

Abstract
Previously human cytochrome P450 3A4 was efficiently and specifically photolabeled by the photoaffinity ligand lapachenole. One of the modification sites was identified as cysteine 98 in the B-C loop region of the protein. Loss of CO binding capacity and subsequent decrease of catalytic activity were observed in the labeled CYP3A4, which suggested that aromatic substitution on residue 98 triggered a critical conformational change and subsequent loss of enzyme activity. To test this hypothesis, C98A, C98S, C98F and C98W mutants were generated by site-directed mutagenesis and expressed functionally as oligohistidine-tagged proteins. Unlike the mono-adduction observed in the wild-type protein, simultaneous multiple adductions occurred when C98F and C98W were photolabeled under the same conditions as the wild-type enzyme, indicating a substantial conformational change in these two mutants compared with the wild-type protein. Kinetic analysis revealed that the C98W mutant had a drastic 16-fold decrease in catalytic efficiency (Vmax/Km) for 1′-OH midazolam formation, and about an 8-fold decrease in catalytic efficiency (Vmax/Km) for 4-OH midazolam formation, while the C98A and C98S mutants retained the same enzyme activity as the wild-type enzyme. Photolabeling of C98A and C98S with lapachenole resulted in monoadduction of only Cys-468, in contrast to the labeling of Cys-98 in wild-type CYP3A4, demonstrating the marked selectivity of this photoaffinity ligand for cysteine residues. The slight increases in the midazolam binding constants (Ks) in these mutants suggested negligible perturbation of the heme environment. Further activity studies using different P450:reductase ratios suggested that the affinity of P450 to reductase was significantly decreased in the C98W mutant, but not in the C98A and C98S mutants. In addition, the C98W mutant exhibited a 41% decrease in the maximum electron flow rate between P450 and reductase as measured by reduced nicotinamide adenine dinucleotide phosphate consumption at a saturating reductase concentration. In conclusion, our data strongly suggest that cysteine 98 in the B-C loop region significantly contributes to conformational integrity and catalytic activity of CYP3A4, and that this residue or residues nearby might be involved in an interaction with P450 reductase.
Introduction
Cytochrome P450 (P450)1 3A4, the major P450 isoform present in human liver, is involved in the metabolism of more than 50% of clinically used drugs, making it one of the single most important human xenobiotic-metabolizing enzymes [1]. The large binding pocket in CYP3A4 can accommodate a wide variety of structurally diverse substrates while maintaining remarkable regioselectivity and stereoselectivity with many compounds. For example, the enzyme catalyzes the 1′- and 4-hydroxylation of midazolam [2], the 2β-, 6β-, and 15β-hydroxylation of testosterone, the 6β- and 16α-hydroxylation of progesterone [3], and the M1-, M17- and M21-oxidation of cyclosporine A [4]. The size of the active site may also be responsible for the phenomenon of cooperativity that could be clinically significant due to the role it can play in enhancing drug-drug interactions [5]. Because of its general importance in drug metabolism and carcinogen bioactivation, elucidation of the key structural elements responsible for substrate recognition leading to oxidation by CYP3A4 is of considerable interest.
Despite the wealth of information on the importance, regulation, and substrate specificity of the cytochrome P450 3A subfamily, structure-function analysis of these enzymes is only now being rigorously approached. A major step toward this goal has recently been achieved with the determination of high-resolution structures by X-ray crystallography of modified forms of human CYP3A4 [6,7]. However, X-ray crystallography, as important as it is, primarily provides information on static CYP3A4 structures. Several other techniques also have been employed to characterize P450 active sites and substrate binding sites for enzyme-substrate complexes, including spectroscopic analysis [8,9], site-directed mutagenesis [10-12], photoaffinity labeling [13,14] and mechanism-based inhibition [15,16]. Unlike cytochromes P450 of family 2, CYP3A enzymes within or across species exhibit few dramatic substrate specificity differences that could provide obvious leads for site-directed mutagenesis of particular residues. Most of our present knowledge has come from studies using a combination of site-directed mutagenesis and molecular modeling based upon alignment of sequences with bacterial P450s [17,18]. Additional studies by Halpert and colleagues with the use of alanine-scanning mutagenesis identified some amino acid residues necessary for substrate specificity and flavonoid activation of CYP3A4 [19].
Photoaffinity labeling, like mechanism-based inhibition, has the advantage of providing direct information concerning active site topology, and photoaffinity ligands (PALs) do not require enzymatic activity to be utilized as active site probes [20]. In our previous work [21,22], CYP3A4 was efficiently and specifically photolabeled by the substrate and PAL, lapachenole. Cysteine 98 in the B-C loop region was subsequently identified as one of the modification sites using a combination of techniques, including tandem mass spectrometry. The labeled protein showed loss of CO binding capacity and a dramatic decrease of catalytic activity, which strongly suggested that large aromatic substitutions on residue 98 triggered critical enzyme conformational changes and subsequent loss of catalytic activity [21].
In the present work, we have tested this hypothesis by generating different substitutions on position 98 of CYP3A4 using site-directed mutagenesis. One goal was to use a mutational substitution to mimic the labeling effects of lapachenole on the wild-type protein. Our results strongly suggest that cysteine 98 in the B-C loop region significantly contributes to conformational integrity and catalytic activity of CYP3A4, and that this residue or residues nearby might be involved in an interaction with P450 reductase.
Experimental Procedures
Materials
MDZ, NADPH, 3-(3-cholamidopropyl) dimethylammonio-1-propanesulfonate (CHAPS), HEPES, EDTA, reduced glutathione (GSH), imidazole, Triton X-100 and CF3CO2H (TFA) were purchased from Sigma (St. Louis, MO). Lapachenole was chemically synthesized in our laboratory (22). 1′-OH MDZ and 4-OH MDZ were obtained from GENTEST (Bedford, MA). L-α-dilaurylphosphatidylcholine (DLPC), L-α-dioleoylphosphatidylcholine (DOPC), and L-α-dilaurylphosphatidylserine (DLPS) were purchased from Avanti Polar lipids Inc. (Alabaster, AL). The QuikChange site-directed mutagenesis kit was from Stratagene (La Jolla, CA). Slide-A-Lyzer dialysis cassettes were from Pierce (Rockford, IL). A POROS R2 perfusion column was from Applied Biosystems (Framingham, MA). HPLC solvents were of the highest grade commercially available and were used as received. All other reagents were analytically grade.
Mutagenesis and Expression of CYP3A4
Recombinant CYP3A4 was produced in Escherichia coli XL1-Blue cells using the expression vector pCW 3A4-His6 kindly provided by Dr. Ron Estabrook (University of Texas SW Medical Center, Dallas, Texas). The same plasmid pCW 3A4-His6 was used as the template for amplification reactions with the QuikChange mutagenesis kit (Stratagene, La Jolla, CA). Oligonucleotide primers used in the mutagenesis procedure were as follows (mismatches indicated by the underlined bases): C98A forward, 5’ CTAGTGAAAGAAGCTTATTCTGTCTTCACAAACC 3’; C98A reverse, 5’ GGTTTGTGAAGACAGAATAAGCTTCTTTCACTAG 3’. C98S forward, 5’ AAACAGTGCTAGTGAAAGAAAGTTATTCTGTCTTCACAAACCG 3’; C98S reverse, 5’ CGGTTTGTGAAGACAGAATAACTTTCTTTCACTAGCACTGTTT 3’. C98F forward, 5’ CTAGTGAAAGAATTCTATTCTGTCTTCACAAACC 3’; C98F reverse, 5’ GGTTTGTGAAGACAGAATAGAATTCTTTCACTAG 3’. C98W forward, 5’ GCTAGTGAAAGAATGGTATTCTGTCTTCACAAACC 3’; C98W reverse, 5’ GGTTTGTGAAGACAGAATACCATTCTTTCACTAGC 3’. Dpn I digested DNA was transformed into XL1-Blue cells, and DNA from several of the resulting colonies was isolated. The cDNA sequence was checked for the presence of the desired mutation and the absence of extraneous mutations (University of Washington Sequencing Facility).
