

Abstract
Produced by the action of lightning in the atmosphere of the pre-biotic earth, nitric oxide (NO) is a free radical molecule that provided the major nitrogen source for development of life. Remarkably, when atmospheric sources of NO became restrictive, organisms evolved the capacity for NO biosynthesis and NO took on bioregulatory roles. We now recognize NO as an ancestral regulator of diverse and important biological functions, acting throughout the phylogenetic tree. In mammals, NO has been implicated as a pivotal regulator of virtually every major physiological system. The bioactivities of NO, and reactive species derived from NO, arise predominantly from their covalent addition to proteins. Importantly, S-nitrosylation of protein cysteine (Cys) residues has emerged as a preeminent effector of NO bioactivity. How and why NO selectively adds to particular Cys residues in proteins is poorly understood, yet fundamental to how NO communicates it bioactivities. Also, evolutionary pressures that have shaped S-nitrosylation as a biosignaling modality are obscure. Considering recently recognized NO signaling paradigms, we speculate on the origin of NO signaling in biological systems and the molecular adaptations that have endowed NO with the ability to selectively target a subset of protein Cys residues that mediate biosignaling.
A history of earthly NO: the origin of biosignaling by protein S-nitrosylation
Nitric oxide (NO) is a free-radical gas, the fifth-smallest molecule, and a common constituent of star-forming gases throughout the cosmos[1]. Generation of NO occurred on the Hadean earth (4.5 – 3.8 Gyr ago) by chemical reactions involving N2 and CO2, driven by atmospheric electrical discharges[2]. Although nitrogen was a principal component of the earth's early atmosphere, the chemical stability of N2 effectively precluded its direct use in the prebiotic soup for synthesis of the nitrogenous building blocks of life (i.e., purines, pyrimidines and amino acids). Thus, N2 needed to be “fixed” into a chemically useful form, nitrate or ammonium, and this occurred through the intermediacy of NO. With the availability of NO-derived nitrogenous substrates, the stage was set for evolution of terrestrial life forms. While NO provided the necessary source of fixed nitrogen to fuel life through most of the Archean period (3.8 – 2.2 Gyr ago), atmospheric simulation experiments suggest that a crisis in the biosphere occurred at the end of this period, when abiotic production of NO is estimated to have waned 100-fold due to diminishing levels of atmospheric CO2[3]. This NO insufficiency is hypothesized to have caused such a severe ecological crisis that it provided the selective pressure for organisms to evolve the ability to perform nitrogen fixation[3] – biological nitrogen fixation represented an evolutionary milestone, as it obviated the dependence of life on the limitations of abiotic nitrogen fixation. Nitrogen bioavailability continues to this day as the most common limiting nutrient for earth's ecosystems, determining the base width of food-chains[3].
Once life appeared on earth, it must have had a dichotomous relationship with NO. Whereas NO formation on the prebiotic earth provided a pivotal source of fixed nitrogen that enabled life to evolve, reactions of NO with biomolecules must have also posed a threat to life. Indeed, like O2, NO is poison to the catalytic activity of transition metal centers in metabolic enzymes required for the anoxic respiration of earth's ancestral prokaryotes. The problem of NO toxicity was likely met by the evolution of multiple chemoprotective strategies. One fascinating biological approach to cope with NO excess is suggested from functional studies of modern day hemoglobin family members (single domain globins and flavohemoglobins) in bacteria, yeast, protozoa and fungi. While hemoglobins are expressed in perhaps all current day microbes[4], it is obvious that these microorganisms have no significant barrier to gas diffusion and hence hemoglobins are not needed to facilitate O2 distribution. Instead, bacterial hemoglobins have been shown to protect against NO toxicity[5] and it was hypothesized that microbial hemoglobins in ancestral progenitors arose for the purpose of NO binding, rather than O2 binding. This view is suggested by the apparent emergence of hemoglobins at a time when the earth was anoxic as well as knowledge that hemoglobins bind NO with an affinity that is far greater than that of O2, often by >6-orders of magnitude. The proposed use of ancient hemoglobins for NO biodefense would likely have extended to scavenging toxic O2 at a later time, when O2 had accumulated significantly on earth due to the emergence and expansion of photosynthetic bacteria. Importantly, bacterial flavohemoglobins have been shown to function in bacteria as enzymes, consuming one mole each of toxic NO and O2, yielding NO3− as a nutrient product[6,7]. With the evolutionary advance toward aerobiosis and multicellularity (at a time when abiotic NO production was relatively insignificant), hemoglobins were primed to accept their currently recognized role for support of aerobic respiration by O2 binding and distribution via cardiovascular systems. Nonetheless, hemoglobin in current day microbes continues to function as a scavenger of NO for host-defense[8]) and, in some cases, as a scavenger of O2 for maintaining anerobiosis[9].
Despite the biohazard that abioticly produced NO posed to early life on earth, the adaptive benefit of NO is evident from the fact that NO producing enzymes (NO synthases; NOSs) emerged in ancestral organisms, apparently via a series of gene duplication and rearrangement events involving pre-existent protein modules that subserved other metabolic functions[10,11]. Whereas lower eukaryotes typically possess a single NOS gene, higher eukaryotes (e.g., mammals) have evolved three distinct NOS genes (eNOS, iNOS and nNOS) that each catalyze an identical chemistry, but differ in their kinetic properties and in amino acid sequences that determine subcellular protein localization, protein-protein interactions, and gating of activity by phosphorylation and binding of calcium/calmodulin (Ca2+/CaM)[11,12]. Interestingly, the nitrogen-source for NO production by all NOSs is the most nitrogen-rich amino acid in proteins, L-arginine.
An early adaptive benefit afforded by biologically-derived NO may have been to increase the chemical diversity of biomolecules that natural selection can operate on. Indeed, the sole recognized function of a NOS in a modern day bacterium is biosynthetic, not cell signaling – NO-dependent nitration of tryptophan was found to be necessary for synthesis of a nitroindole moiety in a phytotoxic peptide that is required for the pathogenicity of certain streptomycete strains[13,14]. Production of excess quantities of NO is a strategy that has been broadly employed across phyla for the global disruption of protein functions during the innate immune response[15]. NO-dependent biochemical modifications of proteins, elicited by intricate regulation of NOS enzyme isoforms, have played progressively more pervasive cell signaling roles with the evolution of higher eukaryotes. Notwithstanding, the relevance of NO mediated cell signaling in lower eukaryotes is apparent from the seminal role played by neuronally-produced NO in the feeding response of molluscs and gastropods[16-18] and in the widespread use of a suppression in endogenous NO production as a morphogenic switch in species that exhibit biphasic life cycles, (i.e., rapid and extreme life cycle transitions from larval to juvenile stages of development), such as that which occurs in sea urchins and gastropods[19].
