNAi knock-down partially inhibit UCP-2 up-regulation
To determine if the Wy14,643-induced UCP-2 up-regulation involved PPARα, cells were treated with a non-competitive PPARα antagonist, MK886 (5 μM) at a concentration that blocks Wy14,643-induced UCP-2 up-regulation at both mRNA and protein levels (Li et al., 2006). In N27 cells, MK886 did not completely block Wy14,643-mediated up-regulation of UCP-2 (Fig 4A). Similarly, Western blots also showed that RNAi knock-down of PPARα, which blocked constitutive PPARα expression (data not shown), did not completely block up-regulation of UCP-2 (Fig 4B). To confirm that MK886 or PPARα-RNAi abolished Wy14,643-mediated PPARα activation, PPARα-driven reporter activity was determined. Both MK886 and PPARα-RNAi diminished reporter activation (Fig. 4C), showing the treatments blocked PPARα activation. It was concluded that Wy14,643 induction of UCP-2 is mediated only in part through a PPARα-dependent pathway and that an alternative pathway contributes to UCP-2 up-regulation.
Fig. 4.
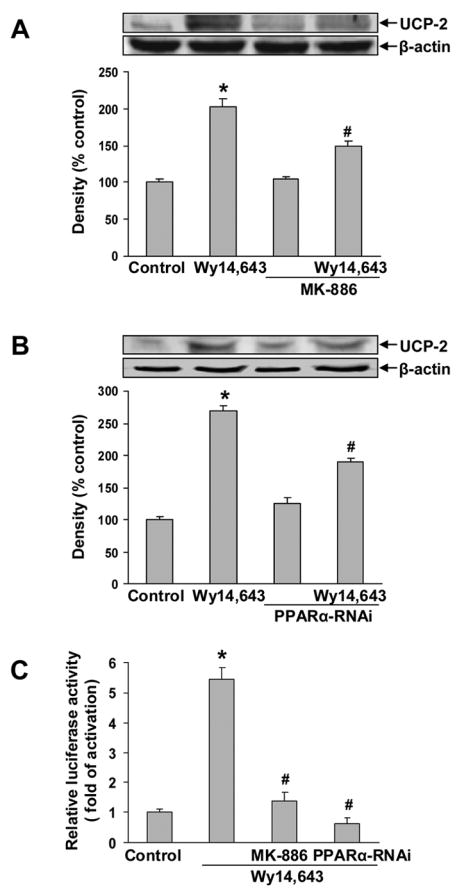
PPARα antagonist and PPARα RNAi partially blocked UCP-2 up-regulation. (A) Cells were pretreated with MK886 (5 μM) 30 min, followed by Wy14,643 (100 μM) for 12 h. UCP-2 expression was determined by Western blotting. Data represent mean ± SEM for three separate experiments. (B) Cells were transfected with PPARα-RNAi 24 h followed by Wy14,643 (100 μM) for 12 h. UCP-2 was detected by Western blotting. Data represent mean ± SEM for three separate experiments. (C) Cells were transfected with PPRE3-TK-LUC and pRL-CMV-RLUC with or without PPARα-RNAi for 24 h, and then were treated with Wy14,643 (100 μM) for 6 h in the presence or absence of MK886 (5 μM) for 30 min. Luciferase activity was normalized to the Renilla luciferase activity. Results are expressed as mean ± SEM of three independent experiments. *Significantly different from control group. #Significantly different from Wy14,643 group, P < 0.05. MK886 x Wy14,643 interaction was significant, P<0.05 (two-way ANOVA). PPARα-RNAi x Wy14,643 interaction was significant, P<0.05 (two-way ANOVA).
ROS generation contributes to UCP-2 up-regulation
Since increased generation of ROS (oxidative stress) can up-regulate UCP-2 expression (Yang et al., 2000), the effect of Wy14,643 on cellular ROS levels was determined. A surge of ROS generation was observed after 4–12 h of incubation with Wy14,643 (Fig. 5A), paralleling the temporal profile of UCP-2 up-regulation. Also the ratio of cellular GSH/GSSG was significantly decreased (Fig. 5B), indicating Wy14,643 produced a significant oxidative stress. To determine the involvement of oxidative stress in up-regulation of UCP-2, cells were loaded with an ROS scavenger (GSH-EE) or pretreated with an antioxidant (Vitamin E). Western blots showed that both GSH-EE and vitamin E significantly reduced the Wy14,643 mediated UCP-2 up-regulation (Fig. 6A, B), thus showing that oxidative stress contributes to UCP-2 up-regulation.
