

Abstract
This review discusses proteomic methods to detect and identify S-nitrosated proteins. Protein S-nitrosation, the post-translational modification of thiol residues to form S-nitrosothiols, has been suggested to be a mechanism of cellular redox signaling by which nitric oxide can alter cellular function through modification of protein thiol residues. It has become apparent that methods that will detect and identify low levels of S-nitrosated protein in complex protein mixtures are required in order to fully appreciate the range, extent and selectivity of this modification in both physiological and pathological conditions. While many advances have been made in the detection of either total cellular S-nitrosation or individual S-nitrosothiols, proteomic methods for the detection of S-nitrosation are in relative infancy. This review will discuss the major methods that have been used for the proteomic analysis of protein S-nitrosation and discuss the pros and cons of this methodology.
Keywords: Reviews, S-Nitrosothiols, Proteomics, Nitrosation, Nitric oxide, Thiols
1. Introduction
Of all the potential post-translational thiol modifications that have been suggested to be involved in the transmission of intracellular signals, S-nitrosation is perhaps the most highly cited [1]. S-Nitrosation involves the modification of a thiol (RSH) to an S-nitrosothiol (RSNO). This can occur on low molecular weight or protein thiols to form a low molecular weight RSNO (lmwtRSNO) or a protein RSNO (pRSNO), respectively. This is thought to occur as a result of biological nitric oxide (NO) formation and has been thought of as a mechanism by which NO can transmit signals both within and between cells and tissues. While a great deal of information regarding S-nitrosation as a signaling paradigm has been obtained, there are still many unanswered questions. The idea that NO itself can trigger an intracellular signal through its interaction with thiols is at first a rather unlikely one. Metal centers (e.g. ferrous hemes) and free radicals (e.g. superoxide) are kinetically preferable targets, and NO does not react with thiols at any biologically meaningful rate [2, 3]. The direct reaction between NO and thiols is a redox reaction generating nitrous oxide, sulfenic acid and/or disulfide, but not RSNO [4, 5]. However, in the presence of oxygen and thiols, NO generates colored RSNO. These compounds were shown to have some synthetic chemical use, for instance as intermediates in the formation of mixed disulfides [6], and had antibacterial properties [7]. It was not until S-nitroso-N-acetyl-penicillamine (SNAP) was shown to have vasodilatory properties [8] that the biological potential of RSNO in mammalian systems became apparent. With the discovery of NO as a physiological mediator of vascular responses [9, 10], as well as many other processes, the role of endogenously produced RSNO, first measured by Stamler et al. [11], as mediators of a sub-set of NO-dependent responses became, and remains, an area of great interest.
While in vivo mechanisms of S-nitrosation remain the subject of debate, and are clearly more complex than a simple association of NO with a thiol, RSNO can be detected in vivo under basal and pathological conditions [11–14]. Much investigation has focused on examining the role of S-nitrosation in modifying specific cellular pathways. For example, the ability of NO to affect apoptosis has been linked to modification of a catalytically important thiol in caspase-3 [15, 16]. Relatively few studies have examined S-nitrosation from a more global perspective by placing individual effects of nitrosative stress in the context of the proteome of S-nitrosated (or otherwise modified) thiols. This article will assess current methods for the detection of global protein S-nitrosation using proteomic techniques.
2. The chemistry of S-nitrosothiols
As the mechanism of thiol nitrosation is important in attempting to understand the S-nitrosated proteome, in this section we will briefly describe mechanisms of S-nitrosothiol formation. There are four major mechanisms of S-nitrosation that have the potential to occur in biological systems. (i) S-Nitrosothiols can be formed from the reaction of nitrous acid (HONO) with thiols (Eqs. (1) and (2)). This is major mechanism
| (1) |
| (2) |
of RSNO synthesis in the test tube [17, 18], and only occurs at pH values significantly below the physiological norm. As the pKa of nitrous acid is 3.37, only vanishingly low levels of nitrous acid are present at physiological pH, and it is thought that HONO itself requires protonation before it can S-nitrosate thiols [19]. While this reaction may play some role in the gastro-intestinal tract, it is not clear that tissue pH could ever drop low enough to make significant levels of RSNO. (ii) It is possible for RSNO to be formed by the direct addition of nitrosonium (NO+) to a thiol at neutral pH (Eq. (3)). Peroxidase
| (3) |
complexes I and II [20, 21] have been shown to be reduced by NO, presumably generating nitrosonium. However, the major limitation of this mechanism is that nitrosonium is unstable in water at neutral pH, immediately hydrolyzing to nitrite, and so the thiol must be in the immediate vicinity of source of nitrosonium. Myeloperoxidase-dependent N-nitrosation has been reported [22], but so far there are no reports of S-nitrosation by peroxidase-mediated mechanisms. (iii) Direct thiol nitrosation by N2O3 occurs at a relatively fast rate forming an S-nitrosothiol and nitrite (Eq. (4)) [23]. In order
| (4) |
to generate N2O3 however, NO needs to be oxidized to NO2, which will then combine with NO to form N2O3. While this chemistry clearly happens in the test tube, the oxidation of NO to NO2 by oxygen occurs by a reaction that is second order in NO and first order in oxygen (Eq. (5)) [23–25], and consequently very slow at biological concentrations of NO. It has been suggested that hydrophobic areas in membranes and proteins could increase the local concentration of both NO and oxygen and so accelerate this reaction [26, 27], thus increasing the probability of its occurrence in physiological systems. Other mechanisms of NO2 formation are possible such as the
| (5) |
oxidation of nitrite by peroxidase complexes I and II [28]. At a locus of inflammation, where NO, nitrite and hydrogen peroxide may be generated in the presence of peroxidases, it is possible to envisage the chemical requirements for N2O3 formation. In addition, N2O3 could be formed from the condensation of nitrous acid (Eq. (6)). As discussed above, the level of HONO will be very small at neutral pH, and as this
| (6) |
reaction depends on the square of the concentration of HONO, it is likely to have little impact at neutral pH. However, this mechanism may well be responsible for the formation of S-nitrosothiols that have been reported at around pH 6.0 [29] and may be a relevant reaction during ischemic injury, where tissue pH may drop to such low values. (iv) The addition of NO to a thiyl radical (Eq. (7)) will form an S-nitrosothiol by a diffusion
| (7) |
limited radical-radical combination reaction [30, 31]. Consequently any process that can give rise to a thiyl radical has the potential to also generate S-nitrosothiols. The fact that glutathione (GSH) is able to repair other free radicals (such as the tyrosyl radical) by hydrogen (or electron) donation suggests that thiyl radicals may be a general feature of oxidative free-radical exposure. Another possible mechanism of RSNO formation that deserves further investigation is the direct addition of NO to a thiol to form a putative RSNOH intermediate, followed by reaction with oxygen (or a one-electron acceptor) to form RSNO and superoxide [32]. In addition it has been shown that peroxynitrite, the addition product of NO and superoxide, can form S-nitrosothiols very inefficiently [33] either directly [34] or through the intermediacy of thiyl radicals.
