

Abstract
We have analyzed the effectiveness of Hsp90 inhibitors in blocking the replication of negative-strand RNA viruses. In cells infected with the prototype negative strand virus vesicular stomatitis virus (VSV), inhibiting Hsp90 activity reduced viral replication in cells infected at both high and low multiplicities of infection. This inhibition was observed using two Hsp90 inhibitors geldanamycin and radicicol. Silencing of Hsp90 expression using siRNA also reduced viral replication. Hsp90 inhibition changed the half-life of newly synthesized L protein (the large subunit of the VSV polymerase) from >1 hour to less than 15 minutes without affecting the stability of other VSV proteins. Both the inhibition of viral replication and the destabilization of the viral L protein were seen when either geldanamycin or radicicol was added to cells infected with paramyxoviruses SV5, HPIV-2, HPIV-3, or SV41, or to cells infected with the La Crosse bunyavirus. Based these results we propose that Hsp90 is a host factor that is important for the replication of many negative strand viruses.
Keywords: Hsp90 inhibitor, chaperone, vesicular stomatitis virus, paramyxovirus, SV5, La Crosse virus, antiviral, polymerase
Introduction
There is significant scientific and public health interest in viruses that utilize negative strand RNA as genomes. Many of these viruses are important pathogens, including some of the most significant recurring infectious disease problems (influenza, respiratory syncytial virus) as well as some of the most alarming emerging infectious diseases (Lassa, Marburg, Hanta, Rift Valley Fever). All of these viruses encode an RNA dependent RNA polymerase (RDRP). The RDRP is required both for viral transcription and for replication of the viral genome (Whelan, Barr, and Wertz, 2004). In addition to RNA polymerization, the RDRPs are also involved in 5’ and 3’ end modification and in recognizing cis-acting sequences that govern transcription and replication. Functionally, RDRPs from different viruses appear to utilize diverse strategies to accomplish these tasks, but a repeating theme is that the RDRP requires either cooperating viral proteins (Gupta, Shaji, and Banerjee, 2003; Whelan, Barr, and Wertz, 2004) or host proteins (Das et al., 1998; Shen and Masters, 2001; Strauss and Strauss, 1999) to properly accomplish replication and/or transcription. Understanding these virus/virus and virus/host interactions that govern negative strand virus replication are important for developing ways to combat important infectious agents by blocking their ability to replicate.
Heat Shock Protein 90 (Hsp90) is a cellular chaperone that has been the focus of intense research for many years (for reviews see (Terasawa, Minami, and Minami, 2005; Young, Moarefi, and Hartl, 2001)). Knockout models have shown that Hsp90 is essential for viability in yeast and Drosophila (Lange et al., 2000; Panaretou et al., 1998; Rutherford and Lindquist, 1998). Hsp90 displays ATP-dependent folding capacity (Panaretou et al., 1998), and is notable among chaperone proteins because unlike its promiscuous cousin chaperone HSP70 (Agashe and Hartl, 2000), Hsp90 appears to have a specific set of client proteins in vivo (Pratt and Toft, 2003). These client proteins include steroid receptors, a number of transcription factors, and several protein kinases (Terasawa, Minami, and Minami, 2005). Hsp90 forms a complex with co-chaperones (i.e. AHA1, Hop/Sti1, FKBP51 and 52) which then act to fold client proteins. Several known oncogenes are client proteins of Hsp90 (Pratt and Toft, 2003), which has sparked interest in using Hsp90 inhibitors to block the folding of these oncogenic proteins in an effort to drive cancer cells into apoptosis (Whitesell and Lindquist, 2005).
Hsp90 involvement in viral replication has been reported for several different viruses; however, these reports have suggested different roles of Hsp90 in a variety of virus and host systems. During vaccinia virus infection, Hsp90 inhibition blocks viral replication (Hung, Chung, and Chang, 2002). The mechanism for this inhibition is not well established, but Hsp90’s localization is restricted to the cytoplasm following VV infection, where it interacts with the viral core protein 4a, presumably as a client protein (Hung, Chung, and Chang, 2002). Hsp90 activity has also been reported to be important for proper cleavage of newly synthesized hepatitis C NSP2/3 protein (Waxman et al., 2001), and is required for the activity of the hepatitis B reverse transcriptase (Hu and Seeger, 1996; Hu, Toft, and Seeger, 1997).
Recently, there have been several reports implicating a role for Hsp90 in the control of viral polymerase function. In a minireplicon system, Hsp90 binds to the PB2 subunit of the influenza virus RNA polymerase, and stimulates viral polymerase activity (Momose et al., 2002). In Herpes virus infection, blocking Hsp90 activity significantly inhibited viral replication (Burch and Weller, 2005; Li et al., 2004), possibly due to the improper localization of the viral polymerase in drug-treated cells (cytoplasmic rather than nuclear). Similarly, it has been shown that Hsp90 activity is important for the proper stability and localization of the RNA polymerase (protein A) of the insect virus flock house virus (Kampmueller and Miller, 2005).
There is still a great deal to be discovered about Hsp90’s role in virus replication, such as whether Hsp90 is likely to play a role in the life cycle of specific viruses or whether it is involved in the life cycle of entire classes of viruses. In an attempt to address this question, we have studied the effects of Hsp90 inhibitors on the replication of several different negative strand RNA viruses from the rhabdovirus, paramyxovirus, and bunyavirus families. We found that replication of viruses from each of these families was impaired by the ansamycin Hsp90 inhibitor geldanamycin, a second Hsp90 inhibitor radicicol, and by siRNA knockdown of Hsp90 protein levels. This inhibition of replication was most apparent at early stages of the virus life cycle and resulted in the selective destabilization of the L protein of the virus polymerase. Based on this and other data in the literature, we propose that viruses from multiple families and classes have all evolved to require the use of Hsp90 for the proper folding of their polymerases. This suggests that the inhibition of Hsp90 activity could act as broad-range antiviral agents.