State-dependent NO production has evolved wide usage for biosignaling in higher eukaryotes and key roles for NO have been implicated in essentially all physiological systems of mammals. Such multifunctionality of NO as a signaling molecule necessitates the highly targeted delivery of NO to specific sites on regulatory proteins. NO is an atypical biosignaling molecule in that its actions depend on covalent chemical modifications of proteins, rather than “traditional” lock-and-key binding to proteins that function as cellular “receptors”. Appreciation of this fundamental difference in the signaling mode of NO has major consequences: (i) the chemical reactivity of NO determines its biological actions, (ii) many competing reactions of NO and NO-derived species are possible in biological systems, (iii) reactions of NO and NO-derived species, and consequently biological activity, are influenced by the changing intracellular milieu. Thus, the rate, site and microenvironment of NO synthesis are potential determinants of the chemical reactions of NO and ensuing biological consequences. But how is NO selectivity achieved in complex biological systems? We consider this question below, in the context of NO signaling via protein S-nitrosylation.
Signaling by protein S-nitrosylation
NO-mediated vasodilation depends principally on nitrosylation of heme-iron in soluble guanylate cyclase (sGC), enzyme activation and intracellular cGMP accumulation. However, unlike vasorelaxation, which is substantially blocked by sGC inhibitors and stimulated by non-hydrolyzable cGMP analogs, many vascular effects of NO are resistant to these drugs. Indeed, many biological actions of NO involve the formation of nitrosothiols[20], addition of NO to the sulfur atom of Cys, a process alternatively termed S-nitrosation or S-nitrosylation. Even for NO-induced vasodilation, the classical cGMP-dependent response, S-nitrosylation contributes by triggering potassium channel opening[21].
S-nitrosylation of Cys residues in target proteins was first recognized in 1992, when micromolar levels of SNO-albumin were reported to circulate in blood[22]. A 2001 report on the proceedings of a meeting that focused on protein S-nitrosylation listed 115 proteins in which the addition of NO to protein Cys-sulfur had ben reported to occur in biological systems[23]. With the advent of proteomic approaches for unbiased identification of SNO-proteins, this number has more than doubled[24-30] and is likely to grow exponentially as new and more sensitive detection technologies are devised. As a group, the reported SNO-proteins subserve a wide range of cell functions, including ion channels, pumps, receptors for signal molecules, transcription factors, enzymes and structural proteins[31]. Importantly, S-nitrosylation fulfills all essential criteria for validation as a cell signaling mechanism -- it is stimulus-evoked [32,33], reversible[34], precisely-targeted [35], spatiotemporally restricted [36,37], and necessary for specific cell responses (for reviews, see [31,38-40]).
Significance of protein S-nitrosylation for cardiovascular regulation
Protein SNOs have been implicated to play broad roles in vascular homeostasis[41]. The source of the NO moiety for S-nitrosylation can be NO itself, other NOx (higher oxides of nitrogen), metal-NO complexes, nitrite (in acidified milieu), or other nitrosothiols [41]. Notably, once formed, the nitroso- moiety of a protein nitrosothiol can be transferred to other thiols through a process termed transnitrosylation. NO groups can be transferred from protein to protein and to low molecular weight thiols (principally glutathione), before undergoing terminal oxidation to nitrite and nitrate. While a protein denitrosylase has not been discovered (analogous to a protein phosphatase), a selective S-nitrosoglutathione (GSNO) reductase (GSNOR/formaldehyde dehdrogenase) was shown by Liu et al. to function across phylogeny to accelerate the metabolism of GSNO[42]. In accord with the existence of a predicted equilibrium between NO in GSNO and SNO-proteins in blood, GSNOR-null mice would be expected to exhibit elevated circulating levels of SNO-proteins. This prediction was substantiated by levels of NO on hemoglobin (Hb), a principal nitrosoprotein in blood where NO is found on both thiol (Cysβ93) and iron[43-45] – notably, targeted deletion of GSNOR was associated with a 2.5-fold increase SNO-Hb, but no change in iron-nitrosyl Hb relative to GSNOR wildtype mice [46]. Accordingly, the phenotype of GSNOR-nullozygous mice can provide a window into the cardiovascular actions transduced by SNO-proteins. A surprising observation was that anesthetized GSNOR-null mice exhibit profound hypotension[46] suggesting that S-nitrosylation can contribute to the vasodilatory activity of NO (opposing the common view that cGMP-dependent signaling is the exclusive mediator of NO-elicited vasodilation). It was also shown that GSNOR-null mice (relative to GSNOR+/+ mice) exhibit a markedly increased sensitivity to tissue damage and death following treatment with bacterial endotoxin or live bacteria[46]. Thus, SNO-proteins apparently contribute to inflammation and mortality in septic shock. These studies with GSNOR-null mice suggest roles for SNO-proteins in both cardiovascular health and disease.
The conundrum: NO signals based on reactivity, but must achieve selectivity
What is the molecular basis for selective NO addition to protein Cys thiols? For the prototype cell signaling modality, phosphorylation of proteins on Ser, Thr and Tyr residues, specificity is dictated by the molecular recognition of substrates by their cognate kinases. Since NO, and reactive NO-derived species, have been presumed to add to protein Cys-sulfur in solution, by a mechanism that is independent of protein-protein interactions, the basis for selective protein S-nitrosylation in biological systems is non-obvious. Even for proteins known to undergo significant S-nitrosylation in biological systems, it is commonly observed that only one of many available Cys residues undergoes detectable levels of NO addition (i.e., the stoichiometry of NO addition per protein monomer is typically unitary). An extreme example is the ryanodine-responsive calcium release channel of skeletal muscle (RyR1), where only 1 of ∼50 Cys residues were found to undergo detectable levels of S-nitrosylation by NO (Cys3635)[35] and thereby mediate channel-opening and increased contractility[47]. In an unbiased proteomic study, 67 proteins in rat cerebellum were found to contain SNO-Cys residues following treatment with a physiological level of GSNO – 56 of these proteins contained a single SNO-Cys[48]. Such findings beg for a rationalization of the selective addition of NO to discrete protein thiols, despite the promiscuous reactivity of NO towards thiol and non-thiol targets.
Recent results from studies of individual SNO-proteins have converged on two fundamental mechanisms by which Cys residues achieve selective S-nitrosylation for NO-based regulation of protein functions. These mechanisms are: (1) concentration of the nitrosylating species at the Cys modification site (i.e., use the law of mass action to drive the reaction forward), and (2) positioning Cys in a favorable chemical context for reaction with NO (i.e., either increase the forward rate constant for nitrosylation, decrease the denitrosylation rate, or both). We consider each of these strategies below.
