

Abstract
IL-17 has been implicated in a number of inflammatory diseases, but the conditions of antigen exposure that drive the generation of Th17 responses have not been well defined. Epicutaneous (EC) immunization of mice with ovalbumin (OVA), which causes allergic skin inflammation with many characteristics of the skin lesions of atopic dermatitis, was found to also drive IL-17 expression in the skin. EC, but not i.p., immunization of mice with OVA drove the generation of IL-17-producing T cells in draining lymph nodes and spleen and increased serum IL-17 levels. OVA inhalation by EC-sensitized mice induced IL-17 and CXCL2 expression and neutrophil influx in the lung along with bronchial hyperreactivity, which were reversed by IL-17 blockade. Dendritic cells trafficking from skin to lymph nodes expressed more IL-23 and induced more IL-17 secretion by naïve T cells than splenic dendritic cells. This was inhibited by neutralizing IL-23 in vitro and by intradermal injection of anti-TGFβ neutralizing antibody in vivo. Our findings suggest that initial cutaneous exposure to antigens in patients with atopic dermatitis may selectively induce the production of IL-17, which, in turn, drives inflammation of their airways.
Keywords: atopic dermatitis, IL-17, lung inflammation
Effector CD4+ T cells are classically divided into two lineages based on distinct cytokine production profiles: the Th1 lineage, which produces IFN-γ, and the Th2 lineage, which produces IL-4 and IL-13. Recently, a lineage of effector CD4+ T cells that produces IL-17, Th17, has been identified (1, 2). IL-17A is the original member of the IL-17 family. IL-17A and IL-17F share the highest degree of homology and some biological functions such as tissue recruitment of neutrophils (3). The differentiation pathway of Th17 cells from naive CD4+ cells is independent of the transcription factors and signaling molecules that are important for the differentiation of the other two T helper lineages. Moreover, the development of Th17 cells is inhibited by IL-4 and IFN-γ (1, 2). Recent studies have described a critical role for TGFβ and IL-6 in Th17 commitment and an important role for the cytokine IL-23 in the expansion of Th17 cells (4–6).
Extensive studies have focused on the role of Th17 cells in autoimmune diseases. Th17 was shown to be important in mouse models of experimental autoimmune encephalitis and collagen-induced arthritis, in which IFN-γ was previously thought to be a major contributor (7). IL-17A is expressed by cells in airways of patients with moderate-to-severe asthma (8). IL-17 mRNA and protein expression levels are elevated in the serum and the sputum of patients with asthma and correlate with sputum neutrophil counts and with airway hyperreactivity to methacholine (9–11). The exact role of IL-17 in Th2-dominated lung inflammation is controversial. IL-17−/−xDO11.10 transgenic mice failed to induce airway inflammation to inhaled ovalbumin (OVA) (12). In another study, administration of IL-17 to s.c.-immunized mice resulted in decreased airway inflammation and decreased Th2 cytokine expression to inhaled OVA (13).
Atopic dermatitis (AD) is a common pruritic inflammatory skin disease that often begins in infancy. The incidence of AD has been steadily rising, and it now affects >15% of children in the U.S. The majority of infants with AD develop asthma and/or allergic rhinitis later in life (14). Epicutaneous (EC) sensitization with allergens is thought to play an important role in the pathogenesis of AD (15, 16). AD skin lesions show infiltration of T cells and eosinophils and increased expression of Th2 cytokines in the acute stage and additional expression of Th1 cytokines in the chronic stage. IL-17 is also expressed in the acute skin lesions (17).
The physiologic conditions of antigen exposure that drive IL-17 production have not been well defined. Cutaneous IL-17 expression in human AD suggested that antigen exposure via skin might selectively drive IL-17 responses. We have developed a mouse model of AD using repeated EC sensitization with OVA to tape-stripped skin (18, 19). We show here that EC sensitization elicits a robust local and systemic Th17 immune response and that IL-17 plays an important role in the development of lung inflammation after skin sensitization with antigen.
Results
EC Sensitization Results in Cutaneous IL-17 Expression and Elicits Th17-Secreting Antigen-Specific Cells in the Draining Lymph Nodes (LN).
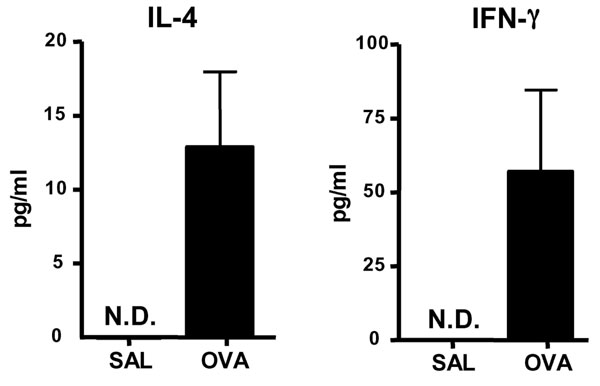
To determine whether Th17 cells are present at sites of EC sensitization, we examined IL-17 mRNA expression. Fig. 1A shows that IL-17A and IL-17F mRNA levels were up-regulated in OVA-sensitized skin, compared with saline controls. As previously described (19, 20), OVA EC sensitization significantly up-regulated mRNA expression of the Th2 cytokines IL-4 and IL-13, but had little effect on IFN-γ expression (Fig. 1B).
