

Summary
The gating ring of cyclic nucleotide modulated channels is proposed to be either a 2-fold symmetric dimer of dimers or a fourfold symmetric tetramer based on high-resolution structure data of soluble cyclic nucleotide binding domains and functional data on intact channels. We addressed this controversy by obtaining structural data on an intact, full-length, cyclic nucleotide-modulated potassium channel MloK1 from M. loti, which also features a putative voltage sensor domain S1-S4. We present here the 3D single particle structure by transmission electron microscopy, and the projection map of membrane-reconstituted 2D crystals of MloK1 in the presence of cAMP. Our data show a four-fold symmetric arrangement of the CNBDs, separated by discrete gaps. A homology model for full-length MloK1 suggests a vertical orientation for the CNBDs. The 2D crystal packing in the membrane-embedded state is compatible with the S1-S4 domains in the vertical “up” state.
Keywords: cyclic nucleotide-modulated potassium channel, MloK1, 2D membrane crystals, voltage sensor
INTRODUCTION
Ion channels are integral membrane proteins that control ion flow across the cell membrane. The activity of an ion channel can be modulated by secondary messenger molecules in order to respond to variations in the cellular physiological conditions. The family of cyclic nucleotide-modulated channels belongs to this category and it comprises the cyclic nucleotide gated channels, known as CNG channels, and the hyperpolarization-activated and cyclic nucleotide gated channels, known as HCN channels. CNG and HCN channels are known to be involved in several crucial physiological pathways, such as the visual and olfactory sensory transduction and the pacemaker activity in heart and brain (Craven and Zagotta, 2006; Kaupp and Seifert, 2001, 2002).
The activity of CNG and HCN channels is increased by binding of ligands (cAMP and cGMP) to an intracellular cyclic nucleotide-binding domain (CNBD). Different gating mechanisms have been proposed for these channels based on the symmetry assumed by the apo and ligand-bound CNBD: 1) dimer of dimers (Kuo et al., 2003; Schumacher et al., 2001; Sun et al., 2002; Zhou et al., 2004), where the four subunits associate and activate as independent dimers, or 2) tetramers (Jiang et al., 2002; Zagotta et al., 2003), where the four subunits activate in either a single concerted step or in four independent steps. These models structurally correspond to a two-fold symmetric tetramer and a four-fold symmetric tetramer, respectively. In an effort to distinguish between these models, structures of the isolated cyclic nucleotide binding-domain have been elucidated. The isolated CNBDs of HCN2 channels form a four-fold symmetric gating ring in the presence of cAMP (Zagotta et al., 2003), while the CNBDs of MloK1 form a dimer, arguing towards the existence of physiological dimers in both the absence and presence of cAMP (Clayton et al., 2004). Electrophysiological experiments on CNG and HCN channels from independent groups showed evidence for both types of association and gating: either as two dimers or independent monomers (Liu et al., 1998; Nache et al., 2006; Nache et al., 2005; Ruiz and Karpen, 1999; Ruiz and Karpen, 1997; Ulens and Siegelbaum, 2003). In order to reconcile all these apparently contradictory data, it was proposed that the CNBDs of cyclic nucleotide-modulated channels bind ligands as two independent dimers, followed by a conformational change from two-fold to four-fold symmetry as the binding signal is relayed to the C-linker for channel opening (Ulens and Siegelbaum, 2003).
As the symmetry arguments are based on biochemical and structural studies performed on isolated CNBDs of cyclic nucleotide-modulated channels, only the structure of a full-length cyclic nucleotide-modulated ion channel will reveal the true symmetry of the CNBD association in the context of the transmembrane portion of the channel. Here we present a structural analysis of a full-length prokaryotic cyclic nucleotide-modulated K+ channel, MloK1, from the symbiotic soil bacterium Mesorhizobium loti. MloK1 is a homolog of eukaryotic CNG and HCN channels, it is a member of the S4 superfamily of eukaryotic K channels, and it was shown previously to bind cyclic nucleotides specifically and be functional in radioactive uptake assays as a K+ channel whose activity is modulated by cAMP and cGMP (Nimigean and Pagel, 2007; Nimigean et al., 2004)
We analyzed the structure of full-length MloK1 by transmission electron microscopy (TEM) imaging of negatively-stained preparations of single-particles and of membrane-reconstituted and two-dimensionally crystallized membrane protein (electron crystallography). We found that the protein is a four-fold symmetric tetramer in the presence of cAMP in both the transmembrane and the cytoplasmic ligand-binding domains, in agreement with the HCN2-CNBD symmetry (Zagotta et al., 2003). We were able to assign an orientation for the CNBDs and the voltage sensor domains. The 2D crystal projection map of MloK1 in a “native” membrane-embedded environment is compatible with the voltage sensor domains arranged vertically, similar to those in the crystal structure of the Kv1.2 voltage-gated K+ channel, supporting their “native-like” arrangement in the three-dimensional (3D) crystal.
RESULTS
Sequence homology of MloK1
MloK1 has a high amino acid sequence homology with eukaryotic cyclic nucleotide-modulated K+ channels, which identifies MloK1 as a good candidate for structural and functional studies on this class of ion channels (Nimigean et al., 2004). The proposed topology of MloK1 and the sequence alignment of the transmembrane domains with eukaryotic voltage gated (Kv1.2 and Shaker) K+ channels are shown in Fig. 1. The membrane portion of MloK1 has a pore region formed by helices S5 and S6, with the characteristic “GYG” signature sequence for K+ selectivity (Heginbotham et al., 1994). The other four transmembrane helices (S1-S4) are homologous to the voltage sensor domains of the voltage-gated K+ channels, including three strategically placed basic residues in S4 (Nimigean et al., 2004) (Fig. 1b). The cytoplasmic portion of MloK1 consists of a conserved CNBD connected by a very short linker (~13 amino acids) to the end of the S6 helix (Nimigean et al., 2004). This linker is an abbreviated version of the extended C-linker domain of eukaryotic CNG and HCN channels, which was shown to play an important role in transducing the ligand-binding signal to the channel gate (Paoletti et al., 1999).
Figure 1.
