

Abstract
Total syntheses of (−)-dictyostatin, 6,16-bis-epi-dictyostatin, 6,14,19-tris-epi-dictyostatin and a number of other isomers and analogs are reported. Three main fragments—top, middle and bottom—were first assembled and then joined by olefination or anionic addition reactions. After appending the two dienes at either end of the molecule, macrolactonization and deprotection completed the syntheses. The work proves both the relative and absolute configurations of (−)-dictyostatin. The compounds were evaluated by cell-based measurements of increased microtubule mass and antiproliferative activity, and in vitro tubulin polymerization assays as well as competitive assays with paclitaxel for its binding site on microtubules. These assays showed dictyostatin to be the most potent of the agents and further showed that the structural alterations caused from 20- to >1000-fold decreases in activity.
Keywords: Microtubule, macrolactone, paclitaxel, discodermolide
INTRODUCTION
Microtubule stabilization by small molecule natural products and analogsi is a clinically proven chemotherapeutic approach to the treatment of solid tumors, and microtubule stabilizers exhibit a diverse assortment of molecular scaffolds. Dictyostatin and discodermolide (Figure 1), the taxanes paclitaxel and docetaxel, the epothilones, the sarcodictyins and eleutherobin, and the ketosteroids 2-ethoxy-7-keto-17β-estradiol and the taccalonolides all bind with varying affinities to the paclitaxel binding site on β-tubulin within microtubules.ii–vii The tau neuronal protein binds onto microtubules in the vicinity of the paclitaxel site, and this site is well-described due to high resolution cryoelectron microscopy analyses of zinc-induced sheets of tubulin polymer stabilized by taxanes or epothilones.viii,ix Drugs binding to the site interact with amino acid residues on the M-loop (for example, Phe270, Thr274 and Arg276) and the H7 alpha helix (for example, Ala231 and His227) of β-tubulin.
Figure 1.
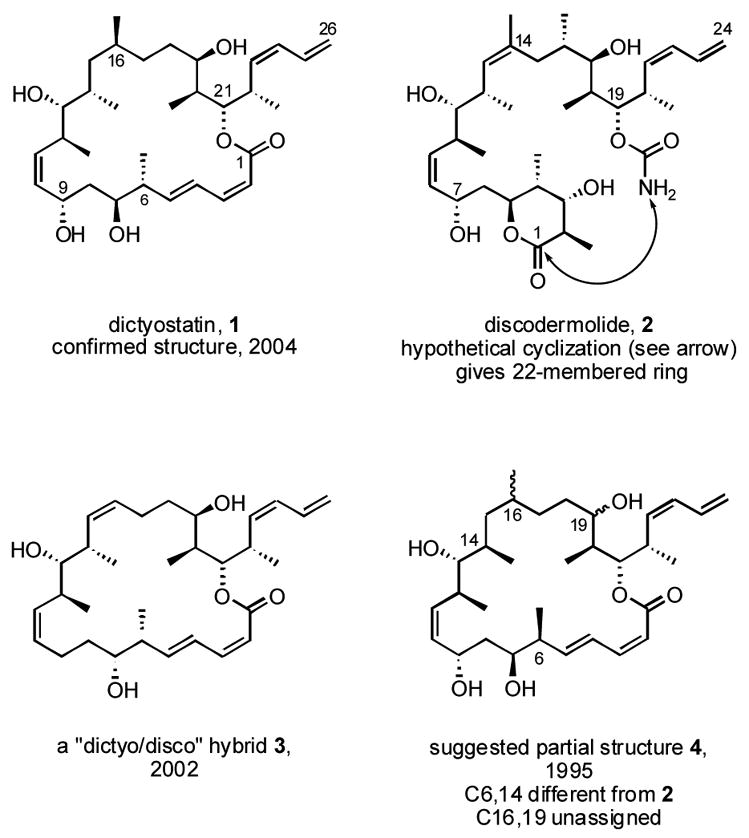
Structures of dictyostatin 1, discodermolide 2 and related compounds
Interest in the dictyostatin family of microtubule stabilizing agents has increased significantly over the last several years. Pettit initially isolated dictyostatin 1 from a marine sponge, suggested the correct two-dimensional structure (constitution), and showed that the compound had potent activity against cancer cells.x Subsequently, Wright and coworkers isolated 1 from a different sponge species and showed that the compound displays potent microtubule-stabilizing actions.xi
Dictyostatin is a structural cousin of the important microtubule stabilizing agent discodermolide 2.xii Dictyostatin’s 26-carbon backbone is two carbons longer than discodermolide’s, and it is joined by a 22-membered macrolactone formed between the carboxyl group on C1 and the alcohol on C21. (The longer chain length means that carbon numbers of dictyostatin are two higher than the comparable carbons of discodermolide.) Dictyostatin also differs from discodermolide by possessing a Z/E diene (C2–C5) instead of a γ-lactone, and it lacks a double bond at C15,16 and a methyl group at C18. Nonetheless, 10 of dictyostatin’s 11 stereocenters are also present in the discodermolide structure.
The three-dimensional structure (configuration) of dictyostatin was uncertain for almost a decade. As an outgrowth of our interest in discodermolide analogs,xiii we made “dictyostatin/discodermolide” hybrids like 3 in 2002,xiv and these compounds exhibited high biological activities in both tubulin and cell assays. An underlying tenet of this work was that a hypothetical cyclization of the carbamate nitrogen of discodermolide 2 onto its lactone carbonyl (C1) would provide a 22-membered ring, the same size as the macrocycle of dictyostatin 1. At this juncture, the lack of knowledge of the complete structure of dictyostatin and the tiny quantities available from isolation were serious impediments to further medicinal chemistry research. With a solid foundation in place from the synthesis of compound 3 and related molecules, we decided to address the dictyostatin structure and supply problems by total synthesis.
In a 1995 patent, Pettit and coworkers suggested a partial configuration for dictyostatin,xv and we selected the absolute configuration depicted in compound 4 because this enantiomer is more closely related to discodermolide. In 4, seven stereocenters have the same configurations as discodermolide, two are different (C6 and C14), and two are not assigned (C16 and C19). In 2004, Paterson and Wright suggested structure 1 for dictyostatin based on detailed NMR studies;xvi in addition to assigning the two missing stereocenters, the configurations of two other centers were inverted. Structure 1 was promptly proved by a pair of total syntheses that appeared in simultaneous communications from Paterson’s groupxvii and ours,xviii and recently Phillips and Ramachandran have also reported total syntheses of 1.xix,xx
In this paper, we provide details of our total synthesis of dictyostatin 1. Along the way, we made several stereoisomers of the natural product as we drew gradually closer to the correct structure. All of these compounds have been characterized by a battery of biological assays. These results, combined with the additional results for new analogs described elsewherexxi,xxii,xxiii and with the known SAR for discodermolide, provide for the first time a good outline of the SAR of dictyostatin. Over 5 mg of synthetic dictyostatin was provided by this work, and this was used for the detailed biological characterization of this molecule. The results of these studies have fully validated the high level of interest in dictyostatin.2
RESULTS
Chemistry
Synthesis of 6,16-bis-epi-dictyostatin 5
Though differing in relative configuration from Pettit’s structure 4 in the center fragment, we initially decided to make compound 5 because a number of key early intermediates were already in hand. We selected the (R) configuration at C16 arbitrarily, and though this proved to be wrong, the molecule—6,16-bis-epi-dictyostatin—ultimately turned out to be structurally much closer to dictyostatin than we had initially thought.
The strategy for the synthesis of this molecule is summarized at a high level in Figure 2. Key targeted bonds were C10,11 (Wittig reaction), C17,18 (Wadsworth-Horner-Emmons reaction) and the O21-C1 (macrolactonization). This divided the molecule into three approximately equal portions, which we call top, middle and bottom. The top and bottom fragments were further subjected to secondary disconnections to excise the two dienes. The removal of these two potentially sensitive groups reduced the convergency, but it also expanded options for fragment coupling based on alkene chemistry. Since speed to the target was more important than efficiency at this stage of the work, we deemed this a worthwhile trade off.
Figure 2.
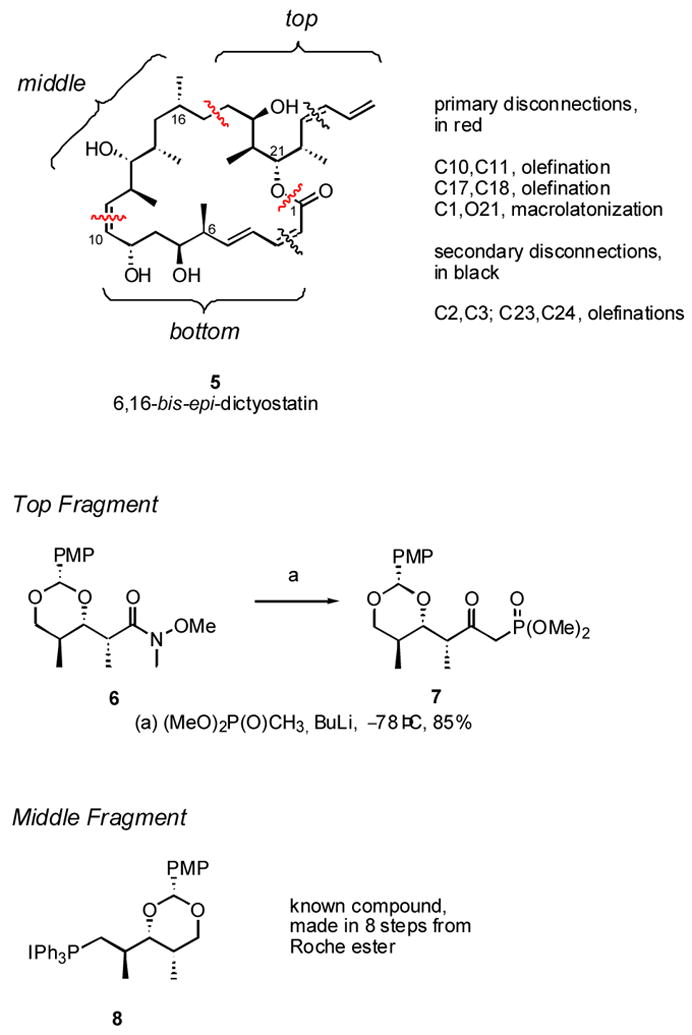
Strategy for synthesis of 5 with top and middle fragments
The selected top and middle fragments are also shown in Figure 2. Like all the work described herein, the early stages of the synthesis of the top and middle fragments borrowed liberally from related work in the discodermolide field. Weinreb amide 6 is readily available in five steps from (2S)-3-hydroxy-2-methylpropionic acid methyl ester (Roche ester),xxiv and this was parlayed into top fragment 7 in 85% yield by reaction with lithiomethyl trimethylphosphonate. This fragment was used for all of the compounds in this paper. Middle fragment 8 was borrowed from our prior work13 because it provides reliable results in Wittig olefinations, and it was readily made in 8 steps and 33% overall yield from the Roche ester. These reactions were readily conducted on multi-gram scale, and the efficiency of the plan was increased because the first few steps of the syntheses of 7 and 8 were the same.
The synthesis of the bottom aldehyde fragment 14 is summarized in Scheme 1. (L)-Malic acid was esterified and selectively reduced with borane dimethylsulfide and NaBH4 in 97% yield to a diol, which was protected as the acetonide 9 with acetone/p-TsOH in 86% yield.xxv The ester in 9 was reduced to an aldehyde with DIBALH in 67% yield, and this was subjected to an Evans syn-aldol reaction to give 10 in 98% yield as a single diastereomer.xxvi The aldol product 10 was reduced to an aldehyde by using Red-Al, and this was homologated by a Horner-Wadsworth-Emmons (HWE) reaction to the α,β-unsaturated ester 11 in 52% yield. The acetonide protecting group was removed with Dowex-H+ resin,xxvii and the resulting triol was triply protected with TBS groups to give 12 in 81% yield. Selective deprotection of the primary TBS group was achieved in 65% yield with HF-pyridine, and the resulting alcohol 13 was oxidized with the Dess-Martin reagent to give aldehyde 14 in 93% yield.
Scheme 1.
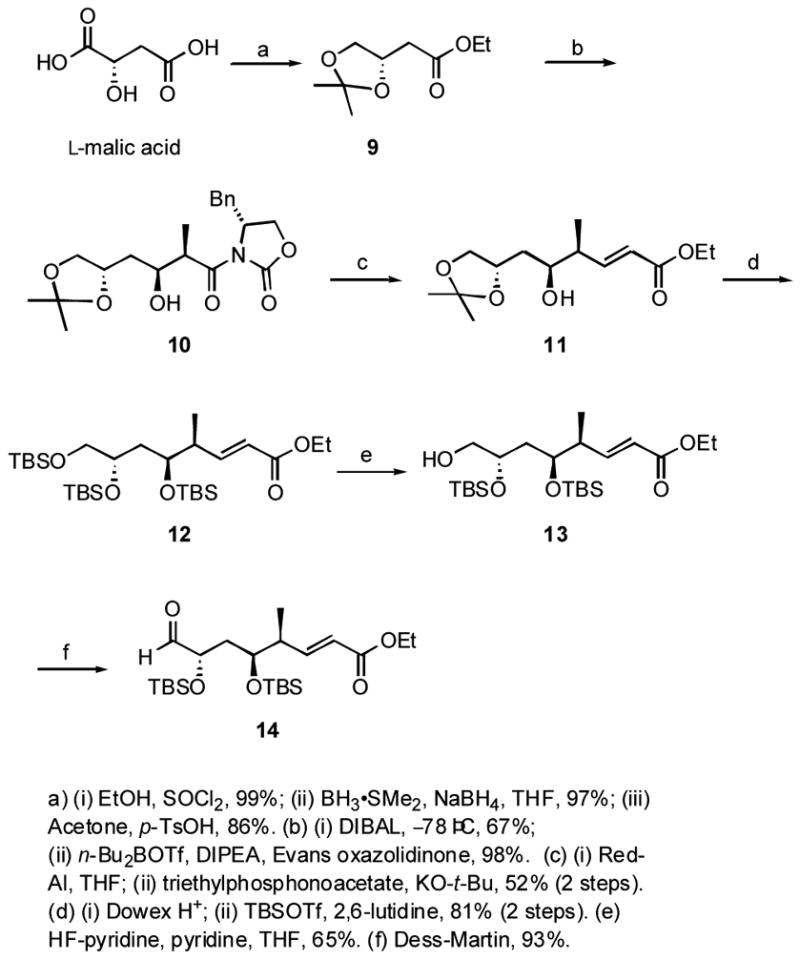
Synthesis at bottom fragment 14
The fragment couplings and associated steps form the intermediate stage of the synthesis, as summarized in Scheme 2. Wittig coupling of 14 was conducted at high concentration (1 M) of the phosphonium salt 8 to give 15 in 75% yield. The formation of the (Z)-alkene at C10–C11 was confirmed by the 10.8 Hz coupling constant between the adjacent vinyl protons. Hydrolysis of the ester functionality in 15 with 1 N KOH, activated ester formation with ethyl chloroformate, and in situ NaBH4 reduction gave the allylic alcohol 16 in 61% yield. Alcohol 16 was protected by a trityl group to give 17 in 99% yield. The PMB acetal in 17 was cleaved with DIBALH to give the primary alcohol 18 in 74% yield, which was oxidized with the Dess-Martin reagent then subjected to HWE reaction to give ester 19 in 93% yield.28a,b The α,β-unsaturated alkene in 19 was reduced selectively with nickel boride to give 20 in 97% yield, and this was subsequently hydrolyzed and coupled with the Evans oxazolidinone by forming the activated ester with pivaloyl chloride to give 21 in 83% yield. Asymmetric methylation gave 22 stereoselectively in 62% yield.xxviiic The optimal conditions were addition of 3 equiv of methyl iodide dropwise over 30 min, and the temperature was kept at −78 °C for 4 h to minimize the formation of the α-dimethylated byproduct.
Scheme 2.
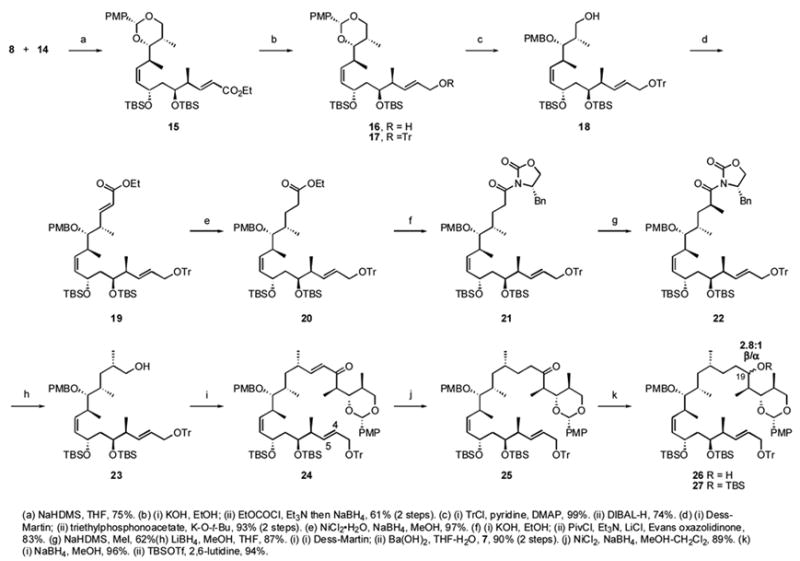
Intermediate stage of 6,16-bis-epi-dictyostatin synthesis
Removal of the Evans auxiliary with LiBH4 produced the primary alcohol 23 in 87% yield (Scheme 2). This was oxidized by the Dess-Martin protocol to the aldehyde, which was subjected to an HWE reaction with upper fragment phosphonate 7 to give the coupled product 24 in 90% yield. The α,β-unsaturated alkene was reduced with nickel boride in 89% yield to give 25 (over-reduction of the C4–C5 alkene was also observed as a minor side reaction). Because the configuration of dictyostatin at C19 was unassigned, the ketone in 25 was reduced unselectively with NaBH4 to give a 2.8:1 mixture of diastereomers, which were separated by silica gel column chromatography. The configuration of the major product 26 was assigned as β by conversion of a small sample to a cyclic PMP acetal between O19 and O21, followed by NOE studies.xxix The secondary hydroxy group of the major β diastereomer 26 was then protected with a TBS group to give 27 in 94% yield.
The final stages of the synthesis of 5 included elaboration of the two dienes and macrocyclization, and these are summarized in Scheme 3. The PMB acetal of 27 was opened with DIBALH to give 28 in 97% yield. Primary alcohol 28 was oxidized to an aldehyde then submitted to a Nozaki-Hiyama reactionxxx with 29 to provide an anti β-hydroxy silane. This crude silane was directly subjected to Peterson-type syn-elimination with excess NaH (20 equiv) in THF to install the (Z)-diene moiety, giving 30 in 72% yield. The trityl group was removed by B-chloro-catecholboranexxxi to give 31 in 61% yield. This moderate yield was due to the partial PMB deprotection at C21 and to the difficulty in chromatographic purification because the product 31 had similar retention characteristics to the TrOH byproduct.
Scheme 3.
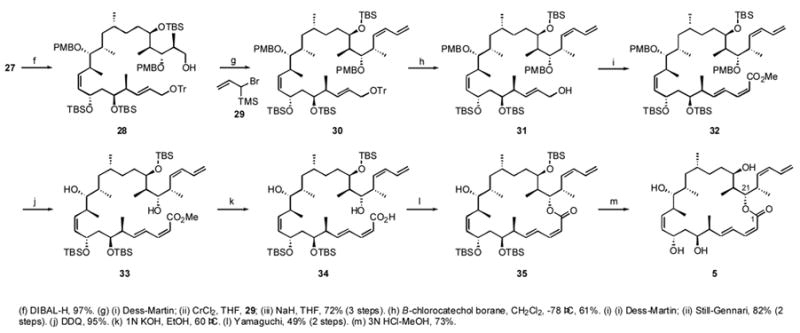
Final stages of the synthesis of 6,16-bis-epi-dictyostatin
The resulting allylic alcohol 31 was oxidized with the Dess-Martin reagent to an aldehyde, which was subjected to a Still-Gennari reactionxxxii to give the (E,Z) doubly unsaturated ester 32 in 82% yield. DDQ deprotection of the two PMB groups at C13 and C21 gave diol 33 in 95% yield. Use of excess DDQ (more than 3 equiv) resulted in the formation of a Diels-Alder adduct with the C2–C5 diene portion, lowering the yield dramatically. Hydrolysis of the methyl ester to acid 34 was achieved by using 1 N KOH in EtOH, setting the stage for the macrocyclization. Finally, Yamaguchi lactonizationxxxiii gave the macrolactone 35 in 49% yield. TBS deprotection with 3 N HCl in MeOH-THF gave the desired product C6,C16-bis-epi-dictyostatin 5 (73%), whose structure was confirmed by 1H-1H COSY, HMQC, HMBC and other NMR experiments. In particular, a key cross peak between H21 and C1 in the HMBC spectrum confirmed the 22-membered lactone.
Synthesis of 6,14-bis-epi-dictyostatin
We next decided to make compounds 36 and 37 (Figure 3), which are two of the four possible diastereomers from the structure proposed by Pettit. These are related to 5, but have the 14S (C13,C14 anti) configuration. Based on some simple computational modeling,xxxiv we also decided to adjust the configuration at C16 from R (α) to S (β). In the end, the two inversions were a wash, one moving us closer to dictyostatin (C16) and the other further away (C14), providing a target 36 that ultimately proved to be 6,14-bis-epi-dictyostatin. Unexpectedly, this target was not accessed in pure form due to an isomerization of the C2,C3 alkene during the macrolactonization (see below). Also, to get more information about the configuration of the alcohol at C19, we decided to take the α epimer at this center through the synthesis, and this provided 6,14,19-tris-epi-dictyostatin 37. The macrolactonization reaction again caused some isomerization, but this time both alkene stereoisomers were isolated.
Figure 3.
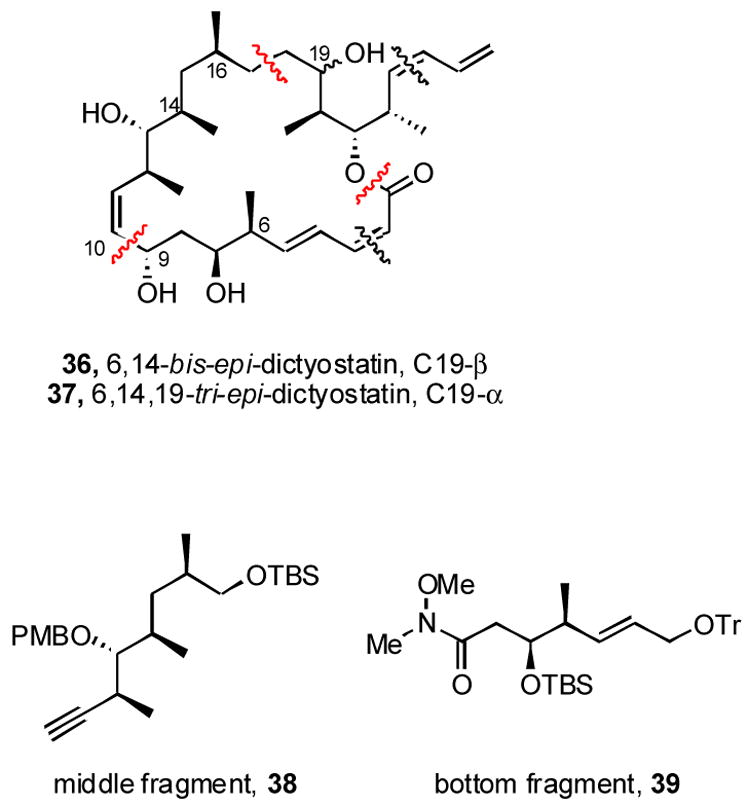
Strategy for synthesis of 6,14-bis-epi-dictyostatin
We initially attempted to couple the middle and bottom fragments by the same strategy that was used to make 5, but the C14 epimer of Wittig reagent 8 (not shown) was a white foam that was not well behaved in the Wittig reaction. We thus reformulated a strategy, summarized in Figure 3, that used the same top fragment 7 as before, but formed the C9,C10 bond (rather than the C10,C11 bond) in an alkyne addition. Middle fragment 38 and bottom fragment 39 were selected as key intermediates in this route, and their syntheses are shown in Schemes 4 and 5, respectively.
Scheme 4.
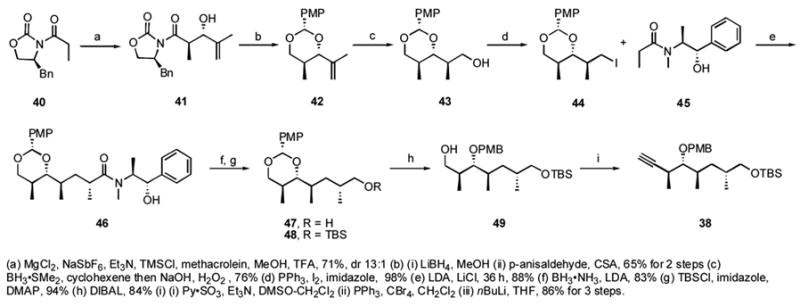
Synthesis of middle fragment 38
Scheme 5.