Results
To determine whether Hsp90 inhibition has an effect on the replication of the prototype negative strand virus vesicular stomatitis virus (VSV) we determined viral growth in cells treated with increasing concentrations of geldanamycin, an ATP-competitor that acts as a specific inhibitor of Hsp90 activity (Roe et al., 1999). We utilized chemically synthesized geldanamycin (provided by the DTP service of NCI) in these experiments because a recent report has shown that geldanamycin purified from S. hygroscopicus contains contaminating activities that inhibit intracellular trafficking but that the NCI synthesized compound does not (Barzilay et al., 2004). BHK cells infected with VSV at an MOI of 0.01 were treated with increasing concentrations of drug, and virus yield at 12hpi was determined by plaque assay. As shown in Figure 1A, geldanamycin was a potent inhibitor of VSV growth, reducing virus replication by 3 orders of magnitude at 100nM and 5 orders of magnitude at 5μM.
Figure 1. Effect of Hsp90 inhibition of VSV replication.

A) viral titers at 12hpi from cells infected with VSV at an MOI=0.01 and treated with increasing concentrations of geldanamycin. B) growth curve of VSV in the presence (△) or absence (■) of geldanamycin (5μM) at high MOI (=10) and C) growth curve at low MOI (=0.1). D) growth curve of VSV in the presence (△) or absence (■) of radicicol (5μM). Virus titers for all panels represent the average of three different experiments. E) Western blot analysis of 25ug of lysate from cells that were mock-transfected, transfected with GAPDH targeting siRNA or transfected with Hsp90 targeting siRNA. Top image shows Hsp90 levels, middle panel shows GAPDH levels, bottom panel shows actin as a loading control. F) growth curve of mock-transfected and siRNA-treated cells infected with VSV at an MOI of 0.01. Error bars reflect standard deviation for three experiments.
To further understand the effect of geldanamycin on virus replication, we carried out single and multiple cycle growth experiments in the absence or presence of geldanamycin at 5μM, a concentration previously shown to have minimal cellular toxicity (Kampmueller and Miller, 2005). As shown in Figure 1B, geldanamycin had a significant effect on slowing virus growth in a single cycle growth experiment (MOI=10). Cells that were infected with VSV and treated with geldanamycin showed a noticeable delay in virus growth, producing lower virus titers between 4 and 16 hpi when compared to cells infected with VSV but not treated with geldanamycin. By 24hpi, virus titers were comparable between drug treated and untreated conditions.
Inhibition of VSV replication by geldanamycin was seen in HeLa cells as well as in BHK cells (data not shown), indicating that this effect was not cell-type specific. Inhibition of viral replication by geldanamycin was more dramatic in multi-cycle growth experiments. Geldanamycin addition delayed the appearance of progeny viruses for 8 hours when compared to infected cells that were not treated with geldanamycin, and inhibited growth by more than two orders of magnitude at 24hpi (Figure 1C). We also determined the effect of radicicol, an Hsp90 inhibitor that is structurally distinct from geldanamycin, on high MOI VSV infection. Radicicol also inhibited VSV replication, decreasing virus titers by more than 2 orders of magnitude at 4hpi and 4 orders of magnitude at 8hpi. This supports the hypothesis that the mechanism of replication inhibition is through the inhibition of Hsp90 function (Figure 1D).
To test this conclusion utilizing a different method, we determined the effect of decreasing the levels of Hsp90 through RNA silencing. Figure 1E shows a representative western blot of HeLa cells that were either mock transfected, transfected with siRNA targeting GAPDH, or transfected with siRNA targeting Hsp90. 48 hours posttransfection, cells transfected with GAPDH siRNA showed a >90% inhibition of GAPDH expression (lane 2). Cells transfected with siRNA targeting the alpha and beta forms of Hsp90 showed an 80-90% drop in Hsp90 levels at the same time posttransfection (lanes 3-6). Cell cultures transfected with either GAPDH or Hsp90 siRNA had fewer cells than contols at 72h posttransfection suggesting that cell proliferation was slowed (Supplemental Figure 1). However, there were no significant differences in cell viability or gross morphological changes in cells transfected with Hsp90 siRNA. To determine the effect of the lower levels of Hsp90 protein levels on VSV infection, cells were infected with VSV at a low MOI (0.01). Analysis of viral growth by plaque assay (figure 1F) showed that cells where Hsp90 was silenced showed a decrease in virus titer compared to a mock-transfected control at both 8 and 24hpi, but that there was a negligible effect of GAPDH silencing on virus titer. These results support the conclusion that Hsp90 is an important host factor in the replication of VSV.
Inhibition of virus replication is at early stages of viral replication
The time-courses shown in Figure 1 indicated that geldanamycin inhibits VSV at early times in the VSV life cycle, and that the inhibitory action diminished at later times postinfection. In a single-cycle growth experiment, drug added coincident with infection had a strong effect on viral replication, but drug added four hours postinfection had almost no effect (data not shown). This suggested that the antiviral activity of geldanamycin was due to an effect on an early, rate-limiting step of viral replication. To determine how the addition of geldanamycin affected viral gene expression, cells were infected with VSV and drug was added at 0, 2 or 4hpi (see figure 2A for time line). At 5hpi, cells were labeled with 35S methionine for 1 hour and lysates were analyzed by SDS-PAGE and phosphorimaging. In cells that were not treated with drug, high level VSV gene expression was evident through the appearance of four major bands representing the L, G, N/P (comigrating), and M proteins (Figure 2B, lane 1). Also apparent was the inhibition of host protein synthesis normally seen following a productive VSV infection. Addition of geldanamycin coincident with virus infection blocked viral gene expression by VSV, but host protein synthesis was still detectable (lane 2). Cells treated with drug 2hpi showed some decrease in viral gene expression, and cells treated with drug at 4hpi showed strong viral gene expression, demonstrating that geldanamycin inhibition of viral gene expression required addition soon after initiating the infection. As shown in Figure 2B, the addition of drug at 4hpi did result in one change from non-treated cells: the viral L protein, the catalytically active component of the RDRP accumulated to much lower levels in these drug-treated cells.
Figure 2. Effect of geldanamycin on viral protein synthesis.