Fig. 5.

Increased intracellular ROS generation produced by Wy14,643. (A) Generation of ROS after treatment with Wy14,643 (100 μM). DCF fluorescence was measured after treatment with Wy14,643 for 0–12 h. (B) Wy14,643 decreased the ratio of GSH/GSSG. Cells were treated with Wy14,643 (100 μM) or DMSO (control) for 12 h and ratio of cellular GSH/GSSG determined. Results are presented as mean ± SEM of three individual experiments. *Significantly different from control group, P < 0.05.
Fig. 6.
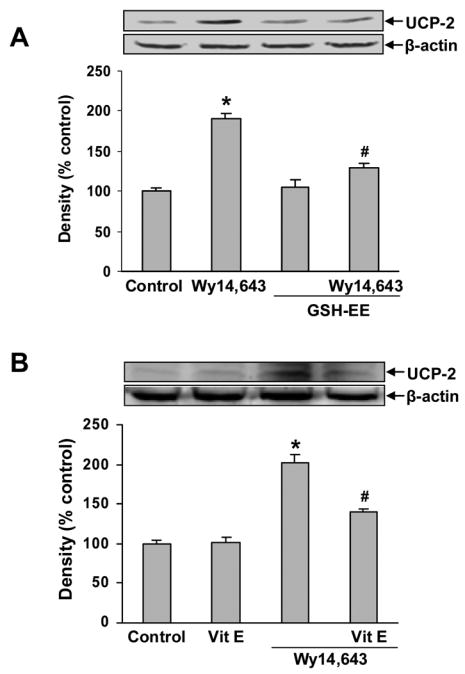
Effect of GSH-EE and vitamin E on UCP-2 expression. (A) Cells were loaded with GSH-EE (2 mM) for 1 h and then treated with Wy14,643 (100 μM) for 12 h. UCP-2 was detected by Western blotting. Data represent mean ± SEM for three separate experiments. (B) Cells were pretreated with vitamin E (100 μM) for 1 h and then Wy14,643 (100 μM) for 12 h. UCP-2 was detected by Western blots analysis. Data represent mean ± SEM for three separate experiments. *Significantly different from control group. #Significantly different from Wy14,643 group, P<0.0.5. GSH-EE x Wy14,643 interaction was significant, P<0.05 (two-way ANOVA). Vit E x Wy14,643 interaction was significant, P<0.05 (two-way ANOVA).
PPARα activation does not stimulate ROS generation
Previous work has shown that Wy14,643 stimulation of ROS generation can be independent of PPARα (Atarod and Kehrer, 2004). To determine if PPARα activation contributed to the ROS generation, the effect of PPARα-RNAi knock-down on ROS production was determined. Knock-down of PPARα did not reduce ROS levels compare to control cells, whereas loading with GSH-EE or pretreatment with vitamin E significantly reduced ROS generation (Fig. 7). In addition, pretreatment with PPARα antagonist MK-886 did not diminish the ROS production (data not shown). It was concluded that the Wy14,643-mediated increase of ROS production did not involve PPARα activation.
Fig. 7.

Effect of GSH-EE and PPARα knock-down on Wy14,643-mediated ROS generation. Cells were transfected with PPARα-RNAi and 24 h later treated with Wy14,643 (100 μM) for 12 h. In the GSH-EE group, cells were loaded with GSH-EE (2 mM) for 1 h or pretreated with vitamin E (100 μM) for 1 h and then Wy14,643 (100 μM) for 12 h. DCF fluorescence levels were then determined. DCF fluorescence is expressed as percent control and each value is the mean of three experiments ± SEM. *Significantly different from control group. #Significantly different from Wy14,643 group, P<0.05.
Blockade of UCP-2 up-regulation requires both PPARα knockdown and decreased ROS generation
To confirm that the Wy14,643 up-regulation of UCP-2 involves both a PPARα-dependent pathway and oxidative stress, cells were transfected with PPARα-RNAi and then loaded with GSH-EE, followed by analysis of UCP-2 expression. Western blots showed that concurrent treatment with PPARα-RNAi and GSH-EE reduced UCP-2 expression to control levels and significantly decreased Wy14,643-mediated mitochondrial uncoupling (Fig. 8A, B). Furthermore, co-treatment with PPARα-RNAi and GSH-EE produced an additive inhibition of Wy14,643-induced UCP-2 expression and mitochondrial uncoupling. These observations provide strong support for involvement of both a PPARα-dependent pathway and an oxidative stress-mediated pathway for up-regulation of UCP-2 expression.