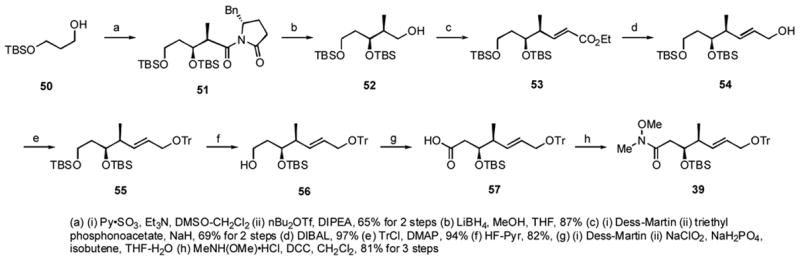
Synthesis of the bottom fragment 39
Synthesis of the middle fragment started with preparation of the Evans anti-aldol product 41,xxxv which was obtained with 13:1 diastereoselectivity in 71% yield (Scheme 4). The auxiliary was then cleaved with LiBH4, and the resulting diol was protected as the PMB acetal using p-anisaldehyde with CSA to give 42 in 65% yield for 2 steps. Hydroboration of alkene 42 with dicyclohexylborane (BH3·SMe2, cyclohexene) afforded the alcohol 43 with the desired anti,anti configuration in 76% yield. This alcohol was converted to the iodide 44 in 98% yield. An attempt to alkylate 44 with Evans auxiliary was unsuccessful; however, Myers asymmetric alkylationxxxvi with 45 gave the alkylated product 46 in 88% yield as a single isomer. Removal of the Myers auxiliary with BH3·NH3 gave alcohol 47 in 83% yield. TBS protection of the resulting primary alcohol gave 48 in 94% yield. DIBALH cleavage of the PMB acetal gave the product 49 in 84% yield. Oxidation to the aldehyde followed by Corey-Fuchs reactionxxxvii gave the alkyne 38 in 86% yield for 3 steps.
To make the bottom fragment 39, 1,3-propanediol was mono-protected to give 50 (Scheme 5), which was oxidized and subjected to Evans syn aldol reaction to give 51 in 65% yield for two steps. Reduction with LiBH4 gave alcohol 52 (87%), which was oxidized to the aldehyde with the Dess-Martin reagent. Subsequent treatment of the aldehyde with triethyl phosphonoacetate gave the Wittig-Horner-Emmons product 53 in 69% for 2 steps. The ester 53 was reduced with DIBALH to give allylic alcohol 54 in 97% yield, and 54 was protected with a trityl group in 94% yield. The primary TBS group of 55 was deprotected by HF-pyridine in pyridine to give 56 in 82% yield. This was oxidized to the aldehyde, then on to the acid 57. Finally, coupling with Weinreb’s salt by using DCCxxxviii gave the bottom fragment 39 in 81% for 3 steps.
The intermediate stage of this synthesis is shown in Scheme 6. Coupling of the middle fragment 38 and bottom 39 fragments parallels related work of Marshall and Jacobsen.xxxix The anion of alkyne 38 was added to bottom fragment 39 to give alkynyl ketone 58 in 98% yield. When 58 was subjected to Noyori reduction conditions,xl a single C9α isomer 59 was formed in 87% yield. About 20 mol% of the (S,S)-Noyori catalyst was required for rapid, clean reaction. The Noyori product 59 was carefully reduced by using the Lindlar catalystxli for 1 h to give the cis-alkene 60 in 90% yield. When the reaction time was extended to 1 day, over-reduction of all multiple bonds occurred. The C7,C9 anti configuration of 60 was confirmed by 13C NMR analysis of the derived acetonide.xlii
Scheme 6.
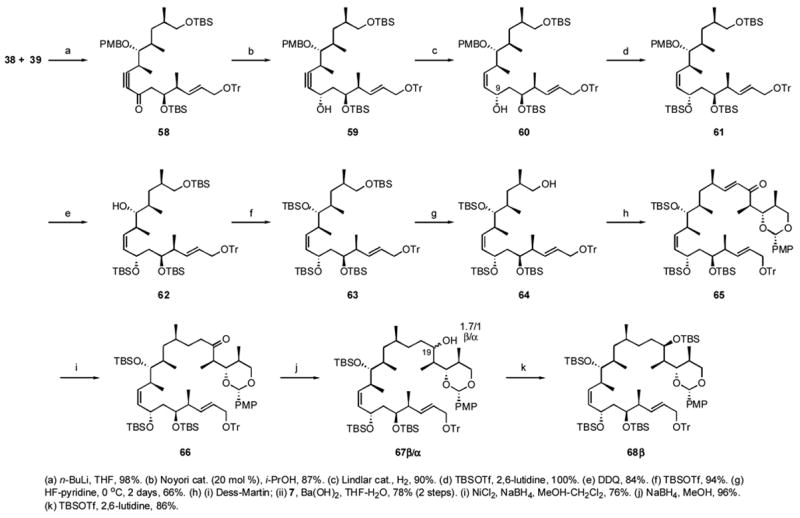
Intermediate stages of the synthesis of 6,14-bis-epi-dictyostatin
The C9 hydroxy group in 60 was protected with a TBS group to give 61 in quantitative yield. The PMB group was then removed with DDQ to give 62 in 84% yield. The resulting secondary hydroxy group was protected again by a TBS group, giving 63 in 94% yield. Selective deprotection of the primary TBS group was accomplished in 66% yield by treatment with HF-pyridine complex in buffered pyridine at 0 °C for 2 days to give 64 along with some unselectively deprotected byproducts.
To append the top fragment, 64 was oxidized to the aldehyde, which then was subjected to an HWE reaction with the phosphonate 7, providing 65 in 78% yield. The alkene in α,β-unsaturated ketone 65 was reduced with nickel boride giving 66 in 76% yield. Some over-reduction of the C4–C5 alkene in the bottom fragment was also observed, but this minor product was separated by chromatography. The C19 ketone was reduced unselectively by NaBH4 yielding a 1.7:1 ratio of diastereomers of 67β/α, with the β isomer as the major (62%), less polar product and the α isomer as the minor (36%), more polar product. These two diastereomers were separated by silica gel column chromatography and pure 67β was advanced first. The newly generated C19 hydroxy group in 67 was protected by a TBS group to give 68β in 86% yield.
The diene elaboration and final stages of the synthesis are shown in Scheme 7. The PMB acetal was cleaved with DIBALH to give the primary alcohol 69β in 97% yield. Oxidation to the aldehyde and subsequent Nozaki-Hiyama and Peterson syn-elimination reactions gave the diene 70β in 85% yield.
Scheme 7.
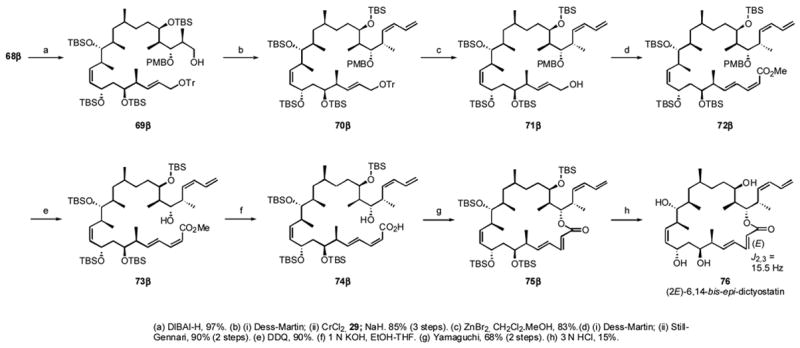
Final stages of the synthesis of 6,14-bis-epi-dictyostatin
Removal of the trityl group in 70β with ZnBr2 in CH2Cl2-MeOHxliii gave 71β in 83% yield, which was oxidized to the aldehyde and the (E,Z)-diene was installed by Still-Gennari reaction in 90% yield. The PMB group in 72β was removed by DDQ to give 73β in 90% yield, and the resulting methyl ester was hydrolyzed with 1 N aqueous KOH in EtOH-THF. Although the resulting acid 74β was exclusively the 2Z,4E-isomer, macrolactonization by the Yamaguchi method produced mainly the 2E,4E-lactone 75β, which was isolated in 68% yield.xliv The isomerization was clearly revealed by a 15.5 Hz coupling constant between H2 and H3 in 75β, whereas the (2,3Z) dictyostatins 1 and 5 have a coupling constant of about 11 Hz. The formation of this geometric isomer might be due to a reversible Michael type addition of 4-DMAP to the activated ester during the lactonization reaction.xlv Final global TBS deprotection yielded 76 in 25% yield.
The C19α epimer was prepared from 67α by using the same reaction pathways detailed in Schemes 6 and 7 (not shown, but fully described in the Supporting Information). In this case, after Yamaguchi lactonization of 74α (Eq 1), two inseparable spots were observed on TLC. In the final global TBS deprotection step, the (2Z,4E)-isomer 37 (less polar, 45%) was isolated along with the isomerized (2E,4E)-isomer 77 (more polar, 15%) in a 3:1 ratio. Coupling constant measurements again secured the alkene geometries.
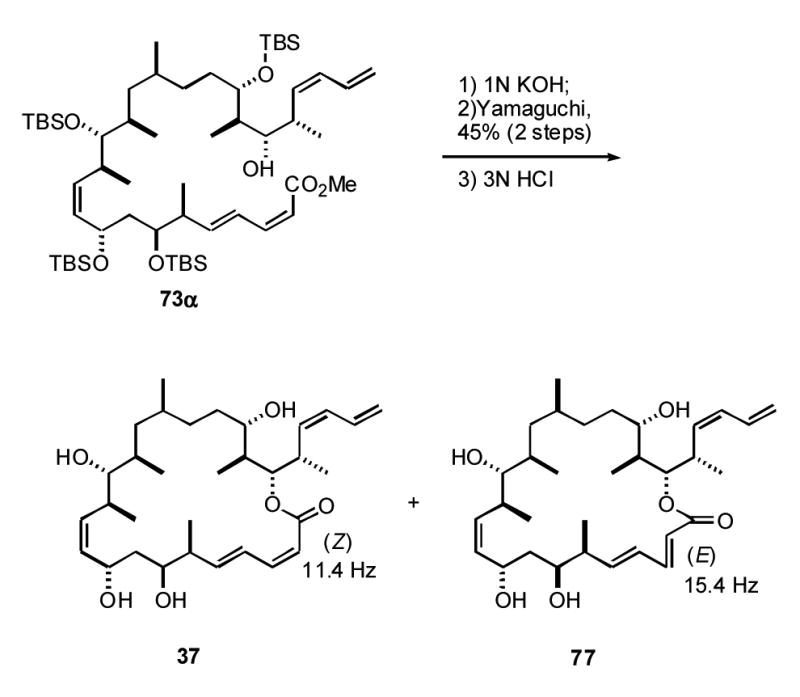 |
Eq 1 |
Synthesis of (−)-dictyostatin
As this work progressed, we compared spectral data of the intermediates and products with both dictyostatin 1 and discodermolide 2 and began to draw conclusions about the structure of dictyostatin. This exercise was greatly assisted by 2D NMR spectroscopy because it was generally possible to assign all or nearly all 1H and 13C resonances with confidence. First, while all of the isomers made to that date were clearly different from dictyostatin in material respects, their spectra were related enough for us to conclude that the two-dimensional structure proposed by Pettit was correct. Second, the spectra from the series of compounds with the β-hydroxy group at C19 clearly resembled both dictyostatin and discodermolide more than the series with the α-hydroxy group, so we concluded that dictyostatin and discodermolide had the same configuration at the C19–C22 stereotetrad.
The comparisons of resonances in the middle and bottom portions of the molecules were very revealing. First, the resonances of compound 5 with the “discodermolide-like” middle fragment closely resembled their counterparts of dictyostatin 1 and differed from those of the 6,14-bis-epi series, for example, compound 36. This suggested that dictyostatin has the same configurations as discodermolide for the C12–C14 stereotriad, and not the configuration assigned in compound 4. Finally, none of the resonances of the spectra in the bottom fragments region closely resembled dictyostatin, and all exhibited significant differences centered in the region of H6/C6 and H7/C7. We thus concluded that this syn configuration as assigned in compound 4 was not correct for dictyostatin, and the now clear stereochemical analogy between the top and middle fragments of dictyostatin and discodermolide suggested that the bottom fragment of dictyostatin probably also had the same configurations as discodermolide.
The only remaining configuration to assign was at the isolated stereocenter at C16, which has no analogy in discodermolide. The need to select between these two isomers was preempted by the appearance of the paper by Paterson and Wright suggesting that dictyostatin was 1 (C16-β) based on detailed NMR analysis and modeling.16 We accordingly decided to focus on making structure 1 as quickly as possible to finally resolve the stereochemical issues.
The strategy for the synthesis of 1 followed closely from the prior work and is summarized in Figure 4. The same disconnections were used as for the 6,14-bis-epi series, providing the standard top fragment 7 along with new middle 78 and bottom 79 fragments.
Figure 4.
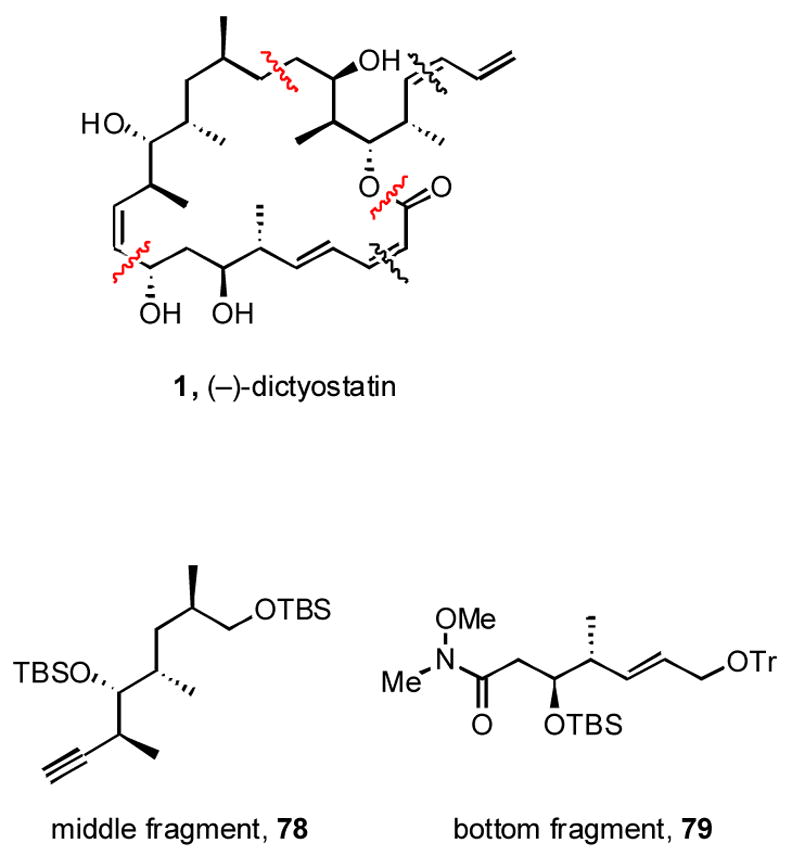
Strategy for synthesis of (−)-dictyostatin 1
Over the course of the work, we developed two routes to bottom fragment 79, and both are summarized in Scheme 8. The first route built on the established route to syn alcohol 52 (see Scheme 5). This compound was converted to the anti-isomer 82 via a tosylation to give 80 and elimination to alkene 81. Hydroboration with 9-BBN and subsequent oxidation provided a 9/1 mixture 82 and 52.xlvi These were readily separable, and 82 was isolated isomerically pure in 72% yield. The remaining sequence of steps to make 79 followed closely after the analogous sequence described in Scheme 5. The fifteen step process starting ultimately from 1,3-propanediol provided key intermediate 79 in 9.5% overall yield.
Scheme 8.
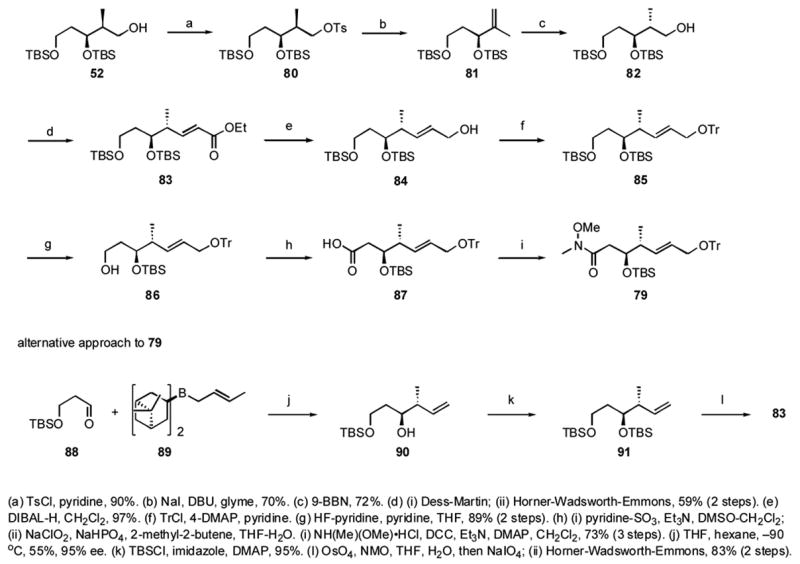
Two routes to the dictyostatin bottom fragment 79.
We found that the indirect process of converting syn 52 to anti 82 could be shortened by beginning with Brown crotylationxlvii of TBS-protected 3-hydroxypropanal 88 with 89, which directly provided anti-alcohol 90. Protection of the alcohol with TBSCl, OsO4-catalyzed dihydroxylation and diol cleavage with periodate, followed by HWE homologation intersected the prior route at unsaturated ester 83. This second generation route reduced the number of steps to 79 from 15 to 11 and improved the overall yield (from 1,3-propanediol) to 27%.
The middle fragment 78 was also prepared in two ways as summarized in Scheme 9. The secondary alcohol of known compound 92,xlviii prepared in four steps from the (S)-Roche ester, was TBS-protected and the Evans auxiliary was removed with LiBH4 to give alcohol 93. Oxidation to the aldehyde and HWE reaction gave the ester 95. Alkene reduction with nickel boride to 96, saponification with LiOH to 97, and coupling with the Evans auxiliary gave amide 98. Asymmetric methylation provided diastereomer 99 exclusively. Removal of the chiral auxiliary by reduction to give 100, TBS protection, and PMB deprotection with DDQ gave primary alcohol 101. Oxidation to the aldehyde and Corey-Fuchs reaction gave the alkyne 78. This route from 92 to 78 proceeded in 16% overall yield. An improved route to 78 involved conversion of 94 to its iodide and asymmetric alkylation with Myer’s auxiliary 45 to give amide 103. Removal of the auxiliary as usual intersected the prior route at 100. This second generation approach to 78 doubled the overall yield from 92 to 31%.
Scheme 9.
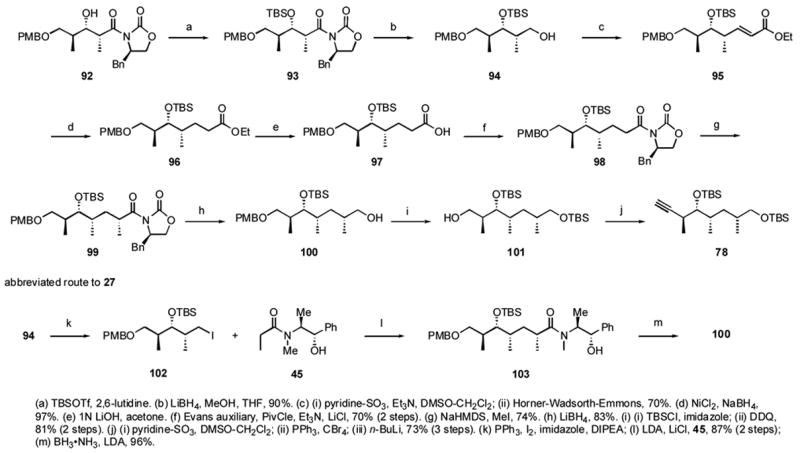
Two routes to the dictyostatin middle fragment 78.
The bottom and middle fragments were then coupled and the synthesis of dictyostatin was advanced as summarized in Scheme 10. The Weinreb amide 79 was reacted with 2 equiv of the anion from alkyne 78 to give the coupling product 104 in high yield. Reduction of 104 with the (S,S)-Noyori catalyst gave one isomer of the alcohol 105, whose alkyne moiety was reduced by Lindlar hydrogenation to alkene 106. The newly generated secondary hydroxyl group was protected with TBSOTf to give 107. Selective deprotection of the primary TBS group with HF-pyridine in buffered pyridine at 0 °C gave 108 in moderate yield. The aldehyde formed by Dess-Martin oxidation was reacted with the phosphonate 7 (Figure 2) under HWE conditions to give the conjugated ketone 109 in good yield. Selective reduction with nickel boride gave the ketone 110, which was again reduced in a purposefully unselective manner with NaBH4 to give a 2.4:1 mixture of the isomers of 111, with the β isomer, necessary for preparation of (−)-dictyostatin, predominating. The isomers of 111 were readily separated by silica gel chromatography. Later, a 5:1 ratio favoring 111 was obtained by use of the bulkier reducing agent LiAl(O-t-Bu)3H, whereas a 1:1 ratio of the α and β isomers was obtained when (L)-Selectride was employed.
Scheme 10.
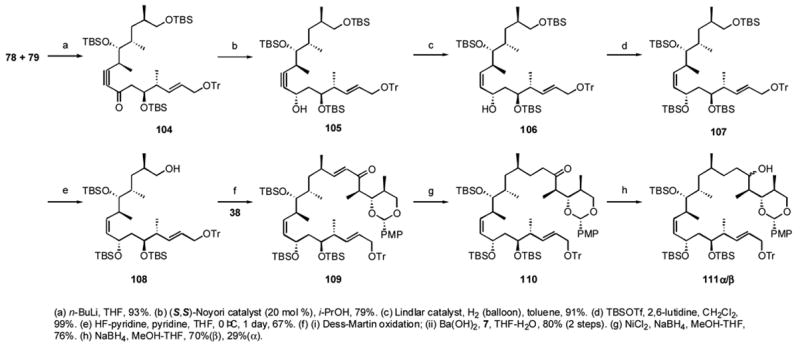
Intermediate stages of the synthesis of (−)-dictyostatin 1
The final stages of the synthesis paralleled the established steps, as summarized in Scheme 11. Alcohol 111β was protected with a TBS group to give 112β, whose PMB acetal was cleaved with DIBALH to give alcohol 113β. Oxidation to the aldehyde followed by Nozaki-Hiyama addition with 29 and Peterson-type elimination installed the (E,Z)-diene to give 114β in high yield. The allylic trityl group was removed with ZnBr2 to give alcohol 115β. Dess-Martin oxidation to the aldehyde and Still-Gennari reaction gave the (E,Z)-conjugated ester 116β. The PMB group was removed with DDQ to give 117β and saponification with aqueous KOH in EtOH-THF to give acid 118β in 61% yield after purification by flash chromatography. Yamaguchi macrolactonization of the purified acid now gave 119β in 78% yield after flash chromatographic purification. Global deprotection with 3 N HCl in MeOH-THF and careful purification gave ~5 mg of (−)-dictyostatin 1. There were at least two minor byproducts from the macrolactonization/deprotection sequence whose yields were estimated to be <15%. Neither sample was isolated in highly pure form, but based on comparison of 1H NMR spectra from the chromatography fractions with spectra of related pure samples, we deduced that one of these side products was the C2,C3 E-isomer (believed to arise during the macrolactonization), while the other was a trans-lactonized product with the lactone formed between O19 and C1 with a free O21 hydroxy group (believed to arise during the deprotection).xlix
Scheme 11.
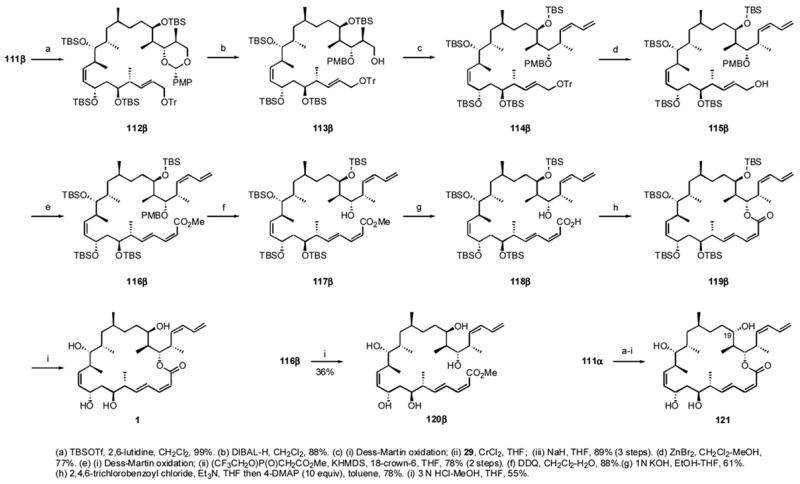
Final stages of the synthesis of dictyostatin
The Paterson group completed an independent total synthesis of 1 roughly concurrently with us, and the two synthetic samples and one natural sample (from Wright) of (−)-dictyostatin were identical in all respects. The [α]25D of our sample was −22.6 (c 0.27, MeOH), in satisfactory agreement with the values of Pettit (−20, c, 0.12, MeOH), Wright (−27.4, c 0.16, MeOH) and Paterson (−32.7, c 0.22, MeOH), so both the relative and absolute configurations of 1 are confirmed. An open-chain analog of 1 was prepared by global desilylation of 116β to give tetraol ester 120β. Finally, the α-epimer at C-19, 111α, was taken through the same sequence of steps as shown in Scheme 11 to provide 19-epi-dictyostatin 121 in comparable overall yield to the β-series (see Supporting Information). Thus, the chapter on the structure assignment of dictyostatin was closed.