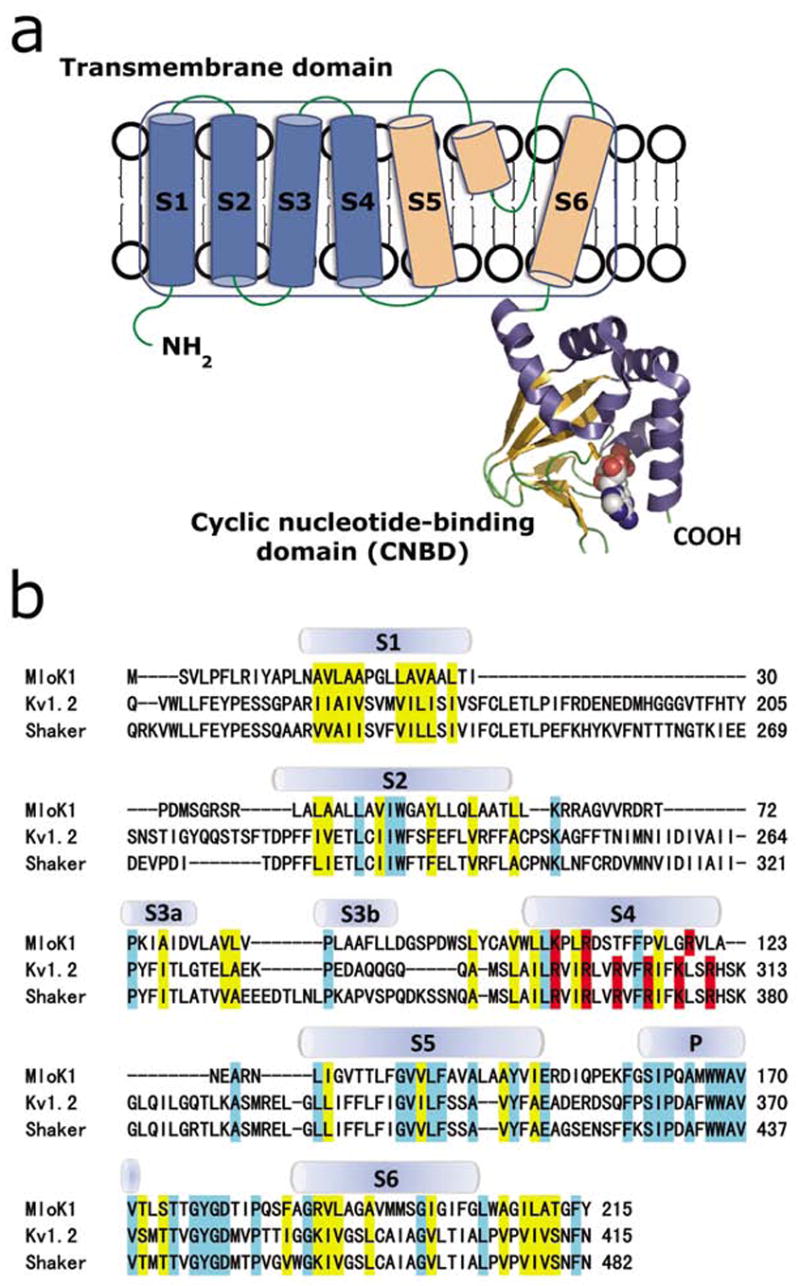
(a) Proposed topology of one MloK1 subunit. Each subunit consists of an N-terminal transmembrane domain and a C-terminal cyclic nucleotide-binding domain (CNBD). Helices S1-S4 (blue) are the putative voltage sensors, and helices S5, S6, and the pore helix (light orange) form the pore region. CNBD is shown in ribbon representation, bound cAMP is shown space-filled in cpk. (b) Sequence alignment of the transmembrane domains of MloK1 (Mesorhizobium loti), Kv1.2 (Rattus norvegicus) and Shaker (Drosophila melanogaster). The secondary structures of Kv1.2 are indicated. Residues of similar chemical characteristics are yellow, identical residues are light blue, and positively charged amino acids (arginine, lysine) in S4 are red.
Single-particle transmission electron microscopy
Monomeric MloK1 has a calculated molecular weight of 37 kDa. The detergent-surrounded tetrameric complex has an estimated molecular weight of ~210 kDa. Detergent-solubilized and purified MloK1 protein was imaged as negatively stained preparation by transmission electron microscopy (TEM), and 14,921 single particles were manually picked with the BOXER program (Ludtke et al., 1999). Of these, 5018 were used to generate a 3D reconstruction to a resolution of 16.3Å (Fig. 2). We imposed C4 symmetrization during the single particle reconstruction as the 2D crystal structure indicated four-fold symmetry (see below). The reconstruction shows a channel which resembles the tetrameric Shaker K+ channel structure (Sokolova et al., 2001) composed of two parts of different sizes, separated by a ~1 nm wide region of lower density (Fig. 2d). The large square domain is hypothesized to be the detergent-surrounded, pore-containing transmembrane region with a height of 5.2 +/− 0.5 nm (which approximates the thickness of the membrane), and a square side length of 8.5 +/− 0.5 nm. The larger transmembrane domain resembles a four-leaf propeller with a concave region in the center. The smaller, “hanging gondola”-like domain is composed of four blobs arranged around the central axis. These domains can be fit inside a square with a height of 4.0 +/− 0.5 nm and a square side length of 6.0 +/− 0.5 nm in the single particle reconstruction. These domains are thought to be the cyclic nucleotide-binding domains. The two domains are connected at four positions over the ~1 nm wide gap by short linkers. The shared four-fold axis passes through the model along the centers of the concave region of the large domain and the hole between the small domains. The resolution of the reconstruction was determined as 16.3Å, based on Fourier-shell correlation (FSC) with the 0.5 criterion (Fig. 2c).
Figure 2.
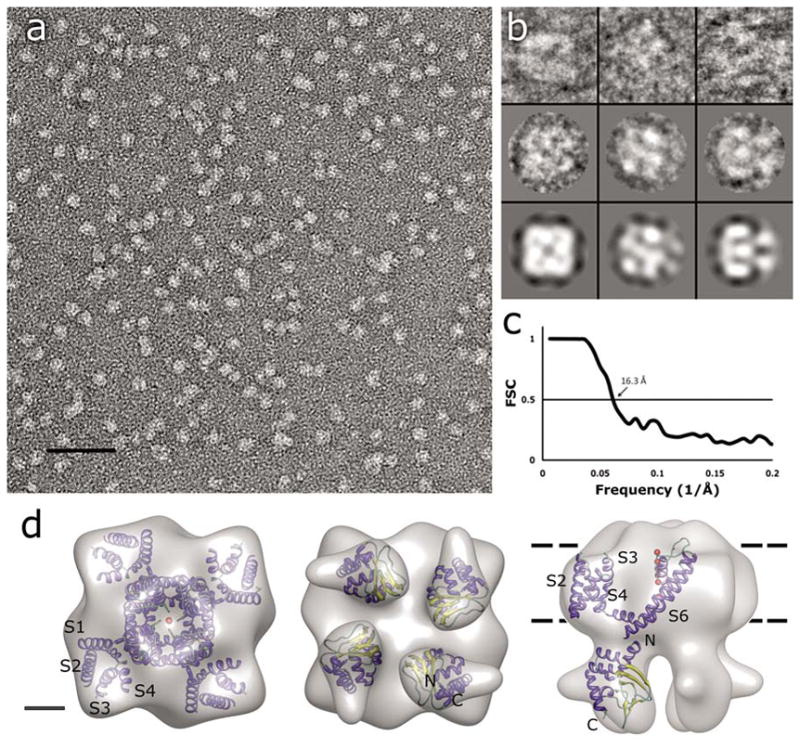
Single-particle TEM imaging of MloK1. (a) Electron micrograph of negatively-stained MloK1 particles. Detergent-solubilized MloK1 are bright on dark background. Scale bar 50 nm. (b) Selected class views and particle images of the MloK1 single particles. The first row is individual particle images from three classes in columns, the second row shows the corresponding class averages, and the last row is the p4-symmetrized 3D reconstruction density projected in the same direction. The width of the square panels is 16 nm. (c) Fourier-shell correlation (FSC) plot of the final 3D reconstruction, indicating a resolution of 16.3 Å (arrow, 0.5 criterion (Böttcher et al., 1997)). (d) The 3D reconstruction from 5018 single-particle images. Left, the proposed top-view from the extracellular side. Center, the bottom view, showing the four CNBDs. Right, the side view. The approximate membrane plane is indicated by the dotted lines. Scale bar 2 nm. Inside each density we show the docking of the high-resolution structures of Kv1.2 (PDB code: 2A79) and the MloK1 CNBD (PDB code: 1VP6) into the MloK1 3D reconstruction. The α-helices are in blue, β-strands in yellow, and loops in green. Red balls represent K ions in the pore.
