

Summary
Bcl-3 is an IκB-family member. Unlike the classical, cytoplasmic IκBs, Bcl-3 does not inhibit RelA- or c-Rel-containing NF-κB transcription factor dimers. Instead, Bcl-3 can enter nuclei and may enhance NF-κB activity, although underlying mechanisms and physiologic functions remain largely unknown. Here we unexpectedly identify Bcl-3 as a regulator of immunologic tolerance to self. In parallel with NF-κB2, Bcl-3 functions within stroma to generate medullary thymic epithelial cells, which are essential for negative selection of autoreactive T cells. Loss of both NF-κB2 and Bcl-3, but not either one alone leads to a profound breakdown in central tolerance resulting in rapid and fatal multi-organ inflammation. These data reveal extensive utilization of the NF-κB system to promote central tolerance in the thymus, in apparent contrast with the well-known roles of NF-κB to promote inflammation and autoimmunity in the periphery.
Introduction
The NF-κB transcription factor family is central to host defense against pathogenic insults. NF-κB functions as a primary intracellular mediator of numerous innate, inflammatory and adaptive immune signals, including signals received through Toll receptors, inflammatory cytokine receptors as well as antigen receptors on T and B cells (Hayden and Ghosh, 2004). More recently NF-κB has been recognized as well for its essential roles during development and maintenance of the immune system, prior to and apparently independent of encounter with pathogens. For example, NF-κB factors are required during the development of lymphocytes, largely, though not only, because they assure the cells’ survival (Claudio et al., 2002; Claudio et al., 2006; Franzoso et al., 1997b; Siebenlist et al., 2005).
NF-κB factors are also required for proper development and function of secondary lymphoid organs as well. Mice deficient in RelB, NF-κB2 or NF-κB inducing kinase (NIK) or which have inactivating mutations in NIK (aly/aly mice) or in IκB kinase (IKK)α(IKK1) display an overlapping spectrum of defects in structure and function of secondary lymphoid organs (Franzoso et al., 1998; Karrer et al., 2000; Matsumoto et al., 1999; Matsushima et al., 2001; Paxian et al., 2002; Senftleben et al., 2001; Weih and Caamano, 2003; Yamada et al., 2000; Yilmaz et al., 2003; Yin et al., 2001). These mutant mice fail to form proper B cell follicles and differentiated follicular dendritic cell networks (FDCs), and, upon challenge, they fail to form proper germinal centers in spleens. They also lack Peyer’s patches and, depending on the knockout they may lack some or all lymph nodes (see below). These deficiencies are due in large part to impaired stromal cell functions (see above references). RelB, NF-κB2, NIK, and IKKαare all components of the non-classical pathway for NF-κB activation. It then follows that the non-classical pathway in stromal cells must be essential for proper lymphoid organogenesis. In this pathway, signal-activated NIK phosphorylates and activates IKKα (IKK1), which in turn phosphorylates p100/NF-κB2 to initiate processing of p100 to p52, thereby liberating p52/RelB dimers to translocate to the nucleus and modulate gene expression (Bonizzi and Karin, 2004; Claudio et al., 2002; Dejardin et al., 2002; Hayden and Ghosh, 2004; Muller and Siebenlist, 2003). p100 is the primary IκB-like inhibitor of RelB in the cytoplasm and its processing relieves inhibition and generates p52/RelB dimers. p100 processing may also liberate other NF-κB heterodimers, such as p50/RelA, which can be trapped by association with the p100 inhibitor in a fashion somewhat analogous to sequestration of p50/RelA by its primary inhibitor IκBα (Basak et al., 2007; Kanno et al., 1994)
Lymphotoxin β receptors (LTβR) are expressed primarily on stromal cells and upon stimulation engage the non-classical pathway for NF-κB activation (Basak et al., 2007; Dejardin et al., 2002; Muller and Siebenlist, 2003). Consistent with a role for LTβR-mediated activation of the non-classical pathway in stromal cells during lymphoid organogenesis, mutant mouse models deficient in LTβR or its main ligand, (LTα)1 (LTβ)2, have defects similar to those described above for mice lacking functional RelB, NIK (aly/aly) or IKKα(Hehlgans and Pfeffer, 2005; Matsumoto et al., 1999; Matsushima et al., 2001; Pfeffer, 2003; Tumanov et al., 2003; Yilmaz et al., 2003). Even though the non-classical pathway is disrupted in NF-κB2 deficient mice, loss of this factor results in a significantly milder phenotype. For example, lymph node structures are present, albeit hypocellular, and mesenteric lymph nodes appear entirely normal in mice lacking NF-κB2 (Franzoso et al., 1998; Weih and Caamano, 2003).
Mice blocked in signaling via the LTβR or via the non-classical pathway share defects in addition to those associated with lymphoid organogenesis. Mice deficient in RelB, NIK (aly/aly) and, possibly to a lesser degree, in LTβR signaling display thymic defects and develop significant multi-organ lymphocytic infiltrations early in life (Boehm et al., 2003; Burkly et al., 1995; Chin et al., 2003; Hehlgans and Pfeffer, 2005; Kajiura et al., 2004; Kinoshita et al., 2006; Pfeffer, 2003; Shinkura et al., 1999; Tsubata et al., 1996; Weih et al., 1995; Wu et al., 1998). In the case of RelB knockouts the inflammatory condition can be fatal as early as 7-8 weeks of age, although there is wide range and mice may survive for many more months. Such pathology is not seen in NF-κB2-deficient mice, which develop only mild organ infiltrations relatively late in life, without any effect on their life span (Caamano et al., 1998; Franzoso et al., 1998; Zhang et al., 2006; Zhu et al., 2006). It is likely that NF-κB2-deficient mice are largely protected from severe disruption of lymphoid organogenesis and profound inflammatory pathology because they retain the ability to activate RelB, even though they lack the non-classical pathway. The reason is that NF-κB2 knockouts are deficient in the p100 precursor, the main inhibitor of RelB; in its absence p50/RelB dimers are free to enter nuclei where they may partially compensate for loss of RelB dimers (including p52/RelB) activated via the non-classical pathway (Claudio et al., 2002; Derudder et al., 2003; Muller and Siebenlist, 2003). Also, other NF-κB dimers may be more readily activated in the absence of p100 (see above) (Basak et al., 2007; Kanno et al., 1994). Therefore, while the actual signal transduction via the non-classical pathway is blocked in NF-κB2-deficient cells, loss of this protein does not completely eliminate all consequences of non-classical signaling.
In addition to the mutant mouse models discussed here, Bcl-3 deficient mice display defects in secondary lymphoid organs (Franzoso et al., 1997a; Paxian et al., 2002; Poljak et al., 1999). These defects are partly similar to, but even milder than those observed in NF-κB2 knockouts. Bcl-3 is structurally related to the IκBα, β and εinhibitors, but is distinct in several critical ways. Bcl-3 is often found in the nucleus, contains domains that can stimulate transcription, interacts predominantly with p50 and p52 homodimers and has been reported to directly or indirectly transactivate or repress gene expression via κB-elements (Bours et al., 1993; Franzoso et al., 1993; Franzoso et al., 1992; Fujita et al., 1993; Hayden and Ghosh, 2004). Bcl-3 has also been implicated in various gene regulatory models and biologic scenarios (Corn et al., 2005; Kashatus et al., 2006; Kim et al., 2005; Leung et al., 2004; Massoumi et al., 2006; Mathas et al., 2005; Mitchell et al., 2002; Riemann et al., 2005; Thornburg et al., 2003; Viatour et al., 2005; Wessells et al., 2004; Westerheide et al., 2001), but its actual physiologic mechanism(s) of action and in vivo targets remain essentially unknown. Nevertheless, the Bcl-3 knockout phenotype does suggest a contribution of this protein in stromal cells of secondary lymphoid organs. Since Bcl-3 is not known to have a direct role in the non-classical NF-κB signal activation pathway, the available evidence suggests two basic models whereby Bcl-3 might influence secondary lymphoid organogenesis: Bcl-3 could modulate NF-κB activity via direct interaction with p52/NF-κB2, or Bcl-3 could function in a pathway separate from, but partially redundant with NF-κB2. To address these possibilities, we generated mice deficient in both proteins. In addition to a profound block in secondary lymphoid organogenesis, the double knockout mice unexpectedly developed severe lymphocytic infiltrates in multiple organs, a condition to which they succumbed within a few weeks of birth. Therefore, Bcl-3 and NF-κB2 have redundant biologic effects that are revealed only in the absence of both proteins. We further demonstrate that autoreactive T cells mediate the lethal inflammatory condition in double knockout mice, and that generation of autoreactive T cells results from defects intrinsic to thymic stromal cells. The results suggest the involvement of multiple components of the NF-κB system to enforce central tolerance to self from within stromal cells.
Results
Novel Phenotypes in Mice with Deficient in NF-κB2 and Bcl-3
We generated NF-κB2, Bcl-3 double knockout mice to investigate whether Bcl-3 interacts with NF-κB2 to modulate its activity or whether these proteins function in distinct pathways that may nevertheless converge on some common targets. Mice deficient in either one of these proteins had fully normal life spans and presented with limited phenotypic defects, mostly resulting from mildly impaired secondary lymphoid organogenesis, though NF-κB2 knockouts (but not Bcl-3 knockouts) in addition developed mild inflammation in some organs later in life (see Introduction). Unexpectedly, loss of both NF-κB2 and Bcl-3 (double knockout (dKO) mice) proved fatal by 4 weeks of age (BL/6 background; 3–7 weeks on mixed background). These dKO mice were also small in size, which became apparent within 2 weeks of birth. By the time of weaning the mutant mice began to develop ruffled fur, scaly skin, a hunched posture and squinting eyes, among other symptoms (Figure 1a). Further analysis revealed an absence of all lymph nodes, including mesenteric lymph nodes, and gradual loss of the thymus, beginning by about 2 weeks after birth (Figure 1b, 1c). Singly deficient mutants had a normal-sized thymus and contained lymph nodes. Although most lymph nodes were reduced in cellularity in NF-κB2 knockouts, mesenteric lymph nodes appeared completely normal in both single knockouts (Franzoso et al., 1998; Franzoso et al., 1997a; Paxian et al., 2002; Poljak et al., 1999; Weih and Caamano, 2003). The loss of NF-κB2 plus Bcl-3 therefore generated a new phenotype, supporting the notion that Bcl-3 and NF-κB2 must have some redundant functions, working independently of each other.
Figure 1.