Biology
The target compounds, 1, 3, 5, 37, 76, 77, 120β, and 121, were screened for cellular and biochemical activity. Their effects of compounds on cellular microtubules were quantified by tubulin immunostaining. In this assay, microtubule stabilizing agents cause a dramatic increase in tubulin immunoreactivity due to formation of bright polymer bundles with intense staining.13c Human cervical carcinoma HeLa cells were treated for 21 h with ten two-fold serial dilutions of test compounds in collagen-coated 384 well microplates, fixed and immunostained with an anti-α-tubulin antibody followed by a FITC conjugated secondary antibody as previously described.2 Paclitaxel and discodermolide 2 were included as known microtubule stabilizing agent controls. Fluorescence images of 1,000 individual cells were acquired on an ArrayScan II (Cellomics, Inc) automated microscope and analyzed for tubulin staining intensity as described.2,49 Extrapolation of minimum detectable effective concentrations (MDECs)50 from ten-point concentration-dependence curves revealed dictyostatin 1 caused microtubule bundling at low nanomolar concentrations, analogs 3, 120β and 121 at submicromolar concentrations (about 20-fold less potent than the parent compound), and compounds 5, 76, 37, and 77 to have little effect on microtubule morphology (Table 1).
Table 1.
Biological activities of dictyostatin (1) and analogs as compared to discodermolide (2) and paclitaxel.
| Cellular | in Vitro | |||||
|---|---|---|---|---|---|---|
| a MDEC for Tubulin Polymer Increase, nM ± SD (N) | b GI50, nM (fold-resistance) (N=4) | c% Tubulin Polymerized by 50 μM Test Agent (N≥3) | % Inhibition of Binding of [3H]Paclitaxel to Tubulin Polymer (N≥3) | |||
| Test Agent | 1A9 | 1A9/Ptx10 | 1A9/Ptx22 | |||
| 1 | 5.4±1.9 (4) | 0.69±0.80 | 3.2 ± 2.4 (5) | 1.3±1.0 (2) | 99±4 | 75±5 (3) |
| 2 | 65±0 (2) | 1.7±1.2 | 6.2±3.6 (4) | 7.0±8.4 (4) | 98±5 | 76±6 (4) |
| 3 | 328±142 (2) | 693±580 | 18400±2010 (27) | 625±9 | 52±2 | 27±8 (6) |
| 5 | 4375±625 (2) | 20900±400 | >50000 (>2) | 11560±1180 | 2±2 | 0±1 (3) |
| 37 | >5000 (4) | 28000±1000 | 26000±500 | 30000±1000 | 5±1 | 0±1 (3) |
| 76 | >5000 (4) | 310±40 | 780±200 (3) | 790±560 (3) | 1±1 | 0±0 (3) |
| 77 | >5000 (4) | 25000±2000 | 25000±1000 | 30000±1000 | 5±4 | 0±1 (3) |
| 120β | 219±36 (4) | 56±16 | 79±13 | 85±2 | 39±7 | 42±1 (3) |
| 121 | 284±108 (4) | 21±14 | 120±60 (6) | 43±12 (2) | 30±2 | 7±2 (3) |
| paclitaxel | 5.2±0.4 (4) | 0.71±0.11 | 64±8 (90) | 51±9 (72) | 89±6 | -- |
Minimum detectable effective concentration of the test agent in HeLa cells after 21 h of continuous exposure.
Fifty percent growth inhibitory concentration after 72 h of continuous exposure to the test agent.
Bovine brain tubulin (10 μM) in 0.2 M monosodium glutamate, 15 min at 20 °C, centrifugation and Lowry determination of remaining soluble tubulin.
Percent competition at 37 °C by 4 μM test agent with 2 μM [3H]paclitaxel for binding to microtubules formed from 2 μM bovine brain tubulin and 20 μM dideoxyGTP.
These same analogs were examined for their antiproliferative activities against cultures of human ovarian carcinoma 1A9 cells and their paclitaxel-resistant mutants, 1A9/Ptx10 and 1A9/Ptx22. Each of these resistant lines contains single mutations in the major β-tubulin gene that confer to the cells, which do not express drug efflux pumps, appreciable tolerance to paclitaxel.51 Paclitaxel had subnanomolar potency against the parental 1A9 cells, but the mutant cells showed ca. 90- and 70-fold resistance to the drug (Table 1). The antiproliferative activities, shown as fifty percent growth inhibitory concentrations (GI50s), of the agents paralleled the activities noted in the cellular microtubule assays, but the GI50 values were typically two- to seven-fold lower than the MDECs. Although not a potent agent, compound 3 showed an interesting property in that the 1A9/Ptx10 cells, which express β-tubulin with a Phe270->Val alteration, were 27-fold resistant to it as compared to the parental 1A9 cells. This suggested that the C6 and/or C16 methyl substituent in 1 could be in close proximity to Phe270 in the taxane binding site.
The abilities of the new compounds to cause tubulin polymerization were determined under reaction conditions with purified bovine brain tubulin (1 mg/mL) in the presence or absence of microtubule-associated proteins (MAPs, 0.75 mg/mL) and GTP (100 μM). Test agents were initially screened at 10 and 40 μM. In these experiments, test agent-induced assembly of soluble tubulin into polymer, with respect to the presence and absence of cofactors and at different temperatures, was monitored by turbidimitry in temperature-controlled spectrophotometers. The initial temperature was set at 0 or 2.5 °C (depending upon the instrument used), then rapidly raised to 10 °C, 20 °C, and 30 °C in succession. This permitted the determination of both the temperature at which a test agent induced assembly and the extent of agent-induced assembly. The temperature increases were followed by a rapid decrease in temperature back to the initial temperature to determine the cold-stability of the polymer formed.
As we have recently reported, the effects of dictyostatin 1 and discodermolide 2 were similar, and both molecules were far more potent than paclitaxel.2 Compounds 3, 120β and 121 all showed some activity in this assay, while the remaining agents were inactive. An example concentration-dependent trace, wherein a single temperature jump from 2.5 °C to 30 °C was used, for compound 3 is shown in Figure 5. All test agents at 50 μM were then incubated with 10 μM tubulin for 15 min at 20 °C, reaction mixtures were centrifuged, and the percent remaining soluble tubulin was measured. Again, compound 1 was by far the most potent, but compounds 3, 120β and 121 showed moderate activity (Table 1).
Figure 5.
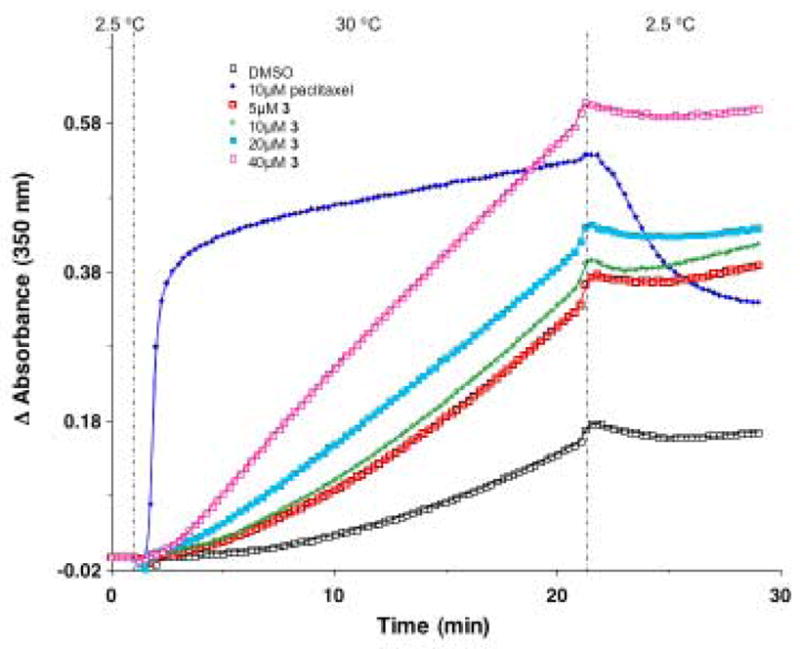
Assembly of bovine brain tubulin induced by compound 3 in comparison to paclitaxel.
Finally, all compounds were tested at 4 μM for their ability to compete with 2 μM [3H]paclitaxel for its binding to 2 μM preassembled bovine brain microtubules. Compounds 120β, 3 and 121 showed competitive activity in this assay, although the latter compound did not compete as well as one might expect based on results from the other assays.
CONCLUSIONS
In summary, we have provided here the full details of synthesis of dictyostatin 1, its open chain methyl ester analog, assorted C6, C16 and C19 epimers, and two macrolactone isomers with (E,E) geometry at C2–C5. The biological evaluation of the compounds revealed a wide range of activities resulting from these very small structural changes. The SAR determined includes the following. The macrolactone is important but not a full requisite for microtubule stabilization and antiproliferative actions. The β configuration of the hydroxyl at C19 is preferred, and the configuration of the C6 and C14 methyl groups are important. The natural E:Z geometry of the macrocyclic diene also appears to be crucial. Finally, either the C6 or C16 methyl appears to interact with a phenylalanine in the paclitaxel binding site. This SAR provided a starting point for preparation of more potent dictyostatin analogs, as described elsewhere21,23,53
EXPERIMENTAL
Ethyl (4R,5S,2E)-5,7-bis(tert-butyldimethylsilyloxy)-4-methylhept-2-enoate (83)
From 82: A solution of triethyl phosphonoacetate (3.5 mL, 17.6 mmol) was added to a cooled (0 °C), stirred suspension of NaH (0.43 g, 17.0 mmol, 95% dispersion in mineral oil) in THF (46 mL) dropwise over 10 min. The mixture was brought to room temperature with a water bath (30 min) and then cooled back to −78 °C, and the aldehyde (2.73 g, 7.58 mmol) in THF (5 mL) was added. The resulting mixture was stirred for 1 h at 0 °C, then pH 7 phosphate buffer solution (10 mL) and Et2O (50 mL) were added. The mixture was allowed to warm to room temperature, and the phases were separated. The organic phase was washed with saturated NH4Cl solution (30 mL) and brine (30 mL), dried with MgSO4, filtered and concentrated to give oily crude product. Purification by flash chromatography (EtOAc/hexane 1:9) afforded pure ester 83 (2.92 g, 59% for 2 steps) as a colorless oil: IR (CHCl3) 2956, 2930, 2857, 1724, 1651, 1472, 1463, 1367, 1256, 1180, 1098, 1036, 836, 775 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.88 (dd, J = 15.8, 7.6 Hz, 1H), 5.74 (d, J = 15.8 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 3.79 (ddd, J = 6.7, 4.7, 4.4 Hz, 1H), 3.59 (m, 2H), 2.43 (m, 1H), 1.53 (m, 2H), 1.22 (t, J = 7.1 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H), 0.83 (s, 18H), 0.02 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 166.4, 150.9, 121.3, 71.8, 59.9, 59.5, 42.0, 36.8, 25.82, 25.78, 26.1, 18.1, 18.0, 14.4, 14.2, −4.6, −4.7, −5.4; LRMS (EI) 415 (M–CH3)+, 373, 303, 147; HRMS (EI) calcd for C21H44O4Si2 415.2710 (M–CH3)+, found 415.2712; [α]20D +3.8 (c 0.21, CHCl3).
(4R,5S,2E)-5,7-bis(tert-Butyldimethylsilyloxy)-4-methylhept-2-en-1-ol (84)
DIBALH (26.5 mL, 26.5 mmol, 1.0 M solution in hexane) was added to the ester 83 (3.14 g. 7.30 μmol) in CH2Cl2 (35 mL) at −78 °C dropwise and stirred for 1 h. The reaction mixture was quenched by adding EtOAc (5 mL) and saturated sodium potassium tartrate solution (20 mL), followed by vigorous stirring for 4 h. The aqueous phase was extracted with CH2Cl2 (3 × 30 mL), and the combined organic layers were washed with brine (10 mL). After drying over MgSO4 and evaporation under vacuum, flash column chromatography (hexane/EtOAc 4:1) provided 2.75 g of alcohol 84 (97%) as a colorless oil: IR (CHCl3) 3349, 2956, 2928, 2857, 1471, 1462, 1255, 1099, 836, 774 cm−1; 1H NMR (300 MHz, CDCl3) δ 5.57 (m, 2H), 4.03 (m, 2H), 3.70 (ddd, J = 9.7, 6.0, 3.8 Hz, 1H), 3.59 (m, 2H), 2.27 (m, 1H), 2.00 (s, 1H), 1.53 (q, J = 6.5 Hz, 2H), 0.96 (d, J = 6.9 Hz, 3H), 0.85 (s, 9H), 0.84 (s, 9H), 0.00 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 134.7, 129.2, 72.4, 63.6, 60.1, 41.8, 36.3, 25.9, 18.2, 18.0, 15.1, 10.7, −4.6, −5.4; LRMS (EI) 370 (M–H2O)+, 303, 171, 147; HRMS (EI) calcd for C20H42O2Si2 370.2723 (M–H2O)+, found 370.2725; [α]20D − 3.0 (c 0.57, CHCl3).
((4R,5S,2E)-5,7-bis(tert-Butyldimethylsilyloxy)-4-methylhept-2-enyloxy)triphenylmethane (85)
Trityl chloride (4.1 g, 14.7 mmol) and DMAP (1.8 g, 14.7 mmol) were added to a solution of alcohol 84 (2.75 g, 7.1 mmol) in pyridine (71 mL). The mixture was heated to reflux for 18 h, cooled to ambient temperature and added to a solution of saturated CuSO4 (200 mL). The mixture was extracted with Et2O (2 × 20 mL), and the combined organic extracts were washed saturated CuSO4 (2 × 20 mL). The organic layer was separated, dried (MgSO4), filtered, and concentrated in vacuo. Flash column chromatography (EtOAc/hexane 1:19) provided 85 (4.46 g, quantitative) as a pale yellow oil: IR (CHCl3) 2954, 2856, 1471, 1448, 1254, 1095, 835, 773, 705 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.56 (m, 6H), 7.32 (m, 9H), 5.79 (dd, J = 15.6, 6.7 Hz, 1H), 5.65 (dd, J = 15.7, 5.0 Hz, 1H), 3.85 (m, 1H), 3.74 (m, 1H), 3.66 (d, J = 4.9 Hz, 1H), 2.43 (m, 1H), 1.70 (q, J = 6.5 Hz, 2H), 1.21 (d, J = 6.9 Hz, 3H), 0.99 (s, 9H), 0.97 (s, 9H), 0.154 (s, 3H), 0.150 (s, 3H), 0.13 (s, 3H), 0.12 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 144.4, 134.2, 128.7, 127.7, 126.9, 126.8, 86.8, 72.6, 65.1, 60.2, 42.1, 36.6, 26.0, 18.3, 18.1, 15.3, −4.4, −5.3; LRMS (ESI) 653.3 [M+Na]+; HRMS (ESI) calcd for C39H58O3Si2Na 653.3822 [M+Na]+, found 653.3851; [α]20D − 1.9 (c 0.42, CHCl3).
(3S,4R,5E)-3-(tert-Butyldimethylsilyloxy)-4-methyl-7-(trityloxy)hept-5-en-1-ol (86)
F-pyridine in pyridine (40 mL, prepared by slow addition of 12 mL pyridine to 3 mL HF-pyridine complex, followed by dilution with 25 mL THF) was added to a solution of TBS ether 85 (4.46 g, 7.07 mmol) in THF (10 mL). The mixture was stirred overnight at room temperature and quenched with saturated NaHCO3 (100 mL). The aqueous layer was separated and extracted with Et2O (3 × 50 mL). The combined organic layers were washed with saturated CuSO4 (3 × 50 mL), dried over MgSO4, and concentrated. Flash column chromatography (EtOAc/hexane 1:4) afforded 3.26 g (89%) of alcohol 86 as a colorless oil: IR (CHCl3) 3407, 2955, 2928, 2856, 1490, 1471, 1448, 1254, 1058, 1031, 836, 773, 705 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.57 (m, 6H), 7.37 (m, 9H), 5.78 (dd, J = 15.6, 6.5 Hz, 1H), 5.73 (dt, J = 15.5, 4.8 Hz, 1H), 3.91 (m, 1H), 3.82 (d, J = 5.9 Hz, 2H), 3.69 (d, J = 4.4 Hz, 2H), 2.51 (m, 1H), 2.22 (br, 1H), 1.77 (m, 2H), 1.13 (d, J = 6.8 Hz, 3H), 1.03 (s, 9H), 0.21 (s, 3H), 0.19 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 144.2, 134.1, 128.6, 127.7, 127.1, 126.9, 86.8, 74.3, 64.9, 60.4, 42.0, 34.8, 25.9, 18.0, 14.5, −4.4, −4.6; LRMS (ESI) 539.2 [M+Na]+; HRMS (ESI) calcd for C33H44O3Si1Na 539.2957 [M+Na]+, found 539.2976; [α]20D − 2.8 (c 2.0, CHCl3).
(3S,4R,5E)-3-(tert-Butyldimethylsilyloxy)-N-methoxy-N,4-dimethyl-7-(trityloxy)hept-5-enamide (79)
Sulfur trioxide pyridine complex (3.02 g, 19.1 mmol) was added to a stirred solution of alcohol 86 (3.26 g, 6.31 mmol) and triethylamine (2.6 mL, 19.1 mmol) in anhydrous CH2Cl2 (6 mL) and DMSO (12 mL) at 0 °C. The reaction mixture was stirred at ambient temperature for 1 h. The mixture was diluted with Et2O (100 mL) and washed with aqueous 0.5 N HCl (50 mL) and brine (10 mL). The separated organic layer was dried over MgSO4. Filtration and concentration, followed by short flash column chromatography (hexane/EtOAc 4:1), provided the crude aldehyde as a colorless oil, which was used without further purification. A solution of the aldehyde in THF (25 mL) and H2O (12 mL) was treated with 2-methyl-2-butene in THF (2 M, 18 mL, 9.0 mmol), NaH2PO4·H2O (2.6 g, 18.8 mmol) and NaClO2 (2.1 g, 18.6 mmol). The reaction mixture was stirred for 2 h, diluted with 1 N HCl (20 mL) and extracted with CH2Cl2 (2 × 40 mL). The combined organic layers were dried over MgSO4, concentrated in vacuo and the crude acid 87 was used for the next reaction without further purification. N,O-Dimethylhydroxylamine hydrochloride (0.62 g, 6.36 mmol), Et3N (0.88 mL, 6.31 mmol), and DMAP (0.63 mmol) were successively added to a solution of the crude acid in CH2Cl2 (10 mL). The reaction mixture was cooled to 0 °C, and DCC (1.30 g, 6.30 mmol) was added. The mixture was stirred at ambient temperature for 15 h and filtered. The filtrate was washed with 0.5 N HCl, saturated aqueous NaHCO3, and brine, dried over anhydrous MgSO4 and concentrated. Purification by column chromatography over silica gel (hexane/EtOAc 4:1) gave the Weinreb amide 79 (2.65 g, 73% for 3 steps) as a colorless oil: IR (CHCl3) 2956, 2929, 2855, 1663, 1448, 1252, 1083, 1032, 836 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.58 m, 6H), 7.37 (m, 9H), 5.89 (dd, J = 15.6, 7.6 Hz, 1H), 5.72 (dt, J = 15.6, 5.2 Hz, 1H), 4.38 (ddd, J = 8.0, 5.0, 3.0 Hz, 1H), 3.74 (s, 3H), 3.70 (d, J = 5.1 Hz, 2H), 3.27 (s, 3H), 2.79 (dd, J = 15.1, 7.4 Hz, 1H), 2.52 (m, 2H), 1.20 (d, J = 6.9 Hz, 3H), 1.02 (s, 9H), 0.22 (s, 3H), 0.16 (s, 3H) ; 13C NMR (75 MHz, CDCl3) δ 172.6, 144.2, 133.3, 128.5, 127.7, 127.5, 126.8, 86.7, 72.4, 64.8, 61.2, 42.4, 36.3, 31.9, 25.8, 18.0, 15.7, −4.6, −5.0; LRMS (ESI) 596.2 [M+Na]+; HRMS (ESI) calcd for C35H47O4NSiNa 596.3172 [M+Na]+, found 596.3165; [α]20D − 14.7 (c 0.65, CHCl3).
(3R,4S)-4,6-bis(tert-Butyldimethylsilyloxy)-3-methylhex-1-ene (91)
A solution of 90 (9.00 g, 36.8 mmol), imidazole (5.06 g, 73.6 mmol) and DMAP (0.45 g, 3.68 mmol) in DMF (37 mL) was treated with TBDMSCl (7.44 g, 47.9 mmol) at 0 °C. The mixture was stirred at room temperature for 12 h, then diluted with ethyl ether and water The aqueous layer was extracted with ethyl ether. The combined organic layers were washed with water and dried over anhydrous MgSO4. The solvent was removed under vacuum to provide the oil. The flash chromatography (hexane/EtOAc 19:1) afforded the title compound (12.54 g, 95% yield): 1H NMR (300 MHz, CDCl3) δ 5.84 (m, 1H), 5.02 (m, 2H), 3.79 (m, 1H), 3.67 (m, 2H), 2.34 (m, 1H), 1.62 (m, 2H), 1.00 (d, J = 6.6 Hz, 3H), 0.92 (brs, 18H), 0.06 (m, 12H).
Ethyl (4R,5S,2E)-5,7-bis(tert-butyldimethylsilyloxy)-4-methylhept-2-enoate (83)
From 91: A mixture of 91 (1.00 g, 2.79 mmol) in acetone (16 mL) and water (2 mL) was treated with OsO4 (2.5 wt% in tert-BuOH, 1.14 mL, 0.11 mmol) at room temperature. After 10 min, NMO (0.43 g, 3.63 mmol) was added. The mixture was stirred at room temperature for 20 h, then NMO (0.23 g) was added. After 3 h, NaIO4 (0.72 g, 3.35 mmol) and water (5 mL) were added. After 4 h, water was added. The aqueous layer was extracted with ethyl ether. The combined organic layers were washed with brine and dried over anhydrous MgSO4. The solvent was removed under vacuum to provide the crude aldehyde, which was used for the next reaction without further purification.
A suspension of NaH (0.28 g, 11.16 mmol) in THF (14 ml) was treated with triethyl phosphinoacetate (2.80 mL, 13.95 mmol) at −5 °C. The mixture was stirred at room temperature for 30 min, and cooled to −5 °C. The crude aldehyde in THF (5 mL) was added dropwise. The mixture was stirred at 0 °C for 2 h, then quenched with sat. aq. NH4Cl. The aqueous layer was extracted with ethyl ether. The combined organic layers were washed with brine and dried over anhydrous MgSO4. The solvent was removed under vacuum to provide the oil. The flash chromatography (hexane:EtOAc 19:1) afforded 83 (1.00 g, 83% yield, two steps). See above for spectroscopic data.
(R)-3-((2R,3S,4S)-5-(4-Methoxybenzyloxy)-3-(tert-butyldimethylsilyloxy)-2,4-dimethylpentanoyl)-4-benzyloxazolidin-2-one (93)
2,6-Lutidine (5.14 mL, 44.2 mmol) and TBSOTf (9.36 mL, 40.8 mmol) were added to a solution of 92 (15.0 g, 33.9 mmol) in CH2Cl2 (340 mL) stirred at 0 °C. The mixture was stirred at 0 °C for 2 h and then quenched by the addition of saturated aqueous NaHCO3. The phases were separated, and the aqueous layer was extracted with CH2Cl2. The combined organic phases were washed with 0.5 M aqueous NaHSO4. The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (hexane/EtOAc 4:1) to give 93 (17.9 g, 95%) as a colorless oil: IR (film) 1781, 1696, 1513, 1383, 1248, 1209, 1110, 1042 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.35-7.28 (m, 7H), 6.85 (d, J = 8.7 Hz, 2H), 4.49 (m, 1H), 4.38 (d, J = 11.7 Hz, 1H), 4.34 (d, J = 11.7 Hz; 1H), 4.03 (m, 3H), 3.81 (m, 1H), 3.77 (s, 3H), 3.54 (dd, J = 9.2, 5.6 Hz, 1H), 3.22 (dd, J = 13.3, 3.1 Hz, 1H), 3.17 (dd, J = 9.1, 5.9 Hz, 1H), 2.72 (dd, J = 13.3, 9.6 Hz, 1H), 1.97 (m, 1H), 1.25 (d, J = 6.5 Hz, 3H), 1.02 (d, J = 7.0 Hz, 3H), 0.91 (s, 9H), 0.07 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 176.4, 159.4, 153.1, 135.8, 131.1, 129.8, 129.3, 129.2, 127.6, 75.6, 72.9, 72.0, 66.1, 55.8, 55.6, 41.9, 39.3, 38.0, 26.4, 18.7, 15.3, 15.2, −3.5, −3.6; HRMS (ESI) calcd for C31H45NO6SiNa 578.2914 [M+Na]+, found 578.2923; [α]20D − 8.1 (c 7.6, CHCl3).