Many of these mechanisms of S-nitrosothiol formation have the potential to be catalyzed by either enzymes or redox-active metal ions. Indeed some metalloproteins (e.g. ceruloplasmin [35]) have been shown to catalyze S-nitrosothiol formation in plasma (albeit in the absence of red cells). A novel heme-mediated mechanism of S-nitosothiol formation was observed in nitrophorins, NO-carrying proteins from blood sucking insects, which contain a heme group ligated to the protein by a cysteinyl residue. Nitrosylation of the ferric heme on the distal side results in heme reduction by the proximal thiolate ligand resulting in a ferrous nitroso heme and a protein thiyl radical, the latter of which can subsequently combine with a second NO (see Eq. (7)) to generate an S-nitrosothiol on the protein [36]. Recent evidence suggest that hemoglobin may act as a nitrite reductase, and in so doing may generate S-nitrosothiols [37–39]. Although the mechanism is as yet unclear, it is possible that this reaction is responsible for the formation of S-nitrosothiols in red cells.
The most important reaction of S-nitrosothiols for this discussion is the reversible transfer of the nitroso group from an S-nitrosothiol to a thiol. This reaction is referred to
| (8) |
as S-transnitrosation and is illustrated in Eq. (8). The reaction rate is dependent on the pKa of the attacking thiol due to the fact that thiols with low pKa values are more extensively ionized at neutral pH values. The biological importance of this reaction is that low molecular weight RSNO are able to modify protein cysteinyl residues, and conversely low molecular weight thiols are able to convert protein RSNO back to the original thiol. The equilibrium distribution of the S-nitroso groups within a cell will then depend on the rates and equilibrium positions of all possible S-transnitrosation reactions, which in turn will be heavily influenced by the pKa values of the thiols involved [40, 41]. As some protein thiol pKa values are very low (as low as 4.5, compared to free amino acid thiol pKa values of 8–9), it is likely that such protein thiols are sensitive targets for modification by transnitrosation if such a thiol is accessible to attack by the donor RSNO.
3. What is meant by “the proteome of S-nitrosated proteins”?
This sounds like a simple question but one that has no simple answer. With currently available methods, it is fair to say that the levels of protein S-nitrosothiols generated in vivo are too low to be subject to proteomic analysis in any meaningful way [14]. Although some studies have reportedly examined the proteome of S-nitrosation from endogenous NO synthase sources [42–44], most have relied on exogenous treatments with NO or related compounds to increase the total intracellular RSNO pool [45–47]. It is possible to take total cellular protein and S-nitrosate almost every free thiol. This could be done by treating the lysate with nitrite at a lowered pH to promote nitrous acid-mediated nitrosation. Alternatively, one could take total cellular protein, and treat it with NO in the presence of oxygen, revealing thiols that are susceptible to nitrosation by the NO/O2 reaction. Thirdly, one could take cell lysate and treat it with a low-molecular weight S-nitrosothiol, such as S-nitrosoglutathione (GSNO), and examine thiols that are susceptible to modification by transnitrosation (Eq. (8)) [48]. Finally, whole cells could be treated in all the above ways to examine the S-nitrosatable proteome in a cellular environment [49–52]. As the mechanism of S-nitrosation in vivo is unknown, it is not clear which of these treatments (if any) more closely resembles physiological S-nitrosation. It is possible (if not probable) that each of these methods would give rise to a different (albeit overlapping) set of modified proteins.
Not only is it the mechanism of S-nitrosation that is important in defining the S-nitrosatable proteome, but also the extent of S-nitrosation. At high (nmol/mg protein) S-nitrosation levels, proteins will be modified that are unlikely to be substantially S-nitrosated in vivo. So to examine the proteome at only one level of nitrosation could lead to a misleading picture, particularly if one is trying to understand the issues surrounding the susceptibility of thiols to S-nitrosation. All current studies are therefore trading off the extent of S-nitrosation with the signal-to-noise ratio of the methods and hoping that the patterns of S-nitrosation observed can be extrapolated back to more physiological levels of protein modification. These studies still have great value and importance as they highlight potential targets of protein modification that can be examined by more direct approaches, such as site-directed mutagenesis.
Finally, S-nitrosation does not take place in isolation but in the complex milieu of a cell or fluid and the extent of S-nitrosation and the stability of S-nitrosothiols are no doubt impacted by other processes. We have demonstrated using bovine aortic endothelial cells, when we titrated the intracellular RSNO tone using S-nitrosocysteine (CysNO, see later), that we could deplete cellular glutathione (GSH) in a concentration-dependent way [53]. As GSH, and associated enzymes, are important in the catabolism of RSNO [54], the proteins that are S-nitrosated in the presence of GSH may be of quite a different character compared to those that are S-nitrosated once GSH has been depleted. The same may be true of other redox couples within the cell involved in the stability of S-nitrosothiols, such as the NADH/NAD+ couple [54].
4. The use of lmwtRSNO transport to titrate the S-nitrosated proteome
Although lmwtRSNO are often used in cell culture as NO donors, several lines of evidence suggest that the interactions between these compounds and cells are much more complex that simple NO release. Firstly, RSNO are relatively stable in metal ion-free buffers in the dark, and in cell culture their decomposition is driven by the presence of cells [55]. We have demonstrated that cystine, present in most cell culture medium, is essential for cell-mediated GSNO metabolism [56]. Secondly, there are several old reports of stereoselectivity of RSNO action, particularly when CysNO is used [57, 58]. Thirdly, the effects of some lmwtRSNO have been shown to be inhibitable by L-leucine and are thought to be mediated through specific transport pathways [59]. These transport mechanisms have recently been fully characterized and involve both the amino acid transport system L (L-AT) [60, 61] and the cystine transporter xc−[62]. One important observation of these studies is that many of the effects of CysNO treatment do not involve the formation of NO, but involve the uptake and metabolism of the nitroso functional group. An understanding of S-nitrosothiol transport allows for the titration of the intracellular RSNO tone, and, in principle, will allow the assessment of the S-nitrosation proteome that forms as a result of transnitrosation processes, at a range of total intracellular RSNO levels. It should then be possible to examine how the pattern of S-nitrosation changes at ever decreasing levels of S-nitrosothiol.