Molecular docking
Two different structures were used for the docking experiments into the calculated single-particle reconstruction. The transmembrane domain of the voltage-gated K+ channel Kv1.2 (PDB code 2A79 (Long et al., 2005)) was used for the large domain, as Kv1.2 was proposed to be in a more “native”-like conformation than KvAP (Lee et al., 2005), due to the presence of lipids during purification and crystallization as well as the constraint of the S1 helix by the T1 domain (Lee et al., 2005). For the smaller ring-like domain, the structure of the CNBD of MloK1 was used (PDB code 1VP6 (Clayton et al., 2004)).
The pore region of Kv1.2, consisting of helices S5 and S6, agreed well with the central region of the reconstructed MloK1 model, and correlated well with the concave region in the center of the reconstruction model (Fig. 2d). The four voltage-sensing subunits (helix S1 to S4) also fit nicely into the four propeller-like regions on the MloK1 model, leaving room for a surrounding detergent belt. The loop between pore helix and helix S5 of Kv1.2 fits the protrusions close to the 4-fold center of the MloK1 reconstruction.
Docking attempts of the MloK1 CNBD dimers (PDB code 1VP6; (Clayton et al., 2004) into the smaller four blobs failed. Similarly, even though the four densities in our single particle reconstruction are arranged in a square with similar outer dimensions than the ring-like structure observed by (Zagotta et al., 2003), docking with a ring-like arrangement of four connected CNBDs failed (Fig. 3b). However, we could dock four individual MloK1 CNBDs into the four densities of our reconstruction, resulting in a monomeric arrangement of the CNBD such that its N-terminus is oriented towards the transmembrane part, and the cyclic AMP binding pocket is facing vertically away from the membrane (Fig. 2, and compare Fig. 3a with Fig. 3b). While the low resolution of our reconstruction does not allow a safe assignment of the orientation of the CNBDs, our data document the presence of narrow gaps between the monomeric CNBDs, and their four-fold symmetric arrangement.
Figure 3.
Arrangement of the CNBDs in the constructed model. (a) A vertical arrangement of the CNBDs fits best into the determined 3D structure. (b) The horizontal arrangement of the CNBDs as in the HCN2 CNBD crystal structure cannot satisfyingly be docked into the 3D volume. S1-S4 helices, blue; S5-S6, yellow, CNBD, red. Scale bar 5 nm.
Electron crystallography of 2D membrane crystals
Purified MloK1 was reconstituted into phospholipid membranes and 2D-crystallized in the membrane-embedded state by temperature-controlled slow detergent dialysis (Jap et al., 1992). 2D crystals of up to 1-μm diameter were obtained in the presence of 200 μM cAMP. Crystals were imaged as negatively stained preparations in the TEM (Renault et al., 2006) (Fig. 4a). The calculated power spectrum of recorded 2D crystal images showed clear diffraction spots up to 2 nm resolution before image unbending (Fig. 4b).
Figure 4.
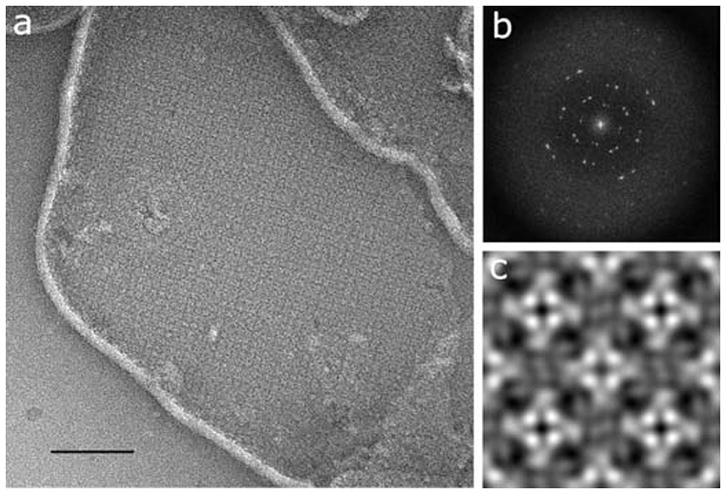
Two-dimensional membrane crystal of MloK1. (a) Electron micrograph of a negatively stained 2D crystal of membrane-reconstituted MloK1. The bright lines are the edges of the membrane crystals; the square lattice of the crystal is clearly visible. Scale bar 200 nm. (b) Typical calculated power spectrum, showing diffraction spots at 2 nm resolution before image processing. (c) The non-symmetrized reconstruction, showing 2×2 unit cells in p4212 symmetry. Unit cell dimensions are a=b=129Å, γ=90°. Protein is white in (a) and (c).
Comparison of phase residuals with the ALLSPACE program (Valpuesta et al., 1994) revealed P4212 symmetry for deeply stained 2D crystals (Fig. 4c and 5a), and P4 symmetry with a second, differently stained tetramer in the corners of the unit cells for unevenly stained sample preparations (Fig. 5b), both at 16.0Å resolution. This indicates a head-to-tail packing of neighboring tetramers, supporting the P4212 symmetry. The unit cell parameters were a=b=129Å, γ=90°. The non-symmetrized unit cell shows a central tetrameric complex with a brighter four-fold symmetric density in the center, with an inner diameter of 2.5 nm and a square side length of 4.5 nm, which is surrounded by a propeller-like density of 7 nm square side length.
Figure 5.