Growth and induction of Escherichia coli were performed as described by Gillam et al. [23]. Solubilized membranes were prepared and the P450s were purified on ProBond nickel resin columns (Invitrogen) under conditions described previously [21]. The column was washed with 20 column volumes of wash buffer: 100 mM Tris-HCl, pH 7.4, 20% glycerol, 40 mM imidazole, 0.05% cholate and 50 μM testosterone. The column was eluted with a minimal volume of elution buffer: 100 mM Tris-HCl, pH 7.4, 20% glycerol, 500 mM imidazole and 0.02% cholate. The eluted protein was dialyzed against 100 mM potassium phosphate, pH 7.4 in 20% glycerol and stored at -80°C. Total protein concentrations were determined by the bicinchoninic acid method. Bovine serum albumin was used as a standard [24]. SDS-polyacrylamide gel eletrophoresis was done according to the procedure of Laemmli [25]. Cytochrome P450 content was determined by reduced carbon monoxide difference spectra using an extinction coefficient of 91 mM-1 cm-1 with the addition of 1% Triton X-100 to protein sample before dilution in solubilization buffer (100 mM potassium phosphate, pH 7.4, 20% glycerol, 0.5% sodium cholate, 1% Emulgen 911 and 1.0 mM EDTA) [26].
Photoaffinity Labeling of CYP3A4
Photoaffinity labeling of CYP3A4 and its mutants was carried out in a photochemical reactor consisting of a Mineralight Model UVGL-15 UV lamp (UVP inc., San Gabriel, CA) as described previously [21]. The focus of the lamp was centered on the sample compartment at a lamp-to-sample distance of 1 cm. The enzyme solution to be photolyzed consisted of CYP3A4 (3 μM), DLPC (220 μg/mL), potassium Hepes (50 mM, pH 7.4), GSH (5 mM), and lapachenole (250 μM) in a final volume of 0.5 mL. The enzyme solution was first incubated at 37 °C for 10 min, and then the sample was taken for 1 min photolysis at room temperature using filtered long-wavelength UV light (360 nm). The same conditions were used for the mutants. Control samples contained lapachenole, but were not photolyzed. The samples were then analyzed by LC/ESI MS as described below.
LC/MS Analysis
Electrospray MS spectra of labeled CYP3A4 WT and mutant proteins were recorded on a Quattro II triple quadrupole mass spectrometer (Micromass, Manchester, UK). Instrument settings were the following: source temperature 100 °C, nebulizing gas flow 20 L/h, N2 drying gas 150 L/h, electrospray voltage 3.8 kV and cone voltage 35 V. Data acquisition was carried out from m/z 500 to 2000 using a 5 sec scanning time. Protein samples (∼200 pmoles) were injected on a POROS R2 perfusion chromatography column operating at a flow rate of 0.3 mL/min and interfaced on-line with the mass spectrometer. A Shimadzu LC10AD solvent delivery module (Shimadzu Scientific Instruments, Columbia, MD) was used to produce the following gradient elution profile: 10-50% solvent B in 5 min, followed by 50% B for 10 min and 50-90% B in 5 min (A = 5% acetonitrile, 0.05% TFA; B = 95 % acetonitrile, 0.05% TFA). Proteolytic digestion and purification of peptide adducts by HPLC were carried out as described previously [21]. Peaks that contained fluorescently-tagged peptide adducts were collected manually, and subjected to MALDI-TOF MS and nano-LC/ESI MS/MS analyses as described [21].
MDZ Hydroxylase Assay
Premixes and working buffers for CYP3A4 were prepared using a procedure described elsewhere [27]. MDZ hydroxylase assay was carried out according to a procedure described previously [28], with minor modifications. The reconstituted system for the assay contained 0.1 μM P450 3A4, 0.2 μM NADPH-P450 reductase, 0.1 μM cytochrome b5, 0.1 mg CHAPS/mL, 0.02 mg/mL liposomes (DLPC, DOPC and DLPS), 30 mM MgCl2 and 3 mM GSH in 50 mM potassium HEPES buffer, pH 7.4. MDZ dissolved in methanol was added to the reconstituted system. After preincubation for 5 min at 37 °C, the reaction was initiated by adding NADPH (1 mM final concentration). The total reaction volume of the assay was 300 μL. The reaction was terminated by addition of 60 μL of 100 mM sodium carbonate after 5 min of incubation at 37 °C. An internal standard solution (D2/37Cl-labeled 1′-OH MDZ, 100 ng/mL, 10 μL) was added. For kinetic studies with variable reductase concentration, the same conditions applied in which 50 μM MDZ was added and the P450:reductase ratio was 10:1, 5:1, 2:1, 1:1, 1:2, 1:5 or 1:20. For all samples, the assay was carried out in triplicate.
The metabolites were extracted with 1 mL of ethyl acetate. The extracts were dried under a nitrogen gas stream and the residue was resuspended in 100 μL 50% methanol. Then 40 μL of the mixture was used for LC/MS quantitative analysis. Samples were injected onto a Zorbax Eclipse XDB-C8 column (5 μm, 2.1 × 50 mm, Agilent) operating at a flow rate of 0.3 mL/min and interfaced on-line with the mass spectrometer. A Micromass Quattro II/MassLynx NT DS in SIR mode was used for detection of the metabolites by monitoring the following masses: 325.9 (MDZ), 341.9 (1′-OH MDZ and 4-OH MDZ), and 345.9 (D2/37Cl-labeled 1′-OH MDZ). The calibration curves for 1′- and 4-OH MDZ were prepared over a concentration range (9 different concentrations) between 0.5 and 250 ng/mL). A Shimadzu LC10AD solvent delivery module (Shimadzu Scientific Instruments, Columbia, MD) was used to produce the following gradient elution profile: 45% solvent B for 0.5 min, 45%-60% B in 1.5 min, followed by 60% B for 0.5 min (A = water, 0.1% acetic acid; B = methanol, 0.1% acetic acid).
Nonlinear regression analysis was performed using the program KaleidaGraph 3.6 (Synergy Software, Inc., Reading, PA) with the Michaelis-Menten equation fitted to the metabolite kinetic data.
Acetaminophen (APAP) Oxidation Assay
CYP3A4 activity was determined by measuring the amount of GS-3-APAP formed as described previously [29], with minor modifications. The reconstituted system for the assay was as described above. Briefly, 1 mM APAP was used for the assay and the reactions were initiated by addition of NADPH (1 mM final concentration). Incubations were carried out at 37 °C for 10 min followed by the addition of 100 μL of 2 M perchloric acid. The amount of GS-3-APAP generated was determined by UV detection at 254 nm compared with the standard.
Spectral Binding Studies
Binding spectra were recorded on a Varian CARY 300 spectrophotometer (Walnut Creek, CA). A solution (1 mL) containing 1 μM protein in 100 mM phosphate, pH 7.4, was divided into two quartz cuvettes (10 mm path length) and a baseline was recorded between 350 and 500 nm. An aliquot of substrate (MDZ) in methanol was then added to the sample cuvette, and the same amount of methanol was added to the reference cuvette. The difference spectra were obtained after the system reached equilibrium (3 min).