Fig. 1.

Expression of IL-17 in EC-sensitized skin and draining LN. (A and B) Expression of mRNA for IL-17A and IL-17F (A) and of mRNA for IL-4, IL-13, and IFN-γ (B) in sensitized skin with OVA or saline controls (n = 5–7 per group). Values for saline-sensitized sites have been set to 1. (C) IL-17A, IL-4, and IFN-γ secretion by OVA stimulated cells from draining LN. Columns and error bars represent mean± SEM (n = 5–7 per group). *, P < 0.05; **, P < 0.01. N.D., not detectable.
We also examined the presence of Th17 antigen-specific cells in the draining LN of BALB/c mice immunized with OVA via the EC route. Fig. 1C shows that cells from inguinal and axillary LN that drain EC-sensitized skin secreted IL-17A after in vitro stimulation with OVA. In contrast, cells from mesenteric LN (MLN) of i.p.-immunized mice secreted very little IL-17A in response to OVA. Cells from draining LN of EC-sensitized mice and MLN of i.p.-immunized mice with OVA secreted comparable amounts of IL-4 and IFN-γ in response to OVA (Fig. 1C). There was little or no detectable secretion of IL-17A, IL-4, or IFN-γ by cells from LN of control mice that were EC-sensitized or i.p.-immunized with saline (Fig. 1C) or by cells from LN of OVA-immunized mice cultured with medium (data not shown). We also examined cytokine production by cells from mediastinal LNs, which serve as draining LNs after i.p. immunization. Cells from these LNs secreted IL-4 and IFN-γ in response to OVA, albeit in lower amounts than MLNs, but no detectable amounts of IL-17A [supporting information (SI) Fig. 7].
EC Sensitization Induces a Systemic Th17 Response.
We examined the presence of Th17 antigen-specific cells in splenocytes from EC-sensitized mice. Fig. 2A shows that splenocytes of EC-sensitized mice secreted ≈20-fold more IL-17A than those of i.p.-immunized mice in response to OVA. In contrast, IL-4 and IFN-γ secretion were comparable between the two groups. Selective induction of Th17 cells by EC sensitization was confirmed by examining the intracellular IL-17A expression. There was a higher percentage of CD4+IL-17+ cells in splenocytes from OVA EC-sensitized mice compared with OVA i.p.-immunized mice (mean ± SEM: 3.7 ± 0.17% vs. 1.29 ± 0.12%, n = 3, P < 0.05). In contrast, the percentage of CD4+ IFN-γ+ was comparable between the two groups (4.57 + 0.55% vs. 4.74 + 0.9%, n = 3, P < 0.05). Splenocytes from saline EC-sensitized mice showed increased percentages of CD4+IL-17+ cells compared with saline i.p.-immunized mice (1.55 ± 0.02% vs. 0.67 ± 0.05%, n = 3, P < 0.05).
Fig. 2.

EC sensitization elicits a systemic Th-17. (A) IL-17A, IL-4, and IFN-γ secretion by OVA-stimulated splenocytes. (B) Serum IL-17A levels in EC versus i.p. sensitized mice. Columns and error bars represent mean± SEM (n = 4–7 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.0001. N.D., not detectable; SAL, saline.
We also examined whether EC sensitization resulted in increased circulating IL-17A levels. Serum IL-17A levels were not detectable in unimmunized mice. OVA EC-sensitized mice had significantly higher serum IL-17A levels than saline controls (Fig. 2B). Serum IL-17A levels rose only slightly in OVA i.p.-immunized mice. It should be noted that EC sensitization, but not i.p. immunization with saline, caused the appearance of moderate amounts of IL-17A in serum. These results suggest that EC sensitization elicits a robust systemic IL-17 response.
Inhalation Allergen Challenge Elicits IL-17 Expression in the Lungs of EC-Sensitized Mice.
We previously showed that allergen inhalation challenge of EC-sensitized mice results in airway inflammation (18, 21). We asked whether Th17 cells are recruited to the lungs of EC-sensitized mice after antigen airway challenge. Fig. 3A shows that lungs from OVA EC-sensitized mice exhibited a significant ≈5-fold increase in IL-17A and IL-17F mRNA expression compared with lungs of saline controls. Induction of IL-17 expression after airway antigen challenge was selective to the EC route of sensitization, because lungs from i.p.-immunized mice exhibited no increase in IL-17A or IL-17F mRNA expression. In contrast to the robust expression of IL-17, lungs of EC-sensitized mice expressed significantly less Th1, Th2 cytokines, and CCL11 than lungs from i.p.-immunized mice, 2-fold versus 40-fold for IL-4, 9-fold versus 75-fold for IL-13, no increase versus 3-fold for IFN-γ mRNA, and 3-fold versus 30-fold for CCL11 (Fig. 3A).
Fig. 3.