Concentrating the nitrosylating species at the Cys modification site: Proximity based NO signaling
NOS-substrate complexes harness the law of mass action for targeting NO to specific protein Cys residues
NO, and reactive NO-derived species, have been considered to diffuse randomly from their sites of synthesis until consumed by chemical reactions. In some cases, these reactions will include S-nitrosylation of one or more proteins that contributes to NO-mediated bioactivities. While simple random diffusion of NO has often been generally assumed, modeled[49] and hailed for its relevance to NO biology, recent findings show that NO reactivity is often channeled to biologically-relevant thiols on recognized NO-signaling proteins.
Efficient signal transduction events are dependent on correct localization and assembly of signaling proteins within discrete subcellular microdomains. The existence of multimeric protein complexes imposes spatial and temporal confinement to signaling systems, allowing tight regulation of the cell's biochemical and signaling properties. The fundamental importance of the subcellular organization of NOSs within cells is implicit in a report by Barouch et al.[50] showing that eNOS and nNOS co-exist in cardiac myocytes, but reside within distinct subcellular compartments (the plasma membrane and sarcoplasmic reticulum respectively). Surprisingly, eNOS-derived NO was found to inhibit myocardial contractility, whereas nNOS-derived NO enhanced contractility. This finding suggests that spatial confinement of eNOS and nNOS in cardiomyocytes can effectively couple NO to discrete signaling pathways that determine downstream effects. Selective S-nitrosylation of a protein thiol may thus be achieved by positioning the thiol in close proximity to a cellular source of NO reactivity. The relevant source of NO reactivity can take multiple forms. For each NOS isoform there are examples where NO reactivity has been shown to be selectively channeled to a particular protein thiol by a mechanism that requires either direct binding of an NO-acceptor protein (as for iNOS/cycloxygenase-2[51] and eNOS/Hsp90[52,53]) or indirect binding via a scaffold protein (as for the nNOS/NMDA receptor/Dexras complex[36,54]). NOS/target protein interactions are complex and suggest that coordinated evolution of NOS and target proteins occurred for enhancement of signaling selectivity. We briefly describe an interacting protein (or protein complex) for each of the three NOS isoforms, as prototypic examples of site-specific S-nitrosylation in NO regulated biosignaling systems.
iNOS and Cyclooxygenase-2
Cycloxygenase-2 (COX-2) is a pivotal enzyme for the metabolism of arachidonic acid to biologically important prostanoids. Like iNOS, COX-2 is induced in response to inflammatory stimuli and prior studies have revealed a complex synergistic relationship between the two enzymes in immunoactivated cells[55]. Recently Snyder and colleagues demonstrated by co-immunoprecipitation that iNOS and COX-2 exhibit a direct and exclusive interaction[51]. As a consequence, iNOS was shown to selectively S-nitrosylate COX-2 on Cys526, resulting in an increased Vmax and enhanced prostaglandin E2 production. The physical interaction of iNOS and COX-2 was demonstrated in this study to be an absolute requirement for S-nitrosylation of COX-2. Notably, when protein-protein contact was precluded by incubation with a blocking peptide, iNOS continued to produce robust quantities of NO, but failed to cause S-nitrosylation or activation of COX-2. These findings suggest that specific nitrosylation of the regulatory thiol on COX-2 is a consequence of evolutionary adaptations that engender a productive protein-protein interaction for NO group transfer. Notably, a tyrosyl radical at Tyr384 in COX-2 (Tyr 385 in COX-1[56]) is critical for enzyme activity and can be avidly quenched by reaction with NO radical in vitro (1−2 × 109 M−1s−1)[57] -- inasmuch as iNOS activates COX-2 in cells, it is likely that the interaction between iNOS and COX-2 serves to channel NO for reaction with Cys526. While an elevated concentration of nitrosylating species is a presumed basis for S-nitrosyation site-specificity in COX-2, undefined features of the thiol environment are likely to also contribute. It would not be surprising if nitrosylation-promoting allosterically-elicited structural changes arise as a consequence of the interaction between iNOS and COX-2.
eNOS and Heat Shock Protein 90
Heat shock protein 90 (Hsp90) is an intracellular chaperone involved in the trafficking and regulation of a variety of signaling proteins, including Src and its family members, Raf and Mitogen Activated Protein Kinase Kinase (MAPKK/MEK)[58]. It was shown that eNOS co-immuoprecipitates with a 90 kDa tyrosine-phosphorylated protein that was identified as Hsp90[52,53]. In endothelial cells, external stimuli such as VEGF, histamine, estrogen and shear-stress promote the association of eNOS with Hsp90, resulting in enhanced eNOS activity[52]. Notably, the eNOS-Hsp90 association was found to be an absolute requirement for Akt-mediated stimulation of eNOS[59]. In response to external stimuli such as VEGF, Hsp90 binding to eNOS was shown to disrupt a pre-existing eNOS-caveolin-1 inhibitory complex, allowing Ca2+/CaM to bind eNOS and expose of a Ser residue for Akt-dependent phosphorylation. Phosphorylation of eNOS at Ser1177 by Akt enhances electron flux from the reductase domain to the oxygenase domain of eNOS, thereby accelerating NO production. Thus, Hsp90 serves two key functions for eNOS activation: (1) it provides a scaffold for recruitment of Akt to eNOS for phosphorylation of Ser1177, and (2) it stabilizes Ca2+/CaM binding[60].
Importantly, the specific interaction of Hsp90 with eNOS positions Hsp90 in proximity to a source of NO. Consquently, Hsp90 was shown to undergo S-nitrosylation, resulting in an enhanced intrinsic ATPase activity[61]. The site of nitrosylation was mapped to Cys597 in the C-terminal domain of Hsp90, a region implicated in the eNOS-Hsp90 interaction. S-nitrosylation of Cys597 was shown to dampen eNOS activity, apparently by triggering a conformational change in the Hsp90 structure that disrupts the eNOS-Hsp90 complex[61]. Evolution of proximity-dependent selective S-nitrosylation of Hsp90 provides an elegant regulatory mechanism by which eNOS-derived NO may protect endothelial cells from overwhelming amounts of NO.
nNOS, the NMDA Receptor and Dexras
nNOS-derived NO synthesis in neurons is coupled to calcium release by the NMDA-type glutamate receptor[62,63]. NMDA receptors (NMDAR) are located at the postsynaptic density (PSD) of excitatory synapses and mediate long-term potentiation and memory formation in some brain regions[64]. The association of nNOS with the NMDAR is indirect, mediated by binding to each of two distinct PDZ domains on the scaffold-protein PSD95 (which possesses a total of three tandem PDZ domain repeats)[65,66]. The NMDAR-PSD95-nNOS ternary complex is necessary for an efficient increase in nNOS activity, following NMDAR activation. As the NMDAR is a calcium channel, it is understood that the transient flux of calcium through the NMDAR must be in close proximity to nNOS for efficient Ca2+/CaM binding and NO production. Notably, with prolonged NMDAR activation, nNOS-derived NO has been shown to selectively S-nitrosylate Cys399 on the NMDAR itself, resulting in channel inactivation and hence, NMDAR desensitization[67,68]. At the organism level, S-nitrosylation of Cys399 in the NMDA receptor may have evolved as a protective mechanism to limit NMDAR-mediated excitotoxic brain damage that can occur after cerebral ischemia-repurfusion injury (i.e., strokes).