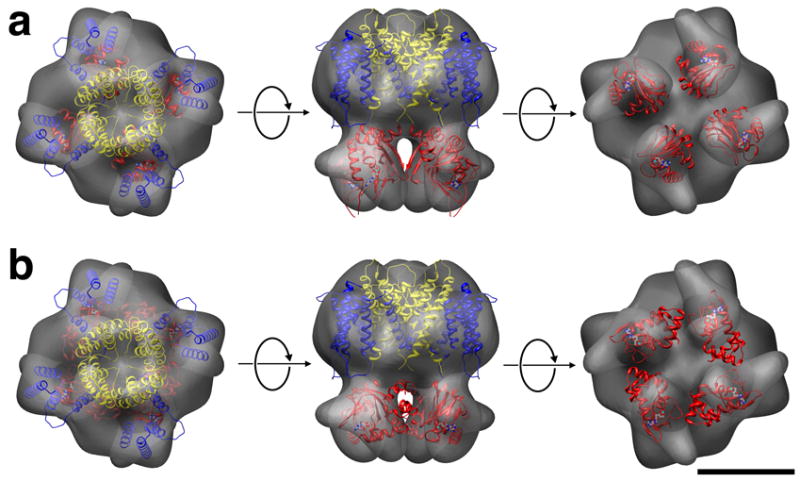
Projection maps of evenly (a) and unevenly (b) stained 2d crystals of MloK1 after image processing. A difference map (c) between the two shows the contours of one surface only. Insets in (a) to (c) are cartoons that depict the staining level. (d) Arrangement of the single particle reconstruction in the 2d crystal-like alternative orientations with overlaid X-ray structures of Kv1.2 (S1-S4, red; S5-S6, yellow) and MloK1 CNBD (blue).
We interpreted the map in Fig. 5a as showing both surfaces equally well stained, and the map in Fig. 5b showing one surface stained more strongly than the other surface. We therefore used the two maps to calculate a difference map, which shows the surface contours of one surface only (Fig. 5c). This map revealed for each tetramer four bright, elongated densities in a propeller-like fashion. These were in good agreement with the shape and arrangement of the four densities ascribed to the CNBD’s in the single particle reconstruction (Fig. 2d, 3, and 5d).
The non-symmetrized 2D crystal projection map shows for the MloK1 CNBDs four symmetrically arranged densities, indicating a tetrameric arrangement of the CNBDs. This calculated projection map corresponds to an average over a large number of potentially differently oriented crystal unit cells. This averaging effect could have produced a four-fold symmetric result from two-fold symmetric unit cells that are randomly oriented in the membrane plane (either vertically or horizontally). However, two facts show that the observed four-fold symmetry is not an averaging artifact. First, the final projection map is strongly contrasted and shows excellent phase residuals up to 16 Å resolution, documenting a low variation among the contributing individual unit cell images. Second, a single-particle image processing of the unit cell images of the 2D crystal, allowing each unit cell to occupy either a vertical or horizontal orientation in the image, still produced the same final four-fold symmetric projection map. This was done by transforming the 2D crystal image into a stack of single particles representing unit cells, and subjecting this to single particle image processing. This was done without applying any symmetry and instead allowing a 90-degree rotation of the individual particle images (Zeng et al., 2007). To minimize the influence of any reference, this single particle processing was done using a maximum likelihood algorithm, which also allowed obtaining a higher resolution than cross-correlational alignment would have produced. The obtained projection map again showed four-fold symmetry (Suppl. Fig. S3), and clearly established the four-fold symmetry of the membrane embedded MloK1 tetramers in the presence of cAMP.
Model building
Models for full-length MloK1 with the voltage sensor paddles in the “vertical”, “tilted”, and “horizontal” states were constructed by satisfying spatial constraints utilizing the program MODELLER (Sali and Blundell, 1993). Initial model restraints were derived from the known structure of the CNBD for MloK1 (PDB code 1VP6; (Clayton et al., 2004; Zagotta et al., 2003) coupled with homology modeling using either the predicted sequence alignment to the transmembrane domain of Kv1.2 (“vertical” state (Long et al., 2005)) or KvAP (“horizontal” state (Jiang et al., 2003), and “tilted” state (Lee et al., 2005)). Additional spatial constraints were derived from the 3-D single particle reconstruction and the 2-D crystal projection map. The resulting “vertical” and “horizontal” models could both be satisfyingly docked into the 3-D single particle reconstruction, yet the “tilted” model had two helices protruding from the 3D reconstruction (Suppl. Fig. S1). However, while the intermediate or “tilted” state could be oriented within the unit cell to match the electron density of the 2D projection map, such a physical arrangement would be impossible within the given unit cell parameters as it would require the paddles from adjacent MloK1 tetramers to occupy the same space (Suppl. Fig. S2). So, only the “vertical” and the “horizontal” state models fit the electron density profile of the 2D crystal projection maps when overlaid in the unit cell configuration (Fig. 6 and Suppl. Fig. S2). This established the “vertical” (or “up”), as the likely configuration of the voltage sensors for MloK1, as the “horizontal” configuration of KvAP might have been influenced by the absence of stabilizing lipids in the 3D crystal (Lee et al., 2005).
Figure 6.