Fig. 8.
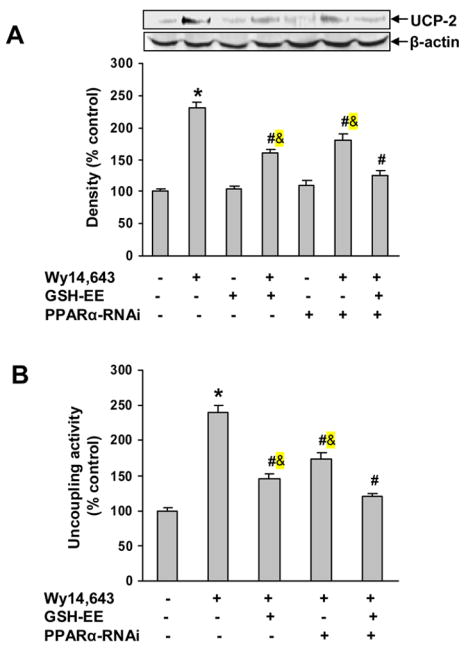
Co-treatment with GSH-EE and PPARα knock-down blocked Wy14,643-induced UCP-2 up-regulation and mitochondrial uncoupling. Cells were transfected with PPARα RNAi and 24 h later treated with Wy14,643 (100 μM) for 12 h. In the GSH-EE group, cells were loaded with GSH-EE (2 mM) for 1 h and then Wy14,643 (100 μM) for 12 h. (A) GSH-EE and PPARα-RNAi blocked Wy14,643-induced UCP-2 expression. Western blots of UCP-2 in cells pretreated with GSH-EE and PPARα-RNAi. Data represent mean ± SEM for three separate experiments. (B) Effect of GSH-EE and PPARα-RNAi on mitochondrial uncoupling. Values are relative O2 consumption (mean ± SEM). *Significantly different from control group. #Significantly different from Wy14,643 group. &Significantly different from Wy14,643+GSH-EE+PPARα-RNAi group. P < 0.05.
PPARα knock-down and decreased ROS generation block cyanide cytotoxicity
Cyanide-mediated cytotoxicity was examined in cells treated with PPARα-RNAi and GSH-EE. Phase contrast microscopy of the cells showed that PPARα knock-down or GSH-EE reduced the Wy14,643 enhancement of cyanide toxicity and combined treatment (GSH-EE/PPARα-RNAi) appeared to block the cell death (Fig. 9A). Quantitation of cell death showed that the co-treatment abolished the cyanide-induced cell death in Wy14,643 treated cells (Fig. 9B), thus demonstrating that Wy14,643-mediated enhancement of cyanide toxicity involves at least two independent pathways, one mediated by PPARα activation and the other through increased oxidative stress.
Fig. 9.
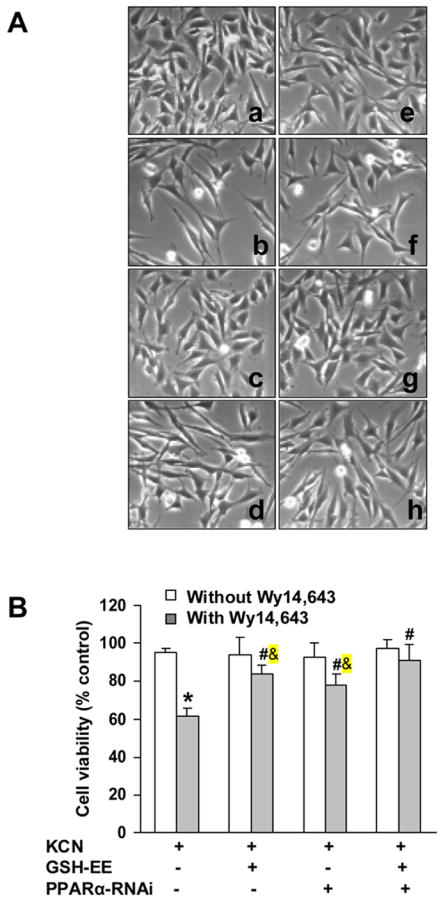
Effect of GSH-EE and PPARα knock-down on Wy14,643-mediated enhancement of cyanide toxicity. Cells were transfected with PPARα-RNAi and 24 h later treated with Wy14,643 (100 μM) for 12 h, followed by KCN (400 μM) for 24 h. In GSH-EE group, cells were loaded with GSH-EE (2 mM) for 1 h and then Wy14,643 (100 μM) for 12 h, followed by KCN for 24 h. (A) Magnification is 200X for phase contrast micrographs of cells: a. KCN; b. Wy14,643+KCN; c. GSH-EE+KCN; d. GSH-EE+Wy14,643+KCN; e. PPARα-RNAi+KCN; f. PPARα-RNAi+Wy14,643+KCN; g. GSH-EE+PPARα-RNAi+KCN; h. GSH-EE+PPARα-RNAi+Wy14,643+KCN. (B) Viability of cells was assessed by MTT assay. Values were expressed as percent viability relative to vehicle-treated control cultures. *Significantly different from control group. #Significantly different from Wy14,643+KCN group. &Significantly different form Wy14,643+GSH-EE+PPARα-RNAi+KCN group. P<0.05.