CAPON (carboxy-terminal PDZ ligand of nNOS), is an adaptor protein that competes with PSD95 for binding to the PDZ domain of nNOS[69]. Thus, CAPON serves to recruit nNOS away from the NMDA receptor and suppresses nNOS activity by limiting access to activating fluxes of calcium[69]. A secondary role for CAPON involves its N-terminal phosphotyrosine-binding (PTB) domain. This PTB domain has been shown to bind the monomeric G-protein, Dexras1[70] positioning NO in a manner that favors S-nitrosylation of Dexras on a single cysteine residue (Cys11), resulting in Dexras1 activation (i.e., exchange of bound GDP for GTP)[36,71]. It is inferred that modification of Dexras by S-nitrosylation of Cys11 induces a conformational change within the protein that triggers dissociation of GDP and binding of GTP, promoting activation[71]. The physiological implications of Dexras1 S-nitrosylation are apparent in nNOS null mice (nNOS-/-), where there is a loss of Dexras1 activation. Notably, H-Ras and four other Ras family members were not affected by nNOS gene knockout, despite the susceptibility of these Ras members to undergo efficient S-nitrosylation and activation by NO donors in vitro[36].
Taken together, the above examples reveal that the assembly of NOS signaling complexes to mediate targeting of NO to precise sites of S-nitrosylation is a general strategy for selective NO-based cell signaling. A related strategy for selective S-nitrosylation, is targeted transnitrosation – this is where NO group transfer occurs as a consequence of the specific binding of a pre-existing SNO-protein and a cognate NO-accepting binding partner. Such targeted transnitrosation was shown to occur natively for NO group transfer from hemoglobin SNO-Cysβ93 to the anion exchange protein (AE1/band3) in human red blood cells[72] and for transfer of NO from SNO-Cys73 of thioredoxin to Cys167 in caspase-3[73]. Selective thiol-thiol NO group transfer could be recapitulated synthetically using a SNO-peptide inhibitor of caspase-3 that selectively transnitrosates the active site thiol in caspase-3 (Cys167), thereby inhibiting caspase activity[74]. This synthetic thiol-targeting strategy raises the possibility for development of NO-donor drugs that precisely bind and target a desired protein thiol for therapeutic S-nitrosylation.
Further, in an elegant series of studies by Snyder and colleagues, an NO triggered signaling cascade was discovered that is initiated by S-nitrosylation of GAPDH (on SNO-Cys149), resulting in the specific binding and stabilization of an otherwise labile protein mediator of apoptotic cell death, the E3 ubiquitin ligase, Siah1[75]. The discovery of this pathway is compelling, as it explains the previously confounding association of nuclear localization of GAPDH with apoptosis[76] – although GAPDH does not possess a nuclear localization signal (NLS) and theoretically should not enter the nucleus, nuclear access to GAPDH is engendered by the NLS on its binding partner, Siah1[75]. Biological importance of the SNO-GAPDH/Siah1 pathway is indicated by its link to neurodegenerative conditions[77-79] and that endogenous levels of SNO-Cys149-GAPDH are present in untreated mouse brains[30,80]. Such S-nitrosylation-dependent protein-protein association may not be uncommon, as indicated by a productive two-hybrid screen that identified NO-dependent protein-protein interactions[81].
Interestingly, multiple SNO-proteins are known to undergo selective S-nitrosylation on non-solvent exposed hydrophobic Cys residues[35,82,83]. Selective NO addition to Cys-sulfur in a hydrophobic millieu has been rationalized as arising from reaction with higher oxides of nitrogen (e.g., N2O3), efficient nitrosylating agents whose formation occurs orders of magnitude faster in hydrophilic milieu due to the concentration of precursor species (NO and O2) in hydrophobic compartments[83]. As shown by Nudler and colleagues for serum albumin[83], localized formation of an nitrosylating species by this mechanism can selectively target NO to acceptor thiols in hydrophobic cores of proteins. Thus, some proteins may effectively autocatalyze their own S-nitrosylation by providing an environment for formation of N2O3 within the protein and in the immediate vicinity of a target thiol.
Together, the above findings indicate that the incorporation into a defined signaling complex of an acceptor protein-thiol and a nitrosating species generator (i.e., NOS, a nitrosothiol, or other thiol-reactive species that derives from NO), constitutes a conserved strategy for targeting NO to specific protein thiols that mediate biosignaling. Contextual features of the acceptor thiol are also presumed to be critical, both for the efficiency of S-nitrosylation and the chemical/structural alterations that mediate the consequent protein function changes and hence, signaling. We next consider contextual features that may be relevant for S-nitrosylation efficiency.
Positioning Cys in a favorable chemical context for reaction with NO: a chemical basis for selective S-nitrosylation
Limited knowledge about the chemical “rules” for S-nitrosylation can be gleaned from the relatively sparse number of endogenous protein SNO-Cys sites for which structural information is available. Notwithstanding, some reasonable inferences can be drawn. Protein S-nitrosylation in aqueous media involves the addition of a nitrosating species to Cys-thiolate (S−). Accordingly, the degree of thiol ionization (nucleophilicity) will be an important determinant of S-nitrosylation susceptibility. Whereas Cys on its own has a pKa of 8.3 and therefore would be < 10.0% in the thiolate form at a physiological pH of 7.2, the environment of some protein Cys residues can possess markedly lower pKa, rendering > 99% thiolate in biological media. Thus, although the majority of thiol groups in a protein are relatively unreactive, thiols can be highly reactive in some contexts. Dominating factors that promote thiol ionization are proximity to basic residues (Arg, His, and Lys), Zn+-coordination and electrostatic interactions with appropriately-oriented and positioned aromatic amino acid side-chains. It is notable that among ∼30 thiols in eNOS, auto-nitrosylation was detected on one alone, a zinc-coordinated thiol[84]. As described below, the positioning of thiols in a positively-charged environment and electrostatic interactions with aromatic rings (factors that increase nucleophilicity) seem at first blush to correlate with susceptibility for S-nitrosylation.