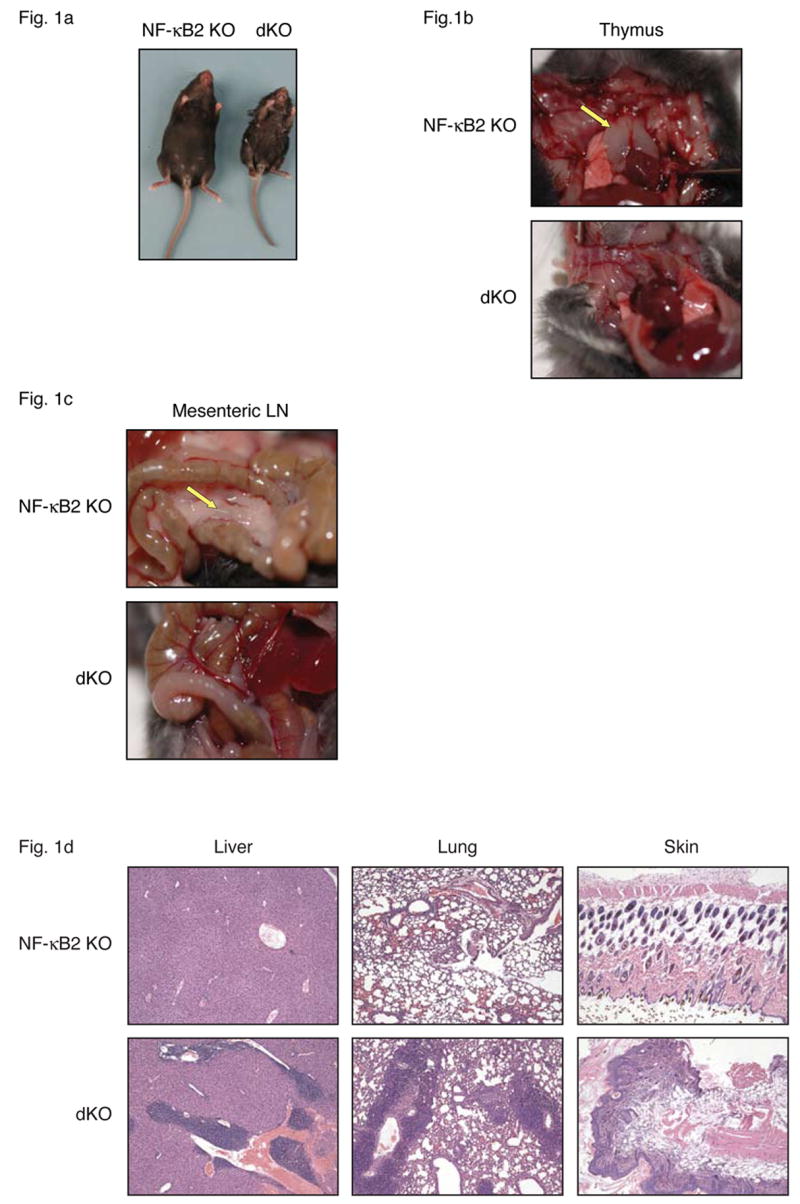
Bcl-3, NF-κB2 double knockout (dKO) mice lack lymph nodes and develop severe multi-organ inflammation. (a) Gross appearance of NF-κB2 KO and dKO mice at almost 4 weeks of age. The dKO mice lack thymi (b) and mesenteric lymph nodes (c) at time of death. Arrows indicate location of thymic lobes and mesenteric lymph node (mLN) in NF-κB2 KO mice. (d) Tissue sections from liver, lung and skin of nearly 4-week-old NF-κB2 KO and dKO mice were stained with hematoxylin and eosin (H&E).
The rapid death of NF-κB2 and Bcl-3 doubly deficient mice was surprising and implied activities of these proteins in addition to those involved in secondary lymphoid organogenesis. The appearance of the mice suggested an inflammatory condition, which was confirmed by tissue analyses. Examination of paraffin-embedded, hematoxylin and eosin (H&E) stained tissue sections from liver, lung and skin revealed inflammatory infiltrates (Figure 1d), which contained many T cells (not shown). We therefore examined the state of splenic T cells in 3–4 week-old double knockout mice. Splenocytes from these mice contained relatively more T cells because mature B cell numbers were severely reduced (Figure 2a; the majority of CD4-CD8- cells are B cells). Among T cells there was a dramatic increase in the relative amounts of CD44+CD62L- effector/memory cells in both the CD4 and CD8 compartments when compared to NF-κB2 single knockout controls (Bcl-3 single knockouts were similar to the NF-κB2 knockout controls; not shown). The double knockout T cell population also contained more activated cells as judged by increased expression of the activation markers CD69 and CD25 (not shown). The increase in effector/memory (and activated) T cells attested to an ongoing immune reaction in these mice.
Figure 2.

Increased numbers of effector/memory and autoreactive T cells in spleens of Bcl-3, NF-κB2 double knockout (dKO) mice. (a) Splenocytes from 26-day-old NF-κB2 KO and dKO mice were examined with flow cytometry for the expression of CD44 and CD62L on CD4+ and CD8+ T cells, as shown. (b) CD4+ T cells purified from spleens of 26-day-old Bcl-3 KO, NF-κB2 KO and dKO mice were cultured with or without irradiated syngeneic antigen-presenting cells (APCs) (see Methods) and pulsed with [3H]thymidine. Data are the mean of triplicates.
Pathology of Double Knockout Mice Mediated by T cells
The presence of lymphocytic infiltrates in multiple organs and the pronounced increase in effector/memory T cells suggested autoimmune reactivity. To test whether T cells from doubly-deficient mutants could react with selfantigens, CD4+ T cells isolated by negative selection were incubated with wild-type antigen-presenting cells (APCs) isolated from syngeneic mice (Figure 2b). T-depleted, irradiated splenocytes served as APCs in this ‘syngeneic’ mixed lymphocyte reaction (MLR). In the absence of APCs the T cells did not proliferate, as judged by thymidine incorporation, save for a very low level of proliferation in T cells from the Bcl-3, NF-κB2 double knockout mice. In the presence of (selfantigen-presenting) syngeneic APCs, T cells isolated from double knockouts (dKOs) proliferated at a significantly higher rate than T cells from the two single knockouts. Double knockout T cells did not intrinsically proliferate better, since non-specific stimulation with anti-CD3 antibodies did not result in their preferential growth (not shown). Together these data provided further evidence for the presence of significant numbers of autoreactive T cells in doubly-deficient mice.
To demonstrate more directly that autoreactive T cells were responsible for the pathology of double knockouts we isolated CD4+ T cells from these mutant mice and injected them intravenously into sublethally irradiated Rag1 knockout mice, which bear no T or B cells of their own. The recipients showed no obvious ill effects after transfer of CD4+ T cells from NF-κB2- or Bcl-3 single knockouts, but all mice receiving CD4+ T cells from the double knockouts succumbed to the inflammatory condition 8 to 12 weeks post transfer (Figure 3a). We observed lymphocytic infiltrates in these mice, especially in lung and liver, although the inflammation appeared less than in the original doubly-deficient mutant mice (not shown).
Figure 3.
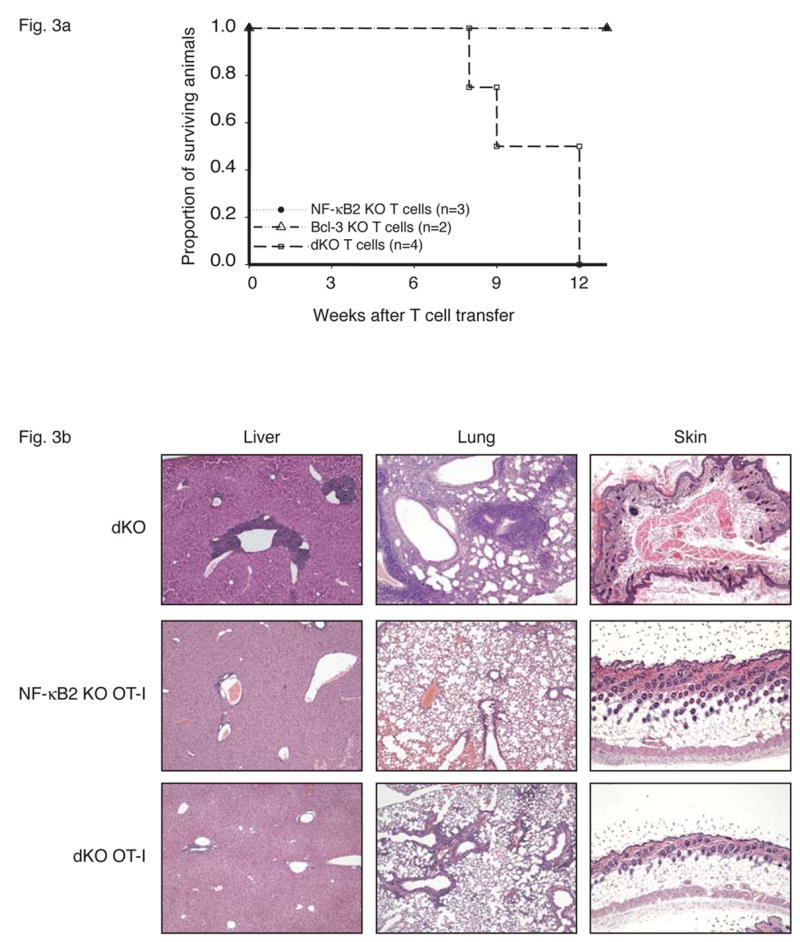
T cells mediate fatal multi-organ inflammation in Bcl-3, NF-κB2 double knockout (dKO) mice. (a) CD4+ T cells were purified from nearly 4-week-old Bcl-3 KO, NF-κB2 KO or dKO mice and transferred into sublethally irradiated rag1 −/− mice (5×106 T cells per mouse). Recipient mice were monitored daily and the surviving proportion was plotted. (p= 0.03 for transfer of dKO T cells when compared to transfer of Bcl-3 KO and NF-κB2 KO T cells). (b) The T cell receptor OT-I transgene prevents fatal multi-organ inflammation in dKO mice. Liver, lung and skin sections from nearly 4-week-old dKO and 12-week-old NF-κB2 KO OT-I and dKO OT-I mice were stained with H&E.
To confirm the requirement of (auto-) antigen-specific T cells in the development of multi-organ inflammation, we generated NF-κB2, Bcl-3 double knockouts bearing a transgene that directs expression on T cells of a class-I restricted T cell receptor (TCR) for a peptide from the chicken ovalbumin protein (OT-I transgene) (Clarke et al., 2000). Allelic exclusion in OT-I transgenic animals severely restricts the appearance of endogenous TCRs, and thus the presence of this transgene in double knockouts should largely block the appearance of autoreactive T cells and thus prevent the development of pathology. As predicted, the OT-I transgene ‘rescued’ the double knockouts and these mice showed no serious pathology even by 12 weeks of age, with only sporadic and limited infiltrates in lung and liver at that time (Figure 3b; we never observed skin infiltrates). Also shown for comparison are sections from NF-kB2 KO OT-I and from double knockout mice. The OT-I transgene also prevented the dramatic rise in effector/memory T cells (most T cells on this background are CD8; not shown). Together these data support the notion that autoantigen-specific T cells mediated the pathology of double knockouts.
T Regulatory Cells
T regulatory cells (Tregs) are critical for maintenance of peripheral tolerance to selfantigens (Ziegler, 2006). Because the NF-κB2, Bcl-3 doubly-deficient mice were unable to control autoimmune reactions we postulated that Tregs might be fewer or functionally impaired in these mice. Flow cytometric analysis of splenic T cells from 26 day-old animals showed that double deficiency of Bcl-3 and NF-κB2 did not reduce and possibly even increased their numbers of CD4+CD25+ Foxp3+ Tregs (Figure 4a; right panels). Expression of the transcription factor Foxp3 is considered to be the best marker to identify these cells (Ziegler, 2006). We also analyzed the thymus of double knockouts at 11 days of age (well before the thymus shrinks) for the presence of CD4+CD25+FoxP3+ Tregs, and noted reduced numbers of these cells at this early time relative to NF-κB2 single knockout (Figure 4a, left panels). Because peripheral Treg numbers just prior to death were not reduced, the significance of reduced numbers of Tregs in the thymus shortly after birth is unclear, but may well be significant, especially since the thymus shrinks starting at 2 weeks of age (see Discussion).