(2S,3R,4S)-5-(4-Methoxybenzyloxy)-3-(tert-butyldimethylsilyloxy)-2,4-dimethylpentan-1-ol (94)
Dry MeOH (1.05 mL, 26.0 mmol) then LiBH4 (13 mL, 2.0 M solution in THF, 26 mmol) were added to a stirred solution of 93 (4.79 g, 8.62 mmol) in THF (75 mL) at 0 °C. The resulting mixture was stirred at 0 °C for 45 min and at room temperature for 1 h. The solution was cooled to 0 °C and treated carefully with a 1.0 M aqueous NaOH (50 mL). The phases were separated, and the aqueous phase was extracted with CH2Cl2. The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (hexane/EtOAc 7:3) to give the alcohol 94 (2.98 g, 90%) as a colorless oil: IR (film) 3425, 1613, 1513, 1463, 1249, 1091, 1037 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.26 (d, J = 8.5 Hz, 2H), 6.88 (d, J = 8.5 Hz, 2H), 4.47 (d, J = 11.7 Hz, 1H), 4.40 (d, J = 11.7 Hz, 1H), 3.84 (s, 3H), 3.75 (dd, J = 5.7, 2.9 Hz, 1H), 3.52 (m, 3H), 3.28 (dd, J = 9.1, 7.1 Hz, 1H), 2.10 (br, 1H), 2.05 (m, 1H), 1.93-1.81 (m, 1H), 0.97 (d, J = 7.0 Hz, 3H), 0.90 (s, 9H), 0.87 (d, J = 7.1 Hz, 3H), 0.07 (s, 3H), 0.05 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 159.2, 130.7, 129.3, 113.8, 74.8, 72.8, 72.7, 66.3, 55.4, 39.0, 37.7, 26.2, 18.4, 15.2, 12.0, −4.1; HRMS (ESI) calcd for C18H31O3SiNa 323.2042 [M+Na]+, found 323.2035; [α]20D − 0.76 (c 2.9, CHCl3).
(4S,5R,6S,2E)-Ethyl-7-(4-methoxybenzyloxy)-5-(tert-butyldimethylsilyloxy)-4,6-dimethylhept-2-enoate (95)
The procedure for 84 was used with the aldehyde from 94 (17.5 g, 31.6 mmol), with Py·SO3 (15.2 g, 95.5 mmol) and Et3N (13.3 mL, 95.5 mmol), followed by NaH (0.90 g, 39.7 mmol) and triethylphosphonoacetate (7.2 mL, 40.3 mmol) to yield 8.96 g (63% for 3 steps) of the ester 95 by flash column chromatography (EtOAc/hexane 1:9) as a colorless oil: IR (CHCl3) 2957, 2931, 2856, 1720, 1651, 1613, 1513, 1463, 1366, 1250, 1180, 1093, 1077, 837 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.31-7.27 (m, 2H), 7.03 (dd, J = 15.8, 7.8 Hz, 1H), 6.93-6.91 (m, 2H), 5.83 (dd, J = 15.8, 1.3 Hz, 1H), 4.48-4.40 (m, 2H), 4.23 (q, J = 7.1 Hz, 2H), 3.84 (s, 3H), 3.67 (m, 1H), 3.52 (m, 1H), 3.30 (dd, J = 9.1, 7.2 Hz, 1H), 2.59 (m, 1H), 2.00 (m, 1H), 1.33 (t, J = 7.1 Hz, 3H), 1.09 (d, J = 6.8 Hz, 3H), 1.01 (d, J = 7.0 Hz, 3H), 0.94 (s, 9H), 0.08 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 166.5, 159.0, 152.7, 130.6, 129.0, 120.4, 113.6, 76.8, 72.5, 71.8, 60.0, 55.1, 40.2, 38.0, 26.0, 18.2, 14.8, 14.3, 14.2, −4.0, −4.2; LRMS (ESI) 473.2 [M+Na]+; HRMS (ESI) calcd for C25H42O5SiNa 473.2699 [M+Na]+, found 473.2716; [α]20D − 28.3 (c 0.41, CHCl3).
(4S,5R,6S)-Ethyl-7-(4-methoxy)benzyloxy)-5-(tert-butyldimethylsilyloxy)-4,6-dimethylheptanoate (96)
NiCl2·6H2O (2.4 g, 10.1 mmol) then portionwise NaBH4 (1.50 g, 39.7 mmol) were added to a stirred solution of unsaturated ketone 95 (8.96 g, 19.9 mmol) in MeOH (66 mL), THF (20 mL) at 0 °C. After 1 h, the solvent was evaporated and filtered with Celite using Et2O as an eluent (60 mL). The organic phase was concentrated, and the residue was purified by flash chromatography (EtOAc/hexane 1:9) to yield 8.76 g of 96 (97%) as a colorless oil: IR (CHCl3) 2957, 2856, 1737, 1613, 1513, 1463, 1374, 1249, 1172, 1091, 1038, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.40-7.37 (m, 2H), 7.02-6.99 (m, 2H), 4.59-4.50 (m, 2H), 4.25 (q, J = 7.1 Hz, 2H), 3.91 (s, 3H), 3.66-3.62 (m, 2H), 3.40 (dd, J = 8.8, 7.3 Hz, 1H), 2.52-2.33 (m, 2H), 2.13-2.02 (m, 1H), 1.90-1.82 (m, 1H), 1.78-1.57 (m, 2H), 1.38 (t, J = 7.1 Hz, 3H), 1.09 (d, J = 6.9 Hz, 3H), 1.03 (s, 9H), 1.00 (d, J = 6.5 Hz, 3H), 0.19 (s, 3H), 0.18 (s, 3H); 13 C NMR (75 MHz, CDCl3) δ 173.6, 158.9, 130.7, 129.0, 113.5, 76.8, 72.5, 60.0, 55.0, 38.0, 35.6, 32.5, 29.9, 26.0, 18.3, 14.9, 14.1, 13.7, −3.9, −4.2; LRMS (ESI) 475.3 [M+Na]+; HRMS (ESI) calcd for C25H44O5SiNa 475.2856 [M+Na]+, found 473.2877; [α]20D−6.0 (c 1.9, CHCl3).
(4S,5R,6S)-7-(4-Methoxybenzyloxy)-5-(tert-butyldimethylsilyloxy)-4,6-dimethylheptanoic acid (97)
Aqueous LiOH (1 N, 193 mL, 0.19 mol) was added to a THF-H2O solution of 96 (8.76 g, 19.4 mmol). The resulting solution was warmed to 60 °C and stirred with heating for 6 h. Aqueous 1 N HCl was added to give a neutral pH, and the mixture was extracted with CH2Cl2, dried over MgSO4, filtered and evaporated to yield 8.22 g of crude acid 97, which was used without further purification: 1H NMR (300 MHz, CDCl3) δ 7.24-7.22 (m, 2H), 6.86-6.83 (m, 2H), 4.39 (m, 2H), 3.77 (s, 3H), 3.69 (q, J = 7.0 Hz, 1H), 3.52 (m, 1H), 3.47 (q, J = 7.0 Hz, 1H), 3.19 (t, J = 8.5 Hz, 1H), 2.16 (m, 1H), 1.90 (m, 1H), 1.65-1.51 (m, 2H), 1.21 (t, J = 7.0 Hz, 2H), 0.92-0.85 (m, 12H), 0.81 (d, J = 6.3 Hz, 3H), 0.00 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 181.0, 158.9, 130.6, 129.1, 113.6, 72.5, 65.8, 58.0, 55.1, 37.8, 30.6, 26.1, 18.3, 18.1, 15.2, 14.0, −3.5, −4.1.
(R)-3-((4S,5R,6S)-7-(4-Methoxybenzyloxy)-5-(tert-butyldimethylsilyloxy)-4,6-dimethylheptanoyl)-4-benzyloxazolidin-2-one (98)
A solution of acid 97 (8.22 g, 19.4 mmol) and Et3N (5.40 mL, 38.8 mmol) in 100 mL of dry THF was cooled to −78 °C and treated dropwise with pivaloyl chloride (2.86 g, 23.3 mmol), stirred in the cold for 2 h and warmed to 0 °C prior to the addition of the oxazolidinone (3.5 g, 19.8 mmol) and LiCl (2.46 g, 58.8 mmol). This mixture was stirred overnight at room temperature and diluted with water (200 mL). The separated aqueous phase was extracted with ether (100 mL), and the combined organic layers were dried and evaporated to give a residue that was chromatographed to yield 7.91 g (70% for 2 steps) of imide 98 by flash column chromatography (EtOAc/hexane 1:4) as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.41-7.23 (m, 7H), 6.94-6.91 (m, 2H), 4.71 (m, 1H), 4.51 (d, J = 11.6 Hz, 1H), 4.46 (d, J = 11.6 Hz, 1H), 4.25-4.16 (m, 2H), 3.84 (s, 3H), 3.63-3.58 (m, 2H), 3.37-3.31 (m, 2H), 3.14-3.04 (m, 1H), 2.94-2.86 (m, 1H), 2.79 (dd, J = 13.3, 9.7 Hz, 1H), 2.04 (m, 1H), 1.87-1.60 (m, 3H), 1.03 (d, J = 6.9 Hz, 3H), 0.99-0.97 (m, 12H), 0.14 (s, 3H), 0.12 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 173.1, 158.8, 153.3, 135.2, 130.7, 129.3, 129.0, 128.8, 127.1, 113.5, 77.1, 72.5, 72.4, 65.9, 55.1, 54.9, 37.9, 37.7, 35.6, 33.7, 29.2, 26.0, 18.3, 14.9, 13.9, −3.8, −4.2.
(R)-3-((2R,4S,5R,6S)-7-(4-Methoxybenzyloxy)-5-(tert-butyldimethylsilyloxy)-2,4,6-trimethylheptanoyl)-4-benzyloxazolidin-2-one (99)
NaHMDS (1 M in THF, 14.9 mL, 14.9 mmol) was added dropwise over a 30 min period to a cooled (−78 °C) suspension of imide 98 (7.91 g, 13.6 mmol) in THF (45 mL). After 15 min of stirring, the resulting cold solution was treated with MeI (2.53 mL, 40.8 mmol) and stirred at −78 °C for 3 h before being warmed to 25 °C overnight (12 h). The reaction was quenched with H2O (100 mL), and the aqueous layer was extracted with Et2O (3 × 150 mL). The combined organic extracts were dried (MgSO4), concentrated in vacuo and chromatographed (EtOAc/hexane 1:9) to provide 5.97 g (74%) of 99 as a colorless oil: 1H NMR (300 MHz, CDCl3) δ 7.42-7.26 (m, 7H), 6.95-6.92 (m, 2H), 4.71 (m, 1H), 4.51 (m, 2H), 4.18 (m, 2H), 3.95 (m, 1H), 3.84 (s, 3H), 3.63 (dd, J = 8.9, 3.8 Hz, 1H), 3.57 (dd, J = 6.4, 2.7 Hz, 1H), 3.35 (t, J = 8.5 Hz, 1H), 3.28 (dd, J = 13.3, 3.1 Hz, 1H), 2.83 (dd, J = 13.3, 9.4 Hz, 1H), 2.10-1.95 (m, 2H), 1.68 (m, 1H), 1.38 (ddd, J = 14.1, 9.8, 4.9 Hz, 1H), 1.31 (d, J = 6.8 Hz, 3H), 1.04 (d, J = 6.9 Hz, 3H), 0.98 (s, 9H), 0.95 (d, J = 6.7 Hz, 3H), 0.14 (s, 3H), 0.13 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 176.8, 158.8, 152.8, 135.1, 130.8, 129.3, 128.9, 128.7, 127.1, 113.5, 77.6, 72.6, 72.4, 65.7, 55.0, 38.9, 38.0, 37.6, 35.3, 33.8, 26.0, 18.8, 18.3, 14.9, 13.8, −3.8, −4.2.
(2R,4S,5R,6S)-7-(4-Methoxybenzyloxy)-5-(tert-butyldimethylsilyloxy)-2,4,6-trimethylheptan-1-ol (100)
From 99: n-BuLi (2.5 M in hexane, 17.6 mL, 44 mmol) was added to a solution of diisopropylamine (6.65 mL, 47.4 mmol) in THF (48 mL) at −78 °C. After 5 min, the mixture was warmed to 0 °C over 15 min. Borane-ammonia complex (90%, 1.55 g, 45.2 mmol) was added, and the resulting mixture was stirred at 0 °C for 15 min, warmed to room temperature for 15 min and then cooled to 0 °C. A solution of amide 99 (6.62 g, 11.3 mmol) in THF (35 mL) was added dropwise and the reaction was stirred at 0 °C for 1 h and then at room temperature for 2 h. The mixture was cooled to 0 °C and quenched carefully with saturated aqueous NH4Cl. The mixture was extracted with Et2O, and the combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (step gradient of 4:1 to 7:3 hexane/EtOAc) to afford the alcohol 100 (4.57 g, 96%) as a colorless oil: IR (film) 3410, 1612, 1513, 1249, 1067, 1038 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.26 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 4.44 (d, J = 11.7 Hz, 1H), 4.39 (d, J = 11.7 Hz, 1H), 3.81 (s, 3H), 3.51 (m, 2H), 3.44 (dd, J = 5.6, 3.4 Hz, 1H), 3.37 (dd, J = 10.6, 6.5 Hz, 1H), 3.22 (dd, J = 9.0, 7.0 Hz, 1H), 2.03-1.95 (m, 1H), 1.78-1.62 (m, 2H), 1.53 (br, 1H), 1.41 (ddd, J = 13.5, 7.5, 5.8 Hz, 1H), 0.95 (d, J = 6.9 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H), 0.88 (s, 9H), 0.87 (d, J = 6.9 Hz, 3H), 0.04 (s, 3H), 0.03 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 159.2, 130.9, 129.4, 113.9, 77.5, 72.8, 67.7, 55.4, 38.3, 38.0, 33.6, 33.2, 26.3, 18.6, 18.0, 15.6, 15.5, −3.5, −3.8; [α]20D−6.3 (c 1.7, CHCl3).
From 103: A solution of diisopropylamine (6.65 mL, 47.4 mmol) in THF (48 mL) stirred at −78 °C was treated with n-butyllithium in hexane (2.5 M, 17.6 mL, 44 mmol). The solution was stirred at −78 °C for 5 min and warmed to 0 °C for 15 min. Borane-ammonia complex (90 %, 1.55 g, 45.2 mmol) was added and the resulting mixture was stirred at 0 °C for 15 min, warmed to room temperature for 15 min and then cooled to 0 °C. A solution of amide 103 (6.62 g, 11.3 mmol) in THF (35 mL) was added dropwise and the reaction was stirred at 0 °C for 1 h and at room temperature for 2 h. The mixture was then cooled to 0 °C and quenched carefully with saturated aqueous NH4Cl. The mixture was extracted with diethyl ether and the combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (hexane/EtOAc 4:1 to 7:3) to afford the alcohol (4.57 g, 96%) as a colorless oil.
(2S,3R,4S,6R)-3,7-bis(tert-Butyldimethylsilyloxy)-2,4,6-trimethylheptan-1-ol (101)
TBSCl (4.16 g, 27.6 mmol) was added to a solution of alcohol 100 (5.86 g, 13.8 mmol), imidazole (2.89 g, 41.4 mmol), and DMAP (169 mg, 1.38 mmol) in CH2Cl2 (55 mL). The resulting white suspension was stirred at room temperature for 2 h, and the volatiles were removed under reduced pressure. The residue was dissolved in hexane and brine. The phases were separated, and the organic layer was washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (hexane/EtOAc 19:1) to afford the TBS protected alcohol (7.04 g, 95%) as a colorless oil: IR (film) 1513, 1471, 1463, 1249, 1091, 1039 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.30 (d, J = 8.6 Hz, 2H), 6.91 (d, J = 8.6 Hz, 2H), 4.48 (d, J = 11.9 Hz, 1H), 4.44 (d, J = 11.9 Hz, 1H), 3.82 (s, 3H), 3.60-3.49 (m, 3H), 3.39-3.28 (m, 3H), 2.05-1.95 (m, 1H), 1.80-1.66 (m, 2H), 1.49-1.40 (m, 2H), 1.02 (d, J = 6.9 Hz, 3H), 1.0-0.91 (m, 24 H), 0.10 (s, 3H), 0.09 (s, 3H), 0.08 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 159.2, 131.1, 129.3, 127.9, 77.3, 73.1, 72.8, 68.4, 55.3, 38.9, 38.5, 33.5, 26.4, 26.2, 18.7, 18.6, 18.1, 15.3, 15.1, −3.4, −3.8, −5.2; [α]20D−15.9 (c 0.47, CHCl3).
A solution of above TBS protected alcohol (5.28 g, 9.8 mmol) in CH2Cl2 (332 mL) and pH 7 phosphate buffer solution (33 mL) was treated with DDQ (3.34 g, 14.7 mmol). The reaction was stirred at room temperature for 1 h and was quenched with saturated aqueous NaHCO3 solution. The phases were separated and the aqueous layer was extracted with CH2Cl2. The combined organic extracts were washed with water, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (hexane/EtOAc 97:3 to 93:7) to afford 101 (4.01 g, 98%) as a colorless oil: IR (film) 3353, 1472, 1463, 1388, 1360, 1255, 1091, 1030, 1005 cm−1; 1H NMR (300 MHz, CDCl3) δ 3.60 (d, J = 5.3 Hz, 2H), 3.55-3.45 (m, 2H), 3.32 (dd, J = 9.7, 6.7 Hz, 1H), 2.49 (br, 1H), 1.45 (ddd, J = 13.5, 7.5, 5.3 Hz, 1H), 0.95 (d, J = 7.1 Hz, 3H), 0.92 (s, 9H), 0.89 (s, 9H), 0.93-0.87 (m, 6H), 0.11 (s, 3H), 0.09 (s, 3H), 0.04 (s, 6H); 13 C NMR (75 MHz, CDCl3) δ 80.9, 68.0, 66.2, 38.4, 37.8, 35.4, 33.5, 26.3, 26.1, 18.5, 18.3, 16.2, 15.7, −3.6, −3.9, −5.3; [α]20D−16.1 (c 4.4, CHCl3).
(3S,4R,5S,7R)-4-(tert-Butyldimethylsilyloxy)-7-((tert-butyldimethylsilyloxy)methyl)-3,5-dimethyloct-1-yne (78)
Sulfur trioxide pyridine complex (5.44 g, 34.2 mmol) was added to a solution of 101 (4.78 g, 11.4 mmol) and triethylamine (4.77 mL, 34.2 mmol) in CH2Cl2 (23 mL) and DMSO (46 mL) at 0 °C. The mixture was stirred at 0 °C for 1 h and then diluted with Et2O. The organic phase was washed with cold 0.5 M aqueous NaHSO4 and then with brine. The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by short flash chromatography (hexane/EtOAc 9:1) to afford the crude aldehyde as a golden oil which was used directly in the next reaction without further purification. Carbon tetrabromide (7.56 g, 22.8 mmol) was added to a solution of triphenylphosphine (12.3 g, 45.6 mmol) in CH2Cl2 (56 mL) at 0 °C. The resulting dark-red mixture was stirred at 0 °C for 10 min. A solution of the crude aldehyde and 2,6-lutidine (2.66 mL, 22.8 mmol) in CH2Cl2 (45 mL) was added dropwise. The dark-brown mixture was stirred at 0 °C for 1 h and then quenched with a saturated aqueous NH4Cl. The layers were separated, and the aqueous phase was extracted with CH2Cl2. The combined organic extracts were washed with H2O, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by short flash chromatography (hexane 100%) to afford the dibromoolefin (4.76 g, 73% yield from the alcohol) as a colorless oil that was used without further purification. A solution of the dibromoolefin (4.76 g, 8.2 mmol) in THF (40 mL) stirred at −78 °C was treated with n-BuLi (1.6 M in hexane, 15.4 mL, 24.6 mmol). The solution was stirred at −78 °C for 2 h and then quenched with saturated aqueous NH4Cl. The mixture was allowed to reach room temperature and was diluted with Et2O. The aqueous layer was extracted with Et2O. The combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (hexane:EtOAc 97:3) to afford the pure alkyne 78 (3.26 g, 95%) as a colorless oil: IR (film) 3313, 2100, 1472, 1463, 1252, 1088, 1005 cm−1; 1H NMR (300 MHz, CDCl3) δ 3.53-3.48 (m, 2H), 3.33 (d, J = 9.7, 6.8 Hz, 1H), 2.62 (ddddd, J = 7.2, 7.2, 7.2, 5.1, 2.5 Hz, 1H) 2.03 (d, J = 2.5 Hz, 1H), 1.97-1.80 (m, 1H), 1.73-1.6 (m, 1H), 1.47 (m, 1H), 1.21 (d, J = 7.1 Hz, 3H), 0.99-0.91 (m, 25H), 0.13 (s, 3H), 0.11 (s, 3H), 0.08 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 87.9, 77.8, 70.2, 68.5, 39.2, 33.9, 33.7, 32.3, 26.4, 26.3, 18.6, 17.9, 17.5, 15.7, −3.6, −5.1; LRMS (ESI) [M+Na]+ 435; [α]20D−8.2 (c 3.1, CHCl3).
(2R,4S,5R,6S)-7-(4-Methoxybenzyloxy)-5-(tert-butyldimethylsilyloxy)-N-((1S,2S)-1-hydroxy-1-phenylpropan-2-yl)-N,2,4,6-tetramethylheptanamide (103)
PPh3 (7.05 g, 26.2 mmol), imidazole (1.78 g, 26.2 mmol), diisopropylethylamine (4.6 mL, 26.2 mmol) in benzene (80 mL), diethyl ether (165 mL) and acetonitrile (33 mL) were stirred at room temperature and treated with iodine (6.65 g, 26.2 mmol). The resulting mixture was vigorously stirred until a beige suspension formed. A solution of alcohol 94 (5.0 g, 13.1 mmol) in Et2O (20 mL) was added dropwise to the suspension, and the resulting mixture was stirred at room temperature for 30 min. The reaction was quenched with saturated aqueous NaHCO3 and diluted with Et2O. The aqueous phase was extracted with Et2O, and the combined organic extracts were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was triturated with hexane and the triturate was concentrated under reduced pressure. This procedure was repeated two more times to afford iodide 102 as a colorless oil that was used directly in the next reaction. A solution of n-BuLi in hexane (2.5 M, 21 mL, 52.4 mmol) was added to a suspension of LiCl (7.05 g, 166.4 mmol) and diisopropylamine (7.85 mL, 56.3 mmol) in THF (40 mL) at −78 °C. The suspension was stirred at −78 °C for 5 min, 0 °C for 15 min and then cooled to −78 °C. A solution of (S,S)-pseudoephedrine propionamide (Myers’ auxiliary, 45) (6.09 g, 27.5 mmol) in THF (70 mL) was added dropwise. The resulting mixture was stirred at −78 °C for 1 h, at 0 °C for 15 min and at room temperature for 5 min. The suspension was cooled to 0 °C, and the iodide was added as a solution in THF (6 mL followed by a 6 mL rinse). The reaction mixture was stirred at room temperature for 24 h and quenched with half-saturated aqueous NH4Cl. The aqueous layer was extracted with EtOAc, and the combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue, which was purified by flash chromatography (hexane/EtOAc 1:1) to afford the amide 103 (6.69 g, 87%) as a colorless oil: IR (film) 3387, 1616, 1513, 1463, 1248, 1087, 1037 cm−1; HRMS (ESI) calcd for C34H56NO5Si [M+H]+ 586.3928, found 586.3940; [α]20D +23.2 (c 1.26, CHCl3).