Discussion
The selective PPARα agonist Wy14,643 sensitized N27 cells to cyanide-induced death through up-regulation of UCP-2. PPARα antagonism or RNAi knock-down only partially blocked Wy14,643-mediated up-regulation of UCP-2, suggesting involvement of a process independent of PPARα activation. Wy14,643 stimulated intracellular ROS generation which in turn increased oxidative stress as reflected by a decreased GSH/GSSG ratio. Loading cells with GSH-EE or pretreatment with an antioxidant significantly reduced UCP-2 expression, demonstrating that enhanced ROS generation contributes to UCP-2 up-regulation. Importantly, knock-down of PPARα did not alter the levels of ROS generation, showing that the increased oxidative stress was not mediated by PPARα activation. Furthermore, co-treatment with PPARα RNAi and GSH-EE abolished the Wy14,643-mediated up-regulation of UCP-2 and the cyanide-induced cell death. It was concluded that two pathways mediate the Wy14,643-induced UCP-2 up-regulation and increased vulnerability of the cells to cyanide. The PPARα-dependent pathway appears to involve direct transcriptional activation of the UCP-2 gene, whereas an oxidative stress mediated pathway is independent of PPARα activation.
UCP-2 is an anionic transporter capable of dissipating the mitochondrial proton gradient and in turn reduces the efficiency of ATP synthesis (Dulloo and Samec, 2001). It is widely accepted that constitutive expression of UCP-2 can produce neuro-protection by uncoupling oxidative phosphorylation to reduce mitochondrial ROS generation (Horvath et al., 2003; Conti et al., 2005). We have shown in primary cortical cells that Wy14,643 can induce UCP-2 up-regulation and switch cyanide-induced apoptosis to necrosis (Li et al., 2006). The up-regulation of UCP-2 by Wy14,643 can play an important role in regulating sensitivity of neuronal cells to stimuli that influence mitochondrial function. The present study extends these observations in the dopaminergic N27 cell line by showing that Wy14,643 up-regulates UCP-2 to sensitize the cells to cyanide-induced toxicity. This cell model was then used to explore the mechanisms underlying up-regulation of UCP-2 and associated cell death.
Pathways by which Wy14,643 mediate UCP-2 up-regulation have not been established. Since ucp-2 is a target gene of PPARα, activation of the receptor would lead to increased expression (Ravnskjaer et al., 2005; Nakatani et al., 2002). Wy14,643 can selectively activate PPARα by stimulating formation of a heterodimeric transcription factor complex with retinoid X receptor alpha (RXRα) (Gearing et al., 1993). The heterodimer then binds to specific peroxisome proliferator-response elements of target genes to stimulate transcription. Previous studies have shown in hepatocytes and primary cortical cells that Wy14,643 can increase UCP-2 expression through activation of PPARα (Nakatani et al., 2002; Li et al., 2006). In the present study, Wy14,643 induced a concentration- and time-dependent up-regulation of UCP-2. Wy14,643 did not increase the constitutive expression by PPARα (data not shown) and the luciferase reporter assay showed that Wy14,643 activated PPARα. It is concluded that PPARα activation is involved in Wy14,643-induced UCP-2 up-regulation., possibly through activation of the peroxisome proliferator response element.
Recent reports have shown that PPARα ligands can produce a number of cellular actions independent of PPARα binding, including inhibition of mitochondrial complex I which can lead to enhanced ROS generation (Scatena et al., 2003, 3004). It appears that PPARα agonist-induced UCP-2 up-regulation can also be independent of PPARα. Peters et al. (2001) showed that conjugated linoleic acid, a PPARα ligand, increased UCP-2 levels in both PPARα wildtype+/+ and PPARα null−/− mice, thus demonstrating that up-regulation of UCP-2 can be mediated by a PPARα independent pathway. The present study used two approaches in blocking the PPARα pathway, a selective PPARα antagonist (MK886) and RNAi knock-down. Both treatments inhibited Wy14,643-mediated PPARα activation, but only partially blocked the Wy14,643-induced UCP-2 expression, thereby providing support for involvement of a PPARα-independent mechanism.