Figure 4.

Analysis of T regulatory cells (Tregs). (a) 11-day-old Bcl-3, NF-κB2 double knockout (dKO) mice have reduced numbers of CD4+CD25+Foxp3+ Tregs in thymus, while 26-day-old dKOs have above normal numbers of Tregs in spleen. Thymocytes and splenocytes were gated on CD4+CD8− and CD4+, respectively, and analyzed for expression of Foxp3 and CD25 with flow cytometry. (b) CD4+CD25+ splenic Tregs of dKO mice have near-normal suppressive activity in vitro. Wild-type CD4+ CD25− T cells were stimulated with anti-CD3 and pulsed with [3H]thymidine in the presence of syngeneic antigen-presenting cells (APCs) as well as freshly isolated CD4+CD25+ T cells (Tregs) from spleens of wild-type or dKO mice (see Methods). 2-fold dilutions of suppressive Tregs are shown, starting with a ratio of Tregs to CD25− of 2. Data are the mean of triplicates. Representative examples are shown (a, b).
To test the inhibitory activity of peripheral Tregs, wild-type CD4+ CD25- naïve splenic T cells were activated in the presence of varied amounts of CD4+ CD25+ splenic (Treg) cells from either wild-type or double knockout mice. As shown in Figure 4b, the CD25+ Tregs isolated from doubly-deficient animals did not appear to be significantly impaired in their ability to suppress anti-CD3-induced proliferation of wild-type CD25- T cells. Together these data suggest that the generation and function of peripheral Tregs is not globally defective in double knockouts, although thymus-derived Tregs may be partially impaired (additional support for this notion is presented below).
Primary Defect of NF-κB2, Bcl-3 Double Knockout Mice in Stromal Cells
As demonstrated above, (autoreactive) T cells from double deficient mice could induce serious pathology upon transfer. It remained to be determined however, whether the causative defect resided in T cells, other hematopoietic cells, stromal cells or a combination of cell types, given that two proteins were involved. To address this question we performed reciprocal bone marrow transplants. First we adoptively transferred bone marrow from NF-κB2, Bcl-3 double knockouts into lethally irradiated Rag1 knockouts (Figure 5a). In initial experiments 3 of 14 recipients of double knockout bone marrow developed serious pathology by 12 weeks post transfer, while the remaining recipients showed no significant problems even at 14 weeks. However, all of the mice receiving double knockout bone marrow experienced a transient weight loss after transfer. In control experiments, none of the Rag1 knockout mice receiving bone marrow from wild-type, NF-κB2 or Bcl-3 single knockouts developed pathology or experienced weight loss. Since whole bone marrow was transferred in these experiments, this included some already fully matured T cells. Sufficient numbers of such mature T cells might have been transferred to cause overt problems in 3 out of 14 recipients and at least transient weight loss in all recipients. To more stringently test whether hematopoietic stem cells (as opposed to already mature T cells) from double knockouts could cause pathology, we first depleted bone marrow of CD4+ and CD8+ T cells before its transfer into lethally irradiated mice, in this case NF-κB2 knockouts. None of the recipient mice developed significant pathology or experienced weight loss by 12 weeks post transfer and even later (a few mice were observed for several additional weeks) (Figure 5a). Further analysis of one of the mice receiving T-depleted bone marrow showed no inflammatory infiltrates or increases in effector/memory cells (bottom panels of Figure 5c and data not shown). Together these findings led us to conclude that hematopoietic stem cells from double knockouts were not sufficient to cause the inflammatory condition. However, it remained possible that defects intrinsic to hematopoietic cells were nevertheless necessary to develop such pathology.
Figure 5.
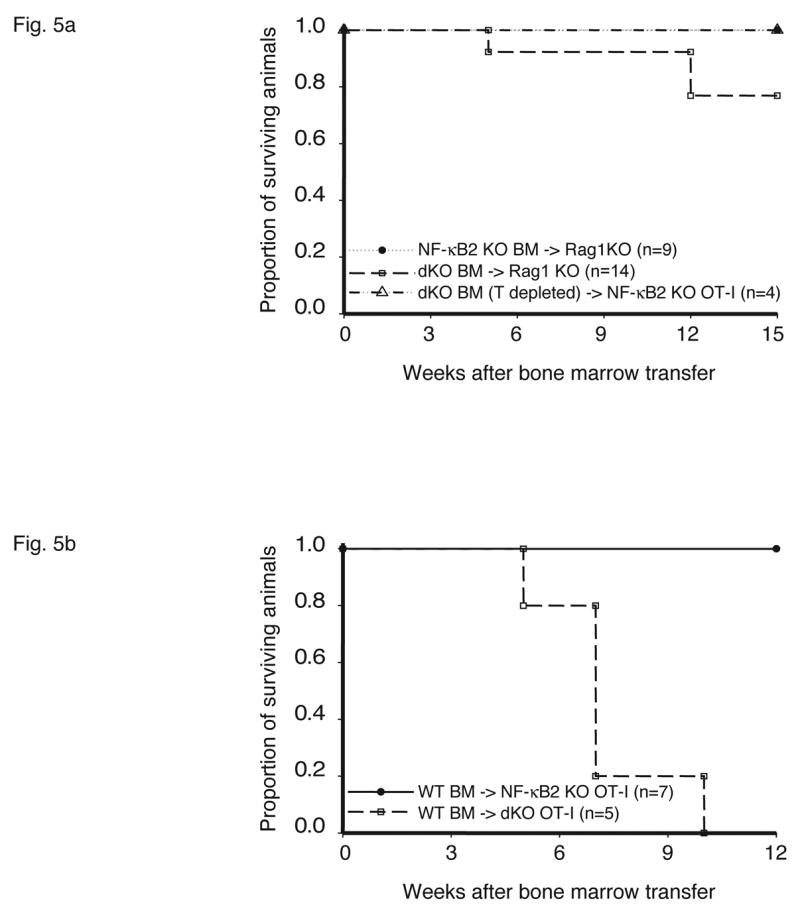
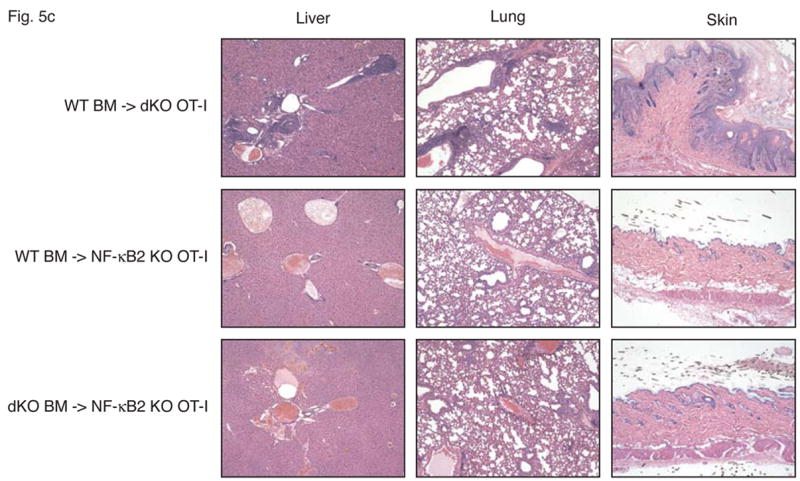
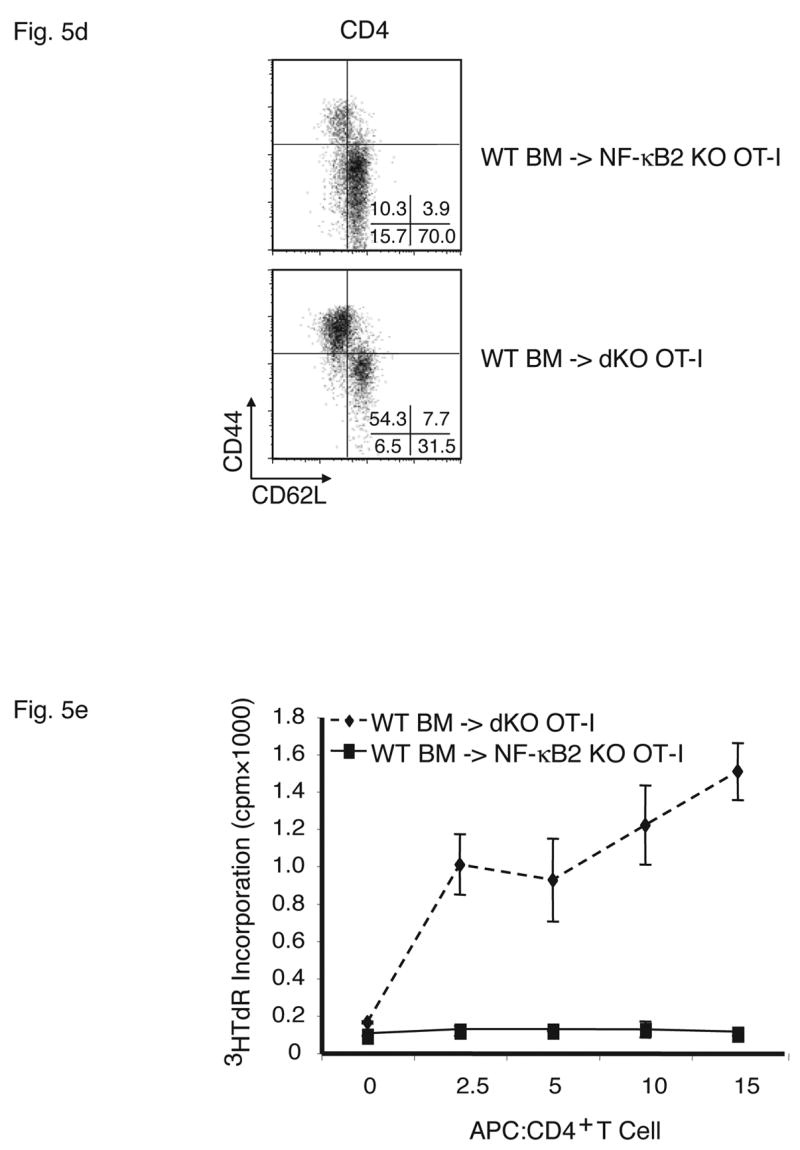
Stromal cells are responsible for fatal multi-organ inflammation and increased numbers of effector/memory T cells in Bcl-3, NF-κB2 double knockout mice (dKO). (a) Bone marrow (BM) cells from NF-κB2 KO and dKO mice were transferred into lethally irradiated rag1 −/− mice. In addition, dKO bone marrow was depleted of CD4+ and CD8+ T cells prior to transfer into NF-κB2 KO OT-I mice, as shown. (b) Wild-type (WT) BM cells were transferred into lethally irradiated NF-κB2 OT-I and dKO OT-I mice. Chimeric mice were monitored and the surviving proportion was plotted (a, b). (c) Liver, lung and skin sections from BM chimeras were stained with H&E. Genotypes of BM donors and recipients were as indicated. (d) T cells from WT BM-reconstituted NF-κB2 KO OT-I and dKO OT-I mice were gated on CD4+ and analyzed for expression of CD44 and CD62L by flow cytometry. (e) T cells from WT BM-reconstituted dKO OT-I mice show elevated autoreactivity. APC = syngeneic antigen presenting cells. The graph shows increasing ratios of APC to constant amounts of CD4+ T cells. See Figure 2b for further details. Representative examples are shown and tissues and cells were from mice 6–7 weeks post transfer (c, d, e).