NADPH Oxidation
CYP3A4 was reconstituted with NADPH-P450 reductase and cytochrome b5 as described above. Reconstituted enzyme systems (570 μL) were preincubated for 5 min at 37 °C in the presence or absence of MDZ (50 μM). Reactions were initiated with the addition of 30 μL of 4.0 mM NADPH, and the decrease in A340 was monitored for 1 min. UV-visible spectra were recorded using a Varian CARY 300 spectrophotometer (Walnut Creek, CA). Rates of NADPH oxidation were calculated using [unk]340 = 6.22 mM-1 cm-1 for NADPH.
H2O2 Formation
Reconstituted systems were prepared as described above. Reactions (570 μL) were initiated by adding 30 μL of 4.0 mM NADPH and were terminated by adding 1.2 mL of cold CF3CO2H (3%, w/v) after 1 min incubation at 37 °C. H2O2 was determined spectrophotometrically by reaction with ferroammonium sulfate and KSCN as described [30].
Molecular Modeling
The Cys-98 of the X-ray crystal structure of CYP3A4 (6, PDB code: 1W0E) was substituted by a tryptophan residue using the mutate feature of DeepView 3.7 [31]. The structure was optimized using the NDDO (neglect of diatomic differential overlap) semiempirical method of PM3 by steepest decent [32,33] with ArgusLab 4.0 (Planaria Software, Seattle, WA).
Results
Photoaffinity Labeling of CYP3A4
In our previous work [21,22], CYP3A4 was efficiently and specifically photolabeled by the substrate, lapachenole. Two protein species were detected by HPLC/ESI MS with molecular weights (MWs) corresponding to the CYP3A4 apoprotein and a monoadduct which incorporated one molecule of lapachenole into one CYP3A4 protein (Figure 1A). The average MWs of apo CYP3A4 and the monoadduct of CYP3A4 were experimentally determined to be 57,280 ± 3 and 57,520 ± 3 Da (Figure 1A), respectively. The mass shift of 240 Da for the adducted CYP3A4 protein suggested that one molecule of lapachenole was bound specifically per CYP3A4 protein and the labeling was irreversible under these severely denaturing HPLC conditions. Unlike the mono-adduction of the wild-type protein, analysis of the labeled C98F and C98W mutants revealed that multiple adductions occurred when either protein was photolabeled with lapachenole under the same conditions as wild-type (Figure 1D and 1E). The average MWs of the apo C98F mutant and three protein adducts were determined to be 57,319 ± 5, 57,559 ± 5, 57,799 ± 5, 58,039 ± 5, respectively. The average MWs of the apo C98W mutant and four protein adducts were determined to be 57,355 ± 3, 57,595 ± 3, 57,835 ± 3, 58,075 ± 3, and 58,315 ± 3, respectively. The observed mass difference between the apoproteins for the wild-type and mutants (the insets, Figure 1) further confirmed the presence of the desired mutants. Furthermore, the mass shift of 240 Da between peaks implies the addition of 1, 2, 3, and 4 molecules of the PAL to the C98W mutant (Figure 1E). All five protein species observed for C98W coeluted at 19.2 min during the 30 min gradient run by reverse phase HPLC on a POROS R2 column. Similar multiple adductions were observed in the labeled C98F mutant. These data strongly suggest that a Cys → Trp or Phe substitution at residue 98 causes a conformational change in the protein, resulting in the availability of several other nucleophilic residues for photoaffinity labeling.
Figure 1.


LC/ESI MS analysis of CYP3A4. Samples were incubated with lapachenole with or without photolyzation and analyzed as described in Experimental Procedures. Deconvoluted spectra of the CYP3A4 wild-type (A), C98A (B), C98S (C), C98F (D), and C98W (E) mutants photolabeled with lapachenole. The insets show deconvoluted spectra of apo-CYP3A4 wild-type, and its mutant (MW = 57280 ± 3 Da, 57248 ± 5 Da, 57262 ± 5 Da, 57319 ± 5 Da, and 57355 ± 3 Da, n = 6 for apo-CYP3A4 wild-type, C98A, C98S, C98F and C98S mutants, respectively).
In marked contrast, only one monoadduct was observed upon photoaffinity labeling of either the C98A or C98S mutant (Figure 1B and 1C). The average MWs of apo C98A mutant and monoadduct were experimentally determined to be 57,248 ± 5 and 57,488 ± 5 Da, respectively, while the average MWs of apo C98S mutant and monoadduct were determined to be 57,262 ± 5 and 57,502 ± 5 Da, respectively. These data suggest that a Cys → Ala or a conservative Cys → Ser substitution at position 98 of CYP3A4 maintains protein conformation. Based on peak heights (Figure 1), a significantly decreased labeling efficiency was also observed in both C98A (29%) and C98S (35%) mutants compared to the wild-type protein (68%).
From the tryptic peptides of the C98S mutant, a single major fluorescent peptide peak F-1 was detected by HPLC (Figure 2), which suggested a fluorescently-tagged peptide adduct in the generated tryptic peptide pool [21]. ESI MS analysis of this collected fluorescent peptide fraction revealed that this doubly charged ion (m/z = 775.9 Da, Figure 3A) was the peptide adduct VLQNFSFKPCK (positions 459-469). The MS/MS spectrum of the doubly charged [M+2H]2+ ion at m/z 775.90 is shown in Figure 3B. In this MS/MS spectrum, the singly charged fragment ions (b3—b7, y3—y10) were observed. The singly charged free peptide ion at m/z 1310.73 and ion resulting from dehydration at m/z 1292.72 were observed together with the lapachenole [M+H]+ ion at m/z 241.09, indicating that some lapachenole was lost during the CID process. The doubly charged free peptide ion at m/z 655.87 was also observed. In the MS/MS analysis, the masses of the C-terminal fragment ions (y3 and y4) were observed to increase 240 Da, and no mass shifts were detected for the observed b ions (b3—b7). These data suggest that the lapachenole modification site was located on the C-terminus of the peptide, associated with Cys-468. Thus, the peptide adduct F-1 was identified as one of the two cysteine monoadducts, VLQNFSFKPCK (positions 459-469) and ECYSVFTNR (positions 97-105), previously characterized after photoaffinity labeling of CYP3A4 WT protein (Figure 3) [21]. However, no adduct was detected to Ser-98 in the C98S mutant. These results clearly showed that a Cys → Ser substitution prevented photoaffinity labeling of position 98 by lapachenole in the C98S mutant. This is also consistent with the results when amino acids cysteine and serine were used to test the reactivity of photolyzed lapachenole, wherein only cysteine was able to react prior to ring closure of the photolysis product (data not shown).
Figure 2.