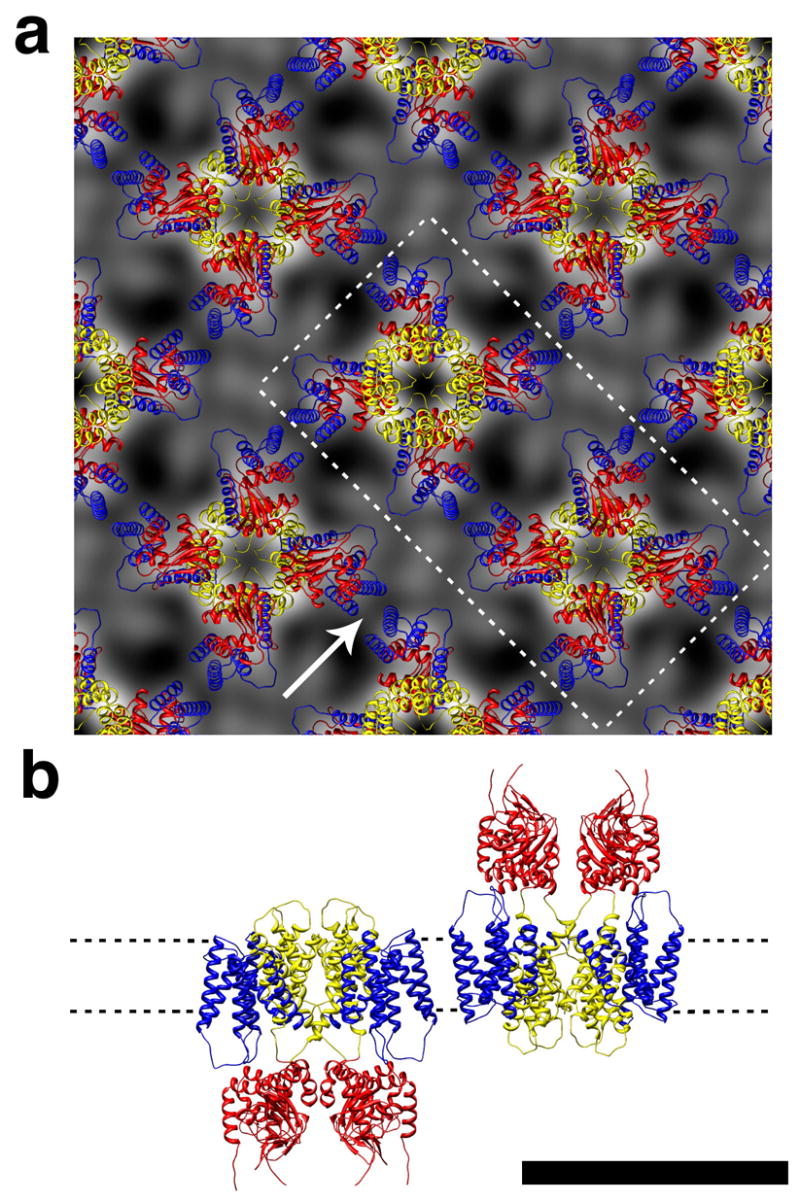
The homology model with “vertically” oriented paddles fit to the 2D crystal projection map. (a) 2×2 unit cell arrangement. Protein is in white. (b) Side view of the two tetramers that are in (a) indicated by the dotted line and seen as indicated by the arrow. Scale bar 10 nm. S1-S4 helices, blue; S5-S6, yellow, CNBD, red.
DISCUSSION
We have determined the 3D structure of full-length MloK1, a prokaryotic cyclic nucleotide-modulated K+ channel, at 16.3Å resolution and the projection map of membrane reconstituted MloK1 within a 2D crystal at 16Å resolution. The structural data show a four-fold symmetric arrangement of the CNBDs in the presence of cAMP, in agreement with the symmetry observed in the HCN2 CNBD structure (Zagotta et al., 2003). In addition, the structure reveals the “native” lipid-embedded arrangement of the voltage sensor domains, which agrees with the structure of Kv1.2 (Long et al., 2005) within the resolution of the data presented here. Although it is unclear whether the open probability of MloK1 is voltage dependent, sequence homology indicates the presence of a voltage sensor-like domain in MloK1, which is clearly defined in the single-particle reconstruction and inferred in the projection structure. Is this a “bona fide” voltage sensor? MloK1 has only three basic residues in S4 as opposed to the 6–9 present in traditional voltage-gated channels. However, it was shown in Shaker channels that the top most four arginines account for most of the gating charge transfer across the membrane during activation, which means that our three basic amino acids should be able to confer voltage-dependent open probability if they are strategically located and if the coupling is intact (Aggarwal and MacKinnon, 1996; Seoh et al., 1996). Furthermore, it was recently shown that a channel from Lysteria monocytogenes with only three charges in its equivalent S4 region displays voltage dependent open probability (Lundby et al., 2006; Santos et al., 2006), and that the MmaK channel, a potassium channel from Magnetospirillum magnetotacticum which displays high sequence homology to MloK1 is sensitive to voltage (Kuo et al., 2007).
The structure of the CNBD domain of HCN2 is also a four fold symmetric tetramer. Although the dimensions of the HCN2 CNBD tetramer arranged horizontally (a square 5.9 nm wide) generally correspond to the outer diameter of the densities attributed to the MloK1 CNBD in both the single-particle reconstruction and the 2D projection structure (approx. 6.5 nm wide with a 2.5 nm diameter gap), they did not satisfyingly dock in either of the two, due to the gaps between the observed CNBD densities (Fig. 3b). Hence, we propose a different, vertical spatial arrangement for the CNBD gating ring of MloK1 (Fig. 3a). However, due to the low resolution of the MloK1 full-length structures, the contact interface and the exact orientation of the CNBDs still needs to be characterized.
One explanation for the HCN2 CNBDs having a different arrangement from MloK1 is that the latter lacks a C-linker domain and it has been shown that all contacts between individual subunits in the HCN2 CNBD tetramer are made by these C-linker domains (Zagotta et al., 2003). In CNG and HCN channels, the hexahelical C-linker was proposed to be the transducer domain, which senses the binding of ligands within individual subunits and then relays the signal to the gate (Gordon and Zagotta, 1995a, b, c; Ildefonse and Bennett, 1991; Johnson and Zagotta, 2001; Paoletti et al., 1999; Zagotta et al., 2003; Zong et al., 1998). MloK1 lacks an extended C-linker region and has only a maximum of 13 amino acids in between the end of S6 and the beginning of the CNBD. Despite missing this structure, MloK1 still forms a four fold symmetric structure and allows cyclic nucleotide modulated ion transport through its pore, suggesting that the extended C-linker is not absolutely necessary for cyclic nucleotide modulation (Nimigean et al., 2004) Due to the fact that it is a central part of gating in CNG channels, the C-linker absence might have interesting consequences for the gating of MloK1.
Interestingly, the X-ray structures of the isolated MloK1 CNBDs suggest that they form physiological dimers with and without cAMP, in contrast with both our findings and with the four fold symmetric tetrameric structure of the HCN2 CNBD. This suggests that the oligomeric state of the isolated MloK1 CNBD domains might have been affected by the 3D crystallization conditions (Clayton et al., 2004) as well as by the lack of the transmembrane part of the protein. On the other hand, the CNBDs of MloK1 as well as those of CNG and HCN channels are homologous to the bacterial transcriptional activator, CAP, and protein kinase A (PKA) and G (PKG), which are physiological homodimers (Shabb and Corbin, 1992), supporting the MloK1 dimer finding. However, the fact that the dimer interface for all the above proteins is different and also different from the HCN2 CNBD tetramer interface implies that the oligomerization of the CNBD-containing proteins is somewhat driven by the effector domains to which the CNBDs are attached. Only structural work on the full-length proteins can clarify the gating mechanisms and symmetry of transitions. Hence, the CNBD gating ring of MloK1 in the presence of cAMP appears to be a four-fold symmetric tetramer due partly to the strong tetramerization signal from the transmembrane domain. Our results are in contrast to the single-particle reconstruction by (Higgins et al., 2002), who determined the 3D structure of the hetero-tetrameric cGMP-gated CNG channel from bovine retina at 35Å resolution and in the absence of ligand, and suggested that the cytoplasmic domains of that channel were arranged as a pair of dimers.