To determine whether stromal cells from doubly-deficient mice were necessary and sufficient to cause multi-organ infiltrates, we performed the reciprocal experiment, transferring wild-type bone marrow into double knockout mice. While the Bcl-3, NF-κB2 double knockout mice could not serve as recipients because their health deteriorated so rapidly after birth, the OT-I transgenic double knockouts were ideal for this purpose because the TCR transgene largely prevented development of an inflammatory condition in these mice (see above). Wild-type bone marrow was transferred into lethally irradiated, Bcl-3, NF-κB2 double knockout OT-I mice, generating chimeric mice in which hematopoietic cells were genetically wild-type (and without transgene), while stromal cells were of double knockout origin. As shown in Figure 5b, all recipient mice succumbed between 5 and 10 weeks post transfer. In control transfers of bone marrow from wild-type mice into OT-I transgenic, NF-κB2 knockouts, no overt pathology was evident in any recipients. Tissue analysis of OT-I double knockouts that had received wild-type bone marrow confirmed severe inflammatory infiltrates in lung, liver and skin (Figure 5c top row of panels). OT-I, NF-κB2 knockout recipients of wild-type bone marrow (controls) did not develop this pathology, although mild infiltration of lung and liver could be observed in a few individuals (Figure 5c, middle row of panels). Consistent with these phenotypes, double knockouts with wild-type hematopoietic cells had dramatically increased CD44+CD62L- effector/memory T cell populations compared with control NF-κB2 single knockouts with wild-type hematopoietic cells (Figure 5d). The latter control chimeric mice variably exhibited somewhat elevated levels of effector/memory cells when compared to normal mice, consistent with variably observed mild inflammation in these chimeric mice, although overt, serious pathology was not observed (Figure 5d and 5c, middle row). Finally, splenic T cells from OT-I transgenic double knockouts reconstituted with wild-type bone marrow exhibited significantly higher autoreactivity than controls as judged by proliferation of T cells in a ‘syngeneic’ mixed lymphocyte reaction (see above) (Figure 5e).
Together these experiments indicated that stromal cells of NF-κB2, Bcl-3 double knockouts were sufficient to cause the T-cell mediated autoimmune condition observed in these mutant mice. Mutant hematopoietic cells, including T cells, do not appear to have intrinsic functional defects responsible for development of pathology. Rather, double knockout stromal cells shape T cells to become mediators of the autoimmune condition. It is noteworthy that loss of NF-κB2 alone resulted in a mild tendency towards development of multi-organ inflammation, especially in the transfer models, although overt pathology was never observed (see Discussion).
Primary Defect in Thymic Stroma Responsible for Pathology in Bcl-3, NF-κB2 Doubly-Deficient Mice
While stromal cells were shown to be critical for development of autoimmune pathology in double knockouts, it remained to be determined whether the defect was mediated by peripheral or by thymic stromal cells, or both. Since the doubly-deficient mutants were missing two genetic loci, it remained possible that two very different stromal cell types were affected. The thymic stroma is considered essential for negative selection of autoreactive T cells, due in part to the generation of tissue-restricted, peripheral selfantigens for presentation by these stromal cells or by cross-primed thymic dendritic cells (Anderson et al., 2005; Gallegos and Bevan, 2006; Kyewski and Klein, 2006). On the other hand, autoreactive Treg cells are not eliminated and may even be positively selected by their recognition of selfantigens generated by and displayed on stromal cells (Aschenbrenner et al., 2007; Cabarrocas et al., 2006; Kyewski and Klein, 2006; Liu, 2006). To rigorously test whether the thymic stroma was sufficient to cause pathology in Bcl-3, NF-κB2 double knockouts, we carried out fetal thymic stroma transplants into athymic nude mice. Day 14.5 embryonic fetal thymi were collected from double knockout mice or NF-κB2-deficient control mice (including NF-κB2−/ −, Bcl-3+/−), and after a week in culture in the presence of 2-deoxyguanosine to eliminate all thymocytes, one or two thymic lobes were transplanted under the renal capsule of nude mice. (The outcome of the experiment was the same regardless of whether thymic lobes originated with dKO or dKO OT-I TCR transgenic animals. The latter mice were included as a source of thymic stroma to ensure that the outcome was not due to a potential transfer of undetectable levels of surviving thymocytes. The transgene should prevent any T cells that might have been derived from the transferred thymocytes from causing pathology.) Between 5 to 8 weeks post transfer, 7 of the 9 nude mice that had been engrafted with double knockout thymus succumbed, while none of the 10 control transfers did (Figure 6a). The remaining 2 recipients of fetal thymi from double knockouts succumbed later, while controls continued to survive, free of overt, serious pathology. Consistent with these results, nude mice with double knockout thymi consistently developed significant inflammatory infiltrates in lung and liver, while those with NF-κB2 knockout thymi only sporadically developed mild infiltrates during this time frame (Figure 6b). Furthermore, splenic T cells in the nude recipients of double knockout thymi displayed a pronounced shift towards the effector/memory (CD44+CD62L-) phenotype when compared to control recipients receiving NF-κB2 knockout thymi, even though these latter transplants already resulted in somewhat elevated levels of the effector/memory cells when compared to normal wild-type mice (Figure 6c). Therefore, loss of both Bcl-3 and NF-κB2 in thymic stromal cells was sufficient to cause pathology, strongly suggesting that a single cell type was impaired by the combined loss.
Figure 6.
Engraftment of athymic nude mice with fetal thymus tissue from Bcl-3, NF-κB2 double knockout (dKO) causes multi-organ inflammation. (a) Nude mice engrafted with (T-depleted) fetal thymus lobes from NF-κB2 KO and dKO mice (with and without OT-I transgene) were monitored and the surviving proportion was plotted. (b) H&E-stained sections from liver and lung of nude mice engrafted 6 weeks prior with fetal thymus from NF-κB2 KO or dKO mice. (c) Splenic T cells from nude mice engrafted 5 weeks prior with fetal thymus from NF-κB2 KO or dKO mice were gated on CD4+ and CD8+and analyzed for expression of CD44 and CD62L by flow cytometry. Representative examples are shown (b, c). (d) Nude mice engrafted with fetal thymus from dKO mice exhibited elevated autoantibodies when compared to nudes engrafted with NF-κB2 KO fetal thymus. Serum was collected from matched nudes between 5 ½ and 7 weeks post transplantation. Serum antibody titers to the indicated autoantigens were determined with ELISA assays (1:100 dilution for anti-IgG, 1:200 dilution for anti-insulin; p=0.047 and p=0.03, respectively). (e) H&E-stained sections from liver and lung of nude mouse engrafted 10 weeks prior with one fetal thymic lobe from a dKO mouse or with the other fetal thymic lobe from the same dKO mouse and one lobe from a WT mouse. (f) Splenic T cells were isolated from the thymic stroma-engrafted nude mouse shown in (e) were gated on CD4+ and CD8+and analyzed for expression of CD44 and CD62L by flow cytometry. The engrafted nude mouse shown in (e,f) is a representative example of 6 other such matched transfers.
Because NF-κB1 and NF-κB2 double deficiency resulted in complete lack of all lymph nodes, similar to that seen in Bcl-3, NF-κB2 doubly deficient mice (Franzoso et al., 1997b; Lo et al., 2006), we transferred fetal thymi from NF-κB1,2 doubly deficient mice into nude mice. These nude recipients succumbed to inflammatory conditions by 5 to 8 weeks post transfer (not shown) (see Discussion).
We also tested for autoantibody production in nude mice which had received thymi from Bcl-3, NF-κB2 doubly deficient mice. The original donor double knockout mice had a clear deficit in mature B cells (see above). Consistent with this deficit in mature B cells we could not detect autoantibodies in the original donors (not shown). The (non-thymic) stromal and hematopoietic cells of nude recipients are genetically wild-type, so the presence of autoreactive T cells should stimulate autoantibody production by B cells. Initial analyses confirmed that nude mice with double knockout thymi did indeed produce some autoantibodies (Figure 6d).
To investigate whether impaired T regulatory cell activity could have contributed to the inflammatory pathology, we co-transferred one WT (or NF-κB2 KO) thymic lobe and one double knockout lobe into nude mice and compared them to matched nude recipients that had received only one double-knockout lobe. The presence of the wild-type thymic lobe largely prevented organ infiltration (Fig. 6e) and it reduced the relative number of CD44+CD62L- effector memory in both the CD4 and CD8 compartments (Fig. 6f) (a representative example of 6 such matched transfers is shown). This result suggests that the wild-type thymus generated a T regulatory activity that controlled the inflammatory potential of the T cells emerging from the mutant thymus. This further suggests the T regulatory activity of mutant thymi was in some way impaired, since it was unable by itself to control organ infiltration.
