

Abstract
Centrosomal abnormalities are frequently observed in cancers and in cells with defective DNA repair. Here, we used light and electron microscopy to show that DNA damage induces centrosome amplification, not fragmentation, in human cells. Caffeine abrogated this amplification in both ATM (ataxia telangiectasia, mutated)- and ATR (ATM and Rad3-related)-defective cells, indicating a complementary role for these DNA-damage-responsive kinases in promoting centrosome amplification. Inhibition of checkpoint kinase 1 (Chk1) by RNA-mediated interference or drug treatment suppressed DNA-damage-induced centrosome amplification. Radiation-induced centrosome amplification was abrogated in Chk1−/− DT40 cells, but occurred at normal levels in Chk1−/− cells transgenically expressing Chk1. Expression of kinase-dead Chk1, or Chk1S345A, through which the phosphatidylinositol-3-kinase cannot signal, failed to restore centrosome amplification, showing that signalling to Chk1 and Chk1 catalytic activity are necessary to promote centrosome overduplication after DNA damage.
Keywords: centrosome, ATM, ATR, Chk1, checkpoint
Introduction
Recent findings that link aberrant centrosome number and chromosome instability (Lingle et al, 2002) have refocused attention on Boveri's idea that centrosomal defects contribute to tumorigenesis (Brinkley, 2001; Nigg, 2002). There are several models that explain centrosome amplification. Numerical centrosome aberrations can arise after failed cytokinesis (Meraldi et al, 2002). Extended G1–S arrest can decouple the centrosome and chromosome cycles in hamster (Balczon et al, 1995; Meraldi et al, 1999), human (Wong & Stearns, 2003) and chicken (Dodson et al, 2004) cells. From the frequent centrosome abnormalities seen in p53-deficient cells (Fukasawa et al, 1996), in cells with DNA repair deficiencies (Griffin et al, 2000; Dodson et al, 2004) and in cells with telomere defects (Guiducci et al, 2001), we have suggested that DNA damage signals have an impact on the centrosome during an extended G2–M arrest (Dodson et al, 2004).
The principal signalling molecules involved in the DNA damage response are the large serine–threonine kinases of the phosphatidylinositol-3-kinase (PI3K) family—ATM (ataxia telangiectasia, mutated) and ATR (ATM and Rad3-related) (Shiloh, 2003). ATM responds primarily to DNA double-strand breaks (DSBs), whereas ATR acts mainly in response to replication fork stalling, although ionizing radiation also activates ATR. Recent work has shown that ATR activity after DSB induction depends on ATM (Jazayeri et al, 2006). The checkpoint kinase 1 (Chk1) is activated by ATM and ATR and is necessary for cell-cycle checkpoints (Bartek & Lukas, 2003). Chk1 is essential in mammalian cells (Liu et al, 2000; Takai et al, 2000). Replication defects and spontaneous DSBs in Chk1-deficient human and chicken cells indicate that Chk1 might be important in the control of DNA replication (Syljuasen et al, 2005; Zachos et al, 2005). Loss of Chk1 function in mammalian and chicken cells also disrupts the G2–M checkpoint, giving rise to frequent aberrant mitoses after genotoxic stresses that might also affect cell viability (Takai et al, 2000; Zhao et al, 2002; Zachos et al, 2003).
Results And Discussion
Early work showed the amplification of microtubule-organizing centres (MTOCs) after irradiation of mouse cells. Electron microscopy showed that a high percentage of these MTOCs did not contain the paired centrioles and pericentrosomal material of the normal centrosome (Sato et al, 1983). Later work on irradiated human U2OS cells (an osteosarcoma cell line) demonstrated MTOC amplification after irradiation (Sato et al, 2000), although the structure of these MTOCs was not determined. We tested whether human lymphoblastoid cells undergo centrosome amplification after γ-irradiation. As shown in Fig 1A,B, we observed a dose-dependent increase in the number of γ-tubulin foci after ionizing radiation treatment. More than two γ-tubulin foci were observed in 12% of cells at 24 h after irradiation, compared with 37% at 48 h after 10 Gy treatment. High levels of cell death precluded analysis at 72 h after ionizing radiation. We observed similar amplification in U2OS cells (see later), Hct116 colon carcinoma cells and in Jurkat T-cell lymphoma cells (data not shown). These γ-tubulin foci also contained pericentrin (Fig 1C), indicating that they were indeed centrosomes.