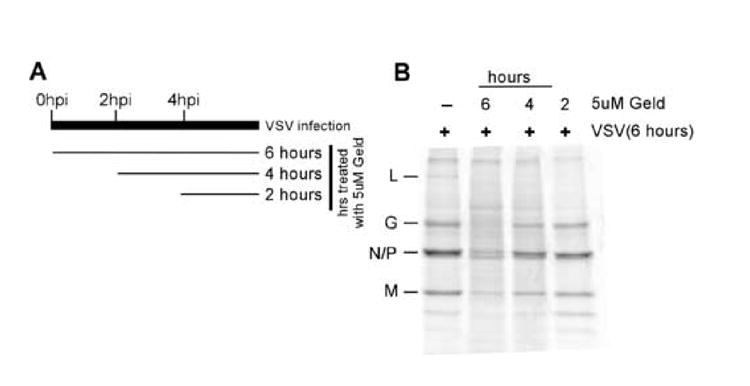
A) Schematic of experimental procedure showing times of geldanamycin treatment after viral infection (top) and total times of geldanamycin treatment (right). B) Phosphorescence image of cell lysates from cells that were infected with VSV and treated with geldanamycin as described, followed by labeling with 35S Methionine for 1 hour.
Inhibition of stability of newly synthesized polymerase
To investigate whether the lower levels of L protein were due to an increased incorporation of L into budded virions, we determined the extent to which newly synthesized L was incorporated into budding virions. At 4hpi, infected cells were treated with 50nM, 500nM or 5μM geldanamycin, proteins were labeled with 35S methionine for 1 hour and the fate of the newly synthesized proteins were determined through analysis of virions budded into the supernatant and labeled protein in the cell after a 1 hour chase with unlabeled methionine. While most proteins produced by VSV were unaffected by addition of geldanamycin, the level of L protein in cell lysates decreased with increasing geldanamycin concentration (Figure 3A). Drug addition at concentrations as low as 50nM decreased the amount of L in lysates, consistent with the disappearance of the L protein being linked to the effect of the drug on viral replication (Figure 1A). In budded virions purified from the supernatant of these same cells the concentration of geldanamycin did not have an effect on the amounts of radiolabeled G,N,P, and M protein were incorporated (Figure 3B), demonstrating that blocking Hsp90 does not affect any of the assembly function of the virus. In the case of the L protein, less radiolabeled L protein was incorporated into virions as geldanamycin concentration increased. This result shows that geldanamycin treatment did not result in the preferential movement of newly synthesized L protein into virions. Some newly synthesized L was found in virions from cells treated with lower concentrations of drug, indicating that some of the newly synthesized L protein remains in the cell long enough to be incorporated into virions.
Figure 3. Assembly of VSV in geldanamycin treated cells.

Cells were infected with VSV for 5 hours, and then proteins were radiolabeled and followed using a one hour pulse and one hour chase protocol. Upon addition of radioactive methionine, different concentrations of geldanamycin were added to individual samples. Following a 1 hour chase in the presence of drug A) intracellular radiolabeling of protein was determined by SDS-PAGE electrophoresis phosphorescence imaging. B) The amount of newly synthesized protein that was incorporated into the virus was determined by virion purification from the media followed by SDS-PAGE and phosphorescence analysis.
To determine how the synthesis and stability of L was changed following Hsp90 inhibition, we performed pulse-chase analysis in the presence and absence of geldanamycin. Radiolabeled cells were chased for 10, 20, or 30 minutes, and then lysates from these cells were analyzed by phosphorimaging (Figure 4A). The resulting image showed that in both geldanamycin-treated and untreated cells infected with virus, significant amounts of 35S methionine were incorporated into the viral L protein. The newly synthesized L protein was stable in untreated cells but disappeared rapidly in cells treated with the drug and was almost completely absent after a 30 minute chase. Quantitation of these data (figure 4B) over multiple experiments showed that the half-life of the L protein was greater than 60 minutes in the absence of drug but was reduced to less than 20 minutes in the presence of drug. This was not seen for the other viral proteins as analysis of the decay of viral M protein (Figure 4C) and the other viral proteins (Figure 4A) showed little evidence of degradation in the presence and absence of drug treatment.
Figure 4. Effect of geldanamycin on viral protein stability.
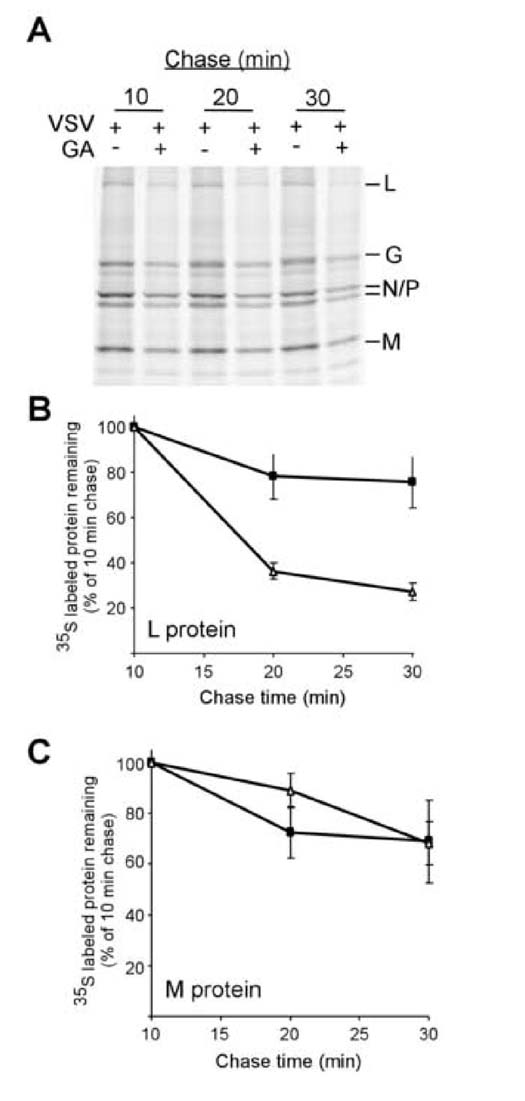
A) BHK cells infected with VSV for 5 hours were starved for methionine, and then protein synthesis and stability were determined by pulse chase analysis (see materials and methods). Results were determined by SDS-PAGE and phosphorescence imaging. Viral proteins are identified to the right of the gel. Levels of protein remaining are quantities for viral L (B) and M (C) proteins presence (△) or absence (■) of 5μM geldanamycin. Values represent the average of three separate experiments +/- standard deviation
L protein is degraded by the proteasome
To investigate a possible mechanism for the observed destabilization of L protein following HSP90 inhibition, we determined whether the L protein was being degraded via the proteasome. This possibility is particularly plausible because other Hsp90 substrates, such as the estrogen receptor, have been shown to be shunted to the proteasome pathway following Hsp90 inhibition (Bagatell et al., 2001; Fan, Park, and Nephew, 2005). To test whether the proteasome is involved in the degradation of newly synthesized L protein, we repeated the experiments shown in Figure 4, but added proteasome inhibitor (proteasome inhibitor II, Calbiochem) to infected cells treated with geldanamycin. At 10 minutes of chase, there were similar levels of incorporation of 35S methionine into L protein in untreated, geldanamycin treated, and geldanamycin + proteasome inhibitor treated conditions (Figure 5A). At 30 minutes chase there was a significant decrease in the amount of L protein in the geldanamycin treated cells similar to that seen in previous experiments. There was a much smaller decrease in the cells treated with both geldanamycin and proteasome inhibitor showing that proteasome inhibition decreased the degradation of the L protein. Addition of proteosome inhibitor alone did not significantly alter the stability of the L protein (Figure 5B). The quantitation of multiple experiments (Figure 5C) showed that the addition of the proteasome inhibitor increased L protein half-life in the presence of geldanamycin from less than 20 minutes to close to that of L in control cells that did not recieve geldanamycin (dotted line, Figure 5B). This argues that the primary cause of the disappearance of the newly synthesized L polymerase following Hsp90 inhibition is the targeting of the newly synthesized polymerase to the proteasome.