5. Challenges for the detection of the S-nitroso functional group
It is relatively simple to detect S-nitrosothiols at levels over ~100 pmol/mg protein using either spectrophotometric, fluorimetric or chemiluminescence methods. In our hands, tri-iodide based chemiluminescence [63, 64] can accurately detect levels of S-nitrosothiol in cell lysate down to 1–2 pmol/mg protein [65]. However, these methods only detect the total S-nitrosothiol content and do not identify which proteins are modified.
Traditional methods for the determination of protein modifications are problematic for the analysis of S-nitrosation. As there are no useful radioactive isotopes of N and O the direct use of radioactivity for the detection of S-nitrosation is not possible. Although an antibody for protein S-nitrosation is commercially available it has so far found use only for immunohistochemistry [66]. Attempts to use this antibody for immunoprecipitation or Western analysis have largely proved unsuccessful. While immunoprecipitation experiments certainly pull down many proteins, our observations have indicated that the RSNO-to-protein molar ratio is unchanged in both the precipitate and the supernatant suggesting a large-degree of non-specific protein association (unpublished data). Consequently, the detection of thiol modifications has to rely on indirect methods in which the nitroso function group is selectively replaced.
5.1. The biotin switch method
The first, and to date the only, method for the specific tagging and detection of S-nitrosated proteins has been generally referred to as the biotin switch method. This method, developed by Jaffrey et al. [42], attempted to replace the S-nitroso functional group with a biotin label that could then be easily detected using streptavidin or an appropriate antibody. The basic principle behind the technique is illustrated in Fig. 1. Protein is first treated, in the presence of SDS, with thiol blocking agents, such as monomethyl thiosulfonate (MMTS), N-ethylmaleimide (NEM) or iodoacetic acid, to chemically inactivate all thiols. The choice of thiol blocking agent is somewhat arbitrary, however, it should be born in mind that MMTS forms a disulfide and so may be more susceptible to later reduction, whereas NEM forms a more stable thioether, and iodoacetate a thioester. After thiol blocking, RSNO are reduced to RSH by incubation with 1 mM ascorbate for 1 hour in the presence of the metal ion chelators EDTA and neocuproine, after which the nascent thiols are labeled with a biotinylation agent specific for thiols. The original method used (N-(6-(biotinamido)hexyl)-3′-(2′-pyridyldithio)-propionamide (biotin-HPDP) which results in a disulfide-linked label. Other available thiol biotinylation agents are based on maleimide or iodinated carboxylates that generate thioether and thioester linkages, respectively. Finally, the biotin-labeled proteins are amenable to selective precipitation for direct mass spectometric analysis, or to on gel detection and isolation of specific proteins using either streptavidin or anti-biotin antibodies.
Fig. 1.
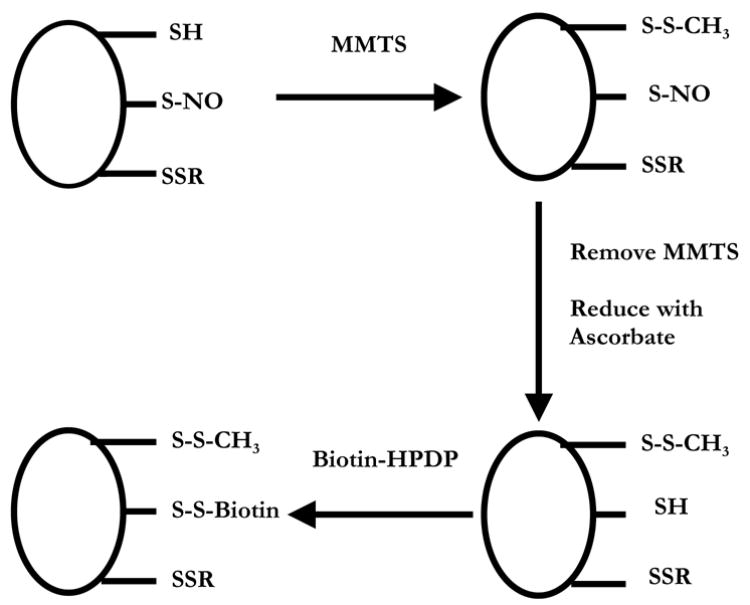
The biotin switch assay (after Jaffrey et al. [42]). Protein thiols (-SH), S-nitrosothiols (-SNO) and disulfides (-SSR) are treated with MMTS to convert all thiols to disulfides (-SSCH3). MMTS is removed and the proteins are incubated with ascorbate to convert –SNO to –SH. The nascent –SH is then labeled with biotin via a disulfide bond using biotin-HPDP.
The first issue that needs to be considered with the biotin switch assay is that it is essentially a difference method in which a very small signal (the RSNO) is being observed in the presence of a very large signal (the protein thiols) by subtracting one from the other. The subtraction is done chemically, by blocking free thiol groups, and the efficiency of this step determines to a large part the signal-to-noise ratio of the assay. As a hypothetical example, if we assume that the level of total cellular protein thiol is about 100 nmol RSH/mg protein (this is a high end estimate for a very cell-type dependent number), then in order to see 100 pmol RSNO/mg protein with a signal-to-noise ratio of 10:1, then 99.99% of protein thiols need to be blocked. This is a simplistic calculation, but it illustrates the relationship between blocking efficiency and the sensitivity of the assay, and highlights the inherent difficulty in detecting levels of RSNO much below 100 pmol/mg protein.