An alternative pathway for UCP-2 up-regulation may involve oxidative stress, since it has been shown that increased ROS can up-regulate UCP-2 (Yang et al., 2000, Ho et al., 2005). The proximal 3.3 kb of the ucp-2 promoter contains nucleotide sequences of ROS-sensitive trans-acting factors, including AP-1 and C/EBP (Cortez-Pinto et al., 1998). Pecqueur et al (2002) showed that UCP-2 is up-regulated when the level of intracellular ROS is high. In hepatocytes, an up-regulation of UCP-2 occurs in response to increased ROS generation, both in vivo and in vitro (Cortez-Pinto et al., 1999; Rashid et al., 1999). It seemed likely that redox regulation of UCP-2 expression was also involved in the response observed in this study. Wy14,643 induced a rapid surge of ROS generation as reflected by a decreased GSH/GSSG ratio, and loading cells with GSH-EE or pretreatment with an antioxidant (vitamin E) inhibited the up-regulation of UCP-2. The results of these experiments show that oxidative stress is involved in up-regulation of UCP-2 by WY14,643.
Previous studies have shown that Wy14,643 can stimulate ROS generation independent of PPARα. In human T-lymphocytic leukemia cells, Wy14,643-mediated ROS generation was not blocked by a PPARα inhibitor (Atarod and Kehrer, 2004). Similarly in this study, RNAi knock-down of PPARα did not diminish the Wy14,643-induced ROS production, showing that the enhanced ROS production is likely independent of PPARα. This is consistent with observations in Kupffer cells from PPARα null−/− mice in which PPARα agonists induced ROS generation independent of PPARα activation (Peters et al., 2000).
The origin of the Wy14,643-mediated oxidative stress has not been well characterized. Atarod and Kehrer (2004) suggest that Wy14,643-induced oxidant production may originate in mitochondria. Interestingly, studies have shown that Wy14,643 can decrease cellular glutathione levels by interfering with its synthesis (Teissier et al., 2004; O’Brien et al., 2001). We have shown that Wy14,643 decreased levels of reduced glutathione which could exacerbate intracellular oxidative stress and increase UCP-2 transcription. In this study, loading the cells with GSH-EE blocked Wy14,643-mediated UCP-2 up-regulation and cyanide-induced cell death, thus providing evidence that intracellular redox conditions play an important role in regulating UCP-2 expression and neurotoxicity. It is important to note that besides UCP-2 up-regulation, Wy14,643 can produce additional actions that may contribute to enhanced sensitivity to cyanide. A recent study has shown that PPARα ligands can directly inhibit complex I in mitochondria, which would lead to increased generation of ROS and mitochondrial dysfunction (reduction of ΔΨm and reduced ATP synthesis) (Scatena et al., 2003; 2004). It is likely these additional mitochondrial actions of Wy14,643 could enhance the sensitivity to KCN.
In conclusion, it was demonstrated that Wy14,643, a selective PPARα agonist, sensitized dopaminergic cells to cyanide-induced death through up-regulation of UCP-2. The Wy14,643-stimulated UCP-2 expression was mediated through both a PPARα-dependent pathway and a redox sensitive pathway involving increased ROS generation which then enhanced the sensitivity of the cells to cyanide.