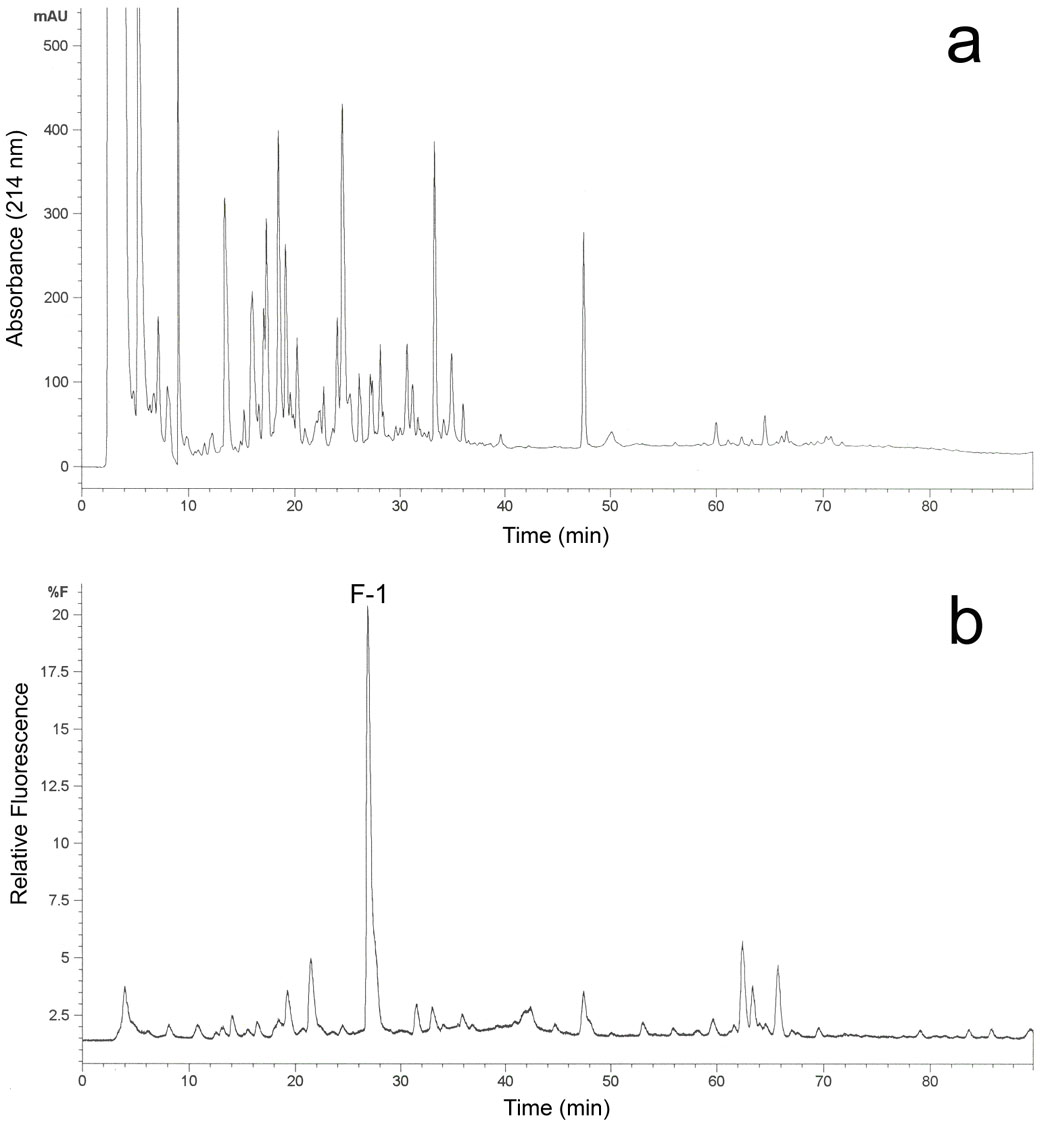
Purification and identification of the C98S mutant tryptic peptide adduct photolabeled with lapachenole. HPLC separation of the proteolytic digests of the lapachenole labeled CYP3A4 monitored by (a) variable wavelength detection at 214 nm and (b) fluorescence detection using 320 nm and 420 nm as excitation and emission wavelengths, respectively. The fraction containing one major fluorescence peak F-1 eluted at 27.2 min was collected and analyzed by nano-LC/ESI MS.
Figure 3.

Nano-LC/ESI MS analysis of the collected fluorescent peptide fraction. (A) MS profile of the doubly charged ion (m/z = 775.9 Da) for the F-1 peptide adduct VLQNFSFKPCK. (B) Nano-LC/ESI MS/MS spectrum of lapachenole-bound CYP3A4 peptide adduct F-1 (experiment performed in an API-US QTOF mass spectrometer).
Determination of Holo- and Apo-P450 content
Total protein concentrations were determined by the bicinchoninic acid method as described above. For CYP3A4 wild-type, C98A, C98S, C98F and C98W, the total protein concentrations were 56.5, 90.4, 66.4, 67.4 and 109.1 μM, respectively. Cytochrome P450 content was 49.5, 88.6, 57.8, 60.5, and 82.9 μM, respectively. SDS-polyacrylamide gel eletrophoresis showed the presence of approximately 10% and 25% impurities in the C98F and C98W mutants (supporting information).
In Vitro MDZ Metabolism
Previously we have identified cysteine 98 as one of the modification sites in the labeled CYP3A4 WT protein [21]. Both a loss of CO binding capacity and a dramatic decrease in catalytic activity were observed in the labeled CYP3A4, which suggested that large aromatic substitutions on residue 98 might trigger a critical conformational change and subsequent loss of enzyme activity [21,22]. To examine the mutational effects on enzyme catalytic activity, we investigated 1′-OH MDZ and 4-OH MDZ formation from MDZ, a common probe for CYP3A4 activity. As shown in Figure 4, the rates of MDZ hydroxylation by the C98A and C98S mutants were not significantly different, whereas rates of hydroxylation by the C98W mutant were dramatically lower compared to the CYP3A4 WT enzyme. Kinetic analysis revealed a marked 16-fold decrease in catalytic efficiency (Vmax/Km) for 1′-OH MDZ formation by the C98W mutant, and a 7.5-fold decrease in catalytic efficiency (Vmax/Km) for 4-OH MDZ formation compared to the WT enzyme (summarized in Table 1). Kinetic parameters determined from the fit of the experimental data to the Michaelis-Menten equation showed two very distinct Km values (4.73 μM and 53.3 μM for 1′-OH and 4-OH MDZ, respectively) for the WT enzyme, as reported by others [34,35]. Approximately a 7-fold decrease was observed in the formation rate of 1′-OH MDZ by the C98W mutant, with a 2-fold increase in the Km. Approximately a 10-fold decrease was observed in the formation rate of 4-OH MDZ by this same mutant with a slight decrease in the Km for this reaction. Therefore, the ratio of catalytic efficiency (Vmax/Km) for the formation of 1′-OH MDZ vs. 4-OH MDZ decreased by a factor of approximately two when comparing the C98W mutant enzyme to the WT enzyme, primarily as a result of an increased Km for 1′-hydroxylation in the mutant form. A moderate decrease of rates of hydroxylation was also observed for the C98F mutant compared to the WT enzyme (Figure 4, Table 1).
Figure 4.

Midazolam (A) 1′- and (B) 4-hydroxylation kinetics by CYP3A4. ●, ▲, ∎, ▼ and ◆ represent wild-type, C98A, C98S, C98F and C98W, respectively. All lines through experimental data points are the fit to the Michaelis-Menten equation.
Table 1.