It is well known that the primary sequence of residues flanking O-phoshorylation sites can be used to predict sites of phosphorylation [85]. Stamler and colleagues proposed an acid-base flanking consensus motif for Cys residues that are susceptible to S-nitrosylation[86], such as SNO-Cysβ93 in human Hb[86] and SNO-Cys3635 in the RYR1[35]. Interestingly, for the regulatory S-nitrosylation site in methionine adenosyltransferase, a three-dimensional version of the SNO consensus sequence (Arg257-Cys121-Asp355) was linked to efficient site-selective S-nitrosylation[87]. While this consensus sequence may hold predictive value for Cys residues that undergo S-nitrosylation by NO or some other NO-donating species, predictive value was not detected from consideration of the linear flanking residues of 68 GSNO-elicited SNO-Cys sites, identified in an unbiased proteomic screen of brain SNO-proteins[30]. Three-dimensional structural information may be needed for many protein S-nitrosylation sites before reliable predictive patterns can be deduced. Nonetheless, some insights may be offered by consideration tubulin S-nitrosylation.
Among 20 Cys residues in tubulin (8 Cys in the α-subunit and 12 Cys in the β-subunit), 4 Cys were identified as sites of endogenous SNO-modification (α-Cys295, α-Cys376, β-Cys12 and β-Cys239) and 3 additional Cys were found to undergo preferential S-nitrosylation following treatment with GSNO (α-Cys347, β-Cys303 and β-Cys354). The crystal stucture of tubulin dimer (PDB coordinates, 1tub) revealed that, with the exception of β-Cys12, none of the NO-targeted Cys lie on the protein surface [88]. Although NO-targeted Cys residues are those that are least exposed to solvent, they are Cys that were found to be most reactive with thiol-modifying reagents[89,90]. Considering over 50 identified SNO-Cys residues in proteins, an apparent correlation with thiol-reactivity was noted[30]. Thus, solvent accessibility of Cys is not predictive of SNO-modification, but a low pKa is apparently key.
The structural basis for high reactivity of endogenous SNO-Cys in tubulin has been considered[30]. Each of the identified SNO-Cys appear to lie in an environment of positive charge. Interestingly, α-Cys295 and α-Cys347 are also within 6 Å of a pair of aromatic residues that can engage in sulfur in electrostatic interactions. Tubulin β-C376 is similarly in proximity for electrostatic interaction with an aromatic ring edge and in hydrogen bond range with a basic residue. It is notable that electronegative Cys-sulfur are commonly found to engage in electrostatic interactions with electropositive hydrogens on aromatic rings – such interactions can contribute to protein structure stability, particularly when the Cys is in a hydrophobic environment[91]. Conversely, disruption of such a thiol-aromatic interaction, as might occur following NO addition, can potentially destabilize protein structure and have functional consequences. We speculate that destabilization of pre-existing aromatic-thiol interactions may be a generic mechanism by which S-nitrosylation alters protein structure and function. Cys-aromatic interactions, along with hydrogen-bonding to basic residues, may be common structural motifs that render Cys residues selectively susceptible to S-nitrosylation.
Did Cys residues evolve in proteins specifically as NO sensors?
It is tempting to speculate that some protein Cys residues evolved for the specific purpose of detecting and responding to NO. A reasonable case in support of this possibility is provided by consideration of argininosuccinate synthetase (AS), a protein that enables the regeneration of the NOS substrate Arg, from the NOS product citrulline[92,93]. The Arg-Cit cycle that AS contributes to provides for a recycling source of Arg substrate for support of high-output NO synthesis. Following treatment of rats or cells with immunostimulants, AS protein expression and activity are elicited in tandem with iNOS. However, with sustained in vivo generation of NO, AS activity becomes inhibited in a thiol-reversible manner owing to specific S-nitrosylation of Cys132 (human AS), one of five Cys residues[34]. This finding suggested that S-nitrosylation of AS provides autoregulatory protection of NO-producing cells, by limiting the possibility for toxic NO overproduction. Conservative mutagenesis of Cys132 was found to have no significant influence on AS catalytic activity, despite ablating AS inhibition by otherwise blocking quantities of NO. Alignment of AS sequences from more than 100 species revealed that Cys132 of huAS is conserved in all mammals and NOS-expressing invertebrates, but absent from yeast and bacteria that do not contain a full-length NOS-encoding gene (Fig 1A). These results are in accord with the view that Cys132 in AS evolved as a ‘NO sensor’ to limit NO overproduction in biological systems.
Figure 1.
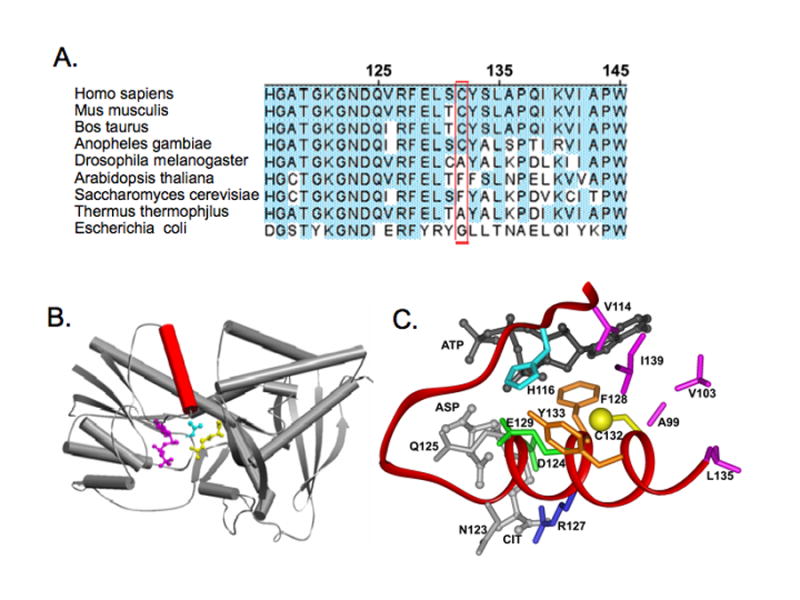
Homology model of human argininosuccinate synthase (AS). A: Alignment of some amino acid sequences of human AS and orthologs, showing Cys132 and phylogenetic substitutions. The depicted result and those extended to 130 additional orthologs, suggest that a Cys132 homolog is present in NOS-expressing species, but absent in non-NOS expressing species. This is in accord with the view that Cys132 evolved as a specific NO-sensor. B: Model structure of AS monomer. The model reveals that Cys132 of human AS resides in the substrate-binding helix, shown in red. Three substrates, ATP, Asp and Cit are shown in pink, cyan and yellow respectively. C: Close up view of the AS active site, showing bound substrates and the Cys132 environment. Sulfur of Cys 132 is depicted as a yellow sphere, and substrates are rendered in ball and stick form, with ATP in black and Asp and Cit in gray. Also shown are hydrophobic residues (pink) surrounding Cys132 (A99, V103, V114, L133, I139). Residues involved in hydrogen-bonding of substrates are indicated (N123, gray; D124, green; Q125, gray; R127, blue) as well as aromatic residues in proximity to Cys132 (F128 and Y133, orange). Residues potentially involved in acid-base catalysis, H116 and E129, are shown in cyan and green respectively. Results suggest that Cys132 is locating on a conserved substrate-binding α-helix. S-nitrosylation of Cys132 would destabilize the helix and have adverse consequences on either binding of substrates or positioning substrates for productive catalysis.