IL-17 expression in allergen-challenged lungs of EC-sensitized and i.p.-immunized mice. (A) IL-17A, IL-17F, IL-4, IL-13, CCL11, and IFN-γ mRNA expression in lung tissues. Values for lungs of saline sensitized mice have been set to 1. (B) IL-17A, IL-13, and IFN-γ secretion by OVA-stimulated lung cells. (C) Intracellular IL-17A staining of CD4+ cells from OVA-stimulated lung cells. (D) IL-17A levels in bronchoalveolar lavage (BAL) fluid. Columns and error bars represent mean ± SEM (n = 5–6 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.0001. Data in C are representative of two experiments. N.D., not detectable; SAL, saline. Numbers in circled areas indicate the percentage of IL-17+CD4+ cells
To confirm the increased IL-17A expression at the protein level, lung cells were stimulated with OVA for 2 days, and the supernatants were examined for their IL-17A content. Fig. 3B shows that lung cells from OVA EC-sensitized mice secreted significantly more IL-17A than those from saline controls. Lung cells from OVA i.p.-immunized mice secreted negligible amounts of IL-17A but secreted significantly more IL-13 and IFN-γ than those from OVA EC-sensitized mice. Recruitment of IL-17-producing CD4+ cells into lungs of sensitized mice was demonstrated by examining the intracellular IL-17A expression in CD4+ cells from lung cell cultures. Fig. 3C shows that there was a higher percentage of lung CD4+IL-17+ cells from OVA EC-sensitized mice than those from OVA i.p.-immunized mice (3.6 ± 1.14% versus 0.37 ± 0.03%, n = 2). Increased production of IL-17A protein in vivo was evidenced by the detection of IL-17A in bronchoalveolar lavage fluid (BALF) from EC-sensitized mice, but not from i.p.-immunized mice (Fig. 3D)
EC Sensitization Results in CXCL2 Expression and Neutrophil Influx in the Lung After Airway Challenge with Antigen.
IL-17A and IL-17F cause neutrophil accumulation by virtue of their ability to induce the expression of neutrophil-attracting chemokines that include CXCL2 (22). Fig. 4A shows that after airway antigen challenge, lungs from OVA EC-sensitized mice exhibited a significant ≈7-fold increase in CXCL2 expression compared with lungs of saline controls. In contrast, there was negligible up-regulation of CXCL2 mRNA expression by lungs from i.p.-immunized mice challenged with OVA.
Fig. 4.

Neutrophil influx in allergen-challenged lungs of EC-sensitized and i.p.-immunized mice. (A) CXCL2 mRNA expression in lung tissues. Values for lungs of saline-sensitized mice have been set to 1. (B) Percentage distribution of cell types in BALF. (C) H&E staining of lung sections from mice EC- and i.p.-sensitized with saline or OVA after OVA-inhalation challenge. Magnification ×200. Further magnification of the black-bordered box shows the predominance of neutrophils (yellow arrows) in lungs from OVA EC-sensitized mice and of eosinophils (red arrows) in lungs from OVA i.p.-sensitized mice. Columns and error bars represent means ± SEM (n = 5–6 per group). *, P < 0.05; **, P < 0.01. SAL, saline.
We next compared the cellular composition of BALF from EC-sensitized versus i.p.-immunized mice after inhalation challenge. The total cells number in BALF from OVA EC-sensitized mice was only half that in BALF from OVA i.p.-immunized mice (3.67 ± 0.05 × 105 versus 7.9 ± 1.7 × 105, n = 3 experiments with six mice each group, P < 0.01). The percentage of neutrophils in BALF of OVA EC-sensitized mice was significantly higher than in BALF of mice i.p.-immunized with OVA, 15 ± 2.8% versus 2.5 ± 0.9%, n = 3, P < 0.01 (Fig. 4B). There was a concomitant decrease in the percentage of eosinophils, with little change in the percentage of lymphocytes and macrophages.
We also compared cellular infiltration in lung tissue from EC-sensitized versus i.p.-immunized mice. Fig. 4C shows that, after inhalation challenge, there was a dense cellular infiltrate in lungs from mice EC-sensitized or i.p.-immunized with OVA but not in lungs from mice EC-sensitized or i.p.-immunized with saline. Examination of the infiltrate revealed abundant neutrophils in lung tissue from EC-sensitized mice compared with lungs from i.p.-immunized mice (Fig. 4C, expanded Inset). In contrast, eosinophils were more predominant in lungs from i.p.-immunized mice compared with lungs from EC-sensitized mice.
IL-17 Mediates Neutrophil Influx to the Lungs and Increased Responsiveness to Methacholine in EC-Sensitized Mice.
To determine the role of IL-17 in the neutrophil influx to the challenged lungs of EC-sensitized mice, we treated EC-sensitized mice with a neutralizing anti-IL-17A mAb. One hundred micrograms of anti-IL-17A mAb or IgG2a isotype control were administered i.v. 7 and 4 days before the first airway challenge. Administration of anti-IL-17A mAb, but not IgG2a isotype control, significantly diminished BALF neutrophilia but had no effect on BALF eosinophilia and lymphocytosis after antigen challenge (Fig. 5A). Anti-IL-17A, but not control mAb, reduced the predominantly neutrophilic cellular infiltration in lung tissue (Fig. 5B) and significantly reduced CXCL2 mRNA but had no significant effect on IL-4 and IL-13 mRNA in lung tissue (Fig. 5C).
Fig. 5.