Figure 5. Geldanamycin destabilization of VSV L protein requires proteosome function.
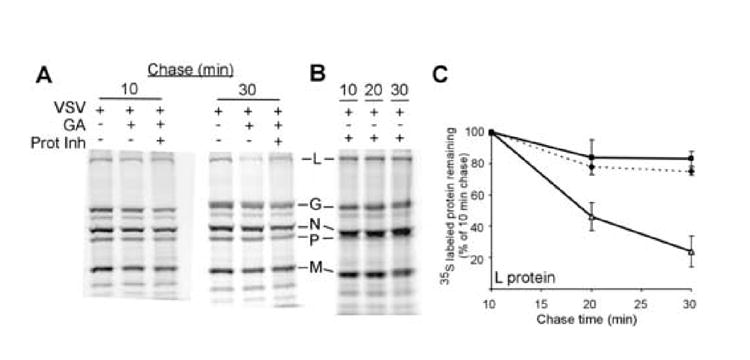
A) BHK cells were infected with VSV, and 5hpi left untreated, treated with geldanamycin, or treated with both geldanamycin and proteosome inhibitor. Pulse-chase results for 10 minute and 30 minute chases are shown. B) Stability of L protein in the proteins presence (△) or absence (■) of 5μM geldanamycin and in the presence of geldanamycin and a proteosome inhibitor (◆, dotted line). Levels are expressed as a percentage of the signal that was present at the 10 minute chase. Values are the average of three separate experiments +/- standard deviation.
Hsp90 inhibitors regulate the replication and stability of multiple negative strand viruses
Based on our findings that Hsp90 inhibitors decreased the replication and destabilized the L polymerase protein of the prototype negative strand virus VSV, we were interested in whether this behavior was unique to VSV or whether it was a more general phenomenon. To test the hypothesis that Hsp90 activity is required for the replication and polymerase stability across negative strand virus families, we initially tested the paramyxoviruses simian virus 5 (SV5) and HPIV-2. To determine the effect of Hsp90 inhibition on viral replication, geldanamycin was added to cells that were infected at a low multiplicity with a GFP-expressing SV5 (rSV5-GFP) or HPIV-2. As shown in Figure 6A, geldanamycin inhibited growth of these viruses, reducing virus yield by more than 3 logs for SV5 and more than 2 logs for HPIV2.
Figure 6. Effect of Geldanamycin on Paramyxovirus growth and L stability.
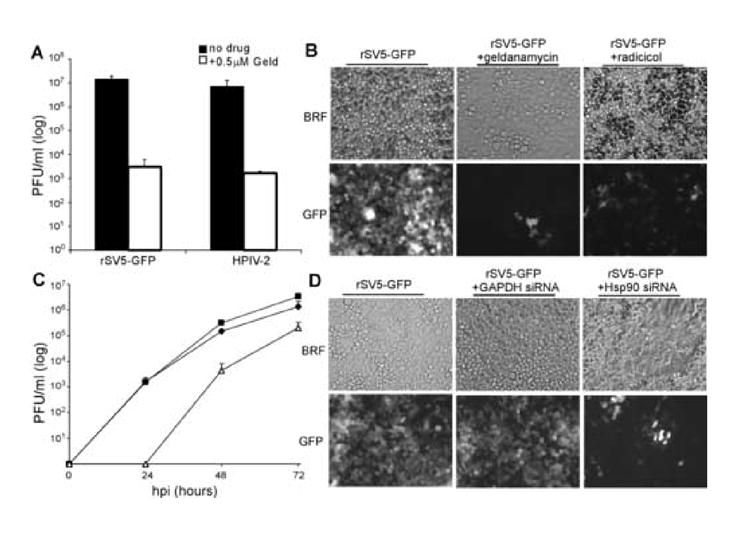
A and B) Cells were infected at an MOI of 0.05 with rSV5-GFP or HPIV-2 and incubated with or without 0.5 μM Geldanamycin. At 72 hpi, virus titers were determined by plaque assay (A) or cells were examined for GFP expression (B). Results are representative of three independent experiments. C and D) Cells that were mock-transfected or transfected with either GAPDH targeting siRNA or Hsp90 targeting siRNA were infected with SV5-GFP at an MOI of 0.05. C) shows SV5-GFP growth curve (done in triplicate) untransfected (■) GAPDH siRNA transfected (◆), and Hsp90 siRNA transfected (■), D) shows representative images showing cell spread of the virus in different transfection conditions.
Geldanamycin and radicicol blocked paramyxovirus spread in a low MOI infection. Cells infected at an MOI of 0.05 with were all infected with rSV5-GFP and expressing GFP by 72hpi (Figure 6B left panels). Consistent with the effect of HSP90 inhibitors on virus production, cells infected with rSV5-GFP but treated with 0.5μM geldanamycin showed dramatically lowered viral spread, with only a few cells showing GFP expression (middle panels). A similar limitation of spread was seen in cells treated with radicicol, with few cells showing the intense GFP signal seen in mock-infected cells (right panels).