(4R,5S,10S,11R,12S,14R,2E)-5,11,15-tris(tert-Butyldimethylsilyloxy)-4,10,12,14-tetramethyl-1-(trityloxy)pentadec-2-en-8-yn-7-one (104)
Alkyne 78 (4.12 g, 10.0 mmol) was dissolved in THF (100 mL), and the mixture was cooled to −78 °C. n-BuLi (6.25 mL, 1.6 M hexane solution) was added slowly. After 5 min, the mixture was warmed to 0 °C, and stirred for 30 min. The mixture was then cooled to −78 °C and amide 79 (6.47 g, 11.3 mmol) in THF (5 mL) was added slowly. After 5 min, the solution was warmed to 0 °C and stirred for 30 min. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was partitioned in a separatory funnel. The aqueous phase was extracted with Et2O (3 × 20 mL). The combined organic extracts were washed with brine and dried over MgSO4. Filtration and concentration under reduced pressure, followed by flash chromatography on silica gel (hexane/EtOAc 19:1), afforded the ynone 104 (9.70 g, 93%) as a pale yellow oil: IR (CHCl3) 2955, 2928, 2856, 2209, 1676, 1471, 1462, 1252, 1085, 836, 774 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.56 m, 6H), 7.36 (m, 9H), 5.80 (dd, J = 15.6, 7.1 Hz, 1H), 5.69 (dt, J = 15.7, 4.8 Hz, 1H), 4.37 (m, 1H), 3.69 (d, J = 4.7 Hz, 2H), 3.61 (m, 1H), 3.58 (dd, J = 9.7, 5.0 Hz, 1H), 3.43 (dd, J = 9.7, 6.5 Hz, 1H), 2.87 (m, 1H), 2.73 (m, 1H), 2.46 (m, 1H), 1.88 (m, 1H), 1.76 (m, 1H), 1.59 (m, 1H), 1.31 (d, J = 7.1 Hz, 3H), 1.15 (d, J = 6.8 Hz, 3H), 1.05 (m, 1H), 1.00 (m, 34H), 0.194 (s, 3H), 0.190 (s, 3H), 0.17 (s, 3H), 0.15 (s, 3H), 0.14 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 186.1, 144.2, 132.9, 128.6, 127.7, 126.9, 96.8, 86.8, 83.1, 71.5, 68.0, 64.9, 50.0, 42.3, 38.1, 34.4, 33.2, 32.1, 26.01, 25.96, 25.85, 18.3, 18.0, 17.9, 17.2, 15.5, 15.4, −3.8, −4.1, −4.6, −4.7, −5.4; LRMS (ESI) 947.5 [M+Na]+; HRMS (ESI) calcd for C56H88O5Si3Na 947.5837 [M+Na]+, found 947.5875; [α]20D−12.0 (c 0.54, CHCl3).
(2E4R,5S,7S,10S,11R,12S,14R,2E)-5,11,15-tris(tert-Butyldimethylsilyloxy)-4,10,12,14-tetramethyl-1-(trityloxy)pentadec-2-en-8-yn-7-ol (105)
Ynone 104 (5.28 g, 5.71 mmol) was taken up in i-PrOH (58 mL). The (S,S)-Noyori catalyst (0.77 g, 1.15 mmol, 20 mol%) was added in one portion, and the solution was stirred overnight. The solvent was removed under vacuum, and the crude residue was purified by flash chromatography on silica gel (hexane/EtOAc 97:3), affording propargylic alcohol 105 (4.18 g, 79%) as a pale yellow oil: IR (CHCl3) 3469, 2955, 2856, 1471, 1448, 1252, 1084, 836, 774 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.55 (m, 6H), 7.36 (m, 9H), 5.71 (m, 2H), 4.59 (m, 1H), 4.03 (quint, J = 3.9 Hz, 1H), 3.65 (d, J = 3.9 Hz, 2H), 3.58 (dd, J = 4.6, 3.2 Hz, 1H), 3.55 (dd, J = 10.1, 5.1 Hz, 1H), 3.38 (dd, J = 9.7, 6.8 Hz, 1H), 2.71 (m, 1H), 2.50 (m, 1H), 2.32 (d, J = 5.4 Hz, 1H), 1.88 (m, 1H), 1.80 (m, 2H), 1.55 (m, 1H), 1.23 (d, J = 7.1 Hz, 3H), 1.11 (d, J = 6.8 Hz, 3H), 0.98 (m, 34H), 0.20 (s, 3H), 0.17 (s, 3H), 0.16 (s, 3H), 0.14 (s, 3H), 0.12 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 144.3, 134.0, 128.6, 127.8, 127.1, 126.9, 88.1, 86.8, 83.0, 72.6, 68.3, 65.8, 65.1, 59.5, 41.9, 40.3, 38.7, 33.5, 33.2, 32.1, 26.0, 25.9, 18.4, 18.1, 17.7, 17.4, 15.7, 15.3, 14.2, −3.9, −4.0, −4.4, −4.5, −5.3; LRMS (ESI) 949.7 [M+Na]+; HRMS (ESI) calcd for C56H90O5Si3Na 949.5994 [M+Na]+, found 949.6018; [α]20D−10.0 (c 1.2, CHCl3).
(2E,4R,5S,7S,8Z,10S,11R,12S,14R)-5,11,15-tris(tert-Butyldimethylsilyloxy)-4,10,12,14-tetramethyl-1-(trityloxy)pentadeca-2,8-dien-7-ol (106)
Lindlar catalyst (ca. 200 mg) was added to a solution of alcohol 105 (4.18 g, 4.51 mmol) in toluene (100 mL). The flask was flushed with H2 via a balloon several times, then stirred under an atmosphere of H2 until starting material was consumed (usually 1 h) as indicated by TLC analysis. The mixture was filtered through a pad of Celite and concentrated under reduced pressure to afford the alkene 106 as a colorless oil (3.82 g, 91%): IR (CHCl3) 3436, 2954, 2926, 2855, 1461, 1378, 1252, 1061, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.56 (m, 6H), 7.34 (m, 9H), 5.73 (m, 2H), 5.60 (t, J = 10.3 Hz, 1H), 5.43 (dd, J = 10.9, 8.4 Hz, 1H), 4.73 (m, 1H), 3.98 (q, J = 5.0 Hz, 1H), 3.68 (d, J = 4.1 Hz, 1H), 3.59 (dd, J = 9.7, 4.7 Hz, 1H), 3.48 (m, 1H), 3.36 (dd, J = 9.0, 7.3 Hz, 1H), 2.79 (m, 1H), 2.58 (m, 1H), 2.23 (br, 1H), 1.78 (m, 1H), 1.71 (m, 1H), 1.66 (m, 2H), 1.50 (m, 1H), 1.11 (d, J = 6.8 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H), 1.00 (m, 34H), 0.22 (s, 3H), 0.18 (s, 3H), 0.14 (s, 6H), 0.13 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 144.3, 135.3, 134.6, 131.5, 128.7, 127.7, 127.0, 126.8, 86.8, 79.6, 73.0, 68.2, 65.0, 64.7, 42.0, 39.6, 38.0, 36.4, 34.9, 33.4, 26.2, 26.0, 25.9, 19.9, 18.4, 18.3, 18.1, 18.0, 15.2, 14.5, −3.4, −3.7, −4.2, −4.4, −4.5, −5.4; LRMS (ESI) 951.7 [M+Na]+; HRMS (ESI) calcd for C56H92O5Si3Na 951.6150 [M+Na]+, found 951.6172; [α]20D 1.0 (c 0.62, CHCl3).
((2E,4R,5S,7S,8Z,10S,11R,12S,14R)-5,7,11,15-tetrakis(tert-Butyldimethylsilyloxy)-4,10,12,14-tetramethylpentadeca-2,8-dienyloxy)triphenylmethane (107)
TBSOTf (2.08 mL, 9.07 mmol) was added to a stirred solution of alcohol 106 (3.82 g, 4.11 mmol) and 2,6-lutidine (1.14 mL, 9.85 mmol) in CH2Cl2 (14 mL) at 0 °C. The reaction mixture was stirred for 1 h at 0 °C. The mixture was quenched by addition of H2O (25 mL). The reaction mixture was extracted with CH2Cl2, and the organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by short column chromatography (hexane/EtOAc 19:1) to yield 107 (4.27 g, 99%) as a colorless oil: IR (CHCl3) 2956, 2929, 2856, 1471, 1462, 1449, 1255, 1089, 1005, 836, 773, 705 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.60 (m, 6H), 7.39 (m, 9H), 5.77 (m, 2H), 5.56 (t, J = 10.8 Hz, 1H), 5.42 (dd, J = 11.0, 8.2 Hz, 1H), 4.69 (m, 1H), 4.07 (m, 1H), 3.71 (d, J = 3.8 Hz, 2H), 3.64 (dd, J = 9.8, 4.8 Hz, 1H), 3.53 (m, 1H), 3.40 (dd, J = 9.6, 7.5 Hz, 1H), 2.74 (m, 1H), 2.55 (m, 1H), 1.89 (m, 3H), 1.59 (m, 3H), 1.12 (d, J = 6.2 Hz, 6H), 1.04 (m, 42H), 0.26 (s, 3H), 0.24 (s, 3H), 0.19 (s, 6H), 0.18 (s, 3H), 0.17 (s, 6H), 0.16 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 144.4, 134.5, 132.9, 132.6, 128.7, 127.7, 126.8, 86.8, 79.9, 72.3, 68.3, 66.5, 65.1, 64.1, 42.4, 41.6, 37.9, 36.0, 35.3, 33.6, 26.3, 26.02, 25.97, 25.7, 19.4, 18.5, 18.4, 18.20, 18.15, 18.1, 15.5, 13.3, −2.9, −3.5, −3.7, −4.1, −4.2, −4.3, −5.3; LRMS (ESI) 1065.9 [M+Na]+; HRMS (ESI) calcd for C62H106O5Si4Na 1065.7015 [M+Na]+, found 1065.7026; [α]20D−10.4 (c 0.53, CHCl3).
(2R,4S,5R,6S,7Z,9S,11S,12R,13E)-5,9,11-tris(tert-Butyldimethylsilyloxy)-2,4,6,12-tetramethyl-15-(trityloxy)pentadeca-7,13-dien-1-ol (108)
HF-pyridine in pyridine (40 mL, prepared by slow addition of 12 mL of pyridine to 3 mL of HF-pyridine complex, followed by dilution with 25 mL of THF) was slowly added to a solution of TBS ether 107 (4.27 g, 4.10 mmol) in THF (5 mL) at 0 °C. The mixture was stirred for 21 h at 0 °C and quenched with saturated aqueous NaHCO3 (100 mL). The aqueous layer was separated and extracted with Et2O (3 × 50 mL). The combined organic layers were washed with saturated aqueous CuSO4 (3 × 50 mL), dried over MgSO4, filtered and concentrated. Flash column chromatography (EtOAc/hexane 1:4) afforded 2.55 g (67%) of the alcohol 108 as a colorless oil: IR (CHCl3) 3350, 2956, 2928, 2856, 1471, 1448, 1254, 1086, 836, 773, 705 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.52 (m, 6H), 7.32 (m, 9H), 5.68 (m, 2H), 5.50 (t, J = 10.6 Hz, 1H), 5.35 (dd, J = 10.9, 8.5 Hz, 1H), 4.61 (t, J = 8.5 Hz, 1H), 4.00 (t, J = 8.1 Hz, 1H), 3.62 (d, J = 3.2 Hz, 2H), 3.58 (dd, J = 10.6, 4.3 Hz, 1H), 3.45 (m, 1H), 3.36 (dd, J = 9.9, 7.3 Hz, 1H), 2.66 (m, 1H), 2.48 (m, 1H), 1.70 (m, 3H), 1.49 (m, 3H), 1.04 (d, J = 6.6 Hz, 6H), 0.97 (s, 18H), 0.93 (m, 6H), 0.87 (s, 9H), 0.18 (s, 3H), 0.16 (s, 3H), 0.11 (s, 6H), 0.10 (s, 3H), 0.08 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 144.3, 134.4, 133.0, 132.1, 128.7, 127.7, 126.8, 86.7, 79.8, 72.3, 67.7, 66.5, 65.1, 42.4, 41.5, 37.3, 35.7, 35.5, 33.3, 26.2, 26.0, 25.9, 19.6, 18.4, 18.14, 18.06, 17.98, 15.7, 13.2, −2.9, −3.6, −3.7, −4.1, −4.2, −4.3; LRMS (ESI) 951.8 [M+Na]+; HRMS (ESI) calcd for C56H92O5Si3Na 951.6150 [M+Na]+, found 951.6162; [α]20D−12.0 (c 0.71, CHCl3).
(2R,4E,6R,8S,9R,10S,11Z,13S,15S,16R,17E)-9,13,15-tris(tert-Butyldimethylsilyloxy)-2-((4S,5S)-2-(4-methoxyphenyl)-5-methyl-1,3-dioxan-4-yl)-6,8,10,16-tetramethyl-19-(trityloxy)nonadeca-4,11,17-trien-3-one (109)
Alcohol 108 (2.55 g, 2.75 mmol) in CH2Cl2 (30 mL) was treated with Dess-Martin periodinane (1.74 g, 4.10 mmol). After 1 h, the mixture was quenched with saturated aqueous NaHCO3 (30 mL) and Na2S2O3 (30 mL). The aqueous layer was extracted with Et2O (2 × 30 mL), and the combined extracts were dried over anhydrous MgSO4. Filtration and concentration followed by short flash column chromatography (hexane/EtOAc 4:1) provided the crude aldehyde as a colorless oil, which was used without further purification. A mixture of ketophosphonate 39 (1.06 g, 2.75 mmol) and Ba(OH)2 (0.38 g, activated by heating to 100 °C for 1-2 h before use) in THF (40 mL) was stirred at room temperature for 30 min. A solution of the above aldehyde in wet THF (4 × 1 mL washings, 40:1 THF/H2O) was then added. After stirring for 12 h, the reaction mixture was diluted with Et2O (30 mL) and washed with saturated aqueous NaHCO3 (50 mL) and brine (50 mL). The organic solution was dried (MgSO4), filtered, and concentrated in vacuo. The residue was chromatographed (hexane/EtOAc 9:1) to yield 109 (2.60 g, 80% for 2 steps) as a colorless oil: IR (CHCl3) 2956, 2928, 2855, 1688, 1618, 1518, 1471, 1461, 1338, 1251, 1080, 1038, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.50 (m, 6H), 7.40 (m, 2H), 7.30 (m, 9H), 6.89 (m, 2H), 6.73 (dd, J = 15.6, 8.5 Hz, 1H), 6.29 (d, J = 15.6 Hz, 1H), 5.66 (m, 2H), 5.46 (t, J = 10.4 Hz, 1H), 5.46 (s, 1H), 5.31 (dd, J = 11.0, 8.4 Hz, 1H), 4.58 (t, J = 8.1 Hz, 1H), 4.12 (dd, J = 11.3, 4.6 Hz, 1H), 3.96 (m, 1H), 3.92 (dd, J = 10.0, 4.2 Hz, 1H), 3.80 (s, 3H), 3.60 (d, J = 2.8 Hz, 2H), 3.56 (m, 1H), 3.39 (t, J = 3.3 Hz, 1H), 2.93 (m, 1H), 2.64 (m, 1H), 2.45 (m, 1H), 2.37 (m, 1H), 2.01 (m, 1H), 1.61 (m, 1H), 1.54 (m, 2H), 1.50 (m, 1H), 1.44 (m, 1H), 1.27 (d, J = 7.0 Hz, 3H), 1.06 (d, J = 6.6 Hz, 3H), 1.02 (d, J = 6.5 Hz, 3H), 0.99 (d, J = 6.6 Hz, 3H), 0.95 (s, 9H), 0.94 (s, 9H), 0.88 (d, J = 6.6 Hz, 3H), 0.84 (s, 9H), 0.79 (d, J = 6.7 Hz, 3H), 0.15 (s, 3H), 0.14 (s, 3H), 0.07 (s, 3H), 0.06 (s, 3H), 0.05 (s, 3H), 0.03 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 200.7, 159.8, 152.3, 144.3, 134.3, 132.8, 132.1, 131.0, 128.6, 127.7, 127.1, 126.8, 126.6, 113.4, 100.8, 86.7, 82.7, 80.0, 72.8, 72.1, 66.4, 65.0, 55.2, 47.1, 42.4, 41.4, 39.3, 35.8, 34.7, 34.6, 32.2, 26.1, 25.92, 25.86, 20.8, 19.7, 18.3, 18.1, 18.0, 15.0, 13.0, 12.4, 10.8, −2.9, −3.7, −3.8, −4.18, −4.25, −4.35; LRMS (ESI) 1209.6 [M+Na]+; HRMS (ESI) calcd for C72H110O8Si3Na 1209.7406 [M+Na]+, found 1209.7474; [α]20 D−6.7 (c 0.11, CHCl3).
(2R,6S,8S,9R,10S,11Z,13S,15S,16R,17E)-9,13,15-tris(tert-Butyldimethylsilyloxy)-2-((4S,5S)-2-(4-methoxyphenyl)-5-methyl-1,3-dioxan-4-yl)-6,8,10,16-tetramethyl-19-(trityloxy)nonadeca-11,17-dien-3-one (110)
NiCl2·6H2O (0.26 g, 1.09 mmol) and NaBH4 (0.17 g, 4.49 mmol) in portions were added to a stirred solution of unsaturated ketone 109 (2.60 g, 2.19 mmol) in 80 mL of 3:2 MeOH/THF at 0 °C. After 1 h, the reaction mixture was evaporated and filtered through Celite eluting with Et2O (30 mL). The organic phase was concentrated, and the residue was purified by flash chromatography (EtOAc/hexane 1:9) to yield 1.98 g of 110 (76%) as a colorless oil: IR (CHCl3) 2955, 2927, 2855, 1711, 1614, 1518, 1461, 1251, 1076, 835, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.46 (m, 6H), 7.27 (m, 11H), 6.85 (m, 2H), 5.60 (m, 2H), 5.43 (s, 1H), 5.40 (m, 1H), 5.27 (m, 1H), 4.52 (m, 1H), 4.11 (dd, J = 11.1, 4.7 Hz, 1H), 3.91 (m, 2H), 3.78 (s, 3H), 3.55 (m 2H), 3.50 (m, 1H), 3.35 (m, 1H), 2.67 (m, 1H), 2.58 (m, 1H), 2.51 (m, 1H), 2.41 (m, 1H), 2.01 (m, 1H), 1.68 (m, 3H), 1.41 (m, 5H), 1.23 (d, J = 7.1 Hz, 3H), 0.96 (d, J = 6.7 Hz, 3H), 0.90 (s, 9H), 0.89 (s, 9H), 0.88 (m, 1H), 0.87 (m, 3H), 0.80 (s, 9H), 0.78 (m, 6H), 0.10 (s, 3H), 0.08 (s, 3H), 0.04 (s, 3H), 0.03 (s, 6H), 0.01 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 211.9, 159.8, 144.5, 144.3, 134.4, 132.9, 132.4, 130.9, 128.6, 127.9, 127.8, 127.7, 127.1, 126.8, 113.4, 100.8, 86.7, 83.1, 79.9, 72.8, 72.2, 66.4, 65.1, 55.1, 48.3, 42.3, 41.5, 41.2, 38.1, 35.7, 35.0, 31.2, 29.8, 29.7, 26.2, 25.92, 25.87, 20.2, 19.4, 18.4, 18.1, 18.0, 15.2, 13.2, 12.1, 9.6, −3.0, −3.5, −3.7, −4.2, −4.28, −4.34; LRMS (ESI) [M+Na]+ 1211.9; HRMS (ESI) calcd for C72H112O8Si3Na [M+Na]+ 1211.7563 (30 mL), found 1211.7616; [α]20D +1.6 (c 0.50, CHCl3).
(2S,3R,6S,8S,9R,10S,11Z,13S,15S,16R,17E)-9,13,15-tris(tert-Butyldimethylsilyloxy)-2-((4S,5S)-2-(4-methoxyphenyl)-5-methyl-1,3-dioxan-4-yl)-6,8,10,16-tetramethyl-19-(trityloxy)nonadeca-11,17-dien-3-ol (111β)
NaBH4 (0.095 g, 2.51 mmol) was added to a solution of ketone 110 (1.98 g, 1.67 mmol) in MeOH (28 mL) at 0 °C. After stirring for 2 h at 0 °C, the reaction mixture was evaporated and water (30 mL) was added. The reaction mixture was extracted with ether (2 × 40 mL), and the organic phase was washed with brine (50 mL), dried over MgSO4 and concentrated in vacuo. The residue was purified by flash chromatography (EtOAc/hexane 1:9) to yield as major product the title compound 111β (1.39 g, 70%, less polar), and as minor product 111α (0.58 g, 28%, more polar) as colorless oils. 111β: IR (CHCl3) 3398, 2954, 2926, 2854, 1517, 1460, 1251, 1072, 835 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.50 (m, 6H), 7.39 (m, 2H), 7.33 (m, 9H), 6.89 (m, 2H), 5.66 (m, 2H), 5.54 (s, 1H), 5.46 (m, 1H), 5.32 (m, 1H), 4.58 (m, 1H), 4.14 (dd, J = 11.3, 4.6 Hz, 1H), 3.95 (m, 1H), 3.87 (m, 1H), 3.80 (s, 3H), 3.72 (d, J = 9.8 Hz, 1H), 3.61 (m, 2H), 3.55 (m, 1H), 3.41 (m, 1H), 3.24 (br, 1H), 2.64 (m, 1H), 2.46 (m, 1H), 2.16 (m, 1H), 1.82 (m, 1H), 1.71 (m, 2H), 1.53 (m, 5H), 1.35 (m, 2H), 1.06 (d, J = 7.2 Hz, 3H), 1.01 (d, J = 6.6 Hz, 3H), 0.95 (s, 9H), 0.93 (s, 9H), 0.90 (m, 9H), 0.85 (s, 9H), 0.78 (d, J = 6.6 Hz, 3H), 0.14 (m, 6H), 0.09 (m, 12H); 13C NMR (75 MHz, CDCl3) δ 160.0, 144.5, 144.3, 134.4, 132.9, 132.4, 130.7, 128.7, 127.9, 127.8, 127.7, 127.6, 127.2, 127.1, 126.8, 113.6, 101.2, 89.1, 86.7, 80.0, 76.9, 73.1, 72.2, 66.5, 65.1, 42.3, 41.5, 41.4, 37.0, 36.7, 35.1, 32.5, 32.1, 30.4, 30.3, 26.2, 25.93, 25.87, 20.4, 19.4, 18.4, 18.1, 18.0, 15.4, 13.2, 11.8, 5.4, −3.0, −3.5, −3.7, −4.2, −4.27, −4.33; LRMS (ESI) 1213.7 [M+Na]+; HRMS (ESI) calcd for C72H114O8Si3Na 1213.7719 [M+Na]+, found 1213.7861; [α]20D + 6.5 (c 0.31, CHCl3). 111α: IR (CHCl3) 3540, 2956, 2929, 2855, 1615, 1518, 1461, 1383, 1251, 1074, 835, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.61 (m, 6H), 7.51 (m, 2H), 7.44-7.32 (m, 9H), 7.00 (m, 2H), 5.77 (m, 2H), 5.61 (s, 1H), 5.55 (m, 1H), 5.45 (m, 1H), 4.71 (m, 1H), 4.24 (dd, J = 11.1, 4.5 Hz, 1H), 4.07 (m, 1H), 4.01 (m, 1H), 3.88 (s, 3H), 3.73-3.60 (m, 4H), 3.54 (m, 1H), 2.76 (m, 1H), 2.56 (m, 1H), 2.49 (m, 1H), 2.24 (m, 1H), 1.94-1.78 (m, 4H), 1.72-1.46 (m, 6H), 1.42-1.31 (m, 2H), 1.22 (d, J = 7.0 Hz, 3H), 1.13 (d, J = 5.9 Hz, 3H), 1.06 (s, 18H), 1.03 (m, 6H), 0.96 (s, 9H), 0.86 (d, J = 6.6 Hz, 3H), 0.27-0.18 (m, 18H); 13C NMR (75 MHz, CDCl3) δ 159.9, 144.4, 144.3, 134.3, 132.9, 132.4, 131.0, 128.6, 127.6, 127.2, 126.8, 113.5, 101.0, 86.7, 82.8, 79.8, 74.8, 73.2, 72.2, 66.4, 65.0, 55.1, 42.3, 41.5, 37.8, 35.9, 34.9, 33.2, 32.4, 30.3, 30.2, 26.2, 25.92, 25.87, 20.4, 19.3, 18.4, 18.1, 18.0, 15.3, 13.2, 11.8, 11.0, −3.0, −3.4, −3.7, −3.9, −4.2, −4.28, −4.34; LRMS (ESI) 1213.9 [M+Na]+; HRMS (ESI) calcd for C72H114O8Si3Na 1213.7719 [M+Na]+, found 1213.7760; [α]20D +2.3 (c 0.75, CHCl3).