The second issue that should be considered is the use of ascorbate as a selective RSNO-reducing agent. Although it has been well established that ascorbate can reduce S-nitrosothiols (although the precise mechanism has been debated) [50, 67–69], the rate of this reaction is not fast. For example, the reduction of GSNO by ascorbate has a second order rate constant of 5 × 10−2 M−1s−1 [68], which corresponds to a pseudo-first order rate constant of 5 × 10−5 s−1 at 1 mM ascorbate, or a half time of almost 4 hours. We have shown that the reduction of S-nitroso bovine serum albumin (BSA-SNO) is considerably slower than the GSNO/ascorbate and will be negligibly reduced after incubation with 1 mM ascorbate for 1 hour [50]. This issue is further illustrated in Fig. 2. Protein RSNO from CysNO-treated bronchial epithelial cells was treated with reducing agent (either ascorbate or DTT), precipitated, and then the RSNO content was monitored by chemiluminescence. It can be seen that while DTT rapidly decreased the RSNO content as expected, ascorbate, even at a concentration as high as 30 mM, had little impact on RSNO levels measured by chemiluminescence, although this treatment was able to garner a positive result by biotin switch assay [50]. Consequently, exposure of RSNO to 30 mM ascorbate for 3 hours only reduces a small amount of total RSNO, but this was enough to see by biotin switch. Taken together, these data indicate that ascorbate is a poor reducer of protein RSNO and high levels and long incubation times are required. However, many investigators have reported success using this assay with original conditions of 1 mM asocorbate and 1 hour incubation time. Some recent data (Xunde Wang, Neil Hogg, Mark Gladwin, manuscript in preparation) suggest that the presence of contaminating transition metal ions, such as copper and iron, play a large role in determining the ability of ascorbate to facilitate RSNO reduction. Regardless of the precise mechanism, these concerns highlight that it is essential to examine the reductive effects of ascorbate by a secondary method (such as chemiluminescence) in order to be sure that the reductive step has, in fact, reduced the levels of RSNO in the sample. Preferably, a concentration-dependent effect of ascorbate should be examined where the loss of RSNO is correlated to an increase in biotin labeling.
Fig. 2.
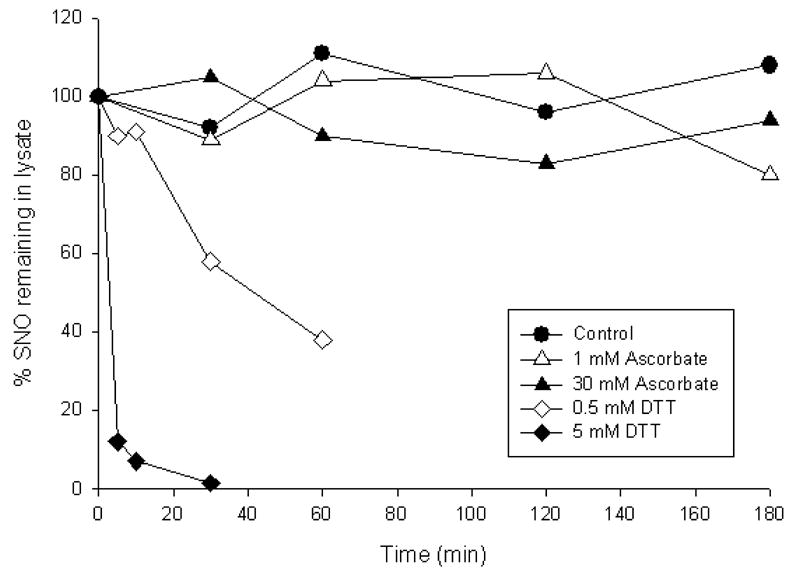
Reduction of protein RSNO by ascorbate and DTT. Human bronchial epithelial cells were incubated with CysNO (50 μM) for 30 minutes. Cells were lyzed in HEPES buffer containing 10 mM NEM, 100 μM DTPA and 10 μM neocuproine. Either DTT or ascorbate was added, and aliquots were taken at the indicated time points. For each aliquot, the protein was precipitated to remove excess reducing agent, and total RSNO was measured by tri-iodide-based chemiluminescence after treatment with sulfanilamide.
A third issue with the biotin switch method is the sensitivity of the assay. We discussed above the importance of thiol blocking with respect to signal-to-noise ratio, but assuming thiol blocking is not a limiting factor, what is the absolute sensitivity of this method? To ascertain this it is important to use an appropriate biotinylated protein standard and to assess the concentration-dependent responsiveness of this standard on Western blots. This has been successfully accomplished by Landar et al. [70] for biotin labeling, using biotinylated cytochorme c (Cyt c). By this method, Cyt c is biotinylated on lysine residues using sulfo-NHS-LC-Biotin, and the extent of biotinylation is monitored spectrophotometrically. The biotinylated Cyt c is then used as a standard on Western blots and is used to quantify biotinylation based on relative band intensities. Such standardization allows a direct comparison between the level of biotinylated protein observed on the gel (calculated from the Cyt c standard) and the amount of RSNO in the experiment (measured by chemiluminescence). The absolute sensitivity of Western blot clearly depends upon many factors, including antibody (or streptavidin) dilution, the development conditions, the chemiluminescence substrates and the length of exposure to film or camera. In our hands, and in agreement with Landar et al. [70], we can detect biotinylated Cyt c down to a level of at least 0.5 pmol biotin. This suggests that if 20 μg of protein is loaded on a gel, the biotin switch assay should be intrinsically sensitive enough to observe an S-nitrosation level of about 25 pmol/mg protein for a single protein. This represents an S-nitrosation level of about 0.1% for a 50 kDa protein, but for a complex protein mixture, this number will increase as the RSNO is distributed across multiple proteins. Experimentally, we have shown that it is difficult to observe significant responsiveness in the biotin switch assay below levels of approximately 1 nmol/mg protein in total cell lysate [50].
The final issue with this assay is that of specificity. While the requirements that allow ascorbate to reduce S-nitrosothiols in a rapid and extensive manner have not yet been fully characterized, the question of whether this reduction is selective for S-nitrosothiols is difficult to assess. However, it is likely that apart from the obvious false positive of endogenous biotin-dependent carboxylases, sulfenic acids will be reduced by ascorbate back to the level of the thiol. While sulfenic acids are a relatively rare and transient thiolic sub-species, their presence in biological systems is becoming increasingly appreciated [71]. A more worrisome issue is the possibility that the ascorbate treatment may reduce disulfides. Two recent studies suggest that ascorbate can give false positives in some proteins due to disulfide reduction or other issues [72, 73], indicating that an ascorbate-dependent increase in band intensity cannot be used as proof positive for the detection of protein RSNO without additional controls.