P450 interaction and metabolic kinetics of the production of midazolam metabolites by CYP3A4 and its mutants.
| Protein | P450 bindinga |
1′-OH |
4-OH |
Ratio | |||||
|---|---|---|---|---|---|---|---|---|---|
| Type | Ks (μM) | Km (μM) | Vmax (nmol/min/nmol) | Vmax/Km (min-1 μM-1) | Km (μM) | Vmax (nmol/min/nmol) | Vmax/Km (min-1 μM-1) | Vmax/Km(1′-OH)/Vmax/Km(4-OH) | |
| WT | I | 5.0±0.7 | 4.73±0.48 | 8.36±0.21 | 1.77±0.18 | 53.34±5.75 | 7.73±0.40 | 0.145±0.017 | 12.2±1.9 |
| C98A | I | 14.4±0.5 | 3.15±0.39 | 8.94±0.24 | 2.84±0.36 | 50.77±4.90 | 9.97±0.45 | 0.196±0.021 | 14.5±2.4 |
| C98S | I | 10.1±0.7 | 4.99±0.52 | 9.19±0.24 | 1.84±0.20 | 52.27±4.45 | 9.30±0.38 | 0.178±0.017 | 10.4±1.5 |
| C98F | I | 9.9±0.3 | 12.72±0.79 | 6.79±0.13 | 0.53±0.03 | 41.18±4.56 | 2.25±0.11 | 0.055±0.007 | 9.8±1.3 |
| C98W | I | 6.1±0.5 | 10.34±5.92 | 1.13±0.19 | 0.11±0.07 | 40.12±4.11 | 0.76±0.03 | 0.019±0.002 | 5.8±3.5 |
Binding constants were determined by the nonlinear plotting of change in absorbance versus [MDZ], which shows the fit to the equation ΔA = ΔAmaxS/(Ks + S), where Ks is the binding constant, and ΔA and ΔAmax are the change in absorbance at a particular substrate concentration (S) and at a saturating substrate concentration, respectively.
In contrast, the C98A and C98S mutants displayed similar kinetics for both 1′- and 4-OH MDZ formation compared to the WT enzyme (Figure 4, Table 1). Only a slight increase of rates of both 1′- and 4-hydroxylation were observed for the C98A and C98S mutants. Overall the kinetic data provide evidence that cysteine 98 in the B-C loop region of CYP3A4 is important for enzyme activity, and that large aromatic substitutions at this position result in a decrease in enzyme catalytic activity.
Spectral Binding Studies
Binding constants (Ks) and maximal absorbance change (ΔAmax) due to MDZ binding were determined using a nonlinear fitting algorithm (KaleidaGraph 3.6, Synergy Software, Inc., Reading, PA) on the average absorbance differences between 390 and 420 nm at different concentrations of midazolam (Figure 5 and Table 1). For the WT enzyme, the Ks for MDZ was found to be very close to the Km value for 1′-OH MDZ formation, but not for 4-OH MDZ formation (Table 1). This supports the previous hypothesis that the binding of MDZ in the orientation for 1′-hydroxylation is responsible for the spectral changes observed upon enzyme-substrate complex formation [34]. The slight increases in the MDZ binding constants (Ks) in C98A, C98S and C98F mutants suggested negligible perturbation of the heme environment. Similar binding constants were observed between the WT and C98W mutant, which suggests that the mutation of C98W did not markedly perturb the active site of the enzyme around the heme.
Figure 5.

Spectral titration of CYP3A4 with increasing concentrations of MDZ. A, B, C, D and E represent wild-type, C98A, C98S, C98F and C98W, respectively; F. The solid lines through the experimental data points show the fit to the equation ΔA = ΔAmaxS/(Ks + S), where Ks is the binding constant, and ΔA and ΔAmax are the change in absorbance at a particular substrate concentration (S) and at a saturating substrate concentration, respectively. ●, ▲, ∎, ▼ and ◆ represent wild-type, C98A, C98S, C98F and C98W, respectively. The MDZ concentration as well as absorbance change have been adjusted for the dilution caused by the addition of MDZ. The protein concentration was 1 μM.
APAP Oxidation Assay
To determine if catalytic activity was similarly affected for substrates other than MDZ, the CYP3A4 oxidation of APAP was assessed in all five enzymes. As shown in Figure 6, at a substrate concentration of 1 mM, the rates of APAP oxidation by C98F and C98W mutant were decreased to approximately 67% and 28% that of the WT enzyme respectively, whereas the oxidation rates were essentially unchanged in both the C98A and C98S mutants compared to the WT enzyme.
Figure 6.
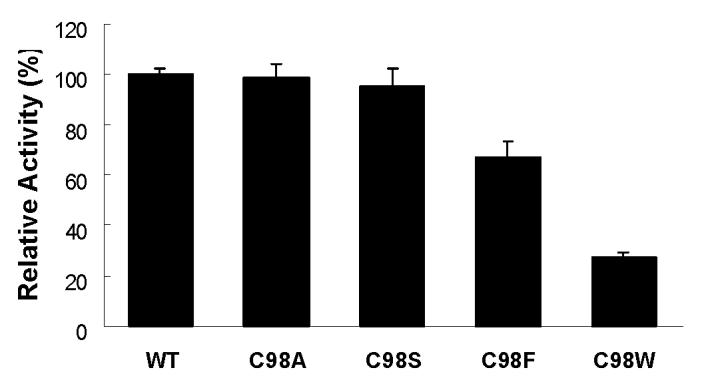
APAP oxidation activity by CYP3A4 wild-type, and its C98A, C98S, C98F and C98W mutants. 1 mM final concentration of APAP was used for the assay.
Reductase Kinetic Studies
A previous molecular modeling study has suggested that the flexible B-C helix loop region of CYP3A4 might serve as an interface with its redox partner, cytochrome P450 reductase [18]. To investigate the mutational effects on the interactions between CYP3A4 and the reductase, MDZ 1′-hydroxylation activity was determined using various ratios of P450:reductase as described in the Experimental Procedures. As depicted in Figure 7, the apparent equilibrium dissociation constants (Kd) between P450 and reductase were determined to be 0.16 ± 0.04, 0.17 ± 0.03, 0.19 ± 0.02, 0.42 ± 0.02 and 1.02 ± 0.04 μM for WT, and its C98A, C98S, C98F and C98W mutants, respectively. A significant decrease (approximately 6.4-fold) of the apparent binding affinity between the C98W mutant and the reductase was observed, while no apparent change was seen in the C98A and C98S mutants compared to the WT enzyme. An approximate 2.6-fold decrease of the P450 binding affinity to reductase was also observed in the C98F mutant. These data suggest that the larger Trp- and Phesubstitutions on position 98 affected P450 interactions with the reductase by decreasing the apparent P450 binding affinity to the coenzyme, while the smaller Ala- and conservative Sersub- stitutions have no significant effects on the interactions.
Figure 7.

Determination of the apparent equilibrium dissociation constant of CYP3A4 to the reductase. ●, ▲, ∎, ▼ and ◆ represent wild-type, C98A, C98S, C98F and C98W, respectively. All lines through experimental data points are the fit to the Michaelis-Menten equation.
NADPH Oxidation and Coupling in CYP3A4 Reactions
Results of mutational effects on NADPH oxidation rates are summarized in Table 2. In the presence of substrate, the rate of NADPH oxidation was significantly decreased in the C98W mutant (41.5%), while increased rates of NADPH oxidation were observed in both the C98A (19.6%) and C98S (7.0%) mutants compared to the WT enzyme.
Table 2.