Our 2D membrane crystals of MloK1 show for the first time structural data for a membrane-embedded conformation of voltage sensors domains. The difference map between an evenly and an unevenly stained 2D crystal image allowed the visualization of the density of only one membrane-distant surface, which mainly shows the projection of the CNBDs of one side of the crystal (Fig. 5a–c). The obtained map shows four densities in propeller like arrangement separated by gaps, in good agreement with the CNBD densities in the single particle 3D reconstruction (Fig. 5d). The S1-S4 region of the MloK1 proteins are not individually assignable in the 2D crystal data, since the staining mostly contours the crystal surface. However, the observed maps and the dimensions of the unit cell are only compatible with the S1-S4 region being located directly above the CNBDs, as also shown in the 3D reconstruction (Fig. 2d, 3).
The Kv1.2 transmembrane region could satisfyingly be docked into the transmembrane region of the MloK1 single particle 3D reconstruction; the propeller density in the MloK1 projection map was in good agreement with the conformations of the voltage sensor domains of Kv1.2. The single-particle reconstruction of MloK1 shows some additional density in the transmembrane region between the four S1-S4 subunits, which could arise from detergent molecules and co-purified lipids that surround and interact with the transmembrane portion of the protein and then conglomerate in the space between the voltage sensors and the pore.
In order to analyze the conformation of the S1-S4 “paddle” domains of MloK1, we constructed homology models of MloK1 based on sequence alignment to Kv1.2, KvAP and the MloK1 CNBD crystal structures. Models were generated such that the “paddle” orientation either corresponds to a “vertical” conformation, as found in the Kv1.2 structure (Long et al., 2005), a “horizontal” conformation, as found in the KvAP crystal structure (Jiang et al., 2003), or the intermediate or “tilted” conformation as described later for KvAP (Lee et al., 2005). While all three models can fit the 3D electron density from the single particle reconstruction at the given resolution (Fig. 3 and Suppl. Fig. S1), only the “vertical” and “horizontal” conformations could fit our 2D membrane protein crystal unit cell without causing steric hindrance (Fig. 6 and Suppl. Fig. S2). Since our 2D crystals in P4212 symmetry are in a head-to-tail packing for neighboring MloK1 tetramers, the “horizontal” conformation of the paddles would be possible although it is generally accepted by now that this extreme, likely non-native, conformation could be the result of crystallization distortions caused by the absence of lipids (Lee et al., 2005). This identified the “vertical” configuration as the state of the paddle in our MloK1 preparation (Fig. 3, 6). Future research using cryo-electron microscopy on these crystals to achieve sub-nanometer resolution should elucidate the precise conformational state of MloK1 in the presence and absence of cAMP, which is currently being pursued.
In conclusion, we determined the structure of a detergent-solubilized cyclic nucleotide-modulated channel at 16.3 Å resolution by single-particle electron microscopy, and the membrane embedded projection map at 16 Å resolution. We found that the channel forms a four-fold symmetric tetramer in the presence of ligands and this symmetry also extends to the CNBDs. The voltage sensor regions are located at the periphery of the core pore-forming region and directly above the CNBDs. The paddles of the MloK1 channel molecules were found in the “vertical” conformation in the 2D membrane crystals.
EXPERIMENTAL PROCEDURES
Expression and purification
MloK1 protein was expressed and purified as previously described (Nimigean et al., 2004). In brief, the C-terminal hexahistidine-tagged MloK1 coding region from M. loti cloned into an Escherichia coli expression vector (pASK90 (Skerra, 1994)) was transformed into E. coli JM83 cells. Transformed cells were grown in Terrific Broth (TB) at 37°C and the expression of the protein was induced for 90 minutes with 0.2 mg/L anhydrotetracycline (Acros organics) when the OD550 = 1. Cells were pelleted, lysed by sonication, solubilized in 50 mM n-decyl maltopyranoside (DM, Anatrace) and purified over a Ni-affinity column (Qiagen) and gel filtration chromatography (Superdex 200, GE). To maintain integrity and function of MloK1, 200 μM cAMP (Fluka) was present throughout the purification process. The same results were obtained when E. coli total lipids (Avanti polar lipids), solubilized in 10 mM DM, were also present during purification.
Transmission electron microscopy (TEM) imaging
Detergent-solubilized and purified MloK1 was diluted with the buffer (100 mM KCl, 20 mM Tris-HCl, 20 mM DM, 200 μM cAMP and pH 7.60) and adsorbed onto carbon-coated copper grids, negatively stained with 2% (w/v) uranyl formate immediately after protein purification, blotted, distilled water-washed and air-dried. Grids were imaged in a JEOL JEM-2100F transmission electron microscope equipped with field-emission gun (FEG), under low-dose conditions. The micrographs were recorded on a 4096×4096 pixel TVIPS F415 CCD camera at 200 kV acceleration voltage, 1.0 to 1.5 μm defocus and 50,000X nominal magnification, resulting in a pixel size of 2 Å.
Single-particle 3D reconstruction
The SPIDER (Frank et al., 1996) and EMAN (Ludtke et al., 1999) program suites were used for processing the single particle images. Raw images were corrected for the contrast transfer function (CTF). 14,921 particle images were manually boxed with a square box size of 80-pixel width. The particle images were centered, classified by multivariate statistical analysis (MSA), and an initial model was built from randomly picked 1,000 particle images by the common-line method to determine the particle Euler angles. This first model was resolution limited to 5 nm, and used for the generation of 40 reference projections. The reconstruction was then iteratively refined, using only the 5018 particles with best correlation to the reconstruction. Particles were reference-based aligned to 190 projections, class average images were calculated, and a C4-symmetrized 3D reconstruction density map was calculated. Fourier shell correlation (FSC) of successive iterations showed no further resolution improvement after 6 iterations (data not shown). The final reconstruction showed a resolution of 16.3 Å, as determined by FSC (using the 0.5 criterion) between reconstructions made from even and odd particles that had been aligned onto the same reference projection set (Fig. 2). For the handedness determination of the single particle 3D reconstruction, a 3D reconstruction of the 2D membrane crystals was determined by electron tomography, using serialEM for the automated data collection (Mastronarde, 2005) and IMOD for the image processing (Kremer et al., 1996). The tomography 3D reconstruction showed 3D densities for the CNBDs (data not shown), which were used for the determination of the correct handedness of the single particle 3D reconstruction.
Model fitting
The atomic coordinates of Kv1.2 (Protein Data Bank (PDB) code: 2A79) and CNBD with ligand of MloK1 (PDB code: 1VP6) were used for docking into the MloK1 3D map. The UCSF CHIMERA graphical package (Pettersen et al., 2004) was used for the manual docking. The docking criterion was supported in the FIT MODELS IN MAP function built in the package. The MloK1 CNBDs were tentatively tetramerized according to the atomic coordinates of the HCN2 CNBDs (PDB code: 1Q5O, residues 501–633), the RMSD of the tetramerized MloK1 CNBDs and HCN2 CNBDs was 1.60 Å. However, the resulting tetrameric structure did not fit the single particle 3D reconstruction.