Consideration of the Cys132 context provides insight into the basis for selective addition of NO to this particular thiol and why S-nitrosylation inhibits AS catalytic function. Homology modeling of huAS, based on crystal structures of two homologous bacteria AS proteins, revealed that Cys132 resides on a conserved α-helix that scaffolds the binding of AS substrates, Asp and Citrulline, by hydrogen bonding (Figs. 1B-C). Cys132-sulfur lies mid-helix, surrounded by a 6.5 angstrom shell of hydrophobic residues with an apparent electrostatic interaction between the Cys-sulfur and a nearby phenyalanine ring. As noted above, Nudler and colleagues demonstrated that hydrophobic protein cores can concentrate lipophilic NO and O2, allowing for the formation of an efficient nitrosating species (i.e., N2O3) precisely at the site of a hydrophobic thiol[94]. Thus, selective targeting of Cys132 for S-nitrosylation is inferred to arise as a consequence of its hydrophobic environment. Further, positive charge on a nearby basic residue (His116) may facilitate thiol deprotonation, fostering thiolate anion formation as an NO target. S-nitrosylation of Cys132 is poised to perturb the conformation of the α-helix on which this residue resides, causing a catalytically unfavorable reorientation of the associated hydrogen-bonded AS substrates, providing a molecular explanation for S-nitrosylation suppresses AS catalytic function. This model offers a conceptual framework for specificity and actions of S-nitrosylation in a relevant NO-based signal transduction system. Assessment of the model's validity and provision of molecular details await future experimentation.
New approaches promise rapid advancement in our understanding of the chemical and biological basis for specificity of protein S-nitrosylation
It is apparent that evolution has worked long and hard to harness the promiscuous reactivity of NO for selective targeting to protein thiols and phylogentically widespread usage as a biosignal. Instability of the S-NO bond has restrained progress toward the discovery of biologically-relevant SNO-Cys residues in proteins and elucidation of the determinants for targeting specificity. New technologies are anticipated to deliver proteomic SNO-Cys site data that should fuel bioinformatic investigations with the potential to elucidate underling structure/function principles governing the selective addition of NO to protein thiols and their consequences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ziurys LM, McGonagle D, Minh Y, Irvine WM. Nitric oxide in star-forming regions: further evidence for interstellar N-O bonds. Astrophys J. 1991;373:535–42. doi: 10.1086/170072. [DOI] [PubMed] [Google Scholar]
- 2.Chyba C, Sagan C. Electrical energy sources for organic synthesis on the early Earth. Orig Life Evol Biosph. 1991;21:3–17. doi: 10.1007/BF01809509. [DOI] [PubMed] [Google Scholar]
- 3.Navarro-Gonzalez R, McKay CP, Mvondo DN. A possible nitrogen crisis for Archaean life due to reduced nitrogen fixation by lightning. Nature. 2001;412:61–4. doi: 10.1038/35083537. [DOI] [PubMed] [Google Scholar]
- 4.Wu G, Wainwright LM, Poole RK. Microbial globins. Adv Microb Physiol. 2003;47:255–310. doi: 10.1016/s0065-2911(03)47005-7. [DOI] [PubMed] [Google Scholar]
- 5.Poole RK, Hughes MN. New functions for the ancient globin family: bacterial responses to nitric oxide and nitrosative stress. Mol Microbiol. 2000;36:775–83. doi: 10.1046/j.1365-2958.2000.01889.x. [DOI] [PubMed] [Google Scholar]
- 6.Hausladen A, Gow AJ, Stamler JS. Nitrosative stress: metabolic pathway involving the flavohemoglobin. Proc Natl Acad Sci U S A. 1998;95:14100–5. doi: 10.1073/pnas.95.24.14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hausladen A, Gow A, Stamler JS. Flavohemoglobin denitrosylase catalyzes the reaction of a nitroxyl equivalent with molecular oxygen. Proc Natl Acad Sci U S A. 2001;98:10108–12. doi: 10.1073/pnas.181199698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poole RK. Nitric oxide and nitrosative stress tolerance in bacteria. Biochem Soc Trans. 2005;33:176–80. doi: 10.1042/BST0330176. [DOI] [PubMed] [Google Scholar]
- 9.Minning DM, Gow AJ, Bonaventura J, Braun R, Dewhirst M, Goldberg DE, Stamler JS. Ascaris haemoglobin is a nitric oxide-activated ‘deoxygenase’. Nature. 1999;401:497–502. doi: 10.1038/46822. [DOI] [PubMed] [Google Scholar]
- 10.Liu Q, Gross SS. Binding sites of nitric oxide synthases. In: Packer L, editor. Methods in Enzymology. Vol. 268. Academic Press; 1996. pp. 311–24. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh DK, Salerno JC. Nitric oxide synthases: domain structure and alignment in enzyme function and control. Front Biosci. 2003;8:d193–209. doi: 10.2741/959. [DOI] [PubMed] [Google Scholar]
- 12.Stuehr DJ. Mammalian nitric oxide synthases. Biochim Biophys Acta. 1999;1411:217–30. doi: 10.1016/s0005-2728(99)00016-x. [DOI] [PubMed] [Google Scholar]
- 13.Buddha MR, Tao T, Parry RJ, Crane BR. Regioselective nitration of tryptophan by a complex between bacterial nitric-oxide synthase and tryptophanyl-tRNA synthetase. J Biol Chem. 2004;279:49567–70. doi: 10.1074/jbc.C400418200. [DOI] [PubMed] [Google Scholar]
- 14.Kers JA, Wach MJ, Krasnoff SB, Widom J, Cameron KD, Bukhalid RA, Gibson DM, Crane BR, Loria R. Nitration of a peptide phytotoxin by bacterial nitric oxide synthase. Nature. 2004;429:79–82. doi: 10.1038/nature02504. [DOI] [PubMed] [Google Scholar]
- 15.Nurnberger T, Brunner F, Kemmerling B, Piater L. Innate immunity in plants and animals: striking similarities and obvious differences. Immunol Rev. 2004;198:249–66. doi: 10.1111/j.0105-2896.2004.0119.x. [DOI] [PubMed] [Google Scholar]
- 16.Teyke T. Nitric oxide, but not serotonin, is involved in acquisition of food-attraction conditioning in the snail Helix pomatia. Neurosci Lett. 1996;206:29–32. doi: 10.1016/0304-3940(96)12434-4. [DOI] [PubMed] [Google Scholar]
- 17.Korneev SA, Piper MR, Picot J, Phillips R, Korneeva EI, O'Shea M. Molecular characterization of NOS in a mollusc: expression in a giant modulatory neuron. J Neurobiol. 1998;35:65–76. doi: 10.1002/(sici)1097-4695(199804)35:1<65::aid-neu6>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi S, Sadamoto H, Ogawa H, Kitamura Y, Oka K, Tanishita K, Ito E. Nitric oxide generation around buccal ganglia accompanying feeding behavior in the pond snail, Lymnaea stagnalis. Neurosci Res. 2000;38:27–34. doi: 10.1016/s0168-0102(00)00136-x. [DOI] [PubMed] [Google Scholar]
- 19.Bishop CD, Brandhorst BP. On nitric oxide signaling, metamorphosis, and the evolution of biphasic life cycles. Evol Dev. 2003;5:542–50. doi: 10.1046/j.1525-142x.2003.03059.x. [DOI] [PubMed] [Google Scholar]