This in-depth discussion of the biotin switch assay has mainly highlighted potential problems and limitations. However, once these limitations are realized, this assay represents a useful addition to the armamentarium of S-nitrosothiol assays [74], and has facilitated the isolation and analysis of the S-nitrosoproteome in a number of studies [49, 51]. A major issue will always be lack of sensitivity, and it is questionable whether any method will be able to achieve sufficient sensitivity to examine the proteome of S-nitrosation from basal or pathological endogenous NO formation. Our experience suggests this is a major challenge, although others have reported successful detection of modified proteins from endogenous sources [42, 44]. In addition, the inherent danger is that this method is used (as it has been many times in the literature) as a stand-alone assay for protein S-nitrosation, which is a function for which it is profoundly unsuited.
5.2. Alternative reducing agents for the biotin switch assay
An alternative approach to the issue of specificity is to ignore it, and ask a different question-which protein thiols are reversibly modified after treatment with NO/RSNO? In other words, bypass specificity by selecting a reducing agent that will reduce many thiol modifications regardless of their nature. To do this we have replaced ascorbate in the biotin-switch assay with DTT, a generic thiol reducing agent. Of course this treatment will also reduce endogenous disulfides, and so controls of untreated samples have to be run in parallel. Fig. 3 illustrates a Western blot of cropped 2D gel of total protein from bronchial epithelial cells treated with and without 10 μM CysNO. As can be seen, several new spots show up in the treated samples that are not present in the control samples. However, this blot was purposely under-developed (increased antibody dilution and short exposure time) to minimize the background contribution and highlight the proteins that are robustly changed by treatment. Development of this blot under optimal conditions would result in many more spots in the control sample. Because relatively few spots are observed it is almost impossible to register these spots with their equivalents on the gel, and so protein identification is a rather hit-and-miss affair. However, this approach does illustrate that CysNO levels as low as 10 μM can robustly modify a few selected thiols in whole cells. While this method may be useful in defining the extent of protein modification it is not so useful for the identification of specifically modified proteins.
Fig. 3.
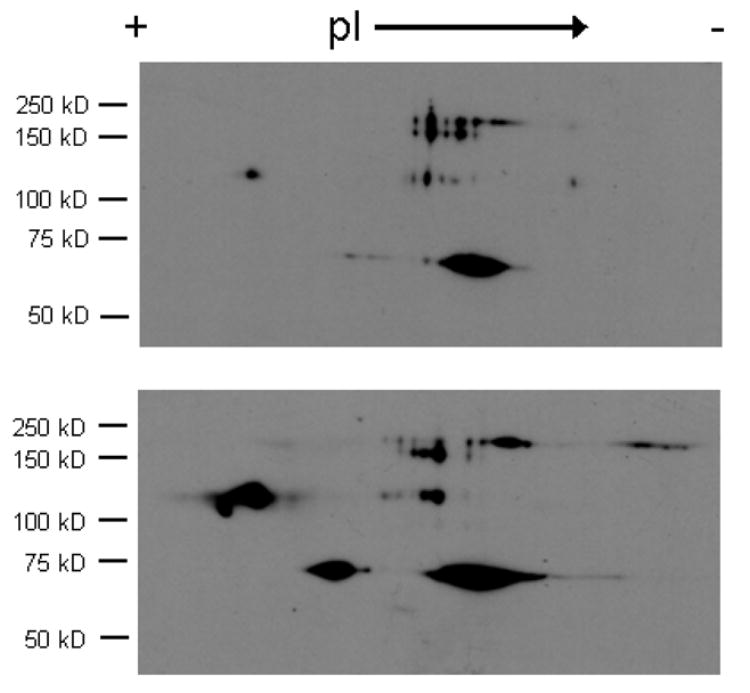
Biotinylation of proteins after DTT reduction. Human bronchial epithelial cells were incubated without (top) or with (bottom) 10 μM CysNO and then subjected to the biotin switch assay using DTT (5 mM) in place of ascorbate. Proteins were separated in 2 dimensions and biotinylation was examined by Western blot analysis.
5.3. 2D-DIGE approaches
One intriguing new approach is the use of difference gel electrophoresis (DIGE) using fluorescent labels [75, 76]. This avoids the necessity of registering a Western blot with a gel, removing the uncertainty from the spot-selection process in situations where only a few proteins are modified. The basic idea (illustrated in Fig. 4) is to label thiols from control and treated samples with two different fluorophores. In this case monofunctional maleimide-conjugated cyanine dyes (Cy3 which has a green fluorescence and Cy5 which has a red fluorescence) have been used. These dyes have been used previously to detect free thiols [77] and redox-sensitive changes in the free thiol pool [78]. Equivalent amounts of the control and the treated samples are then pooled and run in two dimensions. The second dimension SDS-PAGE must be run on a gel cast in low-fluorescence glass plates. Analysis of the gel by fluorescent scanning and image analysis software (e.g. DeCyder from GE Healthcare) will then reveal the thiols that have been modified by a difference in the fluorescent intensity of the two dyes in a single spot. Fig. 4 shows a typical fluorescence scan from a 2D gel using protein from human bronchial epithelial cells exposed to CysNO (10 μM) for 30 minutes, and using DTT (1 mM) as the reducing agent. In order for a protein to appear as a red spot on this gel it must contain no DTT-reducible disulfides and contain a cysteine residue that is modified by CysNO treatment. Most proteins appear as yellow spots as they contain both green and red fluorescence. Computer-aided analysis of the difference in intensity between the red and green fluorescence gives the amount of CysNO-modified thiols. Clearly, if the protein in question has a large number of DTT-reducible disulfides any difference between the red and green fluorescence will be difficult to determine and so the DTT reduction strategy will not be useful to look for modifications of all proteins. In these experiments it is always important to run the experiment with the dyes reversed to make sure that the differences observed are not a function of the label. One major advantage of this approach is that it can be used in combination with robotic spot removal and analysis (e.g. Ettan spot-handling workstation, GE Healthcare), so processing and MALDI identification of target proteins can be automated. In addition, the relative level of thiol modification can be obtained by direct comparison of the fluorescence intensity from both dyes. For quantitative purposes it is possible to label all cellular proteins with a third dye on lysine residues and run this on the same gel. This gives an internal control fluorescence intensity that should be independent of the degree of thiol modification and would aid comparison between gels.
Fig. 4.