Using siRNA to decrease the level of Hsp90 protein in cells also had a significant effect on paramyxovirus replication. Shown in Figure 6C are SV5-GFP virus titers at 24, 48 and 72hpi from HeLa cells that were either mock transfected, transfected with siRNA targeting GAPDH, or transfected with siRNA targeting Hsp90. Silencing GAPDH minimally decreased the replication of SV5-GFP, but silencing Hsp90 reduced SV5-GFP replication significantly. Progeny viruses were not detected at 24hpi in Hsp90-silenced cells infected with SV5-GFP, a 3 log order difference compared to control and GAPDH-silenced cells. Virus replication was observed at both 48 and 72hpi, but at lower levels than was seen in controls. Silencing of Hsp90 expression but not silencing of GAPDH expression inhibited the spread of SV5 GFP (Figure 6D) in these cells.
This inhibition of paramyxovirus growth was accompanied by changes in the stability of the viral polymerase. Figure 7A shows the results of pulse-chase (0.5, 1 and 2 hour chase, followed by immunoprecipitation of indicated protein) experiments that were used to follow the stability of individual SV5 viral proteins. In cells infected with rSV5-GFP but not treated with geldanamycin L, N and P proteins showed very stable profiles, with the polymerase protein L showing a half-life in excess of 1 hour. In geldanamycin-treated cells infected with SV5-GFP, both the N and P proteins remained stable, but the half-life of the L protein dropped to less than half an hour following treatment with geldanamycin, similar to the polymerase destabilization observed in the case of the VSV polymerase.
Figure 7. Hsp90 inhibition destabilizes newly synthesized L proteins from multiple paramyxoviruses.
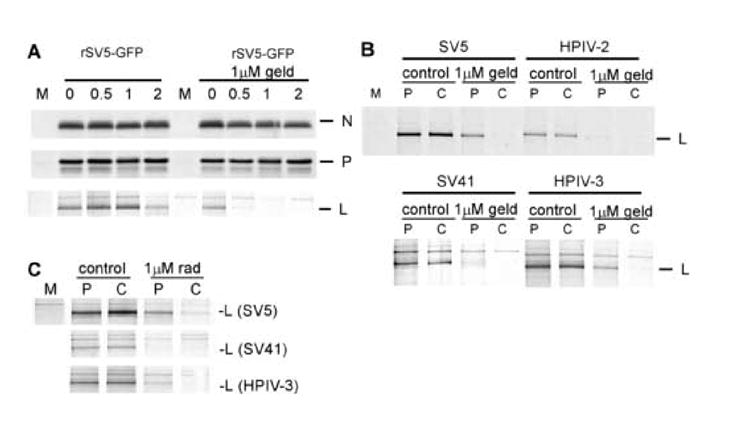
A) Cells were mock infected (M lane) or infected at an MOI of 10with rSV5-GFP. At 13 h pi, cells were treated for 1 hr with or without 1μM Geldanamycin. Cells were radiolabeled for 20 min with 35S-amino acids and incubated in nonradioactive media for the indicated times before lysis and analysis by immunoprecipitation with antibodies specific for NP, P or L. Geldanamycin was included at 1 μM during radiolabeling and chase periods. B) Stability of L proteins. Cells were mock infected or infected at an MOI of 10 with SV5, HPIV-2, SV41 or HPIV-3. 13hpi, cells were treated with 1μM Geldanamycin, radiolabeled for 20 minutes and lysed (Pulse) or chased using unlabeled methionine for 1 hour (Chase). Following cell lysis, samples were immunoprecipitated using L-specific antibodies. C) Experiments similar to B were carried out, but 1μM radicicol was used instead of geldanamycin
In addition to showing antiviral activity against SV5 and HPIV-2, both geldanamycin and radicicol showed antiviral activity against SV41 and HPIV3 (data not shown). Analysis of the viral polymerase of each of these viruses (Figure 7B) directly following pulse labeling (P) for 20 minutes or after a one hour chase (C) showed that 1) all of these polymerase proteins were stable in the absence of geldanamycin, and 2) all displayed a marked decrease in the half-life of the L polymerase protein following geldanamycin addition. 3) The amount of labeled L protein following the 20 minute pulse was reduced in the presence of geldanamycin, suggesting that turnover was occurring during the pulse period. The stability of SV41 L protein appeared to be most significantly decreased in the presence of geldanamycin, suggesting a very rapid turnover of the L protein in this virus following HSP90 treatment. These effects of destabilization of these viral L proteins were attributable to Hsp90 inhibition, as treatment of cells infected with SV5, SV41 or HPIV-3 with radicicol also caused the rapid degradation of newly synthesized viral polymerase (Figure 7C).
HSP90 inhibitors block the replication of La Crosse virus
We also tested the impact of Hsp90 inhibition on the replication of Bunyaviruses, a family of negative-strand viruses containing segmented RNA genomes. In the case of the pediatric encephalitis virus, La Crosse virus (H78 strain (Chandler et al., 1998), a kind gift from B.J. Beaty), virus growth was also inhibited by geldanamycin (Figure 8A) and radicicol (data not shown) at concentrations as low as 100nM. In cells not treated with drug, La Crosse virus infection resulted in the development of noticeable cytopathic effects (Figure 8B, middle panel). Infection with La Crosse in the presence of geldanamycin prevented the development of cytopathic effect (Figure 8B), consistent with the observed inhibition of virus replication. Similar to VSV and paramyxovirus family members, HSP90 inhibition appeared to destabilize the viral polymerase. Pulse-chase analysis of La Crosse virus proteins in infected cells (30 minute pulse followed by a 1 hour chase)) showed that La Crosse L protein is normally stable, but there was a distinct decrease in L protein stability following the addition of geldanamycin (Figure 8C top panel), while other La Crosse virus proteins such as the glycoprotein G1 remained stable (bottom panel).
Figure 8. Geldanamycin inhibition of La Crosse virus replication.