- 20.Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A. 1992;89:444–8. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–3. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 22.Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci U S A. 1992;89:7674–7. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675–83. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 24.Gao C, Guo H, Wei J, Mi Z, Wai PY, Kuo PC. Identification of S-nitrosylated proteins in endotoxin-stimulated RAW264.7 murine macrophages. Nitric Oxide. 2005;12:121–6. doi: 10.1016/j.niox.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Lindermayr C, Saalbach G, Durner J. Proteomic identification of S-nitrosylated proteins in Arabidopsis. Plant Physiol. 2005;137:921–30. doi: 10.1104/pp.104.058719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhee KY, Erdjument-Bromage H, Tempst P, Nathan CF. S-nitroso proteome of Mycobacterium tuberculosis: Enzymes of intermediary metabolism and antioxidant defense. Proc Natl Acad Sci U S A. 2005;102:467–72. doi: 10.1073/pnas.0406133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Keszler A, Broniowska KA, Hogg N. Characterization and application of the biotin-switch assay for the identification of S-nitrosated proteins. Free Radic Biol Med. 2005;38:874–81. doi: 10.1016/j.freeradbiomed.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Ruiz A, Lamas S. Detection and proteomic identification of S-nitrosylated proteins in endothelial cells. Arch Biochem Biophys. 2004;423:192–9. doi: 10.1016/j.abb.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Greco TM, Hodara R, Parastatidis I, Heijnen HF, Dennehy MK, Liebler DC, Ischiropoulos H. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103:7420–5. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 32.Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem. 2002 doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 33.Hoffmann J, Dimmeler S, Haendeler J. Shear stress increases the amount of S-nitrosylated molecules in endothelial cells: important role for signal transduction. FEBS Lett. 2003;551:153–8. doi: 10.1016/s0014-5793(03)00917-7. [DOI] [PubMed] [Google Scholar]
- 34.Hao G, Xie L, Gross SS. Argininosuccinate synthetase is reversibly inactivated by S-nitrosylation in vitro and in vivo. J Biol Chem. 2004 doi: 10.1074/jbc.M404866200. [DOI] [PubMed] [Google Scholar]
- 35.Sun J, Xin C, Eu JP, Stamler JS, Meissner G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc Natl Acad Sci U S A. 2001;98:11158–62. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fang M, Jaffrey SR, Sawa A, Ye K, Luo X, Snyder SH. Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 2000;28:183–93. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- 37.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, Groszmann RJ, Shah VH, Sessa WC. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A. 2006;103:19777–82. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lane P, Hao G, Gross SS. S-nitrosylation is emerging as a specific and fundamental posttranslational protein modification: head-to-head comparison with O-phosphorylation. Sci STKE 2001. 2001:RE1. doi: 10.1126/stke.2001.86.re1. [DOI] [PubMed] [Google Scholar]
- 39.Martinez-Ruiz A, Lamas S. S-nitrosylation: a potential new paradigm in signal transduction. Cardiovasc Res. 2004;62:43–52. doi: 10.1016/j.cardiores.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 40.Gaston BM, Carver J, Doctor A, Palmer LA. S-nitrosylation signaling in cell biology. Mol Interv. 2003;3:253–63. doi: 10.1124/mi.3.5.253. [DOI] [PubMed] [Google Scholar]
- 41.Foster MW, McMahon TJ, Stamler JS. S-nitrosylation in health and disease. Trends Mol Med. 2003;9:160–8. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 42.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–4. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 43.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380:221–6. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 44.Kirima K, Tsuchiya K, Sei H, Hasegawa T, Shikishima M, Motobayashi Y, Morita K, Yoshizumi M, Tamaki T. Evaluation of systemic blood NO dynamics by EPR spectroscopy: HbNO as an endogenous index of NO. Am J Physiol Heart Circ Physiol. 2003;285:H589–96. doi: 10.1152/ajpheart.01010.2002. [DOI] [PubMed] [Google Scholar]
- 45.McMahon TJ, Moon RE, Luschinger BP, Carraway MS, Stone AE, Stolp BW, Gow AJ, Pawloski JR, Watke P, Singel DJ, et al. Nitric oxide in the human respiratory cycle. Nat Med. 2002;8:711–7. doi: 10.1038/nm718. [DOI] [PubMed] [Google Scholar]
- 46.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–28. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 47.Eu JP, Sun J, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/s0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- 48.Hao Gang, D B, Shi Lei, Campagne Fabien, Gross Steven S. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci USA. 2006;103:1012–7. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lancaster JR., Jr Simulation of the diffusion and reaction of endogenously produced nitric oxide. Proc Natl Acad Sci U S A. 1994;91:8137–41. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barouch LA, Harrison RW, Skaf MW, Rosas GO, Cappola TP, Kobeissi ZA, Hobai IA, Lemmon CA, Burnett AL, O'Rourke B, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–9. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 51.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–70. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 52.Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–4. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 53.Venema RC, Sayegh HS, Kent JD, Harrison DG. Identification, characterization, and comparison of the calmodulin-binding domains of the endothelial and inducible nitric oxide synthases. J Biol Chem. 1996;271:6435–40. doi: 10.1074/jbc.271.11.6435. [DOI] [PubMed] [Google Scholar]
- 54.Bredt DS. Targeting nitric oxide to its targets. Proc Soc Exp Biol Med. 1996;211:41–8. doi: 10.3181/00379727-211-43950f. [DOI] [PubMed] [Google Scholar]
- 55.Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci U S A. 1993;90:7240–4. doi: 10.1073/pnas.90.15.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deeb RS, Hao G, Gross SS, Laine M, Qiu JH, Resnick B, Barbar EJ, Hajjar DP, Upmacis RK. Heme catalyzes tyrosine 385 nitration and inactivation of prostaglandin H2 synthase-1 by peroxynitrite. J Lipid Res. 2006;47:898–911. doi: 10.1194/jlr.M500384-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.Eiserich JP, Butler J, van der Vliet A, Cross CE, Halliwell B. Nitric oxide rapidly scavenges tyrosine and tryptophan radicals. Biochem J. 1995;310(Pt 3):745–9. doi: 10.1042/bj3100745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pratt WB. The role of the hsp90-based chaperone system in signal transduction by nuclear receptors and receptors signaling via MAP kinase. Annu Rev Pharmacol Toxicol. 1997;37:297–326. doi: 10.1146/annurev.pharmtox.37.1.297. [DOI] [PubMed] [Google Scholar]