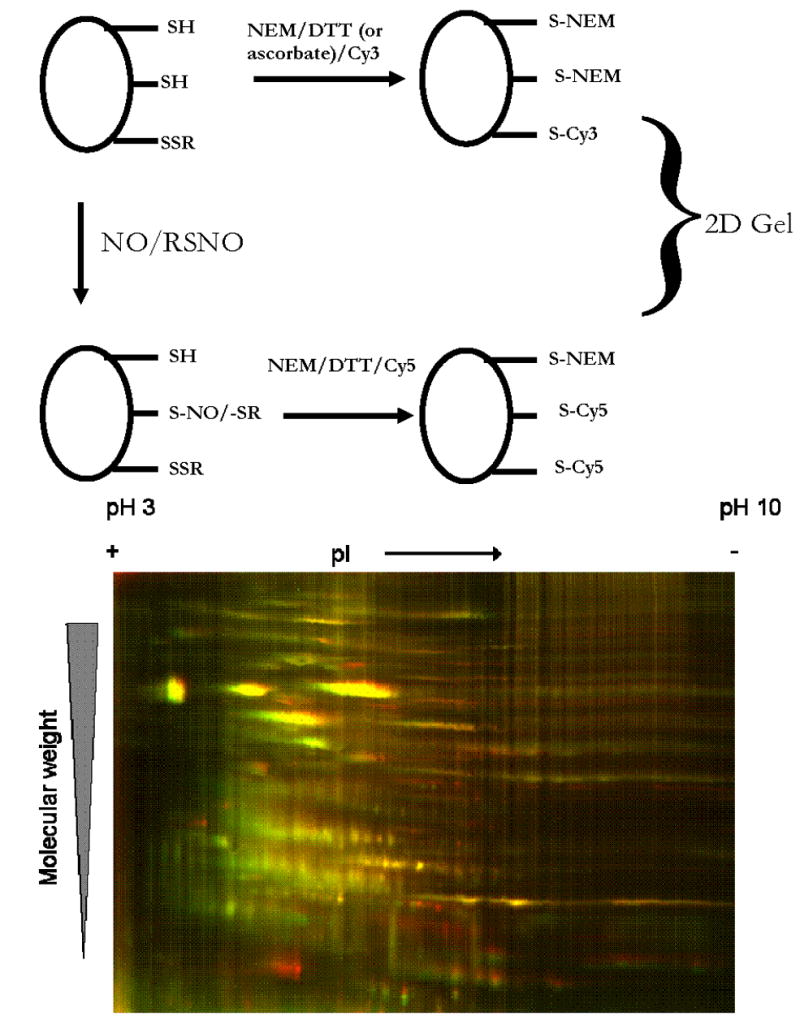
Analysis of protein modification by DIGE. (Top) Schematic of the DIGE labeling scheme using two maleimido-cyanine-based dyes Cy3 (green) and Cy5 (red). RSNO or NO treatment will modify some fraction of the protein thiols as illustrated on the left side of the figure. Both the treated and untreated samples are sequentially incubated with NEM, reducing agent (in this case DTT) and then either Cy3 or Cy5. These samples are pooled at equal protein concentrations are run in 2 dimensions. (Bottom) Human bronchial epithelial cells were treated with or without 5 μM CysNO, and protein was treated as illustrated in the top panel. Pooled protein was run in 2 dimensions and detected by fluorescence scanning.
5.4. Direct peptide capture: Avoiding gels altogether
Recently, two studies have employed direct capture techniques to examine the proteome of S-nitrosated proteins. These techniques have the advantage over gel studies in that there is no danger of generating false positives from proteins that migrate together. The study of Hao et al. [51] used the biotin-switch method on GSNO-treated rat cerebellum lysates to label modified thiols. The total level of protein RSNO in these studies was around 1–2 nmol/mg protein or up to 3% of total thiol. After biotin labeling, the proteins were trypsinized before affinity purification of biotinylated peptides. These peptides were then reduced of the beads with mercaptoethanol and detected and identified by LC-MS/MS techniques. Identified peptides that contained cysteine residues were then putative S-nitrosation targets. This method identified 68 cysteine-containing peptides from 56 proteins. The second study by Greco et al. [49], treated whole cells (human aortic smooth muscle cells) with either CysNO or the NO donor PAPA/NO and achieved levels of intracellular RSNO formation between 0.4 and 3 nmol/mg protein. These authors used a similar approach to Hao et al. [51] but eluted the bound peptides with formic acid rather than reducing them off with mercaptoethanol. The advantage to this is that the biotin remains associated to the peptide and positive identification of the biotinylation site can be obtained by MS analysis. In this study 18 peptides were identified from 16 proteins after CysNO exposure, and 4 from NO exposure, two of which were found in both groups. Interestingly, these authors also identified 18 peptides that showed up as false positives indicating the importance of proper controls.
These methods provide an important addition to gel-based approaches to identify S-nitrosated proteins. While they have not currently been used in a quantitative manner, this is not an intrinsic limitation. As in all other situations positively identified proteins need to be confirmed using methods that do not rely on the biotin switch assay.
6. Conclusion
In this review we have detailed methodologies to identify the S-nitrosated proteome. It is difficult as yet to conclude which strategy will be the most useful, but direct peptide-capture methods have provided the most compelling picture of the S-nitrosated proteome in publications to date. In all of the methods described one has to be aware that the detection methodology is indirect and relies on chemical modification of the S-nitroso group. Consequently, the specificity of this modification needs to be fully assessed and established. Finally it needs to be stated that identification of an S-nitrosated protein does not imply that S-nitrosation affects the activity of this protein. Many thiol nitrosation events may be neutral with respect to protein activity as the cysteine residue in question may play a negligible role in determining protein function. Consequently, proteomic determination alone is not enough to assess the importance of the modification, but rather provides framework by which individual pathways can be assessed at the level of activity.
Acknowledgments
This work was supported by NIH grant GM55792.