NADPH oxidation and H2O2 product formation by CYP3A4 and its mutants.
| nmol/min/nmol of P450 |
||||||
|---|---|---|---|---|---|---|
| CYP3A4 | MDZ | NADPH oxidation | 1′-OH MDZ formation | 4-OH MDZ formation | H2O2 formation | H2O formation a |
| wild type | + | 51.6±2.8 | 7.7±0.6 | 3.8±0.4 | 8.6±0.4 | 15.8 |
| - | 49.7±2.0 | 8.0±0.1 | 20.9 | |||
| C98A | + | 61.7±4.8 | 8.2±0.6 | 4.9±0.3 | 9.7±0.7 | 19.5 |
| - | 50.6±3.4 | 8.4±0.5 | 21.1 | |||
| C98S | + | 55.2±3.1 | 8.3±0.4 | 4.5±0.2 | 8.9±0.7 | 16.8 |
| - | 51.6±2.7 | 7.9±0.2 | 21.9 | |||
| C98F | + | 43.6±3.2 | 5.5±0.4 | 1.3±0.1 | 10.6±0.9 | 13.1 |
| - | 37.7±3.0 | 7.8±0.6 | 15.0 | |||
| C98W | + | 30.2±1.1 | 1.1±0.2 | 0.4±0.1 | 13.8±1.4 | 7.5 |
| - | 26.1±0.2 | 5.8±0.5 | 10.2 | |||
H2O formation was determined by calculating the difference between total NADPH consumption and the sum of H2O2 and hydroxylated products formed. The result was divided by 2 because four electrons would be required to reduce O2 to H2O. No correction was made for O2-, which was not measured. Values represent the means ± SD of three determinations.
All of the CYP3A4 enzymes showed low coupling efficiencies when product formation and NADPH oxidation were compared (Table 2). The low activity C98W mutant showed a decrease in coupling efficiency of over 4-fold (5.0%) compared to the WT enzyme (22.3%) and its C98A (21.2%) and C98S (23.2%) mutants. These results were paralleled by increased rates of H2O2 production in the C98W mutant in the presence of the substrate MDZ as compared to the WT enzyme.
Modeling of the CYP3A4 C98W mutant
Based on the structure constructed from a recent CYP3A4 crystal structure (6, PDB code: 1W0E), the substituted Trp-98 is located within the B-B’ helix loop, a region of CYP3A4 which has previously been shown to have a profound effect on catalysis and substrate recognition [35]. Figure 8 shows that Trp-98 is positioned within approximately 4 Å of Trp-126 in the C-helix in this structure.
Figure 8.

A partial structure of the CYP3A4 C98W mutant. The Cys-98 of the X-ray crystal structure of CYP3A4 (PDB code: 1W0E) was substituted by a tryptophan residue using the mutate feature of DeepView 3.7 as described in Experimental Procedures. The substituted Trp-98 (green) is shown in a ball and stick model with respect to Trp-126 (purple), Cys-468 (green) and other residues (yellow) in the C-helix. The I-helix (cyan) is also shown with respect to a stick model of the heme (red).
Discussion
In the present study, we have shown that a Trp or Phe mutational substitution at position 98 markedly decreased CYP3A4 enzyme activity similar to what was observed previously by photolabeling of this cysteine residue with the bulky aromatic group, lapachenole [21,22]. In contrast, a conservative Ser or Ala mutation at the same position had no significant effect on the enzyme activity and protected the protein from photoaffinity labeling by lapachenole at position 98.
Previous work [21,22] in our laboratory showed the loss of CO binding capacity and a dramatic decrease in enzyme activity upon CYP3A4 labeling with the PAL lapachenole. One of the modification sites was identified as cysteine 98 located in the B-C loop region. Since lapachenole contains a large aromatic structure (Scheme 1), the general hypothesis was that large aromatic substitutions on residue 98 might trigger an enzyme conformational change and subsequent loss of catalytic activity. In this study, tryptophan and phenylalanine were chosen as the substituting residues because of their large aromatic side chains. Multiple adductions occurred when both C98W and C98F mutants were photolabeled with lapachenole, in contrast to the specific monoadduction of WT, and its C98A and C98S mutants. Based on the reactivity of photolyzed lapachenole discussed in the Results, the additional modification sites in C98W and C98F mutants are likely the free cysteine residues in CYP3A4 except Cys-98, for example, the previously identified Cys-468 located on the C-terminus and Cys-239, a residue positioned in the flexible F-G helix loop region of the protein. Although the sites of lapachenole adduction in the C98W and C98F mutants were not identified, the data suggests that a Cys → Trp or Phe substitution, but not a Cys → Ala or Ser substitution, at position 98 caused a conformational change in the protein, resulting in the availability of more nucleophilic residues for photoaffinity labeling. These results also agree with our previous hypothesis that large aromatic substitutions on residue 98 induce a conformational change in CYP3A4.
Scheme 1.
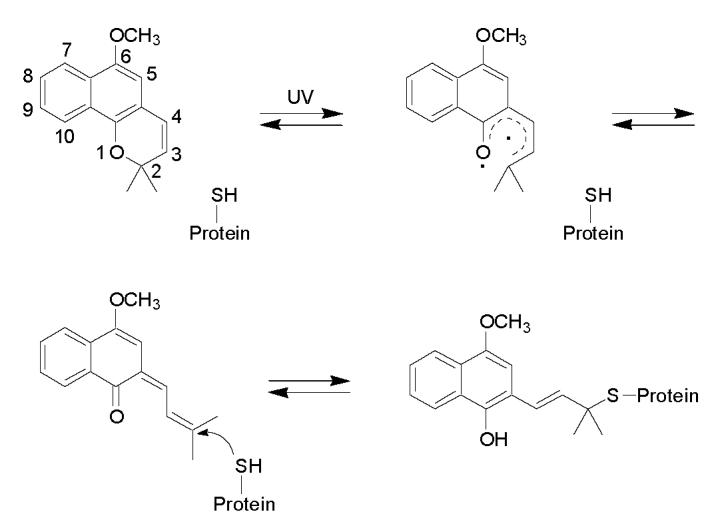
Proposed mechanism of formation of the lapachenole-CYP3A4 adducts. Lapachenole is photoactivated by UV irradiation treatment, and forms Michael addition-type adducts with cysteine residues on the CYP3A4-HT protein.
Along with the protein conformational change in CYP3A4 suggested by mass spectrometry, we investigated the mutational effect on midazolam hydroxylation activities by CYP3A4. Among the various substrates available, MDZ is considered one of the best in vitro and in vivo probes of CYP3A activity [36]. Our data showed two very distinct Km values for the two MDZ metabolites for all three tested enzymes as reported by others [34,37-38], suggesting the existence of two MDZ binding sites in the CYP3A4 protein. The rates of MDZ hydroxylation by the C98A and C98S mutants were not significantly different, whereas rates of hydroxylation by the C98W mutant were dramatically lower compared to the CYP3A4 WT enzyme. A similar decrease of enzyme activity was also observed in the C98W and C98F mutants, but not in the C98A and C98S mutants when APAP was used as the substrate. Because C98A displayed similar enzyme activities compared to the WT enzyme, the data suggests that hydrogen bonding through cysteine 98 in CYP3A4 makes no critical contribution to the structural maintenance and activity of this enzyme. While large aromatic substitutions at position 98 result in a decrease in enzyme catalytic activity, the metabolite ratio of 1′- and 4-OH MDZ formation is also decreased along with the increasing size of substitution. These results suggested that the conformational change induced by a large substitution on position 98 favored 4-OH MDZ formation, compared to 1′-OH MDZ formation.
Despite the decrease of catalytic activity, it is unlikely that residue 98 within the B-C loop region (encompassing SRS-1) would directly interfere with heme binding as suggested by the similar MDZ binding constants (Ks) between the WT enzyme and the mutants. This is consistent with the role of some other residues in the SRS-1 of CYP3A4. Roussel et al. [35] reported that the highly conserved residue S119 in the SRS-1 region is a key determinant of CYP3A4 specificity, and is likely involved in substrate binding and/or substrate recognition. The authors also concluded that although it is possible that substitutions at the residues within SRS-1 region could trigger a protein conformational change, it is very unlikely that these residues would be directly involved in heme binding [35]. This hypothesis is further supported by the recent CYP3A4 crystal structures [6,7], which suggest that small movements of the B-C loop could facilitate substrate entry into the active site and increase the active site volume, and residues localized in the SRS-1 region do not directly affect heme binding.