Effect of anti-IL-17A on cellular composition of bronchoalveolar lavage fluid, CXCL2 and cytokine expression, and Penh to allergen inhalation in EC-sensitized mice. (A) Cells in bronchoalveolar lavage fluid. (B) Expression of mRNA for CXCL2, IL-4, IL-13, and IFN-γ. Values for lungs of saline-sensitized mice have been set to 1. (C) H&E staining of lung sections examined at magnification ×200 magnification. (D) Peak Penh after OVA-inhalation challenge. Columns and error bars represent means ± SEM (n = 5 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.0001. SAL, saline.
We had previously shown that antigen-inhalation challenge of mice EC-sensitized with OVA results in increased responsiveness to methacholine compared with saline controls (18, 21). Fig. 5D shows that, after inhalation challenge, OVA EC-sensitized mice, but not saline controls, developed a robust response to aerosolized methacholine, reflected in a significant increase in enhanced pause (Penh). Penh values represent an estimate for airway obstruction and may indicate airway hyperreactivity and inflammation. The methacholine response of OVA EC-sensitized mice was significantly attenuated by anti-IL-17A mAb treatment (Fig. 5D). These data suggest that IL-17A is critical for the neutrophil influx into the lung and airway and contributes to the increased responsiveness to methacholine after antigen-inhalation challenge of OVA EC-sensitized mice.
Skin-Derived Dendritic Cells (DCs) Express Increased Amounts of IL-23 and Selectively Induce IL-17 Production in Naïve T Cells.
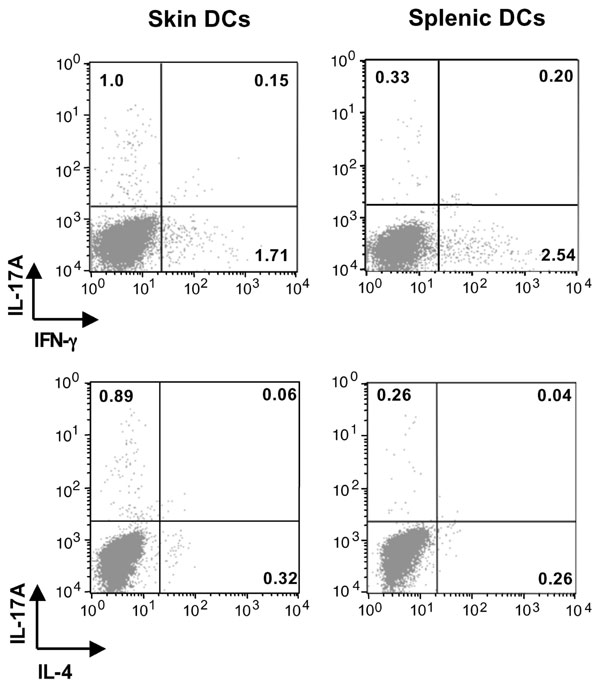
Skin DCs capture antigen and subsequently traffick to regional draining LNs where they present antigens and prime naïve T cells into effector cells (23, 24). To examine the role of skin-derived DCs in the induction of IL-17 after EC sensitization with antigen, we applied FITC to shaved and tape-stripped skin and isolated CD11c+FITC+ from the draining LN 24 h later. We used FITC rather than OVA-FITC because application of FITC results in a higher percentage of FITC+CD11c+ in the draining LN compared with application of OVA-FITC (9.4 ± 1.8% versus 3.9 ± 1.4%). These CD11c+FITC+ DCs, which represent recent emigrants from the skin, were compared with CD11c+ FITC+ DCs isolated from spleens of mice i.p.-injected with FITC for their capacity to induce cytokine secretion by naïve DO11.10 CD4+ cells after stimulation with OVA323–339 peptide. Fig. 6A shows that skin-derived CD11c+FITC+ DCs induced robust IL-17A production by DO11.10 CD4+ T cells. In contrast, splenic CD11c+FITC+ DCs induced only modest IL-17A production. Skin-derived and splenic CD11c+FITC+ DCs elicited comparable production of IL-4, IL-13, and IFN-γ. The selective ability of skin-derived DCs to induce IL-17A production was confirmed by intracellular staining of IL-17A, IFN-γ, and IL-4 in CD4+ T cells (SI Fig. 8). In three experiments, the percentage of CD4+IL-17+ cells in cultures containing skin-derived DCs was 0.93 ± 0.15% compared with 0.28 ± 0.02% cells in cultures containing splenic DCs (P < 0.002). In contrast, the percentage of CD4+IFN-γ+ and of CD4+IL-4+ cells was not significantly different between the two groups (data not shown). There was no difference in the ability of skin-derived and splenic DCs to support the proliferation of DO11.10 CD4+ T cells to either OVA peptide (Fig. 6B) or OVA (data not shown), indicating that the difference in the ability of skin-derived and splenic DCs in inducing IL-17 is not the result of difference in their ability to process or present antigen.
Fig. 6.