Acknowledgments
This work was supported by NIH grant ES04140.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams FS, La Rosa FG, Kumar S, Edwards-Prasad J, Kentroti S, Vernadakis A, Freed CR, Prasad KN. Characterization and transplantation of two neuronal cell lines with dopaminergic properties. Neurochem Res. 1996;21:619–627. doi: 10.1007/BF02527762. [DOI] [PubMed] [Google Scholar]
- Altman FP. Tetrazolium salts and formazans. Prog Histochem Cytochem. 1976;9:1–56. doi: 10.1016/s0079-6336(76)80015-0. [DOI] [PubMed] [Google Scholar]
- Atarod EB, Kehrer JP. Dissociation of oxidant production by peroxisome proliferator-activated receptor ligands from cell death in human cell lines. Free Radic Biol Med. 2004;37:36–47. doi: 10.1016/j.freeradbiomed.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Busquets S, Alvarez B, Van Royen M, Figueras MT, Lopez-Soriano FJ, Argiles JM. Increased uncoupling protein-2 gene expression in brain of lipopolysaccharide-injected mice: role of tumour necrosis factor-alpha? Biochim Biophys Acta. 2001;1499:249–256. doi: 10.1016/s0167-4889(00)00126-9. [DOI] [PubMed] [Google Scholar]
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- Cimini A, Benedetti E, Cristiano L, Sebastiani P, D’Amico MA, D’Angelo B, DiLoreto S. Expression of peroxisome proliferator-activated receptors PPARs and retinoic acid receptors RXRs in rat cortical neurons. Neuroscience. 2005;130:325–337. doi: 10.1016/j.neuroscience.2004.09.043. [DOI] [PubMed] [Google Scholar]
- Conti B, Sugama S, Lucero J, Winsky-Sommerer R, Wirz SA, Maher P, Andrews Z, Barr AM, Morale MC, Paneda C, Pemberton J, Gaidarova S, Behrens MM, Beal F, Sanna PP, Horvath T, Bartfai T. Uncoupling protein 2 protects dopaminergic neurons from acute 1,2,3,6-methyl-phenyl-tetrahydropyridine toxicity. J Neurochem. 2005;93:493–501. doi: 10.1111/j.1471-4159.2005.03052.x. [DOI] [PubMed] [Google Scholar]
- Cortez-Pinto H, Yang SQ, Lin HZ, Costa S, Hwang CS, Lane MD, Bagby G, Diehl AM. Bacterial lipopolysaccharide induces uncoupling protein-2 expression in hepatocytes by a tumor necrosis factor-alpha-dependent mechanism. Biochem Biophys Res Commun. 1998;251:313–319. doi: 10.1006/bbrc.1998.9473. [DOI] [PubMed] [Google Scholar]
- Cortez-Pinto H, Zhi Lin H, Qi Yang S, Odwin Da Costa S, Diehl AM. Lipids up-regulate uncoupling protein 2 expression in rat hepatocytes. Gastroenterology. 1999;116:1184–1193. doi: 10.1016/s0016-5085(99)70022-3. [DOI] [PubMed] [Google Scholar]
- Dulloo AG, Samec S. Uncoupling proteins: their roles in adaptive thermogenesis and substrate metabolism reconsidered. Br J Nutr. 2001;86:123–139. doi: 10.1079/bjn2001412. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Gearing KL, Gottlicher M, Teboul M, Widmark E, Gustafsson JA. Interaction of the peroxisome-proliferator-activated receptor and retinoid X receptor. Proc Natl Acad Sci USA. 1993;90:1440–1144. doi: 10.1073/pnas.90.4.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grav HJ, Tronstad KJ, Gudbrandsen OA, Berge K, Fladmark KE, Martinsen TC, Waldum H, Wergedahl H, Berge RK. Changed energy state and increased mitochondrial beta-oxidation rate in liver of rats associated with lowered proton electrochemical potential and stimulated uncoupling protein 2 UCP-2 expression: evidence for peroxisome proliferator-activated receptor-alpha independent induction of UCP-2 expression. J Biol Chem. 2003;278:30525–30533. doi: 10.1074/jbc.M303382200. [DOI] [PubMed] [Google Scholar]
- Ho PW, Chan DY, Kwok KH, Chu AC, Ho JW, Kung MH, Ramsden DB, Ho SL. Methyl-4-phenylpyridinium ion modulates expression of mitochondrial uncoupling proteins 2, 4, and 5 in catecholaminergic (SK-N-SH) cells. J Neurosci Res. 2005;81:261–268. doi: 10.1002/jnr.20569. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Diano S, Barnstable C. Mitochondrial uncoupling protein 2 in the central nervous system: Neuromodulator and neuroprotector. Biochem Pharmacol. 2003;65:1917–1921. doi: 10.1016/s0006-2952(03)00143-6. [DOI] [PubMed] [Google Scholar]