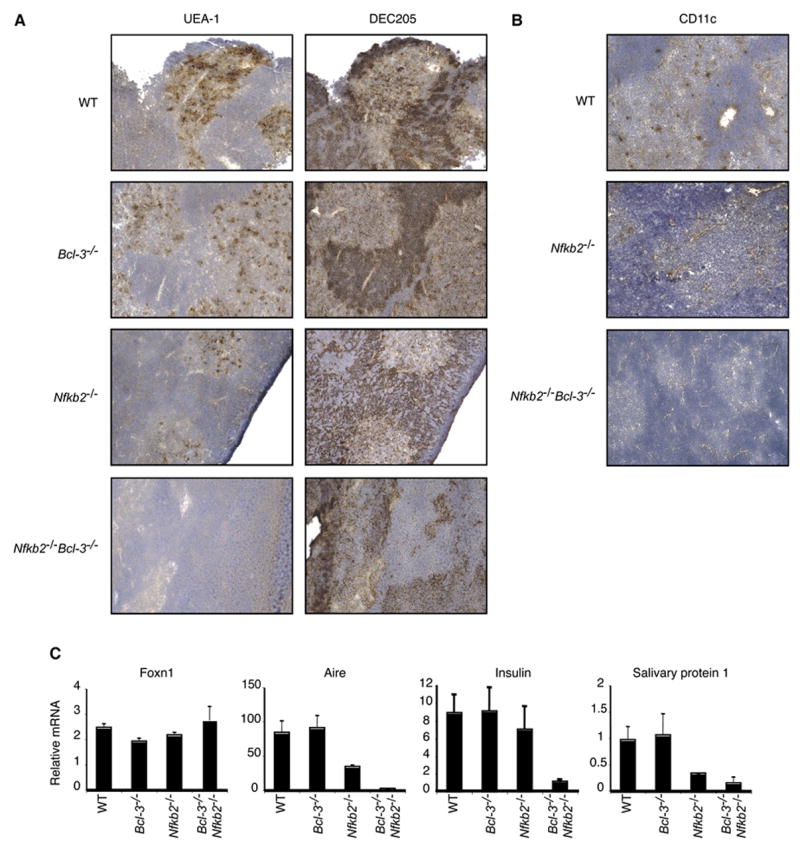
Having shown that the thymic stroma was sufficient to cause multi-organ inflammation, we investigated the thymic structures in the double knockout mice. The thymus contains medullary and cortical epithelium. Medullary thymic epithelial cells (mTECs) express Aire (Autoimmune regulator), which has been implicated in expression of peripheral tissue-restricted autoantigens, such as preproinsulin-2 and salivary protein 1, and in presentation of such autoantigens (Anderson et al., 2005; Gallegos and Bevan, 2006; Kyewski and Klein, 2006). mTECs are thus important for negative selection of at least some autoreactive thymocytes . Thymic sections from 10-day-old mice were stained with the UEA-1 lectin , which detects a subset of mTECs (Surh et al., 1992) and with the DEC205 antibody, which preferentially stains cortical thymic epithelial cells (cTEC), while also weakly staining thymic dendritic cells (DC) in the medulla (Witmer-Pack et al., 1995) (Figure 7a). Additional sections were stained for other markers of medullary (MTS10, K14, ER-TR5 and G8.8 (not shown)) and cortical (ER-TR4) epithelium (Boyd et al., 1993; Farr et al., 1991; Godfrey et al., 1990; Klug et al., 1998; Van Vliet et al., 1985) (Figure S1). The double knockout sections appeared to lack fully differentiated medullary epithelial cells, as no clear staining was seen with UEA-1, ER-TR5, MTS10 or G8.8, while cTECs were readily detected with DEC205 and ER-TR4. There was however some weak staining with K14 in non-cortical and thus presumably medullary regions in double knockout animals. Therefore, mTEC progenitor-like cells may have been present in medullary regions of NF-κB2, Bcl-3 doubly-deficient mutants, but these cells failed to fully differentiate. Of note, NF-κB2 deficient thymi appeared to be partially impaired when compared to wild-type (or Bcl-3) deficient thymi, based on reduced staining with UEA-1, MTS10 and especially with G8.8 and ER-TR5. Partial impairment of the thymus in these mutant mice is consistent with mild inflammatory tendencies later in life and in transfer experiments (as shown here and (Zhu et al., 2006))
In addition to a loss of differentiated mTECs and possibly as a consequence thereof, we also failed to detect thymic dendritic cells in the medulla of double knockout mutant thymic sections (Figure 7b; CD11c staining; the result was confirmed with MIDC-8 staining (not shown)). The absence of thymic dendritic cells may suggest a more general impaired ability to negatively select T cells (see Discussion). Finally, natural killer T (iNKT) cell differentiation and presence in the periphery has been shown to depend on RelB expression in thymic stromal cells (Elewaut et al., 2003; Sivakumar et al., 2003). Consistent with this, we observed a severe loss of iNKT (CD1d-αGalCer tetramer+) cells in liver and spleen of two week old doubly deficient mice (data not shown).
To confirm that differentiated mTECs were not only phenotypically but also functionally absent, we performed quantitative PCR analyses with RNA extracted from 10- to 12-day-old thymi for expression of several genes, including Aire, preproinsulin-2, salivary protein 1, and Foxn1 (Figure 7c). Thymus RNA from NF-κB2, Bcl-3 double knockout mice showed dramatically decreased levels of Aire, insulin and salivary protein 1, while expression of the control gene Foxn1 was essentially at the level of wild-type mice. Thymi from Bcl-3 deficient contained near wild-type levels of mRNAs for Aire, preproinsulin-2 and salivary protein 1, while NF-κB2 deficient mice expressed intermediate levels of these mRNAs, suggesting a partial problem in the latter mutants, consistent with other evidence discussed above. These results show a profound loss of mTECs markers and function in double knockout mice, presumably due to a block in differentiation of these cells, which in turn is likely to impair negative selection of autoreactive T cells that recognize selfantigens such as insulin and salivary protein 1. Loss of differentiated mTECs and thus expression of TSAs most likely also impacted Tregs (see Discussion).
Discussion
The research presented here implicates Bcl-3 and NF-κB in the control of immunological self-tolerance. While Bcl-3 by itself is not absolutely necessary for central tolerance, its importance is revealed in the absence of NF-κB2. Mice lacking both NF-κB2 and Bcl-3, but not mice lacking one of these proteins, fail to survive past 4 weeks after birth, exhibiting severe multi-organ inflammation. Thus NF-κB2 and Bcl-3 have functionally redundant roles in preventing the immune system from attacking its host. Interestingly, these proteins also have redundant functions during lymphoid organogenesis, since only doubly, but not singly deficient mutants lack all lymph nodes.
We investigated the cellular basis of the severe inflammatory condition that occurred in Bcl-3, NF-κB2 doubly deficient mice. First, we established that T cells are responsible. The mutant animals are enriched for effector/memory-type T cells and they contain autoreactive T cells. Furthermore, T cells of mutant animals are sufficient to cause pathology when adoptively transferred into RAG mice. Next we determined that the defect is not intrinsic to T cells, or to a combination of hematopoietic and stromal cells, but instead is intrinsic to stromal cells, which in turn fail to prevent the emergence of autoreactive T cells. This was demonstrated by reciprocal bone marrow transfer experiments. Finally, we showed that mutant stromal cells of the thymus are responsible for the emergence of autoreactive T cells. Pathology is induced in nude mice upon transplantation of mutant thymic stroma. The nude recipients also develop autoantibodies; these are not present in the original doubly-deficient mutant animals, correlating with a paucity of mature splenic B cells in these mice.
Thymi of double knockout mutant animals lack differentiated medullary thymic epithelial cells (mTECs), but do contain non-cortical, medullary-like areas. Wild-type thymus contains clusters or networks of differentiated mTECs that can be identified with a limited set of surface markers, some of which differentiate subclasses of mTECs. mTECs are the source of various peripheral tissue-specific antigens (TSAs) in the thymus, such as insulin and salivary protein 1. mTECs may present these selfantigens directly to newly developing TCR+ T cells or indirectly via cross-priming of local thymic dendritic cells (DCs) (Anderson et al., 2005; Aschenbrenner et al., 2007; Gallegos and Bevan, 2006; Kyewski and Klein, 2006). NF-κB2, Bcl-3 doubly-deficient thymi lack these differentiated mTEC clusters, as judged by the absence of surface markers and as judged also by lack of expression of insulin, salivary protein 1 and Aire. Expression of Aire is largely restricted to mTECs, where it functions as a transcriptional regulator for expression of TSAs. Aire may also have a more general role in presentation of selfantigens (Anderson et al., 2005). Lack of TSAs and Aire in double knockout mice is presumably a consequence of impaired differentiation of mTECs, although this does not rule out the possibility of a more direct role of NF-κB2 and Bcl-3. In addition to mTECs, double knockout mice are missing thymic dendritic cells. These cells normally reside in the medulla and the cortico-medullary junctions; they have been implicated in negative selection of T cells to selfantigens in general, and to TSAs via cross-priming in particular (Aschenbrenner et al., 2007; Gallegos and Bevan, 2006; Gavanescu et al., 2007). Their absence is likely to be secondary to the absence of differentiated mTECs, since we observed normal numbers of thymic dendritic cells in lethally-irradiated Rag1 knockout mice whose hematopoietic compartment had been regenerated upon transfer of double knockout bone marrow (data not shown). We reason that lack of mTEC-dependent local expression of TSAs, coupled with lack of thymic dendritic cells in double knockout mice impairs negative selection of T cells recognizing TSAs, and possibly also other antigens. Defective negative selection could be a primary cause of the observed fatal multi-organ inflammatory pathology.
The lack of differentiated mTEC clusters may also prevent the emergence of T regulatory cells that recognize TSAs. It has been postulated that Tregs are positively selected in thymus by recognition of selfantigens presented by mTECs (Aschenbrenner et al., 2007; Cabarrocas et al., 2006; Kyewski and Klein, 2006; Liu, 2006). If so, our double knockout mutant animals may suffer from holes in the repertoire of their Tregs for this class of selfantigens; such a defect may account for the observed reduction of thymic Tregs shortly after birth. Tregs emerging from the thymus have been referred to as natural Tregs, differentiating them from adaptive Tregs that may develop in the periphery in response to specific antigenic challenges (Bluestone and Abbas, 2003). It has been reported that loss of functional NIK in aly/aly mice and of IKKα in thymic stroma results in a reduction, but not loss of function of peripheral Tregs (Kajiura et al., 2004; Kinoshita et al., 2006). Just prior to death our double knockout mice contained normal numbers of peripheral splenic Tregs with normal levels of non-specific inhibitory activity. Nevertheless, we suspect that the natural Tregs were compromised by holes in their repertoire. In addition it remains possible that Tregs were functionally impaired in vivo at sites of inflammation. The notion that Treg surveillance was somehow impaired in double knockouts is supported by the finding that co-transplantation of wild-type thymus and double knockout thymus significantly reduced inflammatory pathology, presumably due to wild-type thymus-derived regulatory T cells exerting control over T cells emerging from the defective thymus. A deeper understanding of the development and functions of natural and adaptive Tregs will be required to determine if and how Tregs of double knockout mice may have failed to control multi-organ infiltration.
Mice deficient in both NF-κB2 and Bcl-3 bear striking similarities to mice lacking RelB and mice mutated in NIK (aly/aly). These mutant mice fail to develop lymph nodes, lack fully differentiated medullary epithelium and develop multi-organ inflammation, although they differ in the severity of the pathology, with the present double knockouts exhibiting the most severe form (Burkly et al., 1995; Shinkura et al., 1999; Tsubata et al., 1996; Weih et al., 1995). Defects intrinsic to various cell types have been suggested to contribute to the pathology of RelB- deficient mice (Cejas et al., 2005; Martin et al., 2003; Wu et al., 1998; Xia et al., 1999). We found that transplantation of thymic tissue from RelB-deficient thymus caused multi-organ infiltration in nude mice (XZ and US, unpublished observations), similar to thymic tissue from NF-κB2, Bcl-3 double knockout, NF-κB2, NF-κB1 double knockout (this report), aly/aly mice and IKKα-deficient mice (Kajiura et al., 2004; Kinoshita et al., 2006). Therefore, the severe inflammation observed in these mutant mouse models is due first and foremost to defects intrinsic to thymic stroma, and most likely to mTEC precursor cells, regardless of whether other cells were affected and contributed to the final phenotype as well.