Selective induction of IL-17A secretion in DO11.1 CD4+ cells by skin-derived DCs. (A) Secretion of IL-17A, IL-4, IL-13, and IFN-γ by DO11.1 CD4+ cells stimulated with OVA323–339 peptide in the presence of skin- or spleen-derived DCs. (B) Proliferation; columns, and error bars represent means of triplicate wells. (C) Q-PCR analysis of mRNA expression of IL-23/p19, TGFβ, IL-6, and IL-12/p35 in spleen- and skin-derived DCs. Values for splenic DCs have been set to 1. (D) Effect of neutralizing antibodies to IL-23p19, TGFβ, and IL-6 on the capacity of skin-derived DCs to induce secretion of IL-17A, IL-4, and IFN-γ by DO11.1 CD4+ stimulated with OVA323–339 peptide. Except for B, columns and error bars represent means ± SEM of three experiments (n = 3); in B, they represent mean ± SEM of triplicates of one experiment representative of three. *, P < 0.05; **, P < 0.01.; ***, P < 0.0001
TGFβ and IL-6 are important for the induction of IL-17, whereas IL-23 is important for the expansion of Th17 cells (4–6). We compared the expression of these three cytokines in skin-derived and splenic CD11c+FITC+ DCs using quantitative PCR. Fig. 6C shows that skin-derived DCs expressed significantly more the IL-23/p19 mRNA than splenic DCs (3-fold more) but no significantly different levels in TGFβ and IL-6 mRNA. In contrast, skin-derived and splenic DCs expressed comparable levels of IL-12/p35 (Fig. 6C). To determine whether IL-23 plays a role in the induction of IL-17 by skin-derived DCs, we examined the effect of IL-23 neutralizing Ab on IL-17 production by DO11.10 CD4+ T cells stimulated with OVA323–339 peptide in the presence of skin-derived FITC+ DCs. Fig. 6D shows that IL-23-neutralizing Ab significantly inhibited IL-17 production. As expected, neutralizing Abs to TGFβ and IL-6 also significantly inhibited IL-17 production. In contrast, none of the Abs had a significant effect on IL-4 or IFN-γ production (Fig. 6D) or on T cell proliferation (data not shown).
Induction of IL-17 by Skin-Derived DCs Is Inhibited by Intradermal (i.d.) Injection of Anti-TGFβ.
TGFβ is expressed in skin keratinocytes and its expression is up-regulated in human skin after tape-stripping (25, 26). We found a 4-fold increase in TGFβ expression 8 h after tape-stripping of mouse skin (data not shown). We considered the possibility that skin-derived TGFβ may play a role in promoting the ability of skin-derived DCs to induce Th17 cells. To investigate this possibility, anti-TGFβ mAb or control IgG1 mAb was injected i.d. 24 h before FITC painting of the skin. CD11c+FITC+ DCs were isolated 24 h later from draining LNs and examined for their capacity to induce IL-17A production in DO11.10 CD4+ T cells. I.d. injection of anti-TGFβ mAb significantly inhibited the capacity of skin-derived DCs to induce IL-17A production by naïve T cells, but had no effect on their capacity to induce production of IL-4 and IFN-γ or T cell proliferation (SI Fig. 9). I.d. injection of anti-TGFβ mAb had no effect on the recovery of CD11c+FITC+ cells from draining LNs and had no effect on TGFβ mRNA expression by these DCs (data not shown). Tape-striping causes up-regulation of IL-6 mRNA expression in skin (data not shown). However, i.d. injection of anti-IL-6 had no detectable effect on the capacity of skin-derived DCs to induce IL-17A production by naïve T cells (data not shown). These results suggest that skin TGFβ contributes to the ability of skin-derived DCs to polarize naïve T cells toward IL-17 secretion.
Discussion
We demonstrate that EC sensitization selectively induces a systemic Th17 response to antigen and results in airway inflammation characterized by IL-17 expression and neutrophil influx after inhalation challenge. Patients with AD often develop asthma, and our findings indicate that IL-17 links cutaneous sensitization to lung inflammation elicited by inhalation of the same antigen.
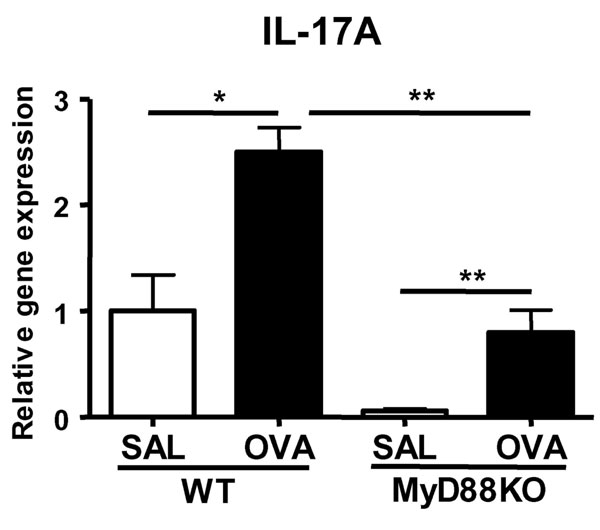
EC-sensitization resulted in a local and systemic Th17 response. OVA-sensitized skin expressed IL-17A and IL-17F mRNA in addition to IL-4 and IL-13 mRNA (Fig. 1 A and B). Increased expression of IL-17 in the skin may be responsible, at least in part, for the presence of neutrophils that we previously reported in OVA-sensitized skin in our model (18). CCR4 is the major skin-homing receptor on mouse T cells (27). We have found impaired expression of IL-17 mRNA in OVA-sensitized skin of CCR4−/− mice (data not shown), suggesting that CCR4 may be expressed on Th17 cells and governs their homing to the skin. Both IL-1 released from keratinocytes after skin injury and Toll-like receptor engagement by products of skin-colonizing microbes may play a role in the induction of a Th17 response to EC sensitization. Indeed, we have found that there was significantly less IL-17A expression in OVA-sensitized skin of MyD88−/− mice (SI Fig. 10).