- Kehrer JP, Biswal SS, La E, Thuillier P, Datta K, Fischer SM, Vanden Heuvel JP. Inhibition of peroxisome-proliferator-activated receptor (PPAR)α by MK886. Biochem J. 2001;356:899–906. doi: 10.1042/0264-6021:3560899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Han JS, Dugan LL. Mitochondrial uncoupling proteins in the central nervous system. Antioxid Redox Signal. 2005;7:1173–1181. doi: 10.1089/ars.2005.7.1173. [DOI] [PubMed] [Google Scholar]
- Koshkin V, Wang X, Scherer PE, Chan CB, Wheeler MB. Mitochondrial functional state in clonal pancreatic beta-cells exposed to free fatty acids. J Biol Chem. 2003;278:19709–19715. doi: 10.1074/jbc.M209709200. [DOI] [PubMed] [Google Scholar]
- Ledesma A, de Lacoba MG, Rial E. The mitochondrial uncoupling proteins. Genome Biol. 2002;3:3015.1–3015.9. doi: 10.1186/gb-2002-3-12-reviews3015. (Reviews) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Prabhakaran K, Mills EM, Borowitz JL, Isom GE. Enhancement of cyanide-induced mitochondrial dysfunction and cortical cell necrosis by uncoupling protein-2. Toxicol Sci. 2005;86:116–124. doi: 10.1093/toxsci/kfi164. [DOI] [PubMed] [Google Scholar]
- Li L, Prabhakaran K, Zhang X, Borowitz JL, Isom GE. PPARalpha-mediated upregulation of uncoupling protein-2 switches cyanide-induced apoptosis to necrosis in primary cortical cells. Toxicol Sci. 2006;93:136–145. doi: 10.1093/toxsci/kfl039. [DOI] [PubMed] [Google Scholar]
- MacManus JP, Graber T, Luebbert C, Preston E, Rasquinha I, Smith B, Webster J. Translation-state analysis of gene expression in mouse brain after focal ischemia. J Cereb Blood Flow Metab. 2004;24:657–667. doi: 10.1097/01.WCB.0000123141.67811.91. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melchor T, Gonzalex-Zulueta M, Nikolich K, Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. 2003;9:1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- Medvedev AV, Snedden SK, Raimbault S, Ricquier D, Collins S. Transcriptional regulation of the mouse uncoupling protein-2 gene. Double E-box motif is required for peroxisome proliferator-activated receptor-gamma-dependent activation. J Biol Chem. 2001;276:10817–10823. doi: 10.1074/jbc.M010587200. [DOI] [PubMed] [Google Scholar]
- Medvedev AV, Robidoux J, Bai X, Cao W, Floering LM, Daniel KW, Collins S. Regulation of the uncoupling protein-2 gene in INS-1 beta-cells by oleic acid. J Biol Chem. 2002;277:42639–42644. doi: 10.1074/jbc.M208645200. [DOI] [PubMed] [Google Scholar]
- Mills EM, Gunasekar P, Borowitz JL, Isom GE. Differential susceptibility of brain areas to cyanide involves different modes of cell death. Toxicol Appl Pharmacol. 1999;156:6–16. doi: 10.1006/taap.1999.8630. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K. Plasma free fatty acids and peroxisome proliferator-activated receptor alpha in the control of myocardial uncoupling protein levels. Diabetes. 2005;54:3496–3502. doi: 10.2337/diabetes.54.12.3496. [DOI] [PubMed] [Google Scholar]
- Nakatani T, Tsuboyama-Kasaoka N, Takahashi M, Miura S, Ezaki O. Mechanism for peroxisome proliferator-activated receptor-alpha activator-induced up-regulation of UCP2 mRNA in rodent hepatocytes. J Biol Chem. 2002;277:9562–9569. doi: 10.1074/jbc.M110132200. [DOI] [PubMed] [Google Scholar]
- Negre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Penicaud L, Casteilla L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1997;11:809–815. [PubMed] [Google Scholar]
- O’Brien ML, Cunningham ML, Spear BT, Glauert HP. Effects of peroxisome proliferators on glutathione and glutathione-related enzymes in rats and hamsters. Toxicol Appl Pharmacol. 2001;171:27–37. doi: 10.1006/taap.2000.9111. [DOI] [PubMed] [Google Scholar]
- Pecqueur C, Alves-Guerra MC, Gelly C, Levi-Meyrueis C, Couplan E, Collins S, Ricquier D, Bouillaud F, Miroux B. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J Biol Chem. 2002;276:8705–8712. doi: 10.1074/jbc.M006938200. [DOI] [PubMed] [Google Scholar]