Given that loss of function of NIK, IKKα, RelB, NF-κB2 plus Bcl-3, or NF-κB2 plus NF-κB1 in transplanted thymic stroma causes serious pathology in nude mice, the non-classical pathway leading to activation of p52/RelB must be crucial for tolerance induction in the thymus. Activation of the non-classical pathway in mTEC precursors most certainly includes signaling via the lymphotoxin β receptor (LTβR), since loss of this signaling route results in multi-organ inflammation as well (Boehm et al., 2003; Chin et al., 2003; Hehlgans and Pfeffer, 2005; Pfeffer, 2003). It has recently been reported that the LTβR activates NF-κB only via the non-classical, not the classical pathway, so loss of NF-κB2 must completely abolish all LTβR signaling to NF-κB (Basak et al., 2007). Therefore, NF-κB2 knockout mice should have the same pathology as that observed in mutant mice lacking other components of the non-classical activation pathway, but they do not. The likely explanation for this much milder condition in NF-κB2 knockouts is that RelB complexes can still be activated in these mice, but not in the other mutant mice. We have shown previously that TNFαstrongly activates p50/RelB in NF-κB2 knockout but not wild-type cells (Muller and Siebenlist, 2003). This cytokine induces the expression of both RelB and NF-κB1 via the classical pathway, and, in the absence of p100/NF-κB2, the resulting p50/RelB heterodimers are no longer inhibited. Therefore, p50/RelB heterodimers are readily activated in NF-κB2 knockout stromal cells, where they may partially compensate for loss of the non-classical signaling path. Based on these considerations we investigated whether Bcl-3 might be required for TNFα-induced activation of p50/RelB in NF-κB2-deficient cells. If so, this could help explain why loss of Bcl-3 in addition to loss of NF-κB2 resulted in fatal inflammatory pathology akin to that seen in RelB knockout mice. However, we were unable to detect any difference in TNFα-induced NF-κB activity in mouse embryo fibroblasts from NF-κB2 single and Bcl-3, NF-κB2 double knockout mice (X.Z., K.B., U.S.; unpublished observation). While Bcl-3 can partly compensate for loss of p52/RelB and signaling via the non-classical pathway in NF-κB2 knockouts, it does not appear to control p50/RelB activity, nor is it known to have any functions within the non-classical pathway. We conclude that in the present biologic context Bcl-3 likely functions by directly regulating target genes, not indirectly via global effects on NF-κB activity. At present the genes involved in the differentiation of mTECs are not known. Future work will have to identify such genes, those among them targeted by Bcl-3, RelB or any of the NF-κB factors, and the molecular mechanisms by which these factors regulate their expression.
To date little is known about physiologic targets of Bcl-3 in any biologic context; furthermore, actual mechanisms of recruitment to and functions within chromatin of potential direct targets remain unknown or unconfirmed (see Introduction). Nevertheless, Bcl-3 does strongly interact with homodimers of p52 and p50 (Franzoso et al., 1993; Franzoso et al., 1992; Fujita et al., 1993; Wessells et al., 2004; Westerheide et al., 2001) In the present biologic context, Bcl-3 must be able to function independent of p52/NF-κB2, but could still function in concert with p50/NF-κB1. There is some indirect support for a model wherein Bcl-3’s contributions to tolerance might be mediated via p50/NF-κB1: RelB, NF-κB1 double knockout mice develop a more severe form of multi-organ inflammation than RelB only-deficient mice (Weih et al., 1997). Therefore, Bcl-3 and p50/NF-κB1 may interact to help differentiate mTECs and induce tolerance.
Loss of NF-κB2 alone does not lead to fatal multi-organ inflammatory pathology, but we did observe a limited increase in effector/memory T cells, especially in transfer experiments, partial impairment of the thymic stroma and mild sporadic organ infiltration after more than 6 months of age. These data are consistent with another study in which NF-κB2 knockout mice developed organ infiltrations around 6 months of age, although the extent of inflammation may have been slightly more apparent (Zhu et al., 2006). The studies described here were done with much younger mice, at a time when NF-kB2 KO mice showed no organ infiltrations at all. In our facility only Bcl-3, NF-κB2 double knockout mice developed a fatal pathology with rapid onset shortly after birth.
It remains to be determined which signal activates Bcl-3 during mTEC differentiation. Past reports suggest that Bcl-3 could be regulated transcriptionally (Ge et al., 2003) and post-transcriptionally, the latter including phosphorylation (Bundy and McKeithan, 1997; Nishikori et al., 2005; Viatour et al., 2005) and ubiquitination (Massoumi et al., 2006). In addition to LTβR signals, differentiation of mTECs must involve at least one other signal. Thymi of TRAF6-deficient mice lack differentiated mTECs and their transplantation into nude mice induces multi-organ inflammation, even though TRAF6 is not a known component of the LTβR pathway (Akiyama et al., 2005). The TRAF6-mediated signal may however contribute indirectly to the non-classical pathway by inducing expression of RelB. Bcl-3 and RelB could both lie downstream of the TRAF6–associated signal. TRAF6 can activate the classical NF-κB pathway, which is known to induce expression of Bcl-3 (Ge et al., 2003).
The present work suggests extensive parallels between thymic stromal cells, specifically mTECs, and stromal cells of secondary lymphoid organs, specifically follicular dendritic cells (FDCs). FDCs and mTECs are highly differentiated cells that serve as antigen-presenting cells for B and T cells, respectively. Based on the knockout data, Bcl-3 and NF-kB2 are critically involved in the differentiation of both cell types, as are the non-classical pathway and RelB (Caamano et al., 1998; Franzoso et al., 1998; Franzoso et al., 1997a; Franzoso et al., 1997b; Lo et al., 2006; Poljak et al., 1999). In addition, differentiation of mTECs and FDCs is partially controlled by LTbR ligands expressed on interacting T or B lymphocytes, respectively (Boehm et al., 2003; Chin et al., 2003; Tumanov et al., 2003).
The results presented here reveal an unanticipated role of Bcl-3 in central tolerance, which emerges in the absence of NF-κB2. Bcl-3 functions redundantly with NF-κB2 to partially compensate for loss of NF-κB2 and thus loss of the non-classical pathway of activation. Based on these data it will be important to investigate whether Bcl-3 is deregulated in human autoimmune diseases. It will also be of interest to explore possible connections between Bcl-3’s roles in tolerance induction and tumorigenesis. Bcl-3 has been implicated as an oncogene in a variety of hematologic and solid tumor malignancies (Kashatus et al., 2006; Massoumi et al., 2006; Mathas et al., 2005; Ohno et al., 1990; Thornburg et al., 2003). Further work is needed to elucidate the precise molecular mechanisms and targets of Bcl-3 underlying its various roles.
Experimental Procedures
Mice
Bcl-3 knockout and NF-κB2 knockout mice were described previously (Franzoso et al., 1998; Franzoso et al., 1997a). Both have been backcrossed to C57 BL/6 for 11 or more generations. Bcl-3, NF-κB2 double knockout (dKO) mice were obtained by cross-breeding single KO mice. Mice were housed in NIAID Institute facilities and all experiments were done with approval of the NIAID Animal Care and Use Committee and in accordance with all relevant institutional guidelines.
Antibodies and Flow Cytometry
Flow cytometric analyses were performed on freshly prepared, erythrocyte-lysed single cell suspensions of thymocytes or splenocytes in PBS/0.5% BSA/2 mM EDTA. Stained cells were analyzed on a FACSCaliburTM (BD Biosciences) using the following: FITC-, PE-, PerCP- or allophycocyanin-conjugated monoclonal antibodies purchased from BD Biosciences (clone name in parenthesis). For surface marker staining, single-cell suspensions from thymus and spleen were preincubated with anti-FcγIII/II receptor (2.4G2) to block Fc receptors, and then stained with anti-CD4 (RM4-5), anti-CD8 (53-6.7), anti-CD25 (PC61), anti-CD44 (IM7), anti- CD62L (MEL-14). For Foxp3 intracellular staining, cells were fixed after surface staining and then stained with anti-Foxp3 (FJK-16s) (eBioscience). Data were analyzed with FlowJo software (Tree Star Inc.).
Cell Purification
Erythrocyte-lysed, single cell suspensions from spleen served as splenocytes. For autoreactive assays CD4+ T cells were purified from splenocytes by negative selection on midi MACS (magnetic cell separation) columns (Miltenyi Biotec) according to the manufacturer’s instructions. Antigen-presenting cells (APCs) were prepared from wild-type splenocytes by depletion of T cells with anti-CD4 and anti-CD8 beads with MACS. For isolation of CD4+CD25+ T cells or CD4+C25- T cells, CD4+ T cells obtained by negative selection with MACS (see above) were incubated with Fc receptor-blocking antibody 2.4G2, then stained with FITC-anti-CD4, PE-anti-CD8 and PerCP-anti-CD25, and finally sorted with a FACSAria cell sorter (BD Biosciences).
Adoptive Transfer of CD4 T Cells
5×106 CD4+ T cells, purified from splenocytes by negative selection (see above), were injected intravenously into sublethally-irradiated (500 rads) male rag1 −/− mice. Mice were weighed weekly and monitored daily.
Bone Marrow Chimeras
Bone marrow cells were harvested by flushing mouse femurs and tibias with cold Hank’s Balance Salt Solution (HBSS). Bone marrow cell suspensions were depleted of T cells by magnetic cell separation with beads conjugated to anti-CD4 and anti-CD8 (Miltenyi Biotec). 3–5×106 erythrocyte-lysed bone marrow cells were injected into the tail veins of lethally irradiated (900 rads) recipient mice in 0.2 ml HBSS. Mice were weighed weekly and monitored daily.
T Cell Assays
For mixed lymphocyte reactions, 1×105 negatively selected CD4+ T cells per well were incubated with 5×105 (unless otherwise specified) syngeneic, T cell-depleted, irradiated splenocytes (3000 rads) serving as (self-) antigen-presenting cells (APCs) in 96-well plates for 72 hours and pulsed with 1μCi of [3H]thymidine per well for final 8 hours. T regulatory cell-mediated suppression assays were performed under the same conditions, except with 5×104 CD4+CD25- T cells as responders, 2.5x105 APCs, anti-CD3 (1 μg/ml) (BD Biosciences), together with 2-fold dilutions of CD4+CD25+ T regulatory cells (as specified).
Autoantibodies
For titers of antibodies to IgG and insulin (Sigma), 96-well plates were coated with these antigens (10 μg/ml in phosphate buffered saline) at 37° C for two hours. Plates were subsequently washed 3 times and then incubated with blocking buffer (BBS, 0.05% Tween 20, 1mM EDTA, 0.25% NaN3) at room temperature for 30 minutes. After three rinses the plates were incubated with 50 μl diluted sera (1:50, 1:100, 1:200) then incubated at room temperature for 2 hours. Antibodies were detected with HorseRadish Peroxidate (HRP)-conjungated rat anti-mouse IgG or IgM (Jackson Immunoresearch).
Histopathology and Immunohistochemistry
Organs/tissues were immersion-fixed in 10% buffered formalin and embedded in paraffin blocks. Sections were stained with hematoxylin and eosin (H&E), and examined by light microscopy. Thymi were embedded in OCT freezing solution. Cryostat sections were fixed for 10 min in ice-cold acetone, then sequentially blocked with peroxidase blocking reagent (DakoCytomation), avidin/biotin blocking kit (Vector Laboratories) and blocking buffer containing 10% rabbit or goat serum, and incubated with biotinylated ulex europaeus agglutinin 1 (UEA-1; Vector Laboratories), or antibody to DEC205 (Serotec), MTS-10 (gift of Dr. Richard Boyd), Keratin 14 (Covance), ER-TR4 and ER-TR5 (gift of Dr. Willem van Ewijk), MIDC-8 (Covance) and CD11c (BD Biosciences) at 4ºC overnight. Staining was developed directly or after incubation with the appropriate biotinylated second step antibody using StreptABComplex/HRP (streptavidin complexed with biotinylated peroxidase), according to the manufacturer’s recommendations (Vector Laboratories).
Fetal Thymus Transplantation
Thymic lobes were isolated from timed embryos 14.5 days after coitus and were cultured for 6–7 days on Nucleopore filters placed in complete medium containing 1.35 mM 2-DG (Sigma), then washed twice for two hours each. Thymic lobes were transplanted under the renal capsule of adult (6–8 weeks of age) male B6/nu mice.