Large aromatic substitutions on position 98 decrease the apparent binding affinity of P450 to the reductase. Since the mono-oxygenation reaction catalyzed by P450s is dependent on the association of P450 with its redox partners, a significant decrease in the P450 binding affinity with reductase would be, at least partially, responsible for the decrease of enzyme catalytic activity observed in the C98W and C98F mutants. In concert with the decreased P450 binding affinity with reductase, a large aromatic substitution at position 98 in CYP3A4 interferes with electron transfer from the reductase to the P450 heme as suggested by NADPH oxidation rates between the WT enzyme and C98W mutant. This is further supported by the results from our molecular modeling study. In the constructed CYP3A4 C98W model, the substituted Trp-98 interacts sterically with the residues in the C-helix. The C-helix of CYP3A4 contains charged residues that are thought to be directly involved in reductase binding [18]. In addition, the substituted Trp-98 positioned within only 4 Å of Trp-126 in the C-helix, a residue that is highly conserved in all members of human CYP1, 3 and 4 families (http://drnelson.utmem.edu/humP450.aln.html). This residue is also conserved in 17 out of 20 members of human CYP2 family. Tryptophan may facilitate electron transfer because of its aromatic character which contains delocalized π electrons to act as donors and acceptors in the transfer [39]. Straub et al. [40,41] reported that the highly conserved residue Trp-120 in CYP2C2 is a potential mediator of electron transfer from the reductase. Interestingly, an alignment of the sequences of CYP3A4 and CYP2C2 shows that Trp-126 in CYP3A4 is equivalent to Trp-120 of CYP2C2 (data not shown). A potential role of Trp-126 in electron transfer in CYP3A4 remains to be elucidated at this point. Additional studies are underway using site-directed mutants to investigate these possible mechanisms.
In summary, we provide evidence that cysteine 98 in the B-C loop region is very important for maintenance of structure and enzyme activity of CYP3A4, and this residue and/or residues nearby are involved in interactions with P450 reductase. Our data support the hypothesis that aromatic substitution on residue 98 of CYP3A4 causes a protein conformational change and subsequent loss of enzyme catalytic activities.
Supplementary Material
Acknowledgment
We thank Drs. Rheem Totah and Nina Isoherranen (Department of Pharmaceutics, University of Washington) for their help in the midazolam hydroxylation assay.
Footnotes
This work was supported by NIH Grant GM32165 (to S.D.N.) and the UW NIEHS sponsored Center for Ecogenetics and Environmental Health: NIEHS P30ES07033.
- P450
- human liver microsomal cytochrome P450
- CYP3A4
- histidine-tagged cytochrome P450 3A4
- b5
- human cytochrome b5
- P450 reductase
- rat NADPH-cytochrome P450 reductase
- MDZ
- midazolam
- APAP
- acetaminophen
- PAL
- photoaffinity ligand
- DLPC
- L-α-dilaurylphosphatidylcholine
- DLPS
- L-α-dilaurylphosphatidylserine
- DOPC
- L-α-dioleoylphosphatidylcholine
- CHAPS
- 3-(3-cholamidopropyl) dimethylammonio-1-propanesulfonate
- GSH
- reduced glutathione
- HPLC/ESI MS
- high-performance liquid chromatography electrospray ionization mass spectrometry
- WT
- wild type
- TFA
- CF3CO2H
- NADPH
- reduced nicotinamide adenine dinucleotide phosphate
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Guengerich FP. In: Cytochrome P450: structure, mechanism, and biochemistry. Oritz de Montellano PR, editor. Plenum Press; New York: 1995. pp. 473–536. [Google Scholar]
- [2].Kronbach T, Mathys D, Umeno M, Gonzalez FJ, Meyer UA. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol. Pharmacol. 1989;36:89–96. [PubMed] [Google Scholar]
- [3].Waxman DJ, Lapenson DP, Aoyama T, Gelboin HV, Gonzales FJ, Korzekwa K. Steroid hormone hydroxylase specificities of eleven cDNA-expressed human cytochrome P450s. Arch. Biochem. Biophys. 1991;290:160–166. doi: 10.1016/0003-9861(91)90602-f. [DOI] [PubMed] [Google Scholar]
- [4].Aoyama T, Yamano S, Waxman DJ, Lapenson DP, Meyer UA, Fischer V, Tyndale R, Inaba T, Kalow W, Gelboin HV, Gonzalez FJ. Cytochrome P-450 hPCN3, a novel cytochrome P-450 IIIA gene product that is differentially expressed in adult human liver. cDNA and deduced amino acid sequence and distinct specificities of cDNA-expressed hPCN1 and hPCN3 for the metabolism of steroid hormones and cyclosporine. J. Biol. Chem. 1989;264:10388–10395. [PubMed] [Google Scholar]
- [5].Tang W, Stearns RA. Heterotropic cooperativity of cytochrome P450 3A4 and potential drug-drug interactions. Curr. Drug. Metab. 2001;2:185–198. doi: 10.2174/1389200013338658. [DOI] [PubMed] [Google Scholar]
- [6].Williams PA, Cosme J, Vinkovi[unk] DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal Structures of Human Cytochrome P450 3A4 Bound to Metyrapone and Progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- [7].Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05 Å resolution. J. Biol. Chem. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- [8].Lee DS, Yamada A, Sugimoto H, Matsunaga I, Ogura H, Ichihara K, Adachi S, Park SY, Shiro Y. Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis. Crystallographic, spectroscopic, and mutational studies. J. Biol. Chem. 2003;278:9761–9767. doi: 10.1074/jbc.M211575200. [DOI] [PubMed] [Google Scholar]
- [9].Davydov R, Kofman V, Fujii H, Yoshida T, Ikeda-Saito M, Hoffman BM. Catalytic mechanism of heme oxygenase through EPR and ENDOR of cryoreduced oxy-heme oxygenase and its Asp 140 mutants. J. Am. Chem. Soc. 2002;124:1798–1808. doi: 10.1021/ja0122391. [DOI] [PubMed] [Google Scholar]
- [10].He YA, He YQ, Szklarz GD, Halpert JR. Identification of three key residues in substrate recognition site 5 of human cytochrome P450 3A4 by cassette and site-directed mutagenesis. Biochemistry. 1997;36:8831–8839. doi: 10.1021/bi970182i. [DOI] [PubMed] [Google Scholar]
- [11].Harlow GR, Halpert JR. Analysis of human cytochrome P450 3A4 cooperativity: construction and characterization of a site-directed mutant that displays hyperbolic steroid hydroxylation kinetics. Proc. Natl. Acad. Sci. U. S. A. 1998;95:6636–6641. doi: 10.1073/pnas.95.12.6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Khan KK, He YQ, Domanski TL, Halpert JR. Midazolam oxidation by cytochrome P450 3A4 and active-site mutants: an evaluation of multiple binding sites and of the metabolic pathway that leads to enzyme inactivation. Mol. Pharmacol. 2002;61:495–506. doi: 10.1124/mol.61.3.495. [DOI] [PubMed] [Google Scholar]