(4S,5S)-4-((2R,3R,6S,8S,9R,10S,11Z,13S,15S,16R,17E)-3,9,13,15-tetrakis(tert-Butyldimethylsilyloxy)-6,8,10,16-tetramethyl-19-(trityloxy)nonadeca-11,17-dien-2-yl)-2-(4-methoxyphenyl)-5-methyl-1,3-dioxane (112β)
TBSOTf (0.40 mL, 1.74 mmol) was added to a stirred solution of alcohol 111β (1.39 g, 1.17 mmol) and 2,6-lutidine (0.27 mL, 2.33 mmol) in CH2Cl2 (23 mL) at 0 °C. After stirring for 1 h at ambient temperature, the reaction mixture was quenched by the addition of water (50 mL) and extracted with CH2Cl2. After drying the organic phases over MgSO4, and concentrated under reduced pressure, the residue was purified by short column chromatography (hexane/EtOAc 9:1) to yield 112β (1.51 g, 99%) as a colorless oil: IR (CHCl3) 2955, 2928, 2855, 1615, 1517, 1461, 1250, 1074, 1039, 835, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.59 (m, 6H), 7.52 (m, 2H), 7.41 (m, 9H), 7.01 (m, 2H), 5.74 (m, 2H), 5.57 (s, 1H), 5.50 (m, 1H), 5.43 (m, 1H), 4.67 (m, 1H), 4.25 (dd, J = 11.3, 4.6 Hz, 1H), 4.04 (m, 1H), 3.94 (s, 3H), 3.78 (m, 1H), 3.70 (m, 3H), 3.49 (m, 1H), 3.16 (m, 1H), 2.72 (m 1H), 2.54 (m, 1H), 2.18 (m, 1H), 2.01 (m, 1H), 1.82 (m, 3H), 1.54 (m, 6H), 1.14 (d, J = 6.9 Hz, 3H), 1.11 (d, J = 6.8 Hz, 3H), 1.10 (d, J = 6.5 Hz, 3H), 1.05 (s, 9H), 1.03 (s, 9H), 1.02 (s, 12H), 0.98 (d, J = 6.3 Hz, 3H), 0.94 (s, 9H), 0.87 (d, J = 6.7 Hz, 3H), 0.24 (s, 3H), 0.22 (s, 3H), 0.17 (m, 18H); 13C NMR (75 MHz, CDCl3) δ 159.6, 144.5, 144.3, 134.4, 133.1, 132.6, 131.5, 128.6, 127.7, 127.6, 127.1, 126.8, 126.7, 113.3, 100.4, 86.7, 81.9, 79.8, 74.9, 73.3, 72.2, 66.4, 65.1, 55.1, 42.3, 41.5, 38.8, 35.9, 34.5, 31.3, 31.2, 30.8, 30.7, 26.3, 25.99, 25.97, 25.91, 22.6, 20.3, 19.2, 18.5, 18.10, 18.05, 15.1, 14.1, 13.1, 12.4, 10.6,, −3.0, −3.2, −3.6, −4.2, −4.25, −4.30; LRMS (ESI) 1327.8 [M+Na]+; HRMS (ESI) calcd for C78H128O8Si4Na 1327.8584 [M+Na]+, found 1327.8693; [α]20D +7.6 (c 0.17, CHCl3).
(2S,3S,4R,5R,8S,10S,11R,12S,13Z,15S,17S,18R,19E)-3-(4-Methoxybenzyloxy)-5,11,15,17-tetrakis(tert-butyldimethylsilyloxy)-2,4,8,10,12,18-hexamethyl-21-(trityloxy)henicosa-13,19-dien-1-ol (113β)
DIBALH (1.0 M in hexane, 11.7 mL, 11.7 mmol) was added dropwise to a stirred solution acetal 112β (1.53 g, 1.17 mmol) in anhydrous CH2Cl2 (2.3 mL) under an atmosphere of N2 at 0 °C. After stirring for additional 30 min at 0 °C, the reaction was quenched by careful addition of saturated aqueous potassium sodium tartrate (30 mL). The resulting mixture was stirred for 3 h at room temperature. The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (20 mL). The combined organic layers were washed with brine and dried over MgSO4 then concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc/hexane 1:9) to yield pure 113β (1.35 g, 88%) as a colorless oil: IR (CHCl3) 3464, 2956, 2929, 2856, 1613, 1514, 1471, 1252, 1087, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.46 (m, 6H), 7.28 (m, 11H), 6.88 (m, 2H), 5.61 (m, 2H), 5.39 (m, 1H), 5.28 (m, 1H), 4.57 (m, 1H), 4.53 (s, 2H), 3.92 (m, 2H), 3.83 (m, 1H), 3.80 (s, 3H), 3.60 (m, 2H), 3.56 (m, 2H), 3.46 (dd, J = 6.2, 4.5 Hz, 1H), 3.37 (m, 1H), 3.03 (m, 1H), 2.86 (m 1H), 2.59 (m, 1H), 2.41 (m, 1H), 1.93 (m, 1H), 1.88 (m, 1H), 1.66 (m, 2H), 1.35 (m, 4H), 1.11 (d, J = 7.0 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H), 0.92 (m, 27H), 0.85 (m, 10H), 0.81 (s, 9H), 0.11 (s, 3H), 0.09 (s, 3H), 0.07 (s, 3H), 0.06 (s, 6H), 0.05 (s, 6H), 0.04 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 159.2, 144.4, 144.3, 134.3, 133.0, 132.4, 130.5, 129.1, 128.6, 127.6, 126.7, 113.8, 86.7, 85.4, 79.8, 75.1, 73.8, 72.2, 66.4, 65.0, 55.0, 42.3, 41.6, 41.5, 40.5, 37.1, 35.8, 34.8, 32.0, 31.9, 30.7, 26.2, 25.94, 25.86, 20.3, 19.2, 18.4, 18.1, 18.0, 15.6, 15.2, 13.2, 10.0, −3.0, −3.4, −3.8, −3.9, −4.2, −4.28, −4.34, −4.4; LRMS (ESI) 1329.8 [M+Na]+; HRMS (ESI) calcd for C78H130O8Si4Na 1329.8741 [M+Na]+, found 1329.8779; [α]20D−8.9 (c 0.46, CHCl3).
((2E,4R,5S,7S,8Z,10S,11R,12S,14S,17R,18R,19S,20S,21Z)-19-(4-Methoxybenzyloxy)-5,7,11,17-tetrakis(tert-butyldimethylsilyloxy)-4,10,12,14,18,20-hexamethyltetracosa-2,8,21,23-tetraenyloxy)triphenylmethane (114β)
Alcohol 113β (1.35 g, 1.03 mmol) in CH2Cl2 (20 mL) was treated with Dess-Martin periodinane (0.66 g, 1.56 mmol). After 1 h, the reaction was quenched with saturated aqueous NaHCO3 (20 mL) and Na2S2O3 (20 mL). The aqueous layer was extracted with Et2O (2 × 20 mL), and the combined extracts were dried over anhydrous MgSO4. Filtration and concentration followed by short flash column chromatography (hexane/EtOAc 9:1) provided the crude aldehyde as a colorless oil, which was used without further purification. CrCl2 (1.06 g, 8.62 mmol) was added to a stirred solution of the crude aldehyde, and 1-bromoallyl trimethylsilane (1.28 g, 5.20 mmol) in anhydrous THF (26 mL) under an atmosphere of N2 at room temperature. After 14 h at ambient temperature, the reaction mixture was diluted with hexane followed by filtration through celite. The solvent was evaporated under reduced pressure, and the residue was purified by short silica gel column chromatography using EtOAc/hexane (1:9) as eluent. This product in THF (40 mL) was cooled to 0 °C, and NaH (95% w/w, 0.52 g, 20.6 mmol) was added in one portion. The ice bath was removed after 15 min, and the mixture was stirred for 2 h at ambient temperature, then cooled to 0 °C. The reaction was quenched with H2O (5 mL) and the mixture extracted with Et2O (2 × 20 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography (hexane/EtOAc 49:1) to obtain 114β (1.17 g, 85% for 3 steps) as a colorless oil: IR (CHCl3) 2956, 2928, 2856, 1614, 1514, 1471, 1462, 1249, 1088, 836, 772 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.46 (m, 6H), 7.27 (m, 11H), 6.86 (m, 2H), 6.58 (ddd, J = 17.0, 10.6, 10.5 Hz, 1H), 6.00 (t, J = 11.0 Hz, 1H), 5.60 (m, 3H), 5.31 (m, 2H), 5.17 (d, J = 16.9 Hz, 1H), 5.09 (d, J = 10.4 Hz, 1H), 4.51 (m, 3H), 3.90 (m, 1H), 3.80 (s, 3H), 3.61 (m, 1H), 3.56 (d, J = 3.7 Hz, 1H), 3.33 (m, 2H), 3.00 (m, 1H), 2.56 (m, 1H), 2.40 (m, 1H), 2.21 (m, 1H), 1.63 (m, 3H), 1.38–1.21 (5H), 1.10 (d, J = 6.7 Hz, 3H), 0.96 (m, 3H), 0.93 (s, 9H), 0.91 (s, 9H), 0.89 (s, 9H), 0.86 (m, 6H), 0.82 (m, 6H), 0.80 (s, 9H), 0.79 (m, 3H), 0.08 (m, 6H), 0.05 (m, 6H), 0.04 (m, 6H), 0.01 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 159.0, 146.2, 144.5, 144.4, 134.6, 134.5, 133.1, 132.7, 132.3, 131.4, 129.0, 128.9, 128.7, 127.7, 126.8, 117.1, 113.7, 86.8, 84.4, 79.9, 75.0, 72.9, 72.3, 66.5, 65.1, 55.2, 42.4, 41.9, 41.6, 40.6, 36.0, 35.6, 35.3, 34.5, 32.5, 31.7, 30.5, 26.3, 26.0, 25.9, 20.2, 19.2, 18.8, 18.5, 18.2, 18.1, 15.1, 13.3, 9.3, −2.9, −3.0, −3.3, −3.6, −3.7, −4.2, −4.3, −4.4; LRMS (ESI) 1351.8 [M+Na]+; HRMS (ESI) calcd for C81H132O7Si4Na 1351.8948 [M+Na]+, found 1351.9012; [α]20D +1.1 (c 1.7, CHCl3).
(2E,4R,5S,7S,8Z,10S,11R,12S,14S,17R,18R,19S,20S,21Z)-19-(4-Methoxybenzyloxy)-5,7,11,17-tetrakis(tert-butyldimethylsilyloxy)-4,10,12,14,18,20-hexamethyltetracosa-2,8,21,23-tetraen-1-ol (115β)
ZnBr2 (0.41 g) in 1.2 mL of 5:1 CH2Cl2/MeOH was added dropwise over 30 min to a stirred solution of 114β (0.24 g, 0.18 mmol) in 1.4 mL of 6:1 CH2Cl2/MeOH at 0 °C. After 4 h, the reaction was quenched with saturated aqueous NaHCO3 (20 mL) and the mixture was extracted with Et2O (2 × 10 mL). The organic phase was separated, dried with MgSO4, filtered and concentrated. The residue was purified by flash chromatography (EtOAc/hexane 1:9) to yield 115β (0.15 g, 77%) as a colorless oil: IR (CHCl3) 3432, 2956, 2856, 1613, 1514, 1471, 1462, 1360, 1250, 1082, 835, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.29 (m, 2H), 6.88 (m, 2H), 6.58 (ddd, J = 16.9, 10.6, 10.6 Hz, 1H), 6.00 (t, J = 11.0 Hz, 1H), 5.63 (m, 3H), 5.38 (t, J = 11.0 Hz, 1H), 5.27 (dd, J = 11.2, 8.3 Hz, 1H), 5.17 (d, J = 16.8 Hz, 1H), 5.10 (d, J = 10.3 Hz, 1H), 4.53 (m, 3H), 4.08 (d, J = 4.4 Hz, 2H), 3.90 (m, 1H), 3.81 (s, 3H), 3.62 (m, 1H), 3.33 (m, 2H), 2.99 (ddd, J = 10.0, 6.8, 3.2 Hz, 1H), 2.57 (m, 1H), 2.39 (m, 1H), 1.63 (m, 3H), 1.42 (m, 3H), 1.28 (m, 5H), 1.11 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H), 0.96 (d, J = 6.9 Hz, 3H), 0.93 (s, 9H), 0.91 (s, 18H), 0.89 (m, 3H), 0.88 (s, 9H), 0.81 (d, J = 6.7 Hz, 3H), 0.80 (d, J = 6.2 Hz, 3H), 0.10 (s, 3H), 0.09 (s, 3H), 0.08 (s, 6H), 0.06 (s, 3H), 0.05 (s, 6H), 0.03 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 159.0, 146.9, 135.2, 134.6, 133.0, 132.7, 132.3, 131.4, 129.2, 129.1, 128.9, 127.93, 127.90, 127.2, 84.4, 80.0, 75.0, 72.8, 72.2, 66.6, 63.9, 55.2, 42.4, 41.8, 41.7, 40.5, 35.9, 35.2, 34.6, 32.6, 31.6, 30.5, 26.3, 25.99, 25.96, 25.93, 20.2, 19.2, 18.8, 18.5, 18.2, 18.1, 15.1, 13.2, 9.2, −3.0, −3.3, −3.6, −3.7, −4.2, −4.4, −4.5; LRMS (ESI) 1109.8 [M+Na]+; HRMS (ESI) calcd for C62H118O7Si4Na 1109.7852 [M+Na]+, found 1109.7897; [α]20D +1.6 (c 0.94, CHCl3).
(2Z,4E,6R,7S,9S,10Z,12S,13R,14S,16S,19R,20R,21S,22S,23Z)-Methyl-21-(4-methoxy-benzyloxy)-7,9,13,19-tetrakis(tert-butyldimethylsilyloxy)-6,12,14,16,20,22-hexamethylhexacosa-2,4,10,23,25-pentaenoate (116β)
Alcohol 115β (127 mg, 0.117 mmol) in CH2Cl2 (4 mL) was treated with Dess-Martin periodinane (75 mg, 0.18 mmol). After 1 h, the reaction was quenched with saturated aqueous NaHCO3 (5 mL) and Na2S2O3 (5 mL). The aqueous layer was extracted with Et2O (2 × 10 mL), and the combined extracts were dried over anhydrous MgSO4. Filtration and concentration followed by short flash column chromatography (hexane/EtOAc 9:1) provided the crude aldehyde as a colorless oil, which was used for the next reaction without further purification. KHMDS (0.28 mL, 0.14 mmol, 0.5M solution in toluene) was added dropwise to a stirred solution of bis(2,2,2-trifluoroethyl)-(methoxycarbonylmethyl) phosphate (0.030 mL, 0.14 mmol) and 18-crown-6 (0.15 g, 0.57 mmol) in THF (2.3 mL) at −78 °C. The aldehyde in THF (0.5 mL) was added and the solution was stirred for 4 h at −78 °C. The reaction was quenched by addition of saturated aqueous NH4Cl (5 mL) and the mixture was diluted with Et2O (20 mL). The organic phase was washed with brine (30 mL), dried with MgSO4, filtered and concentrated. The residue was purified by flash chromatography (EtOAc/hexane 1:19) yielding ester 116β (0.104 g, 86% for 2 steps) as a colorless oil: IR (CHCl3) 2955, 2929, 2856, 1722, 1514, 1471, 1462, 1250, 1174, 1085, 1041, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.39 (dd, J = 15.4, 11.3 Hz, 1H), 7.29 (m, 2H), 6.88 (m, 2H), 6.59 (ddd, J = 16.9, 10.8, 10.6 Hz, 1H), 6.55 (t, J = 11.3 Hz, 1H), 6.01 (t, J = 11.0 Hz, 1H), 6.00 (dd, J = 15.7, 7.0 Hz, 1H), 5.60 (d, J = 11.3 Hz, 1H), 5.59 (t, J = 10.4 Hz, 1H), 5.39 (t, J = 10.4 Hz, 1H), 5.27 (dd, J = 11.0, 8.3 Hz, 1H), 5.18 (d, J = 16.8 Hz, 1H), 5.11 (d, J = 10.3 Hz, 1H), 4.54 (m, 3H), 3.96 (m, 1H), 3.81 (s, 3H), 3.74 (s, 3H), 3.63 (m, 1H), 3.34 (m, 2H), 3.00 (m, 1H), 2.57 (m, 2H), 1.64 (m, 3H), 1.55 (m, 1H), 1.46 (t, J = 5.9 Hz, 2H), 1.26 (m, 5H), 1.11 (d, J = 6.8 Hz, 3H), 1.05 (d, J = 6.7 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.96 (d, J = 7.1 Hz, 3H), 0.94 (s, 9H), 0.92 (s, 9H), 0.91 (s, 9H), 0.87 (s, 9H), 0.83 (d, J = 6.4 Hz, 3H), 0.82 (d, J = 6.0 Hz, 3H), 0.13 (s, 3H), 0.11 (s, 3H), 0.10 (s, 3H), 0.09 (s, 3H), 0.06 (s, 3H), 0.05 (s, 6H), 0.04 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 166.8, 159.0, 147.3, 145.5, 134.6, 132.9, 132.8, 132.4, 131.4, 129.0, 128.9, 126.9, 117.1, 115.5, 113.7, 84.4, 80.0, 75.0, 72.9, 72.1, 66.5, 55.2, 50.9, 43.5, 42.5, 41.8, 40.5, 36.0, 35.3, 34.5, 32.5, 31.6, 30.5, 26.3, 25.99, 25.96, 25.91, 20.2, 19.2, 18.8, 18.5, 18.2, 18.1, 15.0, 13.4, 9.2 −3.0, −3.2, −3.3, −3.6, −3.7, −4.1, −4.4, −4.5; LRMS (ESI) 1163.9 [M+Na]+; HRMS (ESI) calcd for C65H120O8Si4Na 1163.7958 [M+Na]+, found 1163.7985; [α]20D−9.3 (c 1.2, CHCl3).
(2Z,4E,6R,7S,9S,10Z,12S,13R,14S,16S,19R,20R,21S,22S,23Z)-Methyl-7,9,13,19-tetrakis(tert-butyldimethylsilyloxy)-21-hydroxy-6,12,14,16,20,22-hexamethylhexacosa-2,4,10,23,25-pentaenoate (117β)
Ester 116β (81 mg, 71 μmol) was added to CH2Cl2 (2 mL) and H2O (0.1 mL), and DDQ (20 mg, 88 μmol) was added at 0 °C. After 1 h at 0 °C, the reaction was quenched by adding saturated aqueous NaHCO3 (5 mL). The organic phase was washed with saturated aqueous NaHCO3 (3 × 10 mL) and brine, dried over MgSO4, filtered and concentrated. Purification by flash column chromatography (EtOAc/hexane 1:9) furnished 117β (64 mg, 88%) as a colorless oil: IR (CHCl3) 3541, 2956, 2929, 2856, 1722, 1639, 1471, 1462, 1377, 1360, 1254, 1175, 1086, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.34 (dd, J = 15.4, 11.2 Hz, 1H), 6.61 (ddd, J = 16.8, 10.7, 10.6 Hz, 1H), 6.51 (t, J = 11.3 Hz, 1H), 6.06 (t, J = 11.0 Hz, 1H), 5.96 (dd, J = 15.4, 7.1 Hz, 1H), 5.56 (d, J = 11.3 Hz, 1H), 5.39 (t, J = 10.1 Hz, 1H), 5.38 (t, J = 10.3 Hz, 1H), 5.22 (dd, J = 11.0, 8.5 Hz, 1H), 5.17 (d, J = 18.7 Hz, 1H), 5.09 (d, J = 10.1 Hz, 1H), 4.50 (m, 1H), 3.92 (m, 1H), 3.71 (m, 1H), 3.70 (s, 3H), 3.44 (m, 1H), 3.32 (m, 1H), 2.74 (m, 1H), 2.52 (m, 2H), 2.31 (br, 1H), 1.61 (m, 4H), 1.39 (m, 2H), 1.31 (m, 2H), 1.26 (m, 3H), 1.00 (d, J = 6.7 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.92 (d, J = 6.7 Hz, 3H), 0.86 (m, 27H), 0.84 (m, 6H), 0.82 (m, 12H), 0.05 (s, 9H), 0.02 (s, 3H), 0.01 (s, 6H), 0.00 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 166.8, 147.3, 145.5, 135.3, 132.7, 132.6, 132.3, 129.9, 126.8, 117.7, 115.5, 79.9, 77.6, 76.6, 72.1, 66.5, 51.0, 43.5, 42.4, 41.5, 37.7, 36.1, 35.7, 35.0, 32.1, 31.5, 30.6, 26.3, 25.9, 25.9, 20.4, 19.4, 18.5, 18.1, 17.9, 17.7, 15.3, 13.3, 6.9, −3.0, −3.4, −3.7, −4.1, −4.2, −4.4; LRMS (ESI) 1043.6 [M+Na]+; HRMS (ESI) calcd for C57H112O7Si4Na 1043.7383 [M+Na]+, found 1043.7417; [α]20D−25.3 (c 0.61, CHCl3).
(2Z,4E,6R,7S,9S,10Z,12S,13R,14S,16S,19R,20R,21S,22S,23Z)-7,9,13,19-tetrakis(tert-Butyldimethylsilyloxy)-21-hydroxy-6,12,14,16,20,22-hexamethylhexacosa-2,4,10,23,25-pentaenoic acid (118β)
A stirred solution of alcohol 117β (26 mg, 24 μmol) in 2.4 mL of 12:5 EtOH/THF was treated with 1 N aqueous KOH (0.24 mL), and the mixture was refluxed gently for 3 h. The ethanolic solution was concentrated and then diluted with Et2O (4 mL). After the solution was acidified to pH 3 with 1 N aqueous HCl, the organic phase was separated and the aqueous phase was extracted with Et2O (2 × 5 mL). The combined organic phase was dried with MgSO4, filtered, and concentrated. The residue was purified by chromatography (15% EtOAc/hexane) to give 15.7 mg (61%) alcohol: IR (CHCl3) 2956, 2929, 2857, 1693, 1635, 1600, 1471, 1462, 1254, 1088, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.33 (dd, J = 15.2, 11.3 Hz, 1H), 6.61 (t, J = 11.4 Hz, 1H), 6.61 (m, 1H), 6.07 (t, J = 11.0 Hz, 1H), 6.02 (dd, J = 15.8, 7.2 Hz, 1H), 5.58 (d, J = 11.3 Hz, 1H), 5.39 (m, 2H), 5.23 (dd, J = 11.0, 8.2 Hz, 1H), 5.18 (d, J = 16.8 Hz, 1H), 5.09 (d, J = 10.2 Hz, 1H), 4.50 (m, 1H), 3.92 (m, 1H), 3.73 (m, 1H), 3.46 (dd, J = 7.3, 2.6 Hz, 1H), 3.34 (m, 1H), 2.78 (m, 1H), 2.54 (m, 2H), 1.66 (m, 4H), 1.42 (m, 4H), 1.24 (m, 3H), 1.01 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H), 0.88 (m, 30H), 0.84 (m, 15H), 0.09 (s, 3H), 0.07 (s, 3H), 0.06 (s, 6H), 0.02 (s, 6H), 0.01 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 171.2, 148.2, 147.3, 135.2, 133.2, 132.7, 132.3, 123.0, 127.0, 117.7, 115.2, 79.9, 77.6, 76.5, 72.1, 66.4, 43.5, 42.6, 41.6, 37.8, 36.0, 35.8, 34.9, 32.1, 31.5, 30.6, 26.2, 25.93, 25.87, 20.3, 19.4, 18.4, 18.10, 18.05, 17.7, 15.3, 13.6, 6.9 −3.0, −3.4, −3.7, −4.1, −4.19, −4.24, −4.4; LRMS (ESI) 1029.7 [M+Na]+; HRMS (ESI) calcd for C56H110O7Si4Na 1029.7226 [M+Na]+, found 1029.7274; [α]20D−25.7 (c 0.54, CHCl3).