RNA Isolation and Real Time-qPCR
Thymi were frozen, ground on dry ice and powder tissue was used for RNA isolation. 1ml of TRIzol (Invitrogen) was added to 100 mg of powder tissue, homogenized with a needle-syringe, and then 200μl of chloroform was added and mixed by vortex. Aqueous phase was harvested to a new tube after 15 min centrifugation at high speed, and then 500μl of isopropanol was added. RNA preparation was incubated 10–20 min on ice and following by 10 min centrifugation at high speed. RNA Pellet was dissolved in 100μl of DEPC-H2O and the RNA cleanup/Dnase digestion protocol of Qiagene was followed. 2μg RNA was used for cDNA synthesis with superscriptII RT (Invitrogen).
For quantification of gene expression with real time PCR, 1 μl of the cDNA reaction mix was used with the platinum quantitative PCR super Mix-UDG and LUX primers (Invitrogen), following manufactures instructions for the Corbett Rotor-Gene real-time machine.
LUX primers were designed with D-LUX Designer software available on the Invitrogen Web site. The primer sequences were as follows:
| GAPDH: | 5′ CACCATCGTCCCGTAGACAAAATGG[FAM]G 3′
5′ CAAATGGCAGCCCTGGTGA 3′ |
| Foxn-1: | 5′ CCACTCTTCCCAAAGCCCATC 3′
5′CGGAACACTGACTGGAAGGCTTC[FAM]G 3′ |
| Aire: | 5′ CGTGACGGACGACTCTGCTAGTCA[FAM]G 3′
5′ GCAGGATGCCGTCAAATGAGT 3′ |
| Salivary protein-1: | 5′ CAACACTCCTGGCACTCCTTGTG[FAM]TG 3′
5′ CTGTTTGTCTCCGGGTCCTG 3′ |
For quantification of preproinsulin 2, 1 μl of cDNA reaction synthesis was used with platinum SYBR green quantitative PCR super Mix-UDG (Invitrogen) and the following primers:
| Preproinsulin2: | 5′ GCCCTAAGTGATCCGCTACAATC 3′
5′ TCTACAATGCCACGCTTCTGC 3′ |
| β-actin: | 5′GTGGGCCGCTCTAGGCACCAA 3′
5′ CTCTTTGATGTCACGCACGATTTC 3′ |
Supplementary Material
Figure S1. Loss of medullary thymic epithelial markers in NF-κB2, Bcl-3 double knockout mice. Thymic tissue sections of 14 day-old wild-type (WT), NF-κB2 knockout (KO) and NF-κB2, Bcl-3 double knockout (dKO) were stained with MTS10, Keratin 14, ER-TR5 and ER-TR4.
Acknowledgments
We would like to thank Dr. A.S. Fauci for continued support. We are indebted to Dr. R Boyd for MTS10 and to Dr. W. van Ewijk for ER-TR4 and ER-TR5. We thank Dr. B.J. Fowlkes for discussion. This research was supported by the Intramural Research Program of NIAID, NIH.
Footnotes
Competing financial interests
The authors declare that they have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama T, Maeda S, Yamane S, Ogino K, Kasai M, Kajiura F, Matsumoto M, Inoue J. Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science. 2005;308:248–251. doi: 10.1126/science.1105677. [DOI] [PubMed] [Google Scholar]
- Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity. 2005;23:227–239. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Aschenbrenner K, D’Cruz LM, Vollmann EH, Hinterberger M, Emmerich J, Swee LK, Rolink A, Klein L. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat Immunol. 2007;8:351–358. doi: 10.1038/ni1444. [DOI] [PubMed] [Google Scholar]
- Basak S, Kim H, Kearns JD, Tergaonkar V, O’Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A. A Fourth IkappaB Protein within the NF-kappaB Signaling Module. Cell. 2007;128:369–381. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003;3:253–257. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- Boehm T, Scheu S, Pfeffer K, Bleul CC. Thymic medullary epithelial cell differentiation, thymocyte emigration, and the control of autoimmunity require lympho-epithelial cross talk via LTbetaR. J Exp Med. 2003;198:757–769. doi: 10.1084/jem.20030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 directly transactivates through kappa B motifs via association with DNA-binding p50B homodimers. Cell. 1993;72:729–739. doi: 10.1016/0092-8674(93)90401-b. [DOI] [PubMed] [Google Scholar]
- Boyd RL, Tucek CL, Godfrey DI, Izon DJ, Wilson TJ, Davidson NJ, Bean AG, Ladyman HM, Ritter MA, Hugo P. The thymic microenvironment. Immunol Today. 1993;14:445–459. doi: 10.1016/0167-5699(93)90248-J. [DOI] [PubMed] [Google Scholar]
- Bundy DL, McKeithan TW. Diverse effects of BCL3 phosphorylation on its modulation of NF-kappaB p52 homodimer binding to DNA. J Biol Chem. 1997;272:33132–33139. doi: 10.1074/jbc.272.52.33132. [DOI] [PubMed] [Google Scholar]
- Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995;373:531–536. doi: 10.1038/373531a0. [DOI] [PubMed] [Google Scholar]
- Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos–Suarez C, Snapper CM, Bravo R. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabarrocas J, Cassan C, Magnusson F, Piaggio E, Mars L, Derbinski J, Kyewski B, Gross DA, Salomon BL, Khazaie K, et al. Foxp3+ CD25+ regulatory T cells specific for a neo-self-antigen develop at the double-positive thymic stage. Proc Natl Acad Sci U S A. 2006;103:8453–8458. doi: 10.1073/pnas.0603086103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejas PJ, Carlson LM, Kolonias D, Zhang J, Lindner I, Billadeau DD, Boise LH, Lee KP. Regulation of RelB expression during the initiation of dendritic cell differentiation. Mol Cell Biol. 2005;25:7900–7916. doi: 10.1128/MCB.25.17.7900-7916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin RK, Lo JC, Kim O, Blink SE, Christiansen PA, Peterson P, Wang Y, Ware C, Fu YX. Lymphotoxin pathway directs thymic Aire expression. Nat Immunol. 2003;4:1121–1127. doi: 10.1038/ni982. [DOI] [PubMed] [Google Scholar]
- Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78:110–117. doi: 10.1046/j.1440-1711.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- Claudio E, Brown K, Siebenlist U. NF-kappaB guides the survival and differentiation of developing lymphocytes. Cell Death Differ. 2006;13:697–701. doi: 10.1038/sj.cdd.4401894. [DOI] [PubMed] [Google Scholar]
- Corn RA, Hunter C, Liou HC, Siebenlist U, Boothby MR. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. 2005;175:2102–2110. doi: 10.4049/jimmunol.175.4.2102. [DOI] [PubMed] [Google Scholar]
- Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: critical roles for p100. J Biol Chem. 2003;278:23278–23284. doi: 10.1074/jbc.M300106200. [DOI] [PubMed] [Google Scholar]
- Elewaut D, Shaikh RB, Hammond KJ, De Winter H, Leishman AJ, Sidobre S, Turovskaya O, Prigozy TI, Ma L, Banks TA, et al. NIK-dependent RelB activation defines a unique signaling pathway for the development of V alpha 14i NKT cells. J Exp Med. 2003;197:1623–1633. doi: 10.1084/jem.20030141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr A, Nelson A, Truex J, Hosier S. Epithelial heterogeneity in the murine thymus: a cell surface glycoprotein expressed by subcapsular and medullary epithelium. J Histochem Cytochem. 1991;39:645–653. doi: 10.1177/39.5.2016514. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Bours V, Azarenko V, Park S, Tomita-Yamaguchi M, Kanno T, Brown K, Siebenlist U. The oncoprotein Bcl-3 can facilitate NF-kappa B-mediated transactivation by removing inhibiting p50 homodimers from select kappa B sites. Embo J. 1993;12:3893–3901. doi: 10.1002/j.1460-2075.1993.tb06067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-kappa B-mediated inhibition. Nature. 1992;359:339–342. doi: 10.1038/359339a0. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, Grinberg A, Tran T, Scharton-Kersten T, Anver M, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Scharton-Kersten T, Shores EW, Epstein S, Grinberg A, Tran T, Shacter E, Leonardi A, Anver M, et al. Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity. 1997a;6:479–490. doi: 10.1016/s1074-7613(00)80291-5. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997b;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Nolan GP, Liou HC, Scott ML, Baltimore D. The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-kappa B p50 homodimers. Genes Dev. 1993;7:1354–1363. doi: 10.1101/gad.7.7b.1354. [DOI] [PubMed] [Google Scholar]
- Gallegos AM, Bevan MJ. Central tolerance: good but imperfect. Immunol Rev. 2006;209:290–296. doi: 10.1111/j.0105-2896.2006.00348.x. [DOI] [PubMed] [Google Scholar]
- Gavanescu I, Kessler B, Ploegh H, Benoist C, Mathis D. Loss of Aire-dependent thymic expression of a peripheral tissue antigen renders it a target of autoimmunity. Proc Natl Acad Sci U S A. 2007;104:4583–4587. doi: 10.1073/pnas.0700259104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge B, Li O, Wilder P, Rizzino A, McKeithan TW. NF-kappa B regulates BCL3 transcription in T lymphocytes through an intronic enhancer. J Immunol. 2003;171:4210–4218. doi: 10.4049/jimmunol.171.8.4210. [DOI] [PubMed] [Google Scholar]
- Godfrey DI, Izon DJ, Tucek CL, Wilson TJ, Boyd RL. The phenotypic heterogeneity of mouse thymic stromal cells. Immunology. 1990;70:66–74. [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiura F, Sun S, Nomura T, Izumi K, Ueno T, Bando Y, Kuroda N, Han H, Li Y, Matsushima A, et al. NF-kappa B-inducing kinase establishes self-tolerance in a thymic stroma-dependent manner. J Immunol. 2004;172:2067–2075. doi: 10.4049/jimmunol.172.4.2067. [DOI] [PubMed] [Google Scholar]
- Kanno T, Franzoso G, Siebenlist U. Human T-cell leukemia virus type I Tax-protein-mediated activation of NF-kappa B from p100 (NF-kappa B2)-inhibited cytoplasmic reservoirs. Proc Natl Acad Sci U S A. 1994;91:12634–12638. doi: 10.1073/pnas.91.26.12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karrer U, Althage A, Odermatt B, Hengartner H, Zinkernagel RM. Immunodeficiency of alymphoplasia mice (aly/aly) in vivo: structural defect of secondary lymphoid organs and functional B cell defect. Eur J Immunol. 2000;30:2799–2807. doi: 10.1002/1521-4141(200010)30:10<2799::AID-IMMU2799>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Kashatus D, Cogswell P, Baldwin AS. Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev. 2006;20:225–235. doi: 10.1101/gad.1352206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kim B, Cai L, Choi HJ, Ohgi KA, Tran C, Chen C, Chung CH, Huber O, Rose DW, et al. Transcriptional regulation of a metastasis suppressor gene by Tip60 and beta-catenin complexes. Nature. 2005;434:921–926. doi: 10.1038/nature03452. [DOI] [PubMed] [Google Scholar]
- Kinoshita D, Hirota F, Kaisho T, Kasai M, Izumi K, Bando Y, Mouri Y, Matsushima A, Niki S, Han H, et al. Essential role of IkappaB kinase alpha in thymic organogenesis required for the establishment of self-tolerance. J Immunol. 2006;176:3995–4002. doi: 10.4049/jimmunol.176.7.3995. [DOI] [PubMed] [Google Scholar]