Two-dimensional (2D) crystallization and electron crystallography imaging
Detergent solubilized and purified MloK1 was mixed with E.coli total lipid extract and dialyzed against detergent-free buffer (20 mM KCl, 1 mM EDTA, 20 mM Tris, pH 6.7) and in the presence of 200 μM cAMP. Following recommendations from Tom Walz (Harvard Med. School), a temperature profile oscillating 5 times between 37°C and 25°C was used during a 5-day dialysis procedure. Grids of 2D crystals were prepared and imaged as described above. Recorded images were processed with the 2dx software package (Gipson et al., 2007), which is based on the MRC software (Crowther et al., 1996).
Homology modeling
Full-length models of MloK1 were constructed using the program MODELLER9V1 (Sali and Blundell, 1993). Sequence alignment of MloK1 against Kv1.2, KvAP, and the known structure of MloK1’s CNBD provided the initial restraints. The atomic coordinates of the CNBD of MloK1 (PDB code: 1VP6) combined with those of Kv1.2 (PDB code: 2A79) for the transmembrane domain were used as the template for the “vertical” state model, while both the “tilted” and “horizontal” state models used the atomic coordinates of KvAP (PDB codes: 2A0L and 1ORQ, respectively) for the transmembrane domain sequence homology. Additional information from the above mentioned 3-D single particle reconstruction and 2-D projection map, including unit cell parameters, electron density distribution and symmetry, were incorporated as further spatial constraints to optimize the output models. The UCSF CHIMERA graphical package (Pettersen et al., 2004) was used for visualization, model fitting (as described above) and unit cell evaluation.
Supplementary Material
Table 1.
Phase residuals in resolution ranges, obtained during merging of two evenly negatively stained 2D crystal image data. Columns are: Resolution range in Å; IQ=1…8: Intensity Quotient categories of reflections (Henderson et al., 1990); All IQs: Average phase residuals with equal reflection weighting; IQ-wght: Average phase residual with IQ-weighting. In each resolution range, the phase residuals (upper row) and the number of reflections (lower row) are given. This table documents acceptable phase residuals up to 15.4 Å resolution. The projection map data were limited to 16.0 Å resolution.
| PHASE RESIDUALS IN RESOLUTION RANGES | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Res. Range [Å] | IQ= 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | all IQs | IQ-wght |
| 100.0 – 40.8 | 3.0
13 |
9.2
5 |
50.6
6 |
44.4
5 |
172.7
1 |
51.2
3 |
1.9
1 |
142.7
2 |
34.0
36 |
19.8
36 |
| 40.7 – 28.9 | 2.7
1 |
10.6
21 |
8.6
4 |
22.2
2 |
0.0
0 |
158.6
2 |
0.0
0 |
74.8
1 |
22.4
31 |
14.3
31 |
| 28.8 – 23.6 | 2.0
7 |
5.1
1 |
0.0
0 |
25.7
5 |
58.7
2 |
16.7
4 |
7.1
1 |
39.9
6 |
22.2
26 |
11.9
26 |
| 23.5 – 20.4 | 11.0
1 |
12.7
6 |
15.2
5 |
8.1
7 |
29.4
2 |
28.6
2 |
14.8
3 |
66.6
7 |
25.7
33 |
13.7
33 |
| 20.3 – 18.3 | 0.0
0 |
13.0
2 |
16.0
4 |
23.6
1 |
43.1
2 |
4.1
1 |
46.9
1 |
54.6
8 |
36.2
19 |
19.7
19 |
| 18.2 – 16.7 | 0.0
0 |
0.0
0 |
14.0
1 |
23.6
1 |
39.5
2 |
18.7
2 |
49.6
4 |
60.3
6 |
44.6
16 |
32.3
16 |
| 16.6 – 15.4 | 0.0
0 |
0.0
0 |
0.0
0 |
18.4
2 |
31.8
3 |
63.4
1 |
31.2
1 |
52.2
8 |
43.0
15 |
30.7
15 |
| ALL: |
3.0
22 |
10.7
35 |
24.6
20 |
23.3
23 |
50.8
12 |
46.7
15 |
30.0
11 |
60.0
38 |
||
| Overall: Phaseresidual = 30.630 | Number of spots = 176 | |||||||||
Acknowledgments
The single particle 3D reconstruction volume of full-length MloK1 has been deposited in the Electron Microscopy Data Bank (EMDB, http://www.ebi.ac.uk/msd), accession number 5548. This work was in part supported by the National Institutes of Health (NIGMS), the National Science Foundation (NSF-BIO), and the American Heart Association. We thank Tom Walz for suggestions concerning temperature oscillations during dialysis. We also thank Ben Webb for discussions regarding the usage of MODELLER.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aggarwal SK, MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- Böttcher B, Wynne SA, Crowther RA. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature. 1997;386:88–91. doi: 10.1038/386088a0. [DOI] [PubMed] [Google Scholar]
- Clayton GM, Silverman WR, Heginbotham L, Morais-Cabral JH. Structural basis of ligand activation in a cyclic nucleotide regulated potassium channel. Cell. 2004;119:615–627. doi: 10.1016/j.cell.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Craven KB, Zagotta WN. CNG and HCN channels: two peas, one pod. Annu Rev Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Henderson R, Smith JM. MRC image processing programs. J Struct Biol. 1996;116:9–16. doi: 10.1006/jsbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Gipson B, Zeng X, Zhang ZY, Stahlberg H. 2dx-User-friendly image processing for 2D crystals. J Struct Biol. 2007;157:64–72. doi: 10.1016/j.jsb.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Gordon SE, Zagotta WN. A histidine residue associated with the gate of the cyclic nucleotide-activated channels in rod photoreceptors. Neuron. 1995a;14:177–183. doi: 10.1016/0896-6273(95)90252-x. [DOI] [PubMed] [Google Scholar]
- Gordon SE, Zagotta WN. Localization of regions affecting an allosteric transition in cyclic nucleotide-activated channels. Neuron. 1995b;14:857–864. doi: 10.1016/0896-6273(95)90229-5. [DOI] [PubMed] [Google Scholar]
- Gordon SE, Zagotta WN. Subunit interactions in coordination of Ni2+ in cyclic nucleotide-gated channels. Proc Natl Acad Sci USA. 1995c;92:10222–10226. doi: 10.1073/pnas.92.22.10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heginbotham L, Lu Z, Abramson T, MacKinnon R. Mutations in the K+ channel signature sequence. Biophys J. 1994;66:1061–1067. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of Bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- Higgins MK, Weitz D, Warne T, Schertler GF, Kaupp UB. Molecular architecture of a retinal cGMP-gated channel: the arrangement of the cytoplasmic domains. EMBO J. 2002;21:2087–2094. doi: 10.1093/emboj/21.9.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ildefonse M, Bennett N. Single-channel study of the cGMP-dependent conductance of retinal rods from incorporation of native vesicles into planar lipid bilayers. J Membr Biol. 1991;123:133–147. doi: 10.1007/BF01998084. [DOI] [PubMed] [Google Scholar]