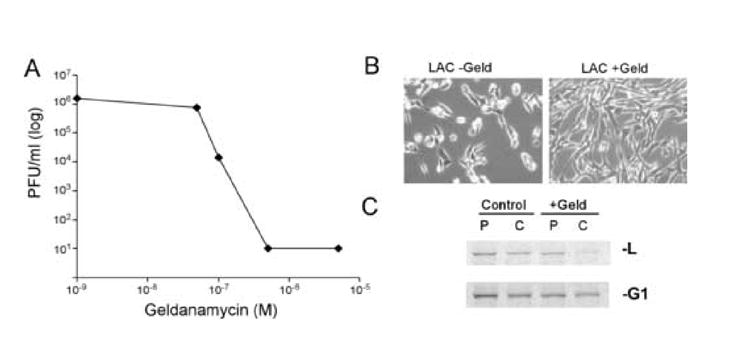
A) La Crosse virus titers at 24hpi from cells infected at an MOI=0.01 and increasing concentrations of geldanamycin at time of infection. B) Light microscope images of cells infected with La Crosse virus in the absence (left) and presence of 0.5uM geldanamycin. C) Protein synthesis and stability of La Crosse L and G1 proteins. 16hpi with Lacrosse virus at an MOI of 1, cells were labeled with 35S-methionine for 20 min and then lysed (P) or incubated in non-radioactive media for 1 hour. Cells were then lysed, and incorporation of radioactivity into viral proteins was determined by SDS-PAGE followed by phosphorescence imaging.
Discussion
The experiments described here demonstrate through both gene-knockdown and cell-permeable inhibitor approaches that Hsp90 activity is important for the rapid growth of negative strand RNA viruses. Mechanistically, geldanamycin and other Hsp90 inhibitors appear to block virus replication by dramatically decreasing the stability of the L subunit of the viral RNA dependent RNA polymerase. This mode of action would explain the effectiveness of Hsp90 inhibition at early stages of viral infection where the level of RNA synthesis and genome replication would be most affected by decreased levels of the viral polymerase. Adding geldanamycin at progressively later times postinfection has progressively less effect on viral gene expression, indicating that accumulation of L protein prior to drug addition can compensate for the rapid turnover of newly synthesized L protein. Our studies show that viruses from the rhabdovirus, paramyxovirus, and bunyavirus families all require Hsp90 activity to achieve normal levels of a stable viral polymerase. Other researchers have shown that the positive-strand RNA virus FHV polymerase (Kampmueller and Miller, 2005) and the influenza virus polymerase (Momose et al., 2002) require Hsp90 activity for proper function. Together, our data and that from other laboratories (Kampmueller and Miller, 2005; Momose et al., 2002) suggest that Hsp90 is a host factor that is of central importance to viral replication for many, if not all, RNA viruses. The RNA polymerases from these different viruses do not share a high degree of sequence homology, so their dependence on Hsp90 points to convergent evolution. To our knowledge we are the first to propose that Hsp90 activity is a conserved requirement for achieving a stable virus polymerase for a wide spectrum of RNA viruses.
For the VSV L protein, its large size (241 kDa) and the existence of several different catalytic activities within the protein (Feller et al., 2000; Grdzelishvili et al., 2006; Li, Fontaine-Rodriguez, and Whelan, 2005; Li, Wang, and Whelan, 2006) implies a number of independently folded domains. This is also likely true of the other viral polymerases affected by Hsp90 inhibition, as the viruses used in this study all have relatively large polymerase proteins (~240kDa) that are likely to have independent functional domains. How Hsp90 is involved in folding these polymerases has yet to be determined, but because Hsp90’s protein folding activity requires the binding and action of additional co-chaperones the folding of viral polymerases is likely to be a coordinated effort of several cellular proteins, of which Hsp90 is the first to be identified.
Despite the attractiveness of hypothesizing that Hsp90 is directly involved in folding viral polymerases, other possibilities exist for how Hsp90 inhibition might destabilize RNA virus polymerases. For viruses of the rhabdovirus and paramyxovirus families, the L polymerase requires the binding of the viral phospho (P) protein for activity (Horikami et al., 1992). Hsp90 may facilitate the L-P interaction to promote the stability of L. Hsp90 could also act to promote the stable formation of L protein multimers (Smallwood, Cevik, and Moyer, 2002). Thus, Hsp90 inhibition could lead to a disruption of L-L interactions resulting in an unstable polymerase that is quickly degraded. Additionally, Hsp90 may be more remotely involved and impact L stability by folding or activating a cellular component that is required for polymerase stability. Further experiments will be required to determine the interaction between Hsp90 and RNA virus polymerases, as well as other cellular factors
The importance of Hsp90 for the replication of multiple viruses opens an interesting possibility for developing new antiviral therapies. There are few compounds that display antiviral activity against major encephalitis-causing and hemorrhagic fever causing negative strand RNA viruses (De Clercq, 2005; Domachowske and Rosenberg, 2005). Our findings suggest that the inhibition of Hsp90 activity should be investigated as a mechanism of limiting the replication and spread of these viruses in vivo. An advantage of this approach is that the lack of Hsp90 activity appears to hamper viral RDRP function. The RDRP represents a rate limiting enzyme step in virus replication that performs many unique and essential replication functions and is synthesized at relatively low levels in infected cells. Additionally, because of the effectiveness of this compound against a wide variety of viruses, Hsp90 inhibitors may be broad spectrum antivirals capable of inhibiting the replication of several different infectious agents.
Already, significant effort has been devoted to developing Hsp90 inhibitors. One of these compounds, 17-AAG (a geldanamycin derivative) has completed phase 1 and is currently in phase 2 clinical trials as an anticancer therapy (Goetz et al., 2005; Whitesell and Lindquist, 2005). Results from phase 1 trials showed that tolerated doses of the drug led to decreases in the levels of Hsp90 client proteins (Banerji et al., 2005; Grem et al., 2005). Thus, this compound has already passed many of the hurdles that confront successful drug development, and it or other Hsp90 inhibitors in development (Whitesell and Lindquist, 2005) are likely to be clinically viable. Regardless of their utility in cancer treatment, we propose that further experimentation should be undertaken to determine wither these compounds have a useful role as antiviral agents. Hsp90 inhibitors may also work synergistically with other antivirals such as ribavirin, a drug that has also been suggested to target the function of the viral polymerase (Graci and Cameron, 2002).
An important corollary to these findings is that these studies highlight the utility of prototype negative strand viruses as tools that can be used to screen for new antiviral compounds. Utilization of viruses that have shown minimal human pathogenicity and have been well characterized offers significant advantages over newly discovered or highly pathogenic viruses for lead compound identification and can serve as an important stepping stone to the discovery of new antiviral compounds that have a wide spectrum of antiviral activities.