- [13].Darbandi-Tonkabon R, Hastings WR, Zeng CM, Akk G, Manion BD, Bracamontes JR, Steinbach JH, Mennerick SJ, Covey DF, Evers AS. Photoaffinity labeling with a neuroactive steroid analogue. 6-azi-pregnanolone labels voltage-dependent anion channel-1 in rat brain. J. Biol. Chem. 2003;278:13196–13206. doi: 10.1074/jbc.M213168200. [DOI] [PubMed] [Google Scholar]
- [14].Kon N, Suhadolnik RJ. Identification of the ATP binding domain of recombinant human 40-kDa 2′,5′-oligoadenylate synthetase by photoaffinity labeling with 8-azido-[alpha-32P]ATP. J. Biol. Chem. 1996;271:19983–19990. doi: 10.1074/jbc.271.33.19983. [DOI] [PubMed] [Google Scholar]
- [15].Lightning LK, Jones JP, Friedberg T, Pritchard MP, Shou M, Rushmore TH, Trager WF. Mechanism-based inactivation of cytochrome P450 3A4 by L-754,394. Biochemistry. 2000;39:4276–4287. doi: 10.1021/bi992412u. [DOI] [PubMed] [Google Scholar]
- [16].Regal KA, Schrag ML, Kent UM, Wienkers LC, Hollenberg PF. Mechanism-based inactivation of cytochrome P450 2B1 by 7-ethynylcoumarin: verification of apo-P450 adduction by electrospray ion trap mass spectrometry. Chem. Res. Toxicol. 2000;13:262–270. doi: 10.1021/tx990195s. [DOI] [PubMed] [Google Scholar]
- [17].Hasemann CA, Kurumbail RG, Boddupalli SS, Peterson JA, Deisenhofer J. Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure. 1995;3:41–62. doi: 10.1016/s0969-2126(01)00134-4. [DOI] [PubMed] [Google Scholar]
- [18].Szklarz GD, Halpert JR. Molecular modeling of cytochrome P450 3A4. J. Computer-Aided Mol. Design. 1997;11:265–272. doi: 10.1023/a:1007956612081. [DOI] [PubMed] [Google Scholar]
- [19].Harlow GR, Halpert JR. Alanine-scanning mutagenesis of a putative substrate recognition site in human cytochrome P450 3A4. J. Biol. Chem. 1997;272:5396–5402. doi: 10.1074/jbc.272.9.5396. [DOI] [PubMed] [Google Scholar]
- [20].Gartner CA. Photoaffinity ligands in the study of cytochrome P450 active site structure. Curr. Med. Chem. 2003;10:671–689. doi: 10.2174/0929867033457890. [DOI] [PubMed] [Google Scholar]
- [21].Wen B, Doneanu CE, Gartner CA, Roberts AG, Atkins WM, Nelson SD. Fluorescent photoaffinity labeling of cytochrome P450 3A4 by lapachenole: identification of modification sites by mass spectrometry. Biochemistry. 2005;44:1833–1845. doi: 10.1021/bi048228c. [DOI] [PubMed] [Google Scholar]
- [22].Gartner CA, Wen B, Wan J, Becker RS, Jones G, II., Gygi SP, Nelson SD. Photochromic agents as tools for protein structure study: lapachenole is a photoaffinity ligand of cytochrome P450 3A4. Biochemistry. 2005;44:1846–1855. doi: 10.1021/bi049231z. [DOI] [PubMed] [Google Scholar]
- [23].Gillam EM, Baba T, Kim BR, Ohmori S, Guengerich FP. Expression of modified human cytochrome P450 3A4 in Escherichia coli and purification and reconstitution of the enzyme. Arch. Biochem. Biophys. 1993;305:123–131. doi: 10.1006/abbi.1993.1401. [DOI] [PubMed] [Google Scholar]
- [24].Smith PK, Krohn RI, Hermanson GT. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- [25].Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- [26].Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. J. Biol. Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- [27].Shaw PM, Hosea NA, Thompson DV, Lenius JM, Guengerich FP. Reconstitution premixes for assays using purified recombinant human cytochrome P450, NADPH-cytochrome P450 reductase, and cytochrome b5. Arch. Biochem. Biophys. 1997;348:107–115. doi: 10.1006/abbi.1997.0378. [DOI] [PubMed] [Google Scholar]
- [28].Isoherranen N, Kunze KL, Allen KE, Nelson WL, Thummel KE. Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab Dispos. 2004;32:1121–1131. doi: 10.1124/dmd.104.000315. [DOI] [PubMed] [Google Scholar]
- [29].Thummel KE, Lee CA, Kunze KL, Nelson SD, Slattery JT. Oxidation of acetaminophen to N-acetyl-p-aminobenzoquinone imine by human CYP3A4. Biochem. Pharmacol. 1993;45:1563–1569. doi: 10.1016/0006-2952(93)90295-8. [DOI] [PubMed] [Google Scholar]
- [30].Atkins WM, Sligar SG. Deuterium isotope effects in norcamphor metabolism by cytochrome P-450cam: kinetic evidence for the two-electron reduction of a high-valent iron-oxo intermediate. Biochemistry. 1988;27:1610–1616. doi: 10.1021/bi00405a033. [DOI] [PubMed] [Google Scholar]
- [31].Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- [32].Stewart JJ. Optimization of parameters for semiempirical methods IV: extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004;10:155–164. doi: 10.1007/s00894-004-0183-z. [DOI] [PubMed] [Google Scholar]
- [33].Pople JA, Segal GA. Approximate self-consistent molecular orbital theory. II. Calculations with complete neglect of differential overlap. J. Chem. Phys. 1965;43:S136–S149. [Google Scholar]
- [34].Gorski JC, Hall SD, Jones DR, VandenBranden M, Wrighton SA. Regioselective biotransformation of MDZ by members of the human cytochrome P450 3A (CYP3A) subfamily. Biochem. Pharmacol. 1994;47:1643–1653. doi: 10.1016/0006-2952(94)90543-6. [DOI] [PubMed] [Google Scholar]
- [35].Roussel F, Khan KK, Halpert JR. The importance of SRS-1 residues in catalytic specificity of human cytochrome P450 3A4. Arch. Biochem. Biophys. 2000;374:269–278. doi: 10.1006/abbi.1999.1599. [DOI] [PubMed] [Google Scholar]
- [36].Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A4. Annu. Rev. Pharmacol. Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- [37].Hosea NA, Miller GP, Guengerich FP. Elucidation of distinct ligand binding sites for cytochrome P450 3A4. Biochemistry. 2000;39:5929–5939. doi: 10.1021/bi992765t. [DOI] [PubMed] [Google Scholar]
- [38].Khan KK, He YQ, Domanski TL, Halpert JR. Midazolam oxidation by cytochrome P450 3A4 and active-site mutants: an evaluation of multiple binding sites and of the metabolic pathway that leads to enzyme inactivation. Mol. Pharmacol. 2002;61:495–506. doi: 10.1124/mol.61.3.495. [DOI] [PubMed] [Google Scholar]
- [39].Davies MD, Qin L, Beck JL, Suslick KS, Koga H, Horiuchi T, Sligar SG. Putidaredoxin reduction of cytochrome P-450cam: dependence of electron transfer on the identity of putidaredoxin′s C-terminal amino acid. J. Am. Chem. Soc. 1990;112:7396–7398. [Google Scholar]
- [40].Straub P, Ramarao MK, Kemper B. Preference for aromatic substitutions at tryptophan-120, which is highly conserved and a potential mediator of electron transfer in cytochrome P450 2C2. Biochem. Biophys. Res. Commun. 1993;197:433–439. doi: 10.1006/bbrc.1993.2498. [DOI] [PubMed] [Google Scholar]
- [41].Straub P, Lloyd M, Johnson EF, Kemper B. Differential effects of mutations in substrate recognition site 1 of cytochrome P450 2C2 on lauric acid and progesterone hydroxylation. Biochemistry. 1994;33:8029–8034. doi: 10.1021/bi00192a006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.