Footnotes
This paper is part of a special issue entitled “Analysis of the L-arginine/NO pathway”, guest edited by D. Tsikas.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stamler JS, Lamas S, Fang FC. Cell. 2001;106:675. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 2.Hogg N, Singh RJ, Kalyanaraman B. FEBS Lett. 1996;382:223. doi: 10.1016/0014-5793(96)00086-5. [DOI] [PubMed] [Google Scholar]
- 3.Folkes LK, Wardman P. Free Radic Biol Med. 2004;37:549. doi: 10.1016/j.freeradbiomed.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 4.DeMaster EG, Quast BJ, Redfern B, Nagasawa HT. Biochemistry. 1995;34:11494. doi: 10.1021/bi00036a023. [DOI] [PubMed] [Google Scholar]
- 5.Pryor WA, Church DF, Govindan CK, Crank G. J Org Chem. 1982;47:159. [Google Scholar]
- 6.Oae S, Fukushima D, Kim YH. J Chem Soc Chem Commun. 1977:407. [Google Scholar]
- 7.Incze K, Farkas J, Mihalyi V, Zukal E. Appl Microbiol. 1974;27:202. doi: 10.1128/am.27.1.202-205.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ignarro LJ, Edwards JC, Gruetter DY, Barry BK, Gruetter CA. FEBS Lett. 1980;110:275. doi: 10.1016/0014-5793(80)80091-3. [DOI] [PubMed] [Google Scholar]
- 9.Palmer RM, Ferrige AG, Moncada S. Nature. 1987;327:524. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 10.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Proc Natl Acad Sci USA. 1987;84:9265. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J. Proc Natl Acad Sci USA. 1992;89:7674. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jourd’heuil D, Gray L, Grisham MB. Biochem Biophys Res Commun. 2000;273:22. doi: 10.1006/bbrc.2000.2892. [DOI] [PubMed] [Google Scholar]
- 13.Gaston B, Sears S, Woods J, Hunt J, Ponaman M, McMahon TJ, Stamler JS. Lancet. 1998;351:1317. doi: 10.1016/S0140-6736(97)07485-0. [DOI] [PubMed] [Google Scholar]
- 14.Giustarini D, Milzani A, Dalle-Donne I, Rossi R. J Chromatogr B. 2006 doi: 10.1016/j.jchromb.2006.09.031. [DOI] [Google Scholar]
- 15.Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS. Science. 1999;284:651. doi: 10.1126/science.284.5414.651. [DOI] [PubMed] [Google Scholar]
- 16.Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. J Cell Biol. 2001;154:1111. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hart TW. Tetrahedron Lett. 1985;26:2013. [Google Scholar]
- 18.Field L, Dilts RV, Ravichandran R, Lanhert PG, Carnahan PG. J Chem Soc Chem Commun. 1978:249. [Google Scholar]
- 19.Williams DLH. Nitrosation reactions and the chemistry of nitric oxide. Ch 7 Elsevier Science; Amsterdam: 2004. [Google Scholar]
- 20.Glover RE, Koshkin V, Dunford HB, Mason RP. Nitric Oxide. 1999;3:439. doi: 10.1006/niox.1999.0256. [DOI] [PubMed] [Google Scholar]
- 21.Abu-Soud HM, Hazen SL. J Biol Chem. 2000;275:37524. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 22.Lakshmi VM, Nauseef WM, Zenser TV. J Biol Chem. 2005;280:1746. doi: 10.1074/jbc.M411263200. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein S, Czapski G. J Am Chem Soc. 1996;118:3419. doi: 10.1021/ja00269a020. [DOI] [PubMed] [Google Scholar]
- 24.Kharitonov VG, Sundquist AR, Sharma VS. J Biol Chem. 1995;270:28158. doi: 10.1074/jbc.270.47.28158. [DOI] [PubMed] [Google Scholar]
- 25.Wink DA, Nims RW, Darbyshire JF, Christodoulou D, Hanbauer I, Cox GW, Laval F, Laval J, Cook JA, Krishna MC. Chem Res Toxicol. 1994;7:519. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Miller MJS, Joshi MS, Thomas DD, Lancaster JR., Jr Proc Natl Acad Sci USA. 1998;95:2175. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nedospasov A, Rafikov R, Beda N, Nudler E. Proc Natl Acad Sci USA. 2000;97:13543. doi: 10.1073/pnas.250398197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Vliet A, Eiserich JP, Halliwell B, Cross CE. J Biol Chem. 1997;272:7617. doi: 10.1074/jbc.272.12.7617. [DOI] [PubMed] [Google Scholar]
- 29.Tsikas D. Nitric Oxide. 2003;9:53. doi: 10.1016/s1089-8603(03)00044-2. [DOI] [PubMed] [Google Scholar]
- 30.Jourd’heuil D, Jourd’heuil FL, Feelisch M. J Biol Chem. 2003;278:15720. doi: 10.1074/jbc.M300203200. [DOI] [PubMed] [Google Scholar]
- 31.Espey MG, Thomas DD, Miranda KM, Wink DA. Proc Natl Acad Sci USA. 2002;99:11127. doi: 10.1073/pnas.152157599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gow AJ, Buerk DG, Ischiropoulos H. J Biol Chem. 1997;272:2841. doi: 10.1074/jbc.272.5.2841. [DOI] [PubMed] [Google Scholar]
- 33.Moro MA, Darley-Usmar VM, Goodwin DA, Read NG, Zamora-Pino R, Feelisch M, Radomski MW, Moncada S. Proc Natl Acad Sci USA. 1994;91:6702. doi: 10.1073/pnas.91.14.6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Vliet A, Chr’t Hoen PA, Wong PSY, Bast A, Cross CE. J Biol Chem. 1998;273:30255. doi: 10.1074/jbc.273.46.30255. [DOI] [PubMed] [Google Scholar]
- 35.Inoue K, Akaike T, Miyamoto Y, Okamoto T, Sawa T, Otagiri M, Suzuki S, Yoshimura T, Maeda H. J Biol Chem. 1999;274:27069. doi: 10.1074/jbc.274.38.27069. [DOI] [PubMed] [Google Scholar]
- 36.Weichsel A, Maes EM, Andersen JF, Valenzuela JG, Shokhireva TK, Walker FA, Montfort WR. Proc Natl Acad Sci USA. 2005;102:594. doi: 10.1073/pnas.0406549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, III, Gladwin MT. Nat Med. 2003;9:1498. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 38.Nagababu E, Ramasamy S, Rifkind JM. Nitric Oxide. 2006;15:20. doi: 10.1016/j.niox.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Angelo M, Singel DJ, Stamler JS. Proc Natl Acad Sci USA. 2006;103:8366. doi: 10.1073/pnas.0600942103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hogg N. Anal Biochem. 1999;272:257. doi: 10.1006/abio.1999.4199. [DOI] [PubMed] [Google Scholar]