Materials and methods
Reagents
Geldanamycin was synthesized at the drug therapeutics program of the NIH. Radicicol and proteosome inhibitor II were obtained from Calbiochem. 35S Methionine was obtained from Amersham. siRNA targeting Hsp90 and polyclonal anti-Hsp90 antibody were obtained from Santa Cruz. siRNA transfection reagent was obtained from Mirus biotechnology. siRNA targeting GAPDH was obtained from Dharmacon, antibody recognizing GAPDH was obtained from chemicon. All other reagents were obtained from Fischer Co. La Crosse virus (H78) was a kind gift of Barry Beaty (Colorado State University)
Virus infection
Cells were plated to reach a density of between 70 and 80% in DMEM+10%FBS. Cells were infected with VSV at an MOI=10 or an MOI=0.01 as described previously (Connor and Lyles, 2002). For dose-response experiments with VSV and La Crosse virus, BHK cells were infected at an MOI of 0.01 and virus budded into culture media was determined by plaque assay using BHK cells. Drugs were added coincident with infection unless otherwise indicated.
siRNA knockdown experiments
For siRNA targeting the human alpha and beta forms of Hsp90, HeLa cells were incubated with 50nM siRNA using Mirus siPORT transfection reagent for 48 hours prior to infection with VSV. For GAPDH siRNA, 75nM siRNA was utilized using the same transfection reagent. For both Hsp90 and GAPDH siRNA tranfections there were no obvious cytotoxicity at 48 hours posttransfection.
Pulse-chase experiments
For VSV, cells were infected with VSV for 5 hours. Cells were labeled with 35S methionine–containing media for 1 hour and then the radioactive media was replaced with media containing cold 35S methionine for an hour or for the times indicated in text describing the figure. For La Crosse virus, a similar procedure was followed, but cells were labeled 16hpi for 30 minutes and chased for 60 minutes. Cells were lysed in RIPA buffer and proteins were separated by SDS-PAGE. For virion purification, media from infected cells was layered on top of a 15% sucrose solution and spun for some 40,000XG for 45 minutes. Pelleted virions were resuspended and proteins were separated by SDS-PAGE.
Paramyxovirus experiments
Infections of HeLa cells at high MOI (10) or low MOI (0.05) with SV5 expressing GFP (rSV5-GFP), HPIV-2, HPIV-3 or SV41 and virus growth analyses were carried out as described previously (Parks, Ward, and Rassa, 2001). Geldanamycin or radicicol was added after attachment of virus to cells, and was maintained throughout growth experiments. GFP expressing cells were analyzed with the Nikon Eclipse microscope and a 20X objective. Images were captured using a QImaging digital camera and processed using QCapture software. Exposure times were manually set to be constant between samples. For radiolabeling experiments, cells were treated with Geldanamycin for 1 hr prior to radiolabeling with Tran35S-label (100 uCi/ml), lysed in SDS and used for immunoprecipitation analysis with polyclonal antibodies specific for the SV5 NP, P, or L proteins as described previously (Parks, Ward, and Rassa, 2001).
Supplementary Material
Acknowledgments
We thank Elizabeth Pettit Kneller and Rebecca F. Connor for helpful comments on this manuscript. This work was supported by grants AI064606 to JHC, AI015892 and AI052304 to DSL, AI42023 to GDP. La Crosse virus (H78) was a kind gift from Barry Beaty (CSU).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Agashe VR, Hartl FU. Roles of molecular chaperones in cytoplasmic protein folding. Semin Cell Dev Biol. 2000;11(1):15–25. doi: 10.1006/scdb.1999.0347. [DOI] [PubMed] [Google Scholar]
- 2.Bagatell R, Khan O, Paine-Murrieta G, Taylor CW, Akinaga S, Whitesell L. Destabilization of steroid receptors by heat shock protein 90-binding drugs: a ligand-independent approach to hormonal therapy of breast cancer. Clin Cancer Res. 2001;7(7):2076–84. [PubMed] [Google Scholar]
- 3.Banerji U, O’Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, Simmons L, Maloney A, Raynaud F, Campbell M, Walton M, Lakhani S, Kaye S, Workman P, Judson I. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol. 2005;23(18):4152–61. doi: 10.1200/JCO.2005.00.612. [DOI] [PubMed] [Google Scholar]
- 4.Barzilay E, Ben-Califa N, Supino-Rosin L, Kashman Y, Hirschberg K, Elazar Z, Neumann D. Geldanamycin-associated inhibition of intracellular trafficking is attributed to a co-purified activity. J Biol Chem. 2004;279(8):6847–52. doi: 10.1074/jbc.M312799200. [DOI] [PubMed] [Google Scholar]
- 5.Burch AD, Weller SK. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J Virol. 2005;79(16):10740–9. doi: 10.1128/JVI.79.16.10740-10749.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandler LJ, Borucki MK, Dobie DK, Wasieloski LP, Thompson WH, Gundersen CB, Case K, Beaty BJ. Characterization of La Crosse virus RNA in autopsied central nervous system tissues. J Clin Microbiol. 1998;36(11):3332–6. doi: 10.1128/jcm.36.11.3332-3336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connor JH, Lyles DS. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J Virol. 2002;76(20):10177–87. doi: 10.1128/JVI.76.20.10177-10187.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das T, Mathur M, Gupta AK, Janssen GM, Banerjee AK. RNA polymerase of vesicular stomatitis virus specifically associates with translation elongation factor-1 alphabetagamma for its activity. Proc Natl Acad Sci U S A. 1998;95(4):1449–54. doi: 10.1073/pnas.95.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Clercq E. Recent highlights in the development of new antiviral drugs. Curr Opin Microbiol. 2005;8(5):552–60. doi: 10.1016/j.mib.2005.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Domachowske JB, Rosenberg HF. Advances in the treatment and prevention of severe viral bronchiolitis. Pediatr Ann. 2005;34(1):35–41. doi: 10.3928/0090-4481-20050101-10. [DOI] [PubMed] [Google Scholar]
- 11.Fan M, Park A, Nephew KP. CHIP (carboxyl terminus of Hsc70-interacting protein) promotes basal and geldanamycin-induced degradation of estrogen receptor-alpha. Mol Endocrinol. 2005;19(12):2901–14. doi: 10.1210/me.2005-0111. [DOI] [PubMed] [Google Scholar]