(8S,10S,14R,20R)-tetrakis(tert-Butyldimethylsilyloxy)-(7R,13S,15S,17S,21S)-pentamethyl-(22S)-((1S)-methylpenta-2,4-dienyl)oxacyclodocosa-3,5,11-trien-2-one (119β)
A solution of 118β (15.7 mg, 15.6 μmol) in THF (2 mL) was treated at 0 °C with Et3N (0.013 mL, 93 μmol) and 2,4,6-trichlorobenzoyl chloride (0.012 mL, 77 μmol). The reaction mixture was stirred at 0 °C for 30 min and then added to 4-DMAP (7.8 mL, 0.02 M solution in toluene) at 25 °C. After stirring for 12 h, the reaction mixture was concentrated, Et2O (10 mL) was added, and the mixture was washed with 1 N HCl (2 × 5 mL) and dried over MgSO4. Purification by flash column chromatography (EtOAc/hexane 1:49) furnished the macrolactone (12 mg, 78%) as a colorless oil: IR (CHCl3) 2955, 2929, 2857, 1716, 1642, 1474, 1225, 1043, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.98 (dd, J = 14.8, 11.3 Hz, 1H), 6.55 (m, 1H), 6.52 (t, J = 11.2 Hz, 1H), 6.04 (t, J = 10.5 Hz, 1H), 6.01 (dd, J = 15.4, 6.4 Hz, 1H), 5.59 (d, J = 11.2 Hz, 1H), 5.58 (m, 1H), 5.38 (t, J = 10.6 Hz, 1H), 5.33 (dd, J = 11.3, 8.1 Hz, 1H), 5.19 (d, J = 16.6 Hz, 1H), 5.11 (d, J = 10.5 Hz, 1H), 5.06 (dd, J = 7.6, 3.7 Hz, 1H), 4.52 (m, 1H), 4.01 (m, 1H), 3.63 (m, 1H), 3.19 (d, J = 6.2 Hz, 1H), 3.03 (m, 1H), 2.58 (m, 1H), 2.52 (m, 2H), 1.81 (m, 4H), 1.45 (m, 3H), 1.25 (m, 3H), 1.09 (m, 3H), 1.02 (d, J = 6.8 Hz, 3H), 1.01 (d, J = 7.0 Hz, 3H), 0.97 (d, J = 6.6 Hz, 3H), 0.95 (d, J = 6.4 Hz, 3H), 0.91 (s, 9H), 0.89 (s, 9H), 0.88 (s, 9H), 0.86 (s, 9H), 0.77 (d, J = 6.4 Hz, 3H), 0.75 (d, J = 6.5 Hz, 3H), 0.10 (s, 3H), 0.07 (s, 3H), 0.06 (s, 3H), 0.04 (s, 3H), 0.033 (s, 6H), 0.026 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 166.5, 143.1, 141.8, 133.9, 132.7, 131.8, 130.2, 129.8, 128.0, 118.4, 118.1, 81.0, 78.0, 70.4, 66.5, 62.5, 43.1, 42.3, 41.4, 39.1, 35.2, 34.8, 34.5, 31.6, 30.3, 29.7, 29.3, 26.2, 26.0, 25.94, 25.85, 20.2, 19.7, 18.5, 18.24, 18.16, 18.08, 16.2, 14.0, 9.9, −2.7, −3.4, −3.5, −3.8, −3.9, −4.2, −4.3; [α]20D−18.1 (c 0.24, CHCl3). A small amount of the impure 2E,4E-isomer was also isolated: 1H NMR (300 MHz, CDCl3) δ 7.35 (dd, J = 11.2, 15.5 Hz, 1H), 6.63 (dt, J = 10.5, 16.8 Hz, 1H), 6.44 (t, J = 11.3 Hz, 1H), 6.01–5.80 (m, 2H), 5.57 (t, J = 10.6 Hz, 1H), 5.50 (d, J = 11.4 Hz, 1H), 5.38 (t, J = 10.3 Hz, 1H), 5.30 (dd, J = 8.5, 11.0 Hz, 1H), 5.22–5.01 (m, 3H), 4.47 (m, 1H), 3.77 (m, 1H), 3.48 (m, 1H), 3.30 (m, 1H), 2.97 (m, 1H), 2.56 (m, 1H), 2.44 (m, 1H), 1.91 (m, 1H), 1.65 (m, 1H), 1.52–0.71 (m, 63H), 0.12–0.04 (m, 24H).
(8S,10S,14R,20R)-Tetrahydroxy-(7R,13S,15S,17S,21S)-pentamethyl-(22S)-((1S)-methylpenta-2,4-dienyl)oxacyclodocosa-3,5,11-trien-2-one (1)
A stirred solution of macrolactone 119β (18 mg, 18 μmol) in THF (3 mL) at 0 °C was treated with 3 N HCl (10 mL, prepared by adding 2.5 mL of conc. HCl to 7.5 mL MeOH). After 24 h at room temperature, the reaction mixture was diluted with EtOAc (4 mL) and H2O (4 mL). The organic phase was saved, and the aqueous phase was extracted with EtOAc (2 × 4 mL). The combined organic phase was washed with saturated aqueous NaHCO3 (10 mL), dried with MgSO4, filtered and concentrated. The residue was purified by flash chromatography (EtOAc/hexane 3:2) to yield 1 as a white solid (5.3 mg, 55%): IR (CHCl3) 3406, 2960, 2924, 2872, 1693, 1637, 1461, 1378, 1274, 1181, 1069, 998, 738 cm−1; 1H NMR (600 MHz, CD3OD) δ 7.21 (dd, J = 15.6, 11.1 Hz, 1H), 6.71 (ddd, J = 16.9, 11.0, 10.6 Hz, 1H), 6.65 (dd, J = 11.3, 11.3 Hz, 1H), 6.17 (dd, J = 15.6, 6.7 Hz, 1H), 6.06 (dd, J = 11.1, 11.1 Hz, 1H), 5.56 (d, J = 11.3 Hz, 1H), 5.55 (dd, J = 11.0, 11.0 Hz, 1H), 5.41 (dd, J = 11.1, 8.8 Hz, 1H), 5.34 (dd, J = 10.7, 10.6 Hz, 1H), 5.25 (dd, J = 16.8, 1.8 Hz, 1H), 5.15 (d, J = 10.1 Hz, 1H), 5.14 (dd, J = 7.0, 5.0 Hz, 1H), 4.65 (ddd, J = 9.5, 9.5, 3.3 Hz, 1H), 4.05 (ddd, J = 10.6, 3.7, 2.8 Hz, 1H), 3.37 (m, 1H), 3.17 (ddq, J = 10.1, 6.8, 6.6 Hz, 1H), 3.10 (dd, J = 8.1, 2.9 Hz, 1H), 2.76 (m, 1H), 2.60 (m, 1H), 1.89 (m, 1H), 1.84 (dddd, J = 12.9, 11.2, 6.4, 5.4 Hz, 1H), 1.60 (m, 1H), 1.58 (m, 1H), 1.54 (m, 1H), 1.50 (ddd, J = 14.1, 10.7, 3.5 Hz, 1H), 1.42 (ddd, J = 14.0, 10.0, 2.7 Hz, 1H), 1.25 (ddd, J = 13.7, 10.6, 3.6 Hz, 1H), 1.15 (d, J = 6.9 Hz, 3H), 1.12 (d, J = 7.0 Hz, 3H), 1.10 (m, 1H), 1.07 (d, J = 6.9 Hz, 3H), 1.01 (d, J = 6.8 Hz, 3H), 0.95 (d, J = 6.5 Hz, 3H), 0.93 (d, J = 6.5 Hz, 3H), 0.90 (m, 1H), 0.71 (dddd, J = 12.9, 12.8, 8.7, 4.9 Hz, 1H); 13C NMR (150 MHz, CD3OD) δ 168.10, 146.42, 144.90, 134.87, 134.54, 133.43, 131.32, 131.27, 128.60, 118.58, 118.04, 80.37, 78.64, 73.73, 70.41, 65.53, 44.07, 42.28, 40.84, 40.65, 35.84, 35.78, 35.33, 32.75, 32.51, 31.23, 21.81, 19.36, 18.08, 15.98, 13.80, 10.41; LRMS (ESI) 555.3 [M+Na]+; HRMS (ESI) calcd for C32H52O6Na 555.3662 [M+Na]+, found 555.3665; [α]20D−22.6 (c 0.27, MeOH).
(2Z,4E,6R,7S,9S,10Z,12S,13R,14S,16S,19R,20S,21S,22S,23Z)-Methyl-7,9,13,19,21-penta-hydroxy-6,12,14,16,20,22-hexamethylhexacosa-2,4,10,23,25-pentaenoate (120β)
3 N HCl (10 mL, prepared by adding 2.5 mL of conc. HCl to 7.5 mL MeOH) was added to a stirred solution of 116β (23 mg, 23 μmol) in THF (3 mL) at 0 °C. After 24 h at room temperature, the reaction mixture was diluted with EtOAc (4 mL) and H2O (4 mL). The organic phase was retained, and the aqueous phase was extracted with EtOAc (2 × 4 mL). The combined organic phase was washed with saturated aqueous NaHCO3 (10 mL), dried with MgSO4, filtered and concentrated. The residue was purified by flash chromatography (EtOAc/hexane 3:2) to yield the product 120β (4.5 mg, 36%) as a colorless oil: IR 1H NMR (600 (CHCl3) 3399, 2917, 2849, 1713, 1635, 1600, 1461, 1439, 1197, 1178, 970, 757 cm−1; MHz, CD3OD) δ 7.36 (dd, J = 15.3, 11.2 Hz, 1H), 6.67 (ddd, J = 16.9, 11.1, 10.6 Hz, 1H), 6.63 (dd, J = 11.3, 11.3 Hz, 1H), 6.14 (dd, J = 15.4, 8.3 Hz, 1H), 6.03 (dd, J = 11.0, 11.0 Hz, 1H), 5.59 (d, J = 11.4 Hz, 1H), 5.43 (dd, J = 10.7, 10.7 Hz, 1H), 5.42 (dd, J = 10.8, 9.2 Hz, 1H), 5.32 (dd, J = 10.4, 10.4 Hz, 1H), 5.17 (dd, J = 16.8, 2.0 Hz, 1H), 5.08 (d, J = 10.2 Hz, 1H), 4.61 (ddd, J = 12.9, 8.5, 4.6 Hz, 1H), 3.80 (ddd, J = 8.9, 4.4, 4.4 Hz, 1H), 3.69 (s, 3H), 3.63 (m, 1H), 3.46 (t, J = 5.8 Hz, 1H), 3.13 (dd, J = 8.0, 3.2 Hz, 1H), 2.93 (m, 1H), 2.71 (m, 1H), 2.38 (m, 1H), 1.73 (m, 1H), 1.56-1.53 (m, 3H), 1.52-1.46 (m, 2H), 1.44-1.36 (m, 3H), 1.09 (d, J = 6.9 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.96 (d, J = 6.6 Hz, 3H), 0.95 (m, 2H), 0.94 (d, J = 6.9 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.8 Hz, 3H); 13C NMR (150 MHz, CD3OD) δ 168.4, 148.5, 146.9, 135.7, 135.4, 133.85, 133.82, 130.6, 128.2, 117.7, 116.2, 79.2, 78.5, 74.8, 72.4, 65.8, 51.5, 45.1, 43.2, 43.0, 41.0, 37.2, 36.5, 34.0, 33.3, 33.1, 31.0, 20.7, 18.6, 18.4, 16.7, 13.8, 7.8; LRMS (ESI) 587.5 [M+Na]+; HRMS (ESI) calcd for C33H56O7Na 587.3924 [M+Na]+, found 587.3953; [α]20D +8.7 (c 0.30, CDCl3).
(4S,5S)-4-((2R,3S,6S,8S,9R,10S,11Z,13S,15S,16R,17E)-3,9,13,15-tetrakis(tert-Butyldimethylsilyloxy)-6,8,10,16-tetramethyl-19-trityloxynonadeca-11,17-dien-2-yl)-2-(4-methoxyphenyl)-5-methyl-1,3-dioxane (112α)
The procedure for 112β was used with 112α (0.58 g, 0.49 mmol), TBSOTf (0.17 mL, 0.74 mmol) and 2,6-lutidine (0.11 mL, 0.97 mmol) to yield 0.62 g (97%) of the product by flash column chromatography (EtOAc/hexane 1:9) as a colorless oil: IR (CHCl3) 2955, 2856, 1615, 1518, 1462, 1385, 1251, 1082, 835, 773, 705 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.55 (m, 6H), 7.49 (m, 2H), 7.40-7.27 (m, 9H), 6.96 (m, 2H), 5.72 (m, 2H), 5.53 (m, 1H), 5.52 (s, 1H), 5.38 (m, 1H), 4.65 (m, 1H), 4.19 (dd, J = 11.0, 4.4 Hz, 1H), 4.03 (m, 1H), 3.95 (d, J = 8.7 Hz, 1H), 3.86 (m, 1H), 3.84 (s, 3H), 3.66 (d, J = 3.7 Hz, 2H), 3.56 (t, J = 11.1 Hz, 1H), 3.50 (m, 1H), 2.71 (m, 1H), 2.52 (m, 1H), 2.12 (m, 1H), 1.90-1.79 (m, 2H), 1.75-1.68 (m, 3H), 1.61-1.37 (m, 6H), 1.08 (d, J = 6.6 Hz, 6H), 1.02-0.91 (m, 45H), 0.81 (d, J = 6.5 Hz, 3H), 0.22-0.13 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 159.7, 144.5, 134.4, 132.8, 132.6, 131.7, 128.7, 127.7, 127.3, 126.8, 113.4, 100.9, 86.7, 81.4, 80.1, 73.4, 72.3, 71.5, 66.5, 65.1, 55.1, 42.4, 41.5, 37.9, 35.5, 35.1, 31.3, 30.8, 30.2, 27.9, 26.3, 26.01, 25.99, 25.93, 25.7, 20.6, 19.5, 18.4, 18.11, 18.07, 15.4, 13.4, 13.3, 12.2, 9.2, −2.9, −3.5, −3.7, −3.9, −4.1, −4.2, −4.3, −4.9; LRMS (ESI) 1328.0 [M+Na]+; HRMS (ESI) calcd for C78H128O8Si4Na 1327.8584 [M+Na]+, found 1327.8624; [α]20D +6.1 (c 0.93, CHCl3).
(2S,3S,4R,5S,8S,10S,11R,12S,13Z,15S,17S,18R,19E)-3-(4-Methoxybenzyloxy))-5,11,15,17-tetrakis(tert-butyldimethylsilyloxy)-2,4,8,10,12,18-hexamethyl-21-trityloxyhenicosa-13,19-dien-1-ol (113α)
The procedure for 113β was used with 112α (0.62 g, 0.47 mmol) and DIBALH (1.0 M in hexane, 4.7 mL, 4.7 mmol) to yield 0.54 g (87%) of the product after flash column chromatography (EtOAc/hexane 1:9) as a colorless oil: IR (CHCl3) 3479, 2955, 2928, 2856, 1613, 1514, 1471, 1251, 1084, 835, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.56-7.52 (m, 6H), 7.38-7.33 (m, 9H), 7.30 (m, 2H), 6.94 (m, 2H), 5.72 (m, 2H), 5.52 (m, 1H), 5.38 (m, 1H), 4.67 (d, J = 10.3 Hz, 1H), 4.65 (m, 1H), 4.60 (d, J = 10.4 Hz, 1H), 4.03 (m, 1H), 3.83 (s, 3H), 3.70 (m, 3H), 3.65 (d, J = 3.7 Hz, 2H), 3.49 (m, 1H), 3.02 (m, 1H), 2.71 (m 1H), 2.52 (m, 1H), 1.91 (m, 2H), 1.80-1.64 (m, 3H), 1.60-1.34 (m, 8H), 1.09-0.90 (m, 54H), 0.21-0.12 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 159.2, 144.5, 144.3, 134.4, 132.8, 132.4, 130.6, 129.0, 128.6, 127.7, 126.8, 113.8, 86.7, 84.7, 80.0, 74.8, 74.5, 72.2, 66.5, 66.2, 65.1, 55.1, 42.3, 41.4, 38.7, 35.5, 35.2, 35.0, 31.5, 30.9, 30.7, 29.8, 26.2, 26.00, 25.95, 25.89, 20.5, 19.4, 18.4, 18.13, 18.08, 18.02, 15.4, 15.2, 13.2, 10.4, −3.0, −3.6, −3.7, −3.8, −4.18, −4.24, −4.3, −4.4; LRMS (ESI) 1329.8 [M+Na]+; HRMS (ESI) calcd for C78H130O8Si4Na 1329.8741 [M+Na]+, found 1329.8782; [α]20D−6.8 (c 0.66, CHCl3).
((2E,4R,5S,7S,8Z,10S,11R,12S,14S,17S,18R,19S,20S,21Z)-19-(4-Methoxybenzyloxy)-5,7,11,17-tetrakis(tert-butyldimethylsilyloxy)-4,10,12,14,18,20-hexamethyltetracosa-2,8,21,23-tetraenyloxy)triphenylmethane (114α)
The procedure for 114β was used with 113α (0.54 g, 0.41 mmol) and Dess-Martin periodinane (0.26 g, 0.61 mmol), 1-bromoallyl trimethylsilane (0.50 g, 2.60 mmol) and CrCl2 (0.42 g, 3.42 mmol), and NaH (95% w/w, 0.21 g, 8.31 mmol) to yield 0.46 g (83% for 3 steps) of the product by flash column chromatography (EtOAc/hexane 1:9) as a colorless oil: IR (CHCl3) 2955, 2928, 2856, 1613, 1514, 1462, 1250, 1069, 835, 773, 705 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.58-7.54 (m, 6H), 7.40-7.35 (m, 9H), 7.33-7.30 (m, 2H), 6.96-6.93 (m, 2H), 6.69 (ddd, J = 16.8, 10.6, 10.5 Hz, 1H), 6.12 (t, J = 11.0 Hz, 1H), 5.80-5.67 (m, 3H), 5.52 (t, J = 10.4 Hz, 1H), 5.40 (m, 1H), 5.28 (d, J = 16.8 Hz, 1H), 5.19 (d, J = 10.2 Hz, 1H), 4.63 (m, 3H), 4.03 (m, 1H), 3.85 (s, 3H), 3.67 (m, 2H), 3.51 (m, 1H), 3.38 (m, 1H), 2.96 (m, 1H), 2.72 (m, 1H), 2.53 (m, 1H), 1.93-1.74 (m, 2H), 1.66-1.37 (m, 7H), 1.31-1.23 (m, 3H), 1.18 (d, J = 6.8 Hz, 3H), 1.09 (m, 6H), 1.03-0.92 (m, 45H), 0.23-0.10 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 159.1, 144.5, 144.4, 134.7, 134.5, 133.0, 132.6, 132.2, 131.4, 129.1, 129.0, 128.7, 127.7, 126.8, 117.5, 113.7, 86.8, 84.6, 79.9, 74.7, 73.6, 72.3, 66.5, 65.1, 55.2, 42.5, 42.4, 41.6, 36.1, 35.9, 34.8, 32.0, 30.8, 29.8, 26.3, 26.02, 25.96, 20.5, 19.3, 18.6, 18.5, 18.2, 18.1, 15.4, 13.3, 10.5, −2.9, −3.4, −3.7, −4.1, −4.2, −4.3; LRMS (ESI) 1352.0 [M+Na]+; HRMS (ESI) calcd for C81H132O7Si4Na 1351.8948 [M+Na]+, found 1351.8987; [α]20D−8.6 (c 1.6, CHCl3).
(2E,4R,5S,7S,8Z,10S,11R,12S,14S,17S,18R,19S,20S,21Z)-19-(4-Methoxybenzyloxy)-5,7,11,17-tetrakis(tert-butyldimethylsilyloxy)-4,10,12,14,18,20-hexamethyltetracosa-2,8,21,23-tetraen-1-ol (115α)
The procedure for 115β was used with 114α (0.46 g, 0.35 mmol) and ZnBr2 (0.41 g in 5.8 mL of 24:5 CH2Cl2/MeOH) to yield 0.21 g (55%) of the product after flash column chromatography (EtOAc/hexane 1:9) as a colorless oil: IR (CHCl3) 3410, 2956, 2929, 2856, 1614, 1514, 1471, 1462, 1251, 1075, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.28 (m, 2H), 6.87 (m, 2H), 6.60 (ddd, J = 16.8, 10.7, 10.6 Hz, 1H), 6.03 (t, J = 11.0 Hz, 1H), 5.67-5.57 (m, 3H), 5.41 (m, 1H), 5.29 (m, 1H), 5.20 (d, J = 18.2 Hz, 1H), 5.11 (d, J = 10.2 Hz, 1H), 4.56 (m, 3H), 4.10 (d, J = 4.4 Hz, 1H), 3.93 (m, 1H), 3.81 (s, 3H), 3.66-3.57 (m, 2H), 3.40 (dd, J = 4.6, 2.6 Hz, 1H), 3.28 (dd, J = 6.2, 4.2 Hz, 1H), 2.85 (m, 1H), 2.60 (m, 1H), 2.39 (m, 1H), 1.79 (m, 1H), 1.70 (m, 1H), 1.66-1.56 (m, 2H), 1.51-1.19 (m, 8H), 1.09 (d, J = 6.8 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.92-0.86 (m, 45H), 0.11-0.00 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 159.0, 135.2, 134.7, 132.8, 132.7, 132.3, 131.4, 129.2, 129.1, 129.0, 117.4, 113.7, 84.6, 80.0, 74.7, 73.6, 72.2, 66.6, 63.9, 63.3, 55.3, 42.4, 41.7, 36.1, 35.8, 34.8, 31.9, 30.8, 29.8, 26.3, 26.0, 25.9, 20.5, 19.4, 19.3, 18.6, 18.5, 18.1, 15.3, 13.3, 10.5, −3.0, −3.4, −3.7, −4.2, −4.3, −4.5; LRMS (ESI) 1109.9 [M+Na]+; HRMS (ESI) calcd for C62H118O7Si4Na 1109.7856 [M+Na]+, found 1109.7902; [α]20D−12.0 (c 1.7, CHCl3).
(2Z,4E,6R,7S,9S,10Z,12S,13R,14S,16S,19S,20R,21S,22S,23Z)-Methyl-21-(4-methoxybenzyloxy)-7,9,13,19-tetrakis(tert-butyldimethylsilyloxy)-6,12,14,16,20,22-hexamethylhexacosa-2,4,10,23,25-pentaenoate (116α)
The procedure for 116β was used with 115α (117 mg, 0.108 mmol) and Dess-Martin periodinane (69 mg, 0.16 mmol), bis(2,2,2-trifluoroethyl)-(methoxycarbonylmethyl) phosphate (0.027 mL, 0.13 mmol), 18-crown-6 (0.14 g, 0.53 mmol) and KHMDS (0.26 mL, 0.13 mmol, 0.5 M solution in toluene) to yield 69 mg (56% for 2 steps) of the product after flash column chromatography (EtOAc/hexane 1:19) as a colorless oil: IR (CHCl3) 2956, 2929, 2856, 1722, 1640, 1514, 1471, 1462, 1250, 1174, 1080, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.44 (dd, J = 15.2, 11.3 Hz, 1H), 7.28 (m, 2H), 6.88 (m, 2H), 6.60 (ddd, J = 16.7, 10.6, 10.5 Hz, 1H), 6.56 (t, J = 11.3 Hz, 1H), 6.04 (dd, J = 15.5, 7.1 Hz, 1H), 6.00 (t, J = 11.0 Hz, 1H), 5.62 (m, 2H), 5.42 (m, 1H), 5.27 (m, 1H), 5.21 (d, J = 16.8 Hz, 1H), 5.11 (d, J = 10.3 Hz, 1H), 4.54 (m, 3H), 3.97 (m, 1H), 3.81 (s, 3H), 3.74 (s, 3H), 3.60 (m, 1H), 3.40 (m, 1H), 3.29 (m, 1H), 2.86 (m, 1H), 2.57 (m, 2H), 1.80-1.67 (m, 3H), 1.55-1.41 (m, 4H), 1.40-1.20 (m, 4H), 1.09 (d, J = 6.8 Hz, 3H), 1.06 (d, J = 6.8 Hz, 3H), 0.99 (d, J = 6.6 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H), 0.95-0.85 (m, 42H), 0.13-0.00 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 166.8, 159.0, 147.3, 145.5, 134.7, 132.9, 132.7, 132.3, 131.4, 129.1, 129.0, 126.9, 117.4, 115.5, 113.7, 84.6, 80.0, 74.7, 73.6, 72.1, 66.5, 55.3, 51.0, 43.5, 42.4, 41.6, 36.1, 35.8, 34.8, 31.9, 30.7, 29.8, 26.3, 26.0, 25.9, 20.5, 19.3, 18.6, 18.5, 18.1, 15.3, 13.4, 10.5, −3.0, −3.3, −3.7, −4.10, −4.15, −4.19, −4.3, −4.4; LRMS (ESI) 1163.8 [M+Na]+; HRMS (ESI) calcd for C65H120O8Si4Na 1163.7958 [M+Na]+, found 1163.8000; [α]20D−16.7 (c 0.33, CHCl3).
(2Z,4E,6R,7S,9S,10Z,12S,13R,14S,16S,19S,20R,21S,22S,23Z)-Methyl-7,9,13,19-tetrakis(tert-butyldimethylsilyloxy)-21-hydroxy-6,12,14,16,20,22-hexamethylhexacosa-2,4,10,23,25-pentaenoate (117α)
The procedure for 117β was used with 116α (68 mg, 60 μmol) and DDQ (15 mg, 66 μmol) to yield 56 mg (92%) of the product after flash column chromatography (EtOAc/hexane 1:9) as a colorless oil: IR (CHCl3) 3499, 2956, 2929, 2856, 1723, 1641, 1471, 1462, 1255, 1175, 1081, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.35 (dd, J = 15.2, 11.3 Hz, 1H), 6.64 (ddd, J = 16.9, 10.6, 10.5 Hz, 1H), 6.52 (t, J = 11.3 Hz, 1H), 6.07 (t, J = 11.0 Hz, 1H), 5.96 (dd, J = 15.5, 7.1 Hz, 1H), 5.56 (d, J = 11.3 Hz, 1H), 5.44-5.33 (m, 2H), 5.26-5.21 (m, 1H), 5.17 (d, J = 16.7 Hz, 1H), 5.07 (d, J = 10.1 Hz, 1H), 4.49 (m, 1H), 3.92 (m, 1H), 3.73-3.67 (m, 5H), 3.34 (m, 1H), 3.25 (br, 1H), 2.73 (m, 1H), 2.52 (m, 2H), 1.82-1.50 (m, 4H), 1.44-1.16 (m, 7H), 1.01 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 7.1 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.90 (d, J = 6.9 Hz, 3H), 0.88-0.81 (m, 42H), 0.08-0.00 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 166.8, 147.3, 145.5, 136.5, 132.8, 132.7, 132.6, 129.6, 126.8, 117.3, 115.5, 79.8, 78.4, 74.2, 72.1, 66.5, 51.0, 43.5, 42.5, 41.4, 36.0, 35.9, 35.8, 35.0, 32.4, 31.9, 30.7, 26.3, 26.0, 25.9, 20.4, 19.4, 18.5, 18.12, 18.08, 17.98, 17.4, 15.3, 13.4, 10.8, −3.0, −3.4, −3.7, −4.1, −4.2, −4.3, −4.4, −4.8; LRMS (ESI) 1043.7 [M+Na]+; HRMS (ESI) calcd for C57H112O7Si4Na 1043.7383 [M+Na]+, found 1043.7435; [α]20D−9.4 (c 0.62, CHCl3).