Draining LN cells and splenocytes from EC-sensitized mice produced IL-17A in response to antigenic stimulation in vitro (Figs. 1C and 2 A and B). In contrast, there was little IL-17A secretion by mesenteric LN or splenocytes from i.p.-immunized mice. Increased IL-17 secretion was not the result of a more potent immune response to EC sensitization, because draining LN cells and splenocytes of EC- and i.p.-sensitized mice secreted comparable amounts of IFN-γ and IL-4 (Figs. 1C and 2A). Because IL-17 production can be down-regulated by both IFN-γ and IL-4 (28), this finding suggests that the selective induction of a Th17 response by EC sensitization is not due to decreased production of Th1 and Th2 cytokines after this mode of sensitization. Serum IL-17A levels were elevated in OVA EC-sensitized mice, but not in OVA i.p.-immunized mice (Fig. 2C), indicating that IL-17A was being produced in vivo. Intriguingly, serum IL-17A levels were elevated in saline EC-sensitized mice, albeit less than in OVA controls (Fig. 2C). It is possible that IL-17A production in saline EC-sensitized mice was in response to skin injury and release of danger signals that may include heat shock proteins and/or microbial flora antigens that entered via tape-stripped skin. In addition, IL-6 and IL-23 expression are strongly up-regulated after tape-stripping (data not shown).
Antigen-inhalation challenge caused increased mRNA expression of IL-17A and IL-17F in lungs of OVA EC-sensitized mice but not in lungs of OVA i.p.-immunized mice (Fig. 3A). Similarly, secretion of IL-17A protein predominated in OVA-stimulated lung cells from EC-sensitized mice (Fig. 3B). In contrast, there was little increase in IL-17A secretion by OVA-stimulated lung cells from i.p. sensitized mice, consistent with a recent report on s.c.-immunized mice challenged by inhalation of antigen (13). The presence of IL-17A in BALF from EC-sensitized mice also indicates that IL-17A protein was produced in vivo (Fig. 3D).
Although draining LN cells and splenocytes from EC-sensitized and i.p.-immunized mice secreted equivalent amounts of IL-4 and IFN-γ in response to OVA stimulation (Figs. 1C and 2A), expression and secretion of Th2 (IL-4 and IL-13) and Th1 (IFN-γ) cytokines was blunted in the lungs of EC-sensitized mice compared with lungs of i.p.-immunized mice (Fig. 3 A and B). This is consistent with the observation that administration of IL-17 in the airway inhibits Th2 cytokine production in the lungs of s.c.-immunized mice (13). In addition, expression of CCL11 mRNA, which depends on IL-4 and IL-13, was also reduced in lungs of EC-sensitized mice and may explain the decreased eosinophil accumulation in BALF of EC-sensitized mice.
The Th17-dominated response of lungs from EC-sensitized mice to antigen inhalation was associated with increased expression of the neutrophil chemokine CXCL2, increased neutrophils in the BALF, and a peribronchial infiltration with neutrophils and a relative paucity of eosinophils (Fig. 4). Neutrophilia in the BALF of EC-sensitized mice was reported by Herrick et al. (29). A role for IL-17 in the recruitment of neutrophils to the airways and lungs of EC-sensitized mice was established by our demonstration that treatment of EC-sensitized mice with anti-IL-17A mAb before inhalation challenge resulted in a significantly decreased mRNA expression of CXCL2 and, more importantly, in a significant reduction in BALF neutrophilia (Fig. 5 A and C). Treatment of EC-sensitized mice with anti-IL-17A mAb was also associated with substantial reduction in peribronchial inflammation and attenuation of the increased responsiveness to methacholine after inhalation challenge (Fig. 5 B and D). Administration of anti-IL-17A mAb did not alter IL-4 and IL-13 expression in the lung (Fig. 5C), possibly because of incomplete IL-17 neutralization. Thus, the decrease in inflammation and responsiveness to methacholine caused by anti-IL-17 mAb was not due to decreased expression of these two cytokines.
Our results suggest that the selective ability of EC sensitization to elicit a Th17 response may be due to the selective ability of skin-derived DCs to induce IL-17A production by naïve T cells. FITC+ DCs isolated from LN that drain FITC-painted skin selectively induce a robust IL-17A response in naïve DO11.1 CD4+ T cells (Fig. 6A). FITC+ DCs are likely to represent recent skin emigrants, although we cannot rule out that some may have been DCs resident in the LNs that acquired FITC draining through afferent lymphatic channels or from bona fide skin DCs emigrants. In contrast, splenic DCs induced markedly less IL-17A production by the same CD4+ T cells. The selective ability of skin-derived DCs to induce IL-17 production was not simply the result of a general enhancement of the activity of these DCs, because skin-derived and splenic DCs caused comparable T cell proliferation and secretion of Th1 and Th2 cytokines (Fig. 6 A and B).