- 12.Feller JA, Smallwood S, Horikami SM, Moyer SA. Mutations in conserved domains IV and VI of the large (L) subunit of the sendai virus RNA polymerase give a spectrum of defective RNA synthesis phenotypes. Virology. 2000;269(2):426–39. doi: 10.1006/viro.2000.0234. [DOI] [PubMed] [Google Scholar]
- 13.Goetz MP, Toft D, Reid J, Ames M, Stensgard B, Safgren S, Adjei AA, Sloan J, Atherton P, Vasile V, Salazaar S, Adjei A, Croghan G, Erlichman C. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol. 2005;23(6):1078–87. doi: 10.1200/JCO.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 14.Graci JD, Cameron CE. Quasispecies, error catastrophe, and the antiviral activity of ribavirin. Virology. 2002;298(2):175–80. doi: 10.1006/viro.2002.1487. [DOI] [PubMed] [Google Scholar]
- 15.Grdzelishvili VZ, Smallwood S, Tower D, Hall RL, Hunt DM, Moyer SA. Identification of a new region in the vesicular stomatitis virus L polymerase protein which is essential for mRNA cap methylation. Virology. 2006;350(2):394–405. doi: 10.1016/j.virol.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 16.Grem JL, Morrison G, Guo XD, Agnew E, Takimoto CH, Thomas R, Szabo E, Grochow L, Grollman F, Hamilton JM, Neckers L, Wilson RH. Phase I and pharmacologic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with solid tumors. J Clin Oncol. 2005;23(9):1885–93. doi: 10.1200/JCO.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 17.Gupta AK, Shaji D, Banerjee AK. Identification of a novel tripartite complex involved in replication of vesicular stomatitis virus genome RNA. J Virol. 2003;77(1):732–8. doi: 10.1128/JVI.77.1.732-738.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horikami SM, Curran J, Kolakofsky D, Moyer SA. Complexes of Sendai virus NP-P and P-L proteins are required for defective interfering particle genome replication in vitro. J Virol. 1992;66(8):4901–8. doi: 10.1128/jvi.66.8.4901-4908.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu J, Seeger C. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc Natl Acad Sci U S A. 1996;93(3):1060–4. doi: 10.1073/pnas.93.3.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu J, Toft DO, Seeger C. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. Embo J. 1997;16(1):59–68. doi: 10.1093/emboj/16.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hung JJ, Chung CS, Chang W. Molecular chaperone Hsp90 is important for vaccinia virus growth in cells. J Virol. 2002;76(3):1379–90. doi: 10.1128/JVI.76.3.1379-1390.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kampmueller KM, Miller DJ. The cellular chaperone heat shock protein 90 facilitates Flock House virus RNA replication in Drosophila cells. J Virol. 2005;79(11):6827–37. doi: 10.1128/JVI.79.11.6827-6837.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lange BM, Bachi A, Wilm M, Gonzalez C. Hsp90 is a core centrosomal component and is required at different stages of the centrosome cycle in Drosophila and vertebrates. Embo J. 2000;19(6):1252–62. doi: 10.1093/emboj/19.6.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Fontaine-Rodriguez EC, Whelan SP. Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity. J Virol. 2005;79(21):13373–84. doi: 10.1128/JVI.79.21.13373-13384.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Wang JT, Whelan SP. A unique strategy for mRNA cap methylation used by vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0509821103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li YH, Tao PZ, Liu YZ, Jiang JD. Geldanamycin, a ligand of heat shock protein 90, inhibits the replication of herpes simplex virus type 1 in vitro. Antimicrob Agents Chemother. 2004;48(3):867–72. doi: 10.1128/AAC.48.3.867-872.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Momose F, Naito T, Yano K, Sugimoto S, Morikawa Y, Nagata K. Identification of Hsp90 as a stimulatory host factor involved in influenza virus RNA synthesis. J Biol Chem. 2002;277(47):45306–14. doi: 10.1074/jbc.M206822200. [DOI] [PubMed] [Google Scholar]
- 28.Panaretou B, Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. Embo J. 1998;17(16):4829–36. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parks GD, Ward KR, Rassa JC. Increased readthrough transcription across the simian virus 5 M-F gene junction leads to growth defects and a global inhibition of viral mRNA synthesis. J Virol. 2001;75(5):2213–23. doi: 10.1128/JVI.75.5.2213-2223.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228(2):111–33. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 31.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42(2):260–6. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 32.Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396(6709):336–42. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- 33.Shen X, Masters PS. Evaluation of the role of heterogeneous nuclear ribonucleoprotein A1 as a host factor in murine coronavirus discontinuous transcription and genome replication. Proc Natl Acad Sci U S A. 2001;98(5):2717–22. doi: 10.1073/pnas.031424298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smallwood S, Cevik B, Moyer SA. Intragenic complementation and oligomerization of the L subunit of the sendai virus RNA polymerase. Virology. 2002;304(2):235–45. doi: 10.1006/viro.2002.1720. [DOI] [PubMed] [Google Scholar]
- 35.Strauss JH, Strauss EG. Viral RNA replication. With a little help from the host. Science. 1999;283(5403):802–4. doi: 10.1126/science.283.5403.802. [DOI] [PubMed] [Google Scholar]
- 36.Terasawa K, Minami M, Minami Y. Constantly updated knowledge of Hsp90. J Biochem (Tokyo) 2005;137(4):443–7. doi: 10.1093/jb/mvi056. [DOI] [PubMed] [Google Scholar]
- 37.Waxman L, Whitney M, Pollok BA, Kuo LC, Darke PL. Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci U S A. 2001;98(24):13931–5. doi: 10.1073/pnas.241510898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whelan SP, Barr JN, Wertz GW. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol. 2004;283:61–119. doi: 10.1007/978-3-662-06099-5_3. [DOI] [PubMed] [Google Scholar]
- 39.Whitesell L, Lindquist SL. Hsp90 and the Chaperoning of Cancer. Nat Rev Cancer. 2005;5(10):761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 40.Young JC, Moarefi I, Hartl FU. Hsp90: a specialized but essential protein-folding tool. J Cell Biol. 2001;154(2):267–73. doi: 10.1083/jcb.200104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.