- 41.Arnelle DR, Stamler JS. Arch Biochem Biophys. 1995;318:279. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 42.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Nat Cell Biol. 2001;3:193. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 43.Dall’agnol M, Bernstein C, Bernstein H, Garewal H, Payne CM. Proteomics. 2006;6:1654. doi: 10.1002/pmic.200500240. [DOI] [PubMed] [Google Scholar]
- 44.Gao C, Guo H, Wei J, Mi Z, Wai PY, Kuo PC. Nitric Oxide. 2005;12:121. doi: 10.1016/j.niox.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 45.Prior IA, Clague MJ. Biochim Biophys Acta. 2000;1475:281. doi: 10.1016/s0304-4165(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 46.Martinez-Ruiz A, Lamas S. Arch Biochem Biophys. 2004;423:192. doi: 10.1016/j.abb.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 47.Yang Y, Loscalzo J. Proc Natl Acad Sci USA. 2005;102:117. doi: 10.1073/pnas.0405989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuncewicz T, Sheta EA, Goldknopf IL, Kone BC. Mol Cell Proteomics. 2003;2:156. doi: 10.1074/mcp.M300003-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Greco TM, Hodara R, Parastatidis I, Heijnen HFG, Dennehy MK, Liebler DC, Ischiropoulos H. Proc Natl Acad Sci USA. 2006;103:7420. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Keszler A, Broniowska KA, Hogg N. Free Radic Biol Med. 2005;38:874. doi: 10.1016/j.freeradbiomed.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 51.Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. Proc Natl Acad Sci USA. 2006;103:1012. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rhee KY, Erdjument-Bromage H, Tempst P, Nathan CF. Proc Natl Acad Sci USA. 2005;102:467. doi: 10.1073/pnas.0406133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Broniowska KA, Zhang Y, Hogg N. J Biol Chem. 2006;281:33835. doi: 10.1074/jbc.M603248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. Nature. 2001;410:490. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 55.Gordge MP, Addis P, Noronha-Dutra AA, Hothersall JS. Biochem Pharmacol. 1998;55:657. doi: 10.1016/s0006-2952(97)00498-x. [DOI] [PubMed] [Google Scholar]
- 56.Zeng H, Spencer NY, Hogg N. Am J Physiol. 2001;281:H432. doi: 10.1152/ajpheart.2001.281.1.H432. [DOI] [PubMed] [Google Scholar]
- 57.Davisson RL, Travis MD, Bates JN, Johnson AK, Lewis SJ. Am J Physiol. 1997;272:H2361. doi: 10.1152/ajpheart.1997.272.5.H2361. [DOI] [PubMed] [Google Scholar]
- 58.Travis MD, Stoll LL, Bates JN, Lewis SJ. Eur J Pharmacol. 1996;318:47. doi: 10.1016/s0014-2999(96)00719-4. [DOI] [PubMed] [Google Scholar]
- 59.Satoh S, Kimura T, Toda M, Maekawa M, Ono S, Narita H, Miyazaki H, Murayama T, Nomura Y. J Neurochem. 1997;69:2197. doi: 10.1046/j.1471-4159.1997.69052197.x. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Hogg N. Free Radic Biol Med. 2005;38:831. doi: 10.1016/j.freeradbiomed.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 61.Li S, Whorton AR. J Biol Chem. 2005;280:20102. doi: 10.1074/jbc.M413164200. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Y, Hogg N. In: Puntarulo S, Boveris A, editors. XII Bienial Meeting of the Society for Free Radical Research International Conference Procedings; Medimond; 2004. p. 167. [Google Scholar]
- 63.Yang BK, Vivas EX, Reiter CD, Gladwin MT. Free Radic Res. 2003;37:1. doi: 10.1080/1071576021000033112. [DOI] [PubMed] [Google Scholar]
- 64.Rassaf T, Bryan NS, Kelm M, Feelisch M. Free Radic Biol Med. 2002;33:1590. doi: 10.1016/s0891-5849(02)01183-8. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y, Hogg N. Am J Physiol Lung Cell Mol Physiol. 2004;287:L467. doi: 10.1152/ajplung.00350.2003. [DOI] [PubMed] [Google Scholar]
- 66.Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. J Biol Chem. 2002;277:9637. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 67.Scorza G, Pietraforte D, Minetti M. Free Radic Biol Med. 1997;22:633. doi: 10.1016/s0891-5849(96)00378-4. [DOI] [PubMed] [Google Scholar]
- 68.Holmes AJ, Williams DLH. J Chem Soc Perkin Trans. 2000;2:1639. [Google Scholar]
- 69.Smith JN, Dasgupta TP. Nitric Oxide. 2000;4:57. doi: 10.1006/niox.2000.0272. [DOI] [PubMed] [Google Scholar]
- 70.Landar A, Oh JY, Giles NM, Isom A, Kirk M, Barnes S, Darley-Usmar VM. Free Radic Biol Med. 2006;40:459. doi: 10.1016/j.freeradbiomed.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 71.Poole LB, Karplus PA, Claiborne A. Annu Rev Pharmacol Toxicol. 2004;44:325. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 72.Landino LM, Koumas MT, Mason C, Alston JA. Biochem Biophys Res Commun. 2006;340:347. doi: 10.1016/j.bbrc.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 73.Huang B, Chen C. Free Radic Biol Med. 2006;41:562. doi: 10.1016/j.freeradbiomed.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 74.Gladwin MT, Wang X, Hogg N. Free Radic Biol Med. 2006;41:557. doi: 10.1016/j.freeradbiomed.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 75.Chan JR, Gharbi S, Gaffney PR, Cramer R, Waterfield MD, Timms JF. Proteomics. 2005;5:2908. doi: 10.1002/pmic.200401300. [DOI] [PubMed] [Google Scholar]
- 76.Viswanathan S, Unlu M, Minden JS. Nat Protoc. 2007;1:1351. doi: 10.1038/nprot.2006.234. [DOI] [PubMed] [Google Scholar]
- 77.Maeda K, Finnie C, Svensson B. Biochem J. 2004;378:497. doi: 10.1042/BJ20031634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan HL, Gharbi S, Gaffney PR, Cramer R, Waterfield MD, Timms JF. Proteomics. 2005;5:2908. doi: 10.1002/pmic.200401300. [DOI] [PubMed] [Google Scholar]