- Klug DB, Carter C, Crouch E, Roop D, Conti CJ, Richie ER. Interdependence of cortical thymic epithelial cell differentiation and T-lineage commitment. Proc Natl Acad Sci U S A. 1998;95:11822–11827. doi: 10.1073/pnas.95.20.11822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyewski B, Klein L. A central role for central tolerance. Annu Rev Immunol. 2006;24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601. [DOI] [PubMed] [Google Scholar]
- Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Liu YJ. A unified theory of central tolerance in the thymus. Trends Immunol. 2006;27:215–221. doi: 10.1016/j.it.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Lo JC, Basak S, James ES, Quiambo RS, Kinsella MC, Alegre ML, Weih F, Franzoso G, Hoffmann A, Fu YX. Coordination between NF-kappaB family members p50 and p52 is essential for mediating LTbetaR signals in the development and organization of secondary lymphoid tissues. Blood. 2006;107:1048–1055. doi: 10.1182/blood-2005-06-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, O’Sullivan B, Low P, Thomas R. Antigen-specific suppression of a primed immune response by dendritic cells mediated by regulatory T cells secreting interleukin–10. Immunity. 2003;18:155–167. doi: 10.1016/s1074-7613(02)00503-4. [DOI] [PubMed] [Google Scholar]
- Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 2006;125:665–677. doi: 10.1016/j.cell.2006.03.041. [DOI] [PubMed] [Google Scholar]
- Mathas S, Johrens K, Joos S, Lietz A, Hummel F, Janz M, Jundt F, Anagnostopoulos I, Bommert K, Lichter P, et al. Elevated NF-kappaB p50 complex formation and Bcl-3 expression in classical Hodgkin, anaplastic large-cell, and other peripheral T-cell lymphomas. Blood. 2005;106:4287–4293. doi: 10.1182/blood-2004-09-3620. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Iwamasa K, Rennert PD, Yamada T, Suzuki R, Matsushima A, Okabe M, Fujita S, Yokoyama M. Involvement of distinct cellular compartments in the abnormal lymphoid organogenesis in lymphotoxin-alpha-deficient mice and alymphoplasia (aly) mice defined by the chimeric analysis. J Immunol. 1999;163:1584–1591. [PubMed] [Google Scholar]
- Matsushima A, Kaisho T, Rennert PD, Nakano H, Kurosawa K, Uchida D, Takeda K, Akira S, Matsumoto M. Essential role of nuclear factor (NF)-kappaB-inducing kinase and inhibitor of kappaB (IkappaB) kinase alpha in NF-kappaB activation through lymphotoxin beta receptor, but not through tumor necrosis factor receptor I. J Exp Med. 2001;193:631–636. doi: 10.1084/jem.193.5.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TC, Teague TK, Hildeman DA, Bender J, Rees WA, Kedl RM, Swanson B, Kappler JW, Marrack P. Stronger correlation of bcl-3 than bcl-2, bcl-xL, costimulation, or antioxidants with adjuvant-induced T cell survival. Ann N Y Acad Sci. 2002;975:114–131. doi: 10.1111/j.1749-6632.2002.tb05946.x. [DOI] [PubMed] [Google Scholar]
- Muller JR, Siebenlist U. Lymphotoxin beta receptor induces sequential activation of distinct NF-kappa B factors via separate signaling pathways. J Biol Chem. 2003;278:12006–12012. doi: 10.1074/jbc.M210768200. [DOI] [PubMed] [Google Scholar]
- Nishikori M, Ohno H, Haga H, Uchiyama T. Stimulation of CD30 in anaplastic large cell lymphoma leads to production of nuclear factor-kappaB p52, which is associated with hyperphosphorylated Bcl-3. Cancer Sci. 2005;96:487–497. doi: 10.1111/j.1349-7006.2005.00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H, Takimoto G, McKeithan TW. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determination and cell cycle control. Cell. 1990;60:991–997. doi: 10.1016/0092-8674(90)90347-h. [DOI] [PubMed] [Google Scholar]
- Paxian S, Merkle H, Riemann M, Wilda M, Adler G, Hameister H, Liptay S, Pfeffer K, Schmid RM. Abnormal organogenesis of Peyer’s patches in mice deficient for NF–kappaB1, NF-kappaB2, and Bcl-3. Gastroenterology. 2002;122:1853–1868. doi: 10.1053/gast.2002.33651. [DOI] [PubMed] [Google Scholar]
- Pfeffer K. Biological functions of tumor necrosis factor cytokines and their receptors. Cytokine Growth Factor Rev. 2003;14:185–191. doi: 10.1016/s1359-6101(03)00022-4. [DOI] [PubMed] [Google Scholar]
- Poljak L, Carlson L, Cunningham K, Kosco-Vilbois MH, Siebenlist U. Distinct activities of p52/NF-kappa B required for proper secondary lymphoid organ microarchitecture: functions enhanced by Bcl-3. J Immunol. 1999;163:6581–6588. [PubMed] [Google Scholar]
- Riemann M, Endres R, Liptay S, Pfeffer K, Schmid RM. The IkappaB protein Bcl-3 negatively regulates transcription of the IL-10 gene in macrophages. J Immunol. 2005;175:3560–3568. doi: 10.4049/jimmunol.175.6.3560. [DOI] [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- Shinkura R, Kitada K, Matsuda F, Tashiro K, Ikuta K, Suzuki M, Kogishi K, Serikawa T, Honjo T. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet. 1999;22:74–77. doi: 10.1038/8780. [DOI] [PubMed] [Google Scholar]
- Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol. 2005;5:435–445. doi: 10.1038/nri1629. [DOI] [PubMed] [Google Scholar]
- Sivakumar V, Hammond KJ, Howells N, Pfeffer K, Weih F. Differential requirement for Rel/nuclear factor kappa B family members in natural killer T cell development. J Exp Med. 2003;197:1613–1621. doi: 10.1084/jem.20022234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh CD, Gao EK, Kosaka H, Lo D, Ahn C, Murphy DB, Karlsson L, Peterson P, Sprent J. Two subsets of epithelial cells in the thymic medulla. J Exp Med. 1992;176:495–505. doi: 10.1084/jem.176.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg NJ, Pathmanathan R, Raab–Traub N. Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer Res. 2003;63:8293–8301. [PubMed] [Google Scholar]
- Tsubata R, Tsubata T, Hiai H, Shinkura R, Matsumura R, Sumida T, Miyawaki S, Ishida H, Kumagai S, Nakao K, Honjo T. Autoimmune disease of exocrine organs in immunodeficient alymphoplasia mice: a spontaneous model for Sjogren’s syndrome. Eur J Immunol. 1996;26:2742–2748. doi: 10.1002/eji.1830261129. [DOI] [PubMed] [Google Scholar]
- Tumanov AV, Kuprash DV, Nedospasov SA. The role of lymphotoxin in development and maintenance of secondary lymphoid tissues. Cytokine Growth Factor Rev. 2003;14:275–288. doi: 10.1016/s1359-6101(03)00026-1. [DOI] [PubMed] [Google Scholar]
- Van Vliet E, Jenkinson EJ, Kingston R, Owen JJ, Van Ewijk W. Stromal cell types in the developing thymus of the normal and nude mouse embryo. Eur J Immunol. 1985;15:675–681. doi: 10.1002/eji.1830150707. [DOI] [PubMed] [Google Scholar]
- Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Weih F, Caamano J. Regulation of secondary lymphoid organ development by the nuclear factor-kappaB signal transduction pathway. Immunol Rev. 2003;195:91–105. doi: 10.1034/j.1600-065x.2003.00064.x. [DOI] [PubMed] [Google Scholar]
- Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, Lira SA, Bravo R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- Weih F, Durham SK, Barton DS, Sha WC, Baltimore D, Bravo R. p50-NF-kappaB complexes partially compensate for the absence of RelB: severely increased pathology in p50(−/−)relB(−/−) double-knockout mice. J Exp Med. 1997;185:1359–1370. doi: 10.1084/jem.185.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessells J, Baer M, Young HA, Claudio E, Brown K, Siebenlist U, Johnson PF. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J Biol Chem. 2004;279:49995–50003. doi: 10.1074/jbc.M404246200. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Mayo MW, Anest V, Hanson JL, Baldwin AS., Jr The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G(1) transition. Mol Cell Biol. 2001;21:8428–8436. doi: 10.1128/MCB.21.24.8428-8436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witmer-Pack MD, Swiggard WJ, Mirza A, Inaba K, Steinman RM. Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. II. Expression in situ in lymphoid and nonlymphoid tissues. Cell Immunol. 1995;163:157–162. doi: 10.1006/cimm.1995.1110. [DOI] [PubMed] [Google Scholar]
- Wu L, D’Amico A, Winkel KD, Suter M, Lo D, Shortman K. RelB is essential for the development of myeloid-related CD8alpha- dendritic cells but not of lymphoid-related CD8alpha+ dendritic cells. Immunity. 1998;9:839–847. doi: 10.1016/s1074-7613(00)80649-4. [DOI] [PubMed] [Google Scholar]
- Xia Y, Chen S, Wang Y, Mackman N, Ku G, Lo D, Feng L. RelB modulation of IkappaBalpha stability as a mechanism of transcription suppression of interleukin-1alpha (IL-1alpha), IL-1beta, and tumor necrosis factor alpha in fibroblasts. Mol Cell Biol. 1999;19:7688–7696. doi: 10.1128/mcb.19.11.7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Mitani T, Yorita K, Uchida D, Matsushima A, Iwamasa K, Fujita S, Matsumoto M. Abnormal immune function of hemopoietic cells from alymphoplasia (aly) mice, a natural strain with mutant NF-kappa B-inducing kinase. J Immunol. 2000;165:804–812. doi: 10.4049/jimmunol.165.2.804. [DOI] [PubMed] [Google Scholar]
- Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer’s patch development: differential regulation of p52-RelB by lymphotoxin and TNF. Embo J. 2003;22:121–130. doi: 10.1093/emboj/cdg004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Wu L, Wesche H, Arthur CD, White JM, Goeddel DV, Schreiber RD. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 2001;291:2162–2165. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- Zhang B, Wang Z, Ding J, Peterson P, Gunning WT, Ding HF. NF-kappaB2 is required for the control of autoimmunity by regulating the development of medullary thymic epithelial cells. J Biol Chem. 2006;281:38617–38624. doi: 10.1074/jbc.M606705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Chin RK, Christiansen PA, Lo JC, Liu X, Ware C, Siebenlist U, Fu YX. NF-kappaB2 is required for the establishment of central tolerance through an Aire-dependent pathway. J Clin Invest. 2006;116:2964–2971. doi: 10.1172/JCI28326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler SF. FOXP3: Of Mice and Men. Annu Rev Immunol. 2006;24:209–226. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Loss of medullary thymic epithelial markers in NF-κB2, Bcl-3 double knockout mice. Thymic tissue sections of 14 day-old wild-type (WT), NF-κB2 knockout (KO) and NF-κB2, Bcl-3 double knockout (dKO) were stained with MTS10, Keratin 14, ER-TR5 and ER-TR4.