(2Z,4E,6R,7S,9S,10Z,12S,13R,14S,16S,19S,20R,21S,22S,23Z)-7,9,13,19-tetrakis(tert-Butyldimethylsilyloxy)-21-hydroxy-6,12,14,16,20,22-hexamethylhexacosa-2,4,10,23,25-pentaenoic acid (118α)
The procedure for 118β was used with 117α (56 mg, 55 μmol) and 1 N aqueous KOH (0.54 mL) to yield 118α, which was used without further purification:IR (CHCl3) 2956, 2929, 2857, 1693, 1634, 1471, 1462, 1254, 1082, 836, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.34 (dd, J = 15.1, 11.4 Hz, 1H), 6.64 (ddd, J = 16.5, 10.6, 10.5 Hz, 1H), 6.61 (t, J = 11.2 Hz, 1H), 6.07 (t, J = 11.0 Hz, 1H), 6.01 (dd, J = 15.5, 7.2 Hz, 1H), 5.58 (d, J = 11.3 Hz, 1H), 5.44-5.34 (m, 2H), 5.23 (dd, J = 11.0, 8.2 Hz, 1H), 5.17 (d, J = 18.0 Hz, 1H), 5.08 (d, J = 10.1 Hz, 1H), 4.50 (m, 1H), 3.92 (m, 1H), 3.69 (m, 1H), 3.35 (m, 1H), 2.75 (m, 1H), 2.54 (m, 2H), 1.74-1.56 (m, 4H), 1.49-1.20 (m, 8H), 1.02 (d, J = 6.8 Hz, 3H), 0.98 (d, J = 7.2 Hz, 3H), 0.94 (d, J = 7.0 Hz, 3H), 0.90-0.82 (m, 45H), 0.09-0.01 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 171.1, 148.2, 147.4, 136.4, 132.7, 132.6, 129.6, 127.0, 117.4, 115.1, 79.8, 78.4, 74.2, 72.1, 66.5, 43.5, 42.6, 41.5, 36.0, 35.9, 35.8, 35.0, 31.9, 30.8, 29.7, 26.3, 26.0, 25.9, 20.4, 19.3, 18.5, 18.13, 18.08, 17.97, 17.4, 15.4, 13.7, 10.8, −3.0, −3.4, −3.7, −4.1, −4.2, −4.3, −4.4, −4.8; LRMS (ESI) 1029.8 [M+Na]+; HRMS (ESI) calcd for C56H110O7Si4Na 1029.7226 [M+Na]+, found 1029.7255; [α]20D−6.5 (c 0.17, CHCl3).
(8S,10S,14R,20S)-tetrakis(tert-Butyldimethylsilyloxy)-(7R,13S,15S,17S,21S)-pentamethyl-(22S)-((1S)-methylpenta-2,4-dienyl)-oxacyclodocosa-3,5,11-trien-2-one (119α)
The procedure for 119β was used with 118α, Et3N (0.046 mL, 33 μmol), 2,4,6-trichlorobenzoyl chloride (0.043 mL, 28 μmol) and 4-DMAP (27 mL, 0.02 M solution in toluene) to yield 42 mg (78% for 2 steps) of 119α after flash column chromatography (EtOAc/hexane 1:49) as a colorless oil: IR (CHCl3) 2956, 2929, 2856, 1704, 1638, 1471, 1462, 1378, 1361, 1255, 1086, 1044, 1004, 835, 773 cm−1; 1H NMR (300 MHz, CDCl3) δ 6.98 (dd, J = 15.3, 11.3 Hz, 1H), 6.58 (ddd, J = 16.9, 10.6, 10.5 Hz, 1H), 6.42 (t, J = 11.4 Hz, 1H), 5.94 (t, J = 9.2 Hz, 1H), 5.92 (dd, J = 9.5, 5.2 Hz, 1H), 5.55 (m, 1H), 5.42 (d, J = 11.6 Hz, 1H), 5.33-5.21 (m, 3H), 5.12 (d, J = 15.1 Hz, 1H), 4.99 (d, J = 9.7 Hz, 1H), 4.54 (m, 1H), 3.99 (m, 1H), 3.44 (m, 1H), 3.17 (m, 1H), 2.99 (m, 1H), 2.54 (m, 1H), 2.19 (m, 1H), 1.99 (m, 1H), 1.61-1.42 (m, 7H), 1.37-1.18 (m, 3H), 1.10 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 7.1 Hz, 3H), 1.00 (d, J = 6.3 Hz, 3H), 0.98 (d, J = 6.4 Hz, 3H), 0.98-0.82 (m, 36H), 0.79 (d, J = 6.6 Hz, 3H), 0.66 (d, J = 6.7 Hz, 3H), 0.11-0.01 (m, 24H); 13C NMR (75 MHz, CDCl3) δ 166.4, 146.5, 143.9, 134.3, 132.7, 132.2, 130.8, 129.8, 127.9, 117.4, 117.1, 81.6, 78.0, 77.1, 73.0, 66.7, 46.9, 45.7, 41.2, 37.5, 35.8, 35.1, 34.5, 31.1, 29.7, 26.2, 26.1, 26.0, 25.9, 20.5, 19.5, 19.1, 18.5, 18.4, 18.2, 17.9, 17.3, 16.9, 7.9, −2.6, −3.4, −3.5, −4.3, −4.4, −4.6; LRMS (ESI) 1011.7 [M+Na]+; HRMS (ESI) calcd for C56H108O6Si4Na 1011.7121 [M+Na]+, found 1011.7164; [α]20D−61.6 (c 2.8, CHCl3).
(8S,10S,14R,20S)-Tetrahydroxy-(7R,13S,15S,17S,21S)-pentamethyl-(22S)-((1S)-methylpenta-2,4-dienyl)-oxacyclodocosa-3,5,11-trien-2-one (121)
3 N HCl (10 mL, prepared by adding 2.5 mL of conc. HCl to 7.5 mL MeOH) was added to a stirred solution of macrolactone 119α (42 mg, 42 μmol) in THF (3 mL) at 0 °C. After 24 h at room temperature, the reaction mixture was diluted with EtOAc (4 mL) and H2O (4 mL). The organic phase was retained, and the aqueous phase was extracted with EtOAc (2 × 4 mL). The combined organic phase was washed with saturated aqueous NaHCO3 (10 mL), dried with MgSO4, filtered and concentrated. The residue was purified by flash chromatography (EtOAc/hexane 3:2) to yield 121 (7.9 mg, 35%) as a colorless oil: IR (CHCl3) 3415, 2961, 2917, 2849, 1681, 1637, 1461, 1279, 1067, 965, 758 cm−1; 1H NMR (600 MHz, CD3OD) δ 7.05 (dd, J = 15.3, 11.3 Hz, 1H), 6.65 (ddd, J = 16.9, 10.2, 10.1 Hz, 1H), 6.53 (dd, J = 11.5, 11.5 Hz, 1H), 5.97 (dd, J = 15.3, 9.5 Hz, 1H), 5.94 (dd, J = 11.0, 11.0 Hz, 1H), 5.60 (dd, J = 10.8, 9.6 Hz, 1H), 5.41 (d, J = 11.5 Hz, 1H), 5.20 (dd, J = 10.5, 10.3 Hz, 1H), 5.11 (dd, J = 16.9, 2.0 Hz, 1H), 5.10 (dd, J = 9.7, 2.1 Hz, 1H), 5.01 (d, J = 10.1 Hz, 1H), 4.60 (ddd, J = 10.1, 9.7, 2.7 Hz, 1H), 3.94 (ddd, J = 11.0, 2.1, 2.0 Hz, 1H), 3.38 (ddd, J = 9.8, 3.0, 2.0 Hz, 1H), 3.09 (ddq, J = 13.0, 7.0, 4.9 Hz, 1H), 3.01 (dd, J = 8.3, 2.7 Hz, 1H), 2.70 (m, 1H), 2.23 (ddd, J = 9.3, 7.0, 2.4 Hz, 1H), 2.07 (ddd, J = 7.0, 2.6, 2.5 Hz, 1H), 1.67 (m, 2H), 1.56 (ddd, J = 14.0, 10.9, 2.9 Hz, 1H), 1.51 (m, 1H), 1.47 (ddd, J = 14.1, 10.5, 1.9 Hz, 1H), 1.17 (d, J = 6.9 Hz, 3H), 1.13 (m, 1H), 1.11 (d, J = 7.1 Hz, 3H), 1.09 (d, J = 7.0 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H), 1.00 (m, 1H), 0.93 (d, J = 6.4 Hz, 3H), 0.92 (m, 1H), 0.78 (m, 1H), 0.76 (d, J = 6.7 Hz, 3H), 0.74 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 168.3, 147.6, 145.3, 135.4, 134.3, 133.5, 131.3, 131.0, 130.1, 118.1, 81.2, 79.9, 77.6, 72.0, 65.1, 45.9, 44.8, 42.4, 38.7, 36.0, 35.6, 31.8, 29.8, 27.8, 22.2, 19.8, 18.4, 17.6, 16.4, 9.1; LRMS (ESI) 555.3 [M+Na]+; HRMS (ESI) calcd for C32H52O6 555.3662 [M+Na]+, found 555.3655; [α]20D−76.2 (c 0.45, MeOH).
Supplementary Material
Supplementary Material Available. Contains detailed descriptions of the synthesis of the dictyostatin epimers and copies of NMR spectra of all compounds submitted for biological testing (59 pages).
Acknowledgments
We thank the National Institutes of Health for support through grant CA078390. We thank Drs. Kenneth Bair and Fred Kinder for the gift of discodermolide, Prof. Ian Paterson and Dr. Robert Britton for conducting some of the 1H NMR and TLC comparison experiments of the dictyostatin samples., and Mr. W.-H. Jung for helpful suggestions on the manuscript.
Footnotes
Dedicated to Professor Hisashi Yamamoto on his receipt of the Tetrahedron Prize
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Billy W. Day, Email: bday@pitt.edu.
Dennis P. Curran, Email: curran@pitt.edu.
References
- i.Paterson I, Anderson EA. Science. 2005;310:451–453. doi: 10.1126/science.1116364. [DOI] [PubMed] [Google Scholar]
- ii.(a) Madiraju C, Edler MC, Hamel E, Raccor BS, Balachandran R, Zhu G, Giuliano KA, Vogt A, Shin Y, Fournier JH, Fukui Y, Brückner AM, Curran DP, Day BW. Biochemistry. 2005;44:15053–15063. doi: 10.1021/bi050685l. [DOI] [PubMed] [Google Scholar]; (b) Buey R, Barasoain I, Jackson E, Meyer A, Giannakakou P, Paterson I, Mooberry S, Andreu JM, Diaz JF. Chem Biol. 2005;12:1269–1279. doi: 10.1016/j.chembiol.2005.09.010. [DOI] [PubMed] [Google Scholar]
- iii.Kowalski RJ, Giannakakou P, Gunasekera SP, Longley RE, Day BW, Hamel E. Mol Pharmacol. 1997;52:613–622. [PubMed] [Google Scholar]
- iv.Lowe J, Li H, Downing KH, Nogales E. J Mol Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- v.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- vi.Hamel E, Sackett DL, Vourloumis D, Nicolaou KC. Biochemistry. 1999;38:5490–5498. doi: 10.1021/bi983023n. [DOI] [PubMed] [Google Scholar]
- vii.(a) Verdier-Pinard P, Wang Z, Mohanakrishnan AK, Cushman M, Hamel E. Mol Pharmacol. 2000;57:568–575. doi: 10.1124/mol.57.3.568. [DOI] [PubMed] [Google Scholar]; (b) Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, Quada JC, Jr, Hemscheidt TK, Mooberry SL. Cancer Res. 2003;63:3211–3220. [PubMed] [Google Scholar]
- viii.Kar S, Fan J, Smith MJ, Goedert M, Amos LA. EMBO J. 2003;22:70–77. doi: 10.1093/emboj/cdg001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ix.Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- x.Pettit GR, Cichacz ZA, Gao F, Boyd MR, Schmidt JM. J Chem Soc, Chem Commun. 1994:1111–1112. [Google Scholar]
- xi.(a) Wright, A. E.; Cummins, J. L.; Pomponi, S. A.; Longley, R. E.; Isbrucker, R. A., PCT application 0162239, 2001. Isbrucker RA, Cummins J, Pomponi SA, Longley RE, Wright AE. Biochem Pharm. 2003;66:75–82. doi: 10.1016/s0006-2952(03)00192-8.
- xii.(a) Myles DC, Doherty AM, editors. Ann Rep Med Chem. Vol. 37. Academic Press; San Diego, CA: 2002. pp. 125–132. [Google Scholar]; (b) Gunasekera SP, Wright AE, Cragg GM, Kingston DGI, Newman DJ, editors. CRC Taylor Francis; New York, NY: 2005. pp. 171–189. [Google Scholar]
- xiii.(a) Choy N, Shin Y, Nguyen PQ, Curran DP, Balachandran R, Madiraju C, Day BW. J Med Chem. 2003;46:2846–2864. doi: 10.1021/jm0204136. [DOI] [PubMed] [Google Scholar]; (b) Minguez JM, Kim SY, Giuliano KA, Balachandran R, Madiraju C, Day BW, Curran DP. Bioorg Med Chem. 2003;11:3335–3357. doi: 10.1016/s0968-0896(03)00186-x. [DOI] [PubMed] [Google Scholar]; (c) Minguez JM, Giuliano KA, Balachandran R, Madiraju C, Curran DP, Day BW. Mol Cancer Therapeut. 2002;1:1305–1313. [PubMed] [Google Scholar]
- xiv.(a) Shin, Y.; Choy, N.; Turner, T. R.; Balachandran, R.; Madiraju, C.; Day, B. W.; Curran, D. P.(b) Experimental details for the syntheses reported in this paper are found in Shin, Y., PhD Thesis, University of Pittsburgh, 2005.For new hybrids, see: Paterson I, Gardner NM. Chem Commun. 2007:49–51. doi: 10.1039/b615122a.
- xv.Pettit, G. R.; Cichacz, Z. A. US Patent 5430053, 1995.
- xvi.Paterson I, Britton R, Delgado O, Wright AE. Chem Commun. 2004:632–633. doi: 10.1039/b316390c. [DOI] [PubMed] [Google Scholar]
- xvii.Paterson I, Britton R, Delgado O, Meyer A, Poullennec KG. Angew Chem Int Ed. 2004;43:4629–4633. doi: 10.1002/anie.200460589. [DOI] [PubMed] [Google Scholar]
- xviii.Shin Y, Fournier JH, Fukui Y, Brückner AM, Curran DP. Angew Chem Int Ed. 2004;43:4634–4637. doi: 10.1002/anie.200460593. [DOI] [PubMed] [Google Scholar]
- xix.(a) O’Neil GW, Phillips AJ. J Am Chem Soc. 2006;128:5340–5341. doi: 10.1021/ja0609708. [DOI] [PubMed] [Google Scholar]; (b) Ramachandran PV, Srivastava A, Hazra D. Org Lett. 2007;9:157–160. doi: 10.1021/ol062737k. [DOI] [PubMed] [Google Scholar]
- xx.Related synthetic studies: Gennari C, Castoldi D, Sharon O. Pure Appl Chem. 2007;79:173–180.Prusov E, Rohm H, Maier ME. Org Lett. 2006;8:1025–1028. doi: 10.1021/ol052917e.Jagel J, Maier ME. Synlett. 2006:693–696.Kangani CO, Brückner AM, Curran DP. Org Lett. 2005;7:379–382. doi: 10.1021/ol0478279.O’Neil GW, Phillips AJ. Tetrahedron Lett. 2004;45:4253–4256.
- xxi.Fukui Y, Brückner AM, Shin Y, Balachandran R, Day BW, Curran DP. Org Lett. 2006;8:301–304. doi: 10.1021/ol0526827. [DOI] [PubMed] [Google Scholar]
- xxii.Shin Y, Fournier JH, Balachandran R, Madiraju C, Raccor BS, Zhu G, Edler MC, Hamel E, Day BW, Curran DP. Org Lett. 2005;7:2873–2876. doi: 10.1021/ol050808u. [DOI] [PubMed] [Google Scholar]
- xxiii.Jung W-H, Harrison C, Shin Y, Fournier J-H, Balachandran R, Raccor BS, Sikorski RP, Vogt A, Curran DP, Day BW. J Med Chem. 2007 doi: 10.1021/jm061385k. ASAP. [DOI] [PubMed] [Google Scholar]
- 24.Smith AB, Beauchamp TJ, LaMarche MJ, Kaufman MD, Qiu YP, Arimoto H, Jones DR, Kobayashi K. J Am Chem Soc. 2000;122:8654–8664. [Google Scholar]
- 25.Saito S, Ishikawa T, Kuroda A, Koga K, Moriwake T. Tetrahedron. 1992;48:4067–4086. [Google Scholar]
- xxvi.Evans DA, Bartroli J, Shih TL. J Am Chem Soc. 1981;103:2127–2129. [Google Scholar]
- 27.Coe JW, Roush WR. J Org Chem. 1989;54:915–930. [Google Scholar]
- xxviii.(a) Paterson I, Yeung K, Watson C, Ward RA, Wallace PA. Tetrahedron. 1998;54:11935. [Google Scholar]; (b) Vanderwal CD, Vosburg DA, Sorensen EJ. Org Lett. 2001;26:4307. doi: 10.1021/ol016994v. [DOI] [PubMed] [Google Scholar]; (c) Evans DA, Ennis MD, Mathre DJ. J Am Chem Soc. 1982;104:1737–1739. [Google Scholar]
-
29.To determine the configuration at C19, the major product from NaBH4 reduction was treated with excess (10.0 equiv) DIBALH to open the PMB acetal, and the resulting primary alcohol was selectively protected with a TBS group. DDQ oxidation to remove the PMB group was attempted; however, mixtures of the PMB acetal (less polar) and the overoxidation product (more polar) were obtained. These were separated by silica gel column chromatography. The H19, H21, H22 protons of the PMB acetal were assigned by a 1H-1H COSY NMR (500 MHz) study. A NOESY NMR (500 MHz) experiment was carried out, and the major isomer was assigned as β from the cross peak between Ha-H19 and Ha-H21.
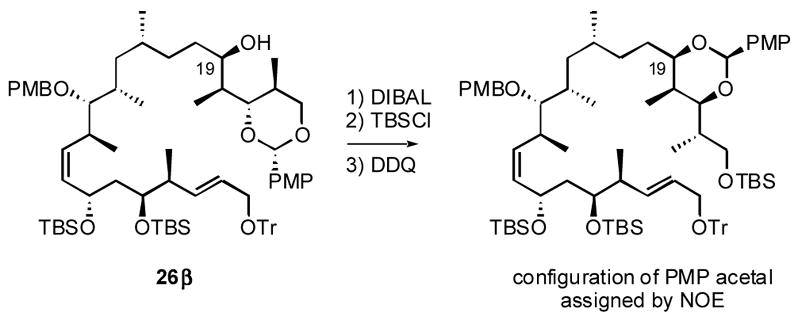
- xxx.(a) Paterson I, Schlapbach A. Synlett. 1995:498–499. [Google Scholar]; (b) Okude Y, Hirano S, Hiyama T, Nozaki H. J Am Chem Soc. 1977;99:3179–3181. [Google Scholar]
- 31.(a) Boeckman RK, Potenza JC. Tetrahedron Lett. 1985;26:1411–1414. [Google Scholar]; (b) King PK, Stroud SG. Tetrahedron Lett. 1985;26:1415–1418. [Google Scholar]
- xxxii.Still WC, Gennari C. Tetrahedron Lett. 1983;24:4405–4408. [Google Scholar]
- xxxiii.Inanaga J, Hirata K, Hiroko S, Katsuki TJ, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 34.Dandapani, S., unpublished results; summarized in Shin, Y., PhD Thesis, University of Pittsburgh, 2005.
- 35.Evans DA, Tedrow JS, Shaw JT, Downey CW. J Am Chem Soc. 2002;124:392–393. doi: 10.1021/ja0119548. [DOI] [PubMed] [Google Scholar]
- 36.(a) Myers A, Yang BY, Chen H, McKinstry L, Kopecky DJ, Gleason J. J Am Chem Soc. 1997;119:6496–6511. [Google Scholar]; (b) Vong BG, Abraham S, Xiang AX, Theodorakis EA. Org Lett. 2003;10:1617–1620. doi: 10.1021/ol034243i. [DOI] [PubMed] [Google Scholar]
- xxxvii.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;36:3769–3772. [Google Scholar]
- 38.Lee K, Kim Y, Oh C, Ham W. Org Lett. 2000;25:4041–4042. doi: 10.1021/ol000289p. [DOI] [PubMed] [Google Scholar]
- 39.(a) Marshall JA, Bourbeau MJ. Org Lett. 2003;5:3197–3199. doi: 10.1021/ol034918h. [DOI] [PubMed] [Google Scholar]; (b) Lebel H, Jacobsen EN. J Org Chem. 1998;63:9624–9625. [Google Scholar]
- 40.(a) Matsumura K, Hashiguchi S, Ikariya T, Noyori R. J Am Chem Soc. 1997;119:8738–8739. [Google Scholar]; (b) Haack KJ, Hashiguchi S, Fujii A, Ikariya T, Noyori R. Angew Chem Int Ed. 1997;36:285–288. [Google Scholar]
- 41.Boland W, Schroer N, Sieler C, Feigel M. Helv Chim Acta. 1987;70:1025–1040. [Google Scholar]
- 42.Alcohol 60 was treated with TBAF to remove both TBS groups. The resulting triol was reacted with excess 2,2-dimethoxypropane (3.0 equiv) to form the acetal, whose HMQC (500 MHz) NMR spectrum showed the two methyl groups of the acetonide at similar chemical shifts (24.5 ppm and 25.1 ppm) and the tertiary carbon at 100.4 ppm. See: Rychnovsky SD, Rogers BN, Richardson TI. Acc Chem Res. 1998;31:9–17.
- 43.(a) Kohli V, Blöcker H, Köster H. Tetrahedron Lett. 1980;21:2683–2686. [Google Scholar]; (b) Lampe TFJ, Hoffmann HMR. Tetrahedron Lett. 1996;37:7695–7698. [Google Scholar]
- xliv.Also isolated was 21% of an impure fraction containing principally the expected 2Z,4E isomer according to 1H NMR analysis. Deprotection of this fraction gave an impure sample of 6,14-bis-epi-dictyostatin 36, again as determined by 1H NMR analysis. Efforts to purify this small sample did not provide a product of suitable quality for biological assays.
- 45.(a) Roush WR, Blizzard TA. J Org Chem. 1984;49:4332–4339. [Google Scholar]; (b) Ghosh AK, Wang Y, Kim JT. J Org Chem. 2001;66:8973–8982. doi: 10.1021/jo010854h. [DOI] [PubMed] [Google Scholar]
- 46.Phukan P, Sasmal S, Maier ME. Eur J Org Chem. 2003:1733–1740. [Google Scholar]
- 47.(a) White JD, Hong J, Robarge LA. J Org Chem. 1999;64:6206–6216. [Google Scholar]; (b) Brown MC, Bhat KS. Org Synth. 1986;108:293–294. [Google Scholar]
- 48.Mickel SJ, Sedelmeier GH, Niederer D, Daeffler R, Osmani A, Schreiner K, Seeger-Weiber M, Berod B, Schaer K, Gamboni R. Org Proced Res Develop. 2004;8:92–101. [Google Scholar]
- 49.This type of translactonized product has been isolated and characterized in the C16-epimeric series. See reference 22.
- 50.Wipf P, Graham TH, Vogt A, Sikorski RP, Ducruet AP, Lazo JS. Chem Biol Drug Des. 2006;67:66–73. doi: 10.1111/j.1747-0285.2005.00313.x. [DOI] [PubMed] [Google Scholar]
- 51.Wipf P, Reeves JT, Balachandran R, Giuliano KA, Hamel E, Day BW. J Am Chem Soc. 2000;122:9391–9395. [Google Scholar]
- 52.Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, Poruchynsky MS. J Biol Chem. 1997;272:17118–171125. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 53.New analogs and associated SAR discussion are found in: Paterson I, Gardner NM, Poullennec KG, Wright AE. Bioorg Med Chem Let. 2007 doi: 10.1016/j.bmcl.2007.02.031. Articles in Press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Available. Contains detailed descriptions of the synthesis of the dictyostatin epimers and copies of NMR spectra of all compounds submitted for biological testing (59 pages).