Skin-derived DCs expressed significantly more IL-23/p19 mRNA than splenic DCs but roughly comparable amounts of TGFβ and IL-6 mRNA (Fig. 6C). This suggests that the selective ability of skin-derived DCs to induce IL-17 may be due to their increased expression of IL-23. Anti-IL-23 antibody inhibited the ability of skin DCs to induce IL-17A secretion by naïve T cells, with no effect on IL-4 or IFN-γ secretion (Fig. 6D) and little effect on proliferation. This finding suggests that increased IL-23 expression by skin-derived DCs contributes to their ability to induce Th17 cells.
The selective ability of skin-derived DCs to induce Th17 cells, was found to depend, in part, on skin TGFβ. Intradermal injection of anti-TGFβ mAb significantly inhibited the ability of FITC+ DCs isolated from lymph nodes that drain FITC-painted skin to induce intracellular expression and secretion of IL-17A (SI Fig. 9A). In human AD lesions, and in our EC-sensitization model, there is evidence of dermal infiltration of eosinophils, which express TGFβ (30, 31). This raises the possibility that introduction of antigen into lesional skin may further promote a Th17 response. The finding that i.d. injection of an anti-IL-6 mAb had no effect on the ability of skin-derived DCs to drive IL-17 production is possibly explained by the fact that IL-6 is also produced by cells in the draining LNs, including DCs. IL-6 in LNs may not be accessible to neutralization by intradermally injected antibody.
AD patients suffer from defects in skin barrier function, associated in some with mutations in the filaggrin gene, a member of the epidermal differentiation complex (32, 33). EC sensitization with environmental allergens is thought to play an important role in allergic sensitization in AD patients (14), and the presence of a mutation in filaggrin significantly increases the risk for asthma in these patients (32). Skin-barrier defects and mechanical skin injury inflicted by scratching in AD are mimicked by tape-stripping in our model. Thus, our findings that sensitization via the skin is a powerful inducer of the Th17 response and that this Th17 response incites airway inflammation in response to inhalation of the same antigen have important implications for patients with AD. They also suggest that IL-17 neutralization may be helpful for the treatment of asthma, which commonly develops in AD patients.
Materials and Methods
Mice and Sensitization.
BALB/c mice were obtained from Charles River Laboratories (Wilmington, MA). DO11.10 TCR transgenic mice on BALB/c background were obtained from The Jackson Laboratory (Bar Harbor, ME). EC sensitization of 8- to 10-week-old female mice was performed as described (18). For i.p. immunization, mice were injected with 50 μg of OVA in alum or alum alone on days 0 and 14.
Cell Preparation and Culture.
Single-cell suspensions from spleen, regional draining LNs, and lung cells were cultured in the presence of OVA (100 μg/ml).
Intracelluar Cytokine Staining and Cytokine ELISA.
These processes were performed as described (2).
Quantitative PCR Analysis of Cytokines.
Quantitative real-time PCR was done as described (20).
Airway Challenge, Analysis of BAL Fluid, and Histological Analysis of Lung.
All processes were performed as described (20). For EC-sensitized mice, Penh was measured as described (21). For blockade of endogenous IL-17, anti-IL-17A mAb, or control rat IgG2a (both from R & D Systems) (100 μg/mouse) was i.v. administered twice during the last week of EC sensitization.
Isolation and Functional Analysis of Skin DC Emigrants and Splenic DCs.
The dorsal skin of anesthetized mice was shaved. FITC (10 mg/ml) was applied in 100-μl aliquots. Cell suspensions of axillary and inguinal LN were prepared 24 h later. FITC+CD11c+ DCs cells were sorted by FACS. Splenic CD11c+ DCs were purified by Auto MACS. In selected experiments, FITC was injected i.p., and FITC+CD11c+ DCs were isolated from spleens 24 h later and used as controls, with results similar to those obtained with total splenic DCs. CD4+ T cells were purified from spleen of DO11.10 T cell receptor mice. FITC+CD11c+ cells or splenic CD11c+ DCs were cocultured in triplicate at a 1:10 ratio with DO11.1 CD4+ T cells stimulated with OVA323–339 peptide (4 μM; Sigma–Aldrich) for 3 days. Wells were pulsed with 1 μCi [3H] thymidine (1 Ci = 37 GB) for proliferation measurement.
Neutralizing antibodies for mouse anti-TGFβ mAb (clone1D11), goat anti-IL-23/p19 (both from R & D Systems), and rat anti-IL-6 mAb (clone MP5–20F3) (eBioscience) were used at 15 μg/ml in vitro. Control cultures received a 1:1:1 mix of mouse IgG1 mAb, goat IgG, and rat IgG1 mAb control. For in vivo neutralization anti-TGFβ mAb (20 μg in 100 μl of PBS) or mouse IgG1 isotype control (both from R & D Systems), were i.d. injected 30 min or 24 h before FITC application, with similar results.
Statistics.
Statistical significance was determined by using the Student's t test for unpaired data and two-way ANOVA for Penh results. A P value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Hans Oettgen for reading the manuscript and for suggestions, Sho Goya for assistance with airway-responsiveness measurement, Dale Umetsu for useful discussions, and Tatyana Sannikova for assistance with i.v. antibody injection. This work was supported by U.S. Public Health Service Grant AR-047417.