- 59.Brouet A, Sonveaux P, Dessy C, Balligand JL, Feron O. Hsp90 ensures the transition from the early Ca2+-dependent to the late phosphorylation-dependent activation of the endothelial nitric-oxide synthase in vascular endothelial growth factor-exposed endothelial cells. J Biol Chem. 2001;276:32663–9. doi: 10.1074/jbc.M101371200. [DOI] [PubMed] [Google Scholar]
- 60.Fontana J, Fulton D, Chen Y, Fairchild TA, McCabe TJ, Fujita N, Tsuruo T, Sessa WC. Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ Res. 2002;90:866–73. doi: 10.1161/01.res.0000016837.26733.be. [DOI] [PubMed] [Google Scholar]
- 61.Martinez-Ruiz A, Villanueva L, Gonzalez de Orduna C, Lopez-Ferrer D, Higueras MA, Tarin C, Rodriguez-Crespo I, Vazquez J, Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci U S A. 2005;102:8525–30. doi: 10.1073/pnas.0407294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bredt DS, Snyder SH. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci U S A. 1989;86:9030–3. doi: 10.1073/pnas.86.22.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 64.Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–7. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 65.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–67. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 66.Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–73. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- 67.Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–32. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 68.Choi YB, Tenneti L, Le DA, Ortiz J, Bai G, Chen HS, Lipton SA. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat Neurosci. 2000;3:15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- 69.Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH. CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron. 1998;20:115–24. doi: 10.1016/s0896-6273(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 70.Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155:123–31. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jaffrey SR, Fang M, Snyder SH. Nitrosopeptide mapping. A novel methodology reveals s-nitrosylation of dexras1 on a single cysteine residue. Chem Biol. 2002;9:1329–35. doi: 10.1016/s1074-5521(02)00293-4. [DOI] [PubMed] [Google Scholar]
- 72.Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–6. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 73.Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat Chem Biol. 2005;1:154–8. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- 74.Mitchell DA, Morton SU, Marletta MA. Design and characterization of an active site selective caspase-3 transnitrosating agent. ACS Chem Biol. 2006;1:659–65. doi: 10.1021/cb600393x. [DOI] [PubMed] [Google Scholar]
- 75.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–74. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 76.Berry MD, Boulton AA. Glyceraldehyde-3-phosphate dehydrogenase and apoptosis. J Neurosci Res. 2000;60:150–4. doi: 10.1002/(SICI)1097-4547(20000415)60:2<150::AID-JNR3>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 77.Hara MR, Thomas B, Cascio MB, Bae BI, Hester LD, Dawson VL, Dawson TM, Sawa A, Snyder SH. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci U S A. 2006;103:3887–9. doi: 10.1073/pnas.0511321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bae BI, Hara MR, Cascio MB, Wellington CL, Hayden MR, Ross CA, Ha HC, Li XJ, Snyder SH, Sawa A. Mutant huntingtin: nuclear translocation and cytotoxicity mediated by GAPDH. Proc Natl Acad Sci U S A. 2006;103:3405–9. doi: 10.1073/pnas.0511316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hara MR, Snyder SH. Nitric Oxide-GAPDH-Siah: A Novel Cell Death Cascade. Cell Mol Neurobiol. 2006;26:525–36. doi: 10.1007/s10571-006-9011-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein s-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–7. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 81.Matsumoto A, Comatas KE, Liu L, Stamler JS. Screening for nitric oxide-dependent protein-protein interactions. Science. 2003;301:657–61. doi: 10.1126/science.1079319. [DOI] [PubMed] [Google Scholar]
- 82.Hao G, Xie L, Gross SS. Argininosuccinate synthetase is reversibly inactivated by S-nitrosylation in vitro and in vivo. J Biol Chem. 2004;279:36192–200. doi: 10.1074/jbc.M404866200. [DOI] [PubMed] [Google Scholar]
- 83.Nedospasov A, Rafikov R, Beda N, Nudler E. An autocatalytic mechanism of protein nitrosylation. Proceedings of the national academy of sciences. 2000;97:13543–8. doi: 10.1073/pnas.250398197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J Biol Chem. 2005;280:19888–94. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]
- 85.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–62. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 86.Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–6. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 87.Perez-Mato I, Castro C, Ruiz FA, Corrales FJ, Mato JM. Methionine adenosyltransferase S-nitrosylation is regulated by the basic and acidic amino acids surrounding the target thiol. J Biol Chem. 1999;274:17075–9. doi: 10.1074/jbc.274.24.17075. [DOI] [PubMed] [Google Scholar]
- 88.Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- 89.Roychowdhury M, Sarkar N, Manna T, Bhattacharyya S, Sarkar T, Basusarkar P, Roy S, Bhattacharyya B. Sulfhydryls of tubulin. A probe to detect conformational changes of tubulin. Eur J Biochem. 2000;267:3469–76. doi: 10.1046/j.1432-1327.2000.01369.x. [DOI] [PubMed] [Google Scholar]
- 90.Kim YJ, Pannell LK, Sackett DL. Mass spectrometric measurement of differential reactivity of cysteine to localize protein-ligand binding sites. Application to tubulin-binding drugs. Anal Biochem. 2004;332:376–83. doi: 10.1016/j.ab.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 91.Reid KSC, Lindley PF, Thornton JM. Sulphur-aromatic interactions in proteins. FEBS Lett. 1985;190:209–13. [Google Scholar]
- 92.Hattori Y, Campbell EB, Gross SS. Argininosuccinate synthetase mRNA and activity are induced by immunostimulants in vascular smooth muscle. Role in the regeneration or arginine for nitric oxide synthesis. J Biol Chem. 1994;269:9405–8. [PubMed] [Google Scholar]
- 93.Xie L, Hattori Y, Tume N, Gross SS. The preferred source of arginine for high-output nitric oxide synthesis in blood vessels. Semin Perinatol. 2000;24:42–5. doi: 10.1016/s0146-0005(00)80054-3. [DOI] [PubMed] [Google Scholar]
- 94.Nedospasov A, Rafikov R, Beda N, Nudler E. An autocatalytic mechanism of protein nitrosylation. Proc Natl Acad Sci U S A. 2000;97:13543–8. doi: 10.1073/pnas.250398197. [DOI] [PMC free article] [PubMed] [Google Scholar]