- Peters JM, Rusyn I, Rose ML, Gonzalez FJ, Thurman RG. Peroxisome proliferator-activated receptor alpha is restricted to hepatic parenchymal cells, not Kupffer cells: implications for the mechanism of action of peroxisome proliferators in hepatocarcinogenesis. Carcinogenesis. 2000;21:823–826. doi: 10.1093/carcin/21.4.823. [DOI] [PubMed] [Google Scholar]
- Peters JM, Park Y, Gonzalez FJ, Pariza MW. Influence of conjugated linoleic acid on body composition and target gene expression in peroxisome proliferator-activated receptor alpha-null mice. Biochim Biophys Acta. 2001;1533:233–342. doi: 10.1016/s1388-1981(01)00155-x. [DOI] [PubMed] [Google Scholar]
- Prabhakaran K, Li L, Mills EM, Borowitz JL, Isom GE. Up-regulation of uncoupling protein 2 by cyanide is linked with cytotoxicity in mesencephalic cells. J Pharmacol Expl Ther. 2005;314:1338–1345. doi: 10.1124/jpet.105.088625. [DOI] [PubMed] [Google Scholar]
- Rashid A, Wu TC, Huang CC, Chen CH, Lin HZ, Yang SQ, Lee FY, Diehl AM. Mitochondrial proteins that regulate apoptosis and necrosis are induced in mouse fatty liver. Hepatology. 1999;29:1131–1138. doi: 10.1002/hep.510290428. [DOI] [PubMed] [Google Scholar]
- Ravnskjaer K, Boergesen M, Rubi B, Larsen JK, Nielsen T, Fridriksson J, Maechler P, Mandrup S. Peroxisome proliferator-activated receptor alpha PPARalpha potentiates, whereas PPARgamma attenuates, glucose-stimulated insulin secretion in pancreatic beta-cells. Endocrinology. 2005;146:3266–3276. doi: 10.1210/en.2004-1430. [DOI] [PubMed] [Google Scholar]
- Rousset S, Alves-Guerra MC, Mozo J, Miroux B, Cassard-Doulcier AM, Bouillaud F, Ricquier D. The biology of mitochondrial uncoupling proteins. Diabetes. 2004;53:S130–S135. doi: 10.2337/diabetes.53.2007.s130. [DOI] [PubMed] [Google Scholar]
- Scatena R, Bottoni P, Vincenzoni F, Messana I, Martorana GE, Nocca G. Bezafibrate induces a mitochondrial derangement in human cell lines: a PPAR-independent mechanism for a peroxisome proliferator. Chem Res Toxicol. 2003;16:1440–1447. doi: 10.1021/tx0341052. [DOI] [PubMed] [Google Scholar]
- Scatena R, Bottoni P, Martorana GE, Ferrari F, De Sole P, Rossi C, Giardina B. Mitochondrial respiratory chain dysfunction, a non-receptor-mediated effect of synthetic PPAR-ligands: Biochemical and pharmacological implications. Biochem Biophys Res Commun. 2004;319:967–973. doi: 10.1016/j.bbrc.2004.05.072. [DOI] [PubMed] [Google Scholar]
- Smith SA, May FJ, Monteith GR, Roberts-Thomson SJ. Activation of the peroxisome proliferator-activated receptor-alpha enhances cell death in cultured cerebellar granule cells. J Neurosci Res. 2001;66:236–241. doi: 10.1002/jnr.1216. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ. Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann Neurol. 2003;53:711–717. doi: 10.1002/ana.10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teissier E, Nohara A, Chinetti G, Paumelle R, Cariou B, Fruchart JC, Brandes RP, Shah A, Staels B. Peroxisome proliferator-activated receptor alpha induces NADPH oxidase activity in macrophages, leading to the generation of LDL with PPAR-alpha activation properties. Circ Res. 2004;95:1174–1182. doi: 10.1161/01.RES.0000150594.95988.45. [DOI] [PubMed] [Google Scholar]
- Vandeputte C, Guizon I, Genestie-Denis I, Vannier B, Lorenzon G. A microtiter plate assay for total glutathione and glutathione disulfide contents in culture/isolated cells: performance study of a new minaturized protocol. Cell Biol Toxicol. 1994;10:415–421. doi: 10.1007/BF00755791. [DOI] [PubMed] [Google Scholar]
- Yang S, Zhu H, Li Y, Lin H, Gabrielson K, Trush MA, Diehl AM. Mitochondrial adaptations to obesity-related oxidant stress. Arch Biochem Biophys. 2000;378:259–268. doi: 10.1006/abbi.2000.1829. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li L, Prabhakaran K, Borowitz JL, Isom GE. Trimethyltin-induced apoptosis is associated with upregulation of inducible nitric oxide synthase and Bax in a hippocampal cell line. Toxicol Appl Pharmacol. 2006;216:34–43. doi: 10.1016/j.taap.2006.05.004. [DOI] [PubMed] [Google Scholar]