Abbreviations
- AD
atopic dermatitis
- BAL
bronchoalveolar lavage
- BALF
BAL fluid
- DC
dendritic cell
- EC
epicutaneous
- LN
lymph node
- OVA
ovalbumin
- Penh
enhanced pause.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0706942104/DC1.
References
- 1.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, et al. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 3.Kolls JK, Kanaly ST, Ramsay AJ. Am J Respir Cell Mol Biol. 2003;28:9–11. doi: 10.1165/rcmb.2002-0255PS. [DOI] [PubMed] [Google Scholar]
- 4.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Veldhoen M, Stockinger B. Trends Immunol. 2006;27:358–361. doi: 10.1016/j.it.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 6.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, Boulet LP, Hamid Q. J Allergy Clin Immunol. 2003;111:1293–1298. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 9.Wong CK, Ho CY, Ko FW, Chan CH, Ho AS, Hui DS, Lam CW. Clin Exp Immunol. 2001;125:177–183. doi: 10.1046/j.1365-2249.2001.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bullens DM, Truyen E, Coteur L, Dilissen E, Hellings PW, Dupont LJ, Ceuppens JL. Respir Res. 2006;7:135–143. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barczyk A, Pierzchala W, Sozanska E. Respir Med. 2003;97:726–733. doi: 10.1053/rmed.2003.1507. [DOI] [PubMed] [Google Scholar]
- 12.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 13.Schnyder-Candrian S, Togbe D, Couillin I, Mercier I, Brombacher F, Quesniaux V, Fossiez F, Ryffel B, Schnyder B. J Exp Med. 2006;203:2715–2725. doi: 10.1084/jem.20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leung DY. J Allergy Clin Immunol. 2000;105:860–876. doi: 10.1067/mai.2000.106484. [DOI] [PubMed] [Google Scholar]
- 15.Jones SM, Sampson HA. Clin Rev Allergy. 1993;11:471–490. [PubMed] [Google Scholar]
- 16.Tan B, Weald D, Strickland I, Friedmann P. Lancet. 1996;347:15–18. doi: 10.1016/s0140-6736(96)91556-1. [DOI] [PubMed] [Google Scholar]
- 17.Toda M, Leung DY, Molet S, Boguniewicz M, Taha R, Christodoulopoulos P, Fukuda T, Elias JA, Hamid QA. J Allergy Clin Immunol. 2003;111:875–881. doi: 10.1067/mai.2003.1414. [DOI] [PubMed] [Google Scholar]
- 18.Spergel J, Mizoguchi E, Brewer J, Martin T, Bhan A, Geha R. J Clin Invest. 1998;101:1614–1622. doi: 10.1172/JCI1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spergel JM, Mizoguchi E, Oettgen H, Bhan AK, Geha RS. J Clin Invest. 1999;103:1103–1111. doi: 10.1172/JCI5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.ElKhal A, Pichavant M, He R, Scott J, Meyer E, Goya S, Geha RS, Umetsu D. J Clin Immunol. 2006;118:1363–1368. doi: 10.1016/j.jaci.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 21.Ma W, Bryce PJ, Humbles AA, Laouini D, Yalcindag A, Alenius H, Friend DS, Oettgen HC, Gerard C, Geha RS. J Clin Invest. 2002;109:621–628. doi: 10.1172/JCI14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, et al. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robert C, Kupper TS. N Engl J Med. 1999;341:1817–1828. doi: 10.1056/NEJM199912093412407. [DOI] [PubMed] [Google Scholar]
- 24.Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, et al. Immunity. 2005;22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 25.Nickoloff BJ, Naidu Y. J Am Acad Dermatol. 1994;30:535–546. doi: 10.1016/s0190-9622(94)70059-1. [DOI] [PubMed] [Google Scholar]
- 26.Streilein JW, Grammer SF, Yoshikawa T, Demidem A, Vermeer M. Immunol Rev. 1990;117:159–183. doi: 10.1111/j.1600-065x.1990.tb00572.x. [DOI] [PubMed] [Google Scholar]
- 27.Baekkevold ES, Wurbel MA, Kivisakk P, Wain CM, Power CA, Haraldsen G, Campbell JJ. J Exp Med. 2005;201:1045–1051. doi: 10.1084/jem.20041059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong C. Nat Rev Immunol. 2006;6:329–333. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- 29.Herrick CA, MacLeod H, Glusac E, Tigelaar RE, Bottomly K. J Clin Invest. 2000;105:765–775. doi: 10.1172/JCI8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiehl P, Falkenberg K, Vogelbruch M, Kapp A. Br J Dermatol. 2001;145:720–729. doi: 10.1046/j.1365-2133.2001.04456.x. [DOI] [PubMed] [Google Scholar]
- 31.Williams TJ. J Clin Invest. 2004;113:507–509. doi: 10.1172/JCI21073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, Goudie DR, Sandilands A, Campbell LE, Smith FJ, et al. Nat Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 33.Smith FJ, Irvine AD, Terron-Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, Liao H, Evans AT, Goudie DR, Lewis-Jones S, et al. Nat Genet. 2006;38:337–342. doi: 10.1038/ng1743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}