- Jap BK, Zulauf M, Scheybani T, Hefti A, Baumeister W, Aebi U, Engel A. 2D crystallization: from art to science. Ultramic. 1992;46:45–84. doi: 10.1016/0304-3991(92)90007-7. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R. X-ray structure of a voltage-dependent K+ channel. Nature. 2003;423:33–41. doi: 10.1038/nature01580. [DOI] [PubMed] [Google Scholar]
- Johnson JP, Jr, Zagotta WN. Rotational movement during cyclic nucleotide-gated channel opening. Nature. 2001;412:917–921. doi: 10.1038/35091089. [DOI] [PubMed] [Google Scholar]
- Kaupp UB, Seifert R. Molecular diversity of pacemaker ion channels. Annu Rev Physiol. 2001;63:235–257. doi: 10.1146/annurev.physiol.63.1.235. [DOI] [PubMed] [Google Scholar]
- Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82:769–824. doi: 10.1152/physrev.00008.2002. [DOI] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- Kuo MM, Saimi Y, Kung C, Choe S. Patch-clamp and phenotypic analyses of a prokaryotic cyclic nucleotide-gated K channel using Escherichia coli as a host. J Biol Chem. 2007 doi: 10.1074/jbc.M703618200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Lee A, Chen J, MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci USA. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DT, Tibbs GR, Paoletti P, Siegelbaum SA. Constraining ligand-binding site stoichiometry suggests that a cyclic nucleotide-gated channel is composed of two functional dimers. Neuron. 1998;21:235–248. doi: 10.1016/s0896-6273(00)80530-9. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Lundby A, Santos JS, Zazueta C, Montal M. Molecular template for a voltage sensor in a novel K+ channel. II. Conservation of a eukaryotic sensor fold in a prokaryotic K+ channel. J Gen Physiol. 2006;128:293–300. doi: 10.1085/jgp.200609573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Nache V, Kusch J, Hagen V, Benndorf K. Gating of cyclic nucleotide-gated (CNGA1) channels by cGMP jumps and depolarizing voltage steps. Biophys J. 2006;90:3146–3154. doi: 10.1529/biophysj.105.078667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nache V, Schulz E, Zimmer T, Kusch J, Biskup C, Koopmann R, Hagen V, Benndorf K. Activation of olfactory-type cyclic nucleotide-gated channels is highly cooperative. J Physiol. 2005;569:91–102. doi: 10.1113/jphysiol.2005.092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimigean CM, Pagel M. Ligand binding and activation in a prokaryotic cyclic nucleotide-modulated channel. J Mol Biol. 2007 doi: 10.1016/j.jmb.2007.1006.1030. in press. [DOI] [PubMed] [Google Scholar]
- Nimigean CM, Shane T, Miller C. A cyclic nucleotide modulated prokaryotic K+ channel. J Gen Physiol. 2004;124:203–210. doi: 10.1085/jgp.200409133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Young EC, Siegelbaum SA. C-Linker of cyclic nucleotide-gated channels controls coupling of ligand binding to channel gating. J Gen Physiol. 1999;113:17–34. doi: 10.1085/jgp.113.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Renault L, Chou HT, Chiu PL, Hill RM, Zeng X, Gipson B, Zhang ZY, Cheng A, Unger V, Stahlberg H. Milestones in electron crystallography. J Comput Aided Mol Des. 2006;20:519–527. doi: 10.1007/s10822-006-9075-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz M, Karpen JW. Opening mechanism of a cyclic nucleotide-gated channel based on analysis of single channels locked in each liganded state. J Gen Physiol. 1999;113:873–895. doi: 10.1085/jgp.113.6.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz ML, Karpen JW. Single cyclic nucleotide-gated channels locked in different ligand-bound states. Nature. 1997;389:389–392. doi: 10.1038/38744. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Santos JS, Lundby A, Zazueta C, Montal M. Molecular template for a voltage sensor in a novel K+ channel. I. Identification and functional characterization of KvLm, a voltage-gated K+ channel from Listeria monocytogenes. J Gen Physiol. 2006;128:283–292. doi: 10.1085/jgp.200609572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410:1120–1124. doi: 10.1038/35074145. [DOI] [PubMed] [Google Scholar]
- Seoh SA, Sigg D, Papazian DM, Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- Shabb JB, Corbin JD. Cyclic nucleotide-binding domains in proteins having diverse functions. J Biol Chem. 1992;267:5723–5726. [PubMed] [Google Scholar]
- Skerra A. Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene. 1994;151:131–135. doi: 10.1016/0378-1119(94)90643-2. [DOI] [PubMed] [Google Scholar]
- Sokolova O, Kolmakova-Partensky L, Grigorieff N. Three-dimensional structure of a voltage-gated potassium channel at 2.5 nm resolution. Structure (Camb) 2001;9:215–220. doi: 10.1016/s0969-2126(01)00578-0. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- Ulens C, Siegelbaum SA. Regulation of hyperpolarization-activated HCN channels by cAMP through a gating switch in binding domain symmetry. Neuron. 2003;40:959–970. doi: 10.1016/s0896-6273(03)00753-0. [DOI] [PubMed] [Google Scholar]
- Valpuesta JM, Carrascosa JL, Henderson R. Analysis of electron microscope images and electron diffraction patterns of thin crystals of phi 29 connectors in ice. J Mol Biol. 1994;240:281–287. doi: 10.1006/jmbi.1994.1445. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Olivier NB, Black KD, Young EC, Olson R, Gouaux E. Structural basis for modulation and agonist specificity of HCN pacemaker channels. Nature. 2003;425:200–205. doi: 10.1038/nature01922. [DOI] [PubMed] [Google Scholar]
- Zeng X, Stahlberg H, Grigorieff N. A maximum-likelihood approach to two-dimensional crystals. 2007 doi: 10.1016/j.jsb.2007.09.013. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Qian Y, Kunjilwar K, Pfaffinger PJ, Choe S. Structural insights into the functional interaction of KChIP1 with Shal-type K(+) channels. Neuron. 2004;41:573–586. doi: 10.1016/s0896-6273(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Zong X, Zucker H, Hofmann F, Biel M. Three amino acids in the C-linker are major determinants of gating in cyclic nucleotide-gated channels. EMBO J. 1998;17:353–362. doi: 10.1093/emboj/17.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.