Figure 1.
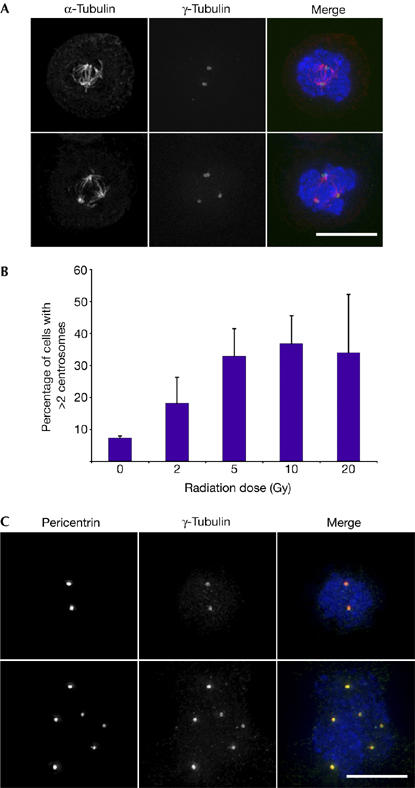
DNA-damage-induced centrosome amplification in human cells. Cells shown were fixed and stained 48 h after 10 Gy ionizing radiation treatment. DNA counterstains (DAPI) are shown in blue. Scale bars, 10 μm. (A) Upper series shows a cell with two centrosomes, and lower series a cell with multiple centrosomes. Cells were stained for γ-tubulin (green) and α-tubulin (red). (B) Dose dependence of centrosome amplification in human lymphoblastoid cells 48 h after treatment with the indicated dose of ionizing radiation. The histogram shows mean+s.d. of three separate experiments in which at least 500 cells were analysed. (C) The upper series shows a cell with two centrosomes, and the lower series a cell with multiple centrosomes. Cells were stained for pericentrin (red) and γ-tubulin (green). DAPI, 4,6-diamidino-2-phenylindole.
As non-centrosomal aggregation of γ-tubulin or centrosome fragmentation can occur after various treatments (Keryer et al, 1984; Hut et al, 2003), we carried out serial-section electron microscopy 48 h after 10 Gy irradiation of human lymphoblastoid cells. Cells on glass coverslips were irradiated, then fixed and stained for pericentrin. Cells with multiple pericentrin signals were photographed and, after embedding and relocation, serially sectioned for electron microscopy. We examined three irradiated control cells with multiple pericentrin spots and observed a total of 31 centrioles, of which 18 were clearly paired (Fig 2Ai,ii). In a similar experiment analysing two A-T cells, we observed 15 centrioles, of which 10 were clearly paired (Fig 2Aiii,iv). As a given section might not contain both centrioles of a centrosome, the number of paired centrioles we describe here is the minimum. These findings show that the additional MTOCs that we observed contained duplicated centrosomes rather than fragments. Next, we used immunofluorescence microscopy to verify the composition of the pericentrin/γ-tubulin structures. As shown in Fig 2B, we found that the centrosome components Aurora A, Cep170 (centrosomal protein 170), Nedd1 (neural precursor cell expressed, developmentally downregulated 1), ninein and PCM1 (pericentriolar material 1) showed typical centrosomal localizations (supplementary Fig S1 online; Dammermann & Merdes, 2002; Meraldi et al, 2002; Guarguaglini et al, 2005). We conclude that centrosome duplication, rather than centrosome splitting or fragmentation, occurs after irradiation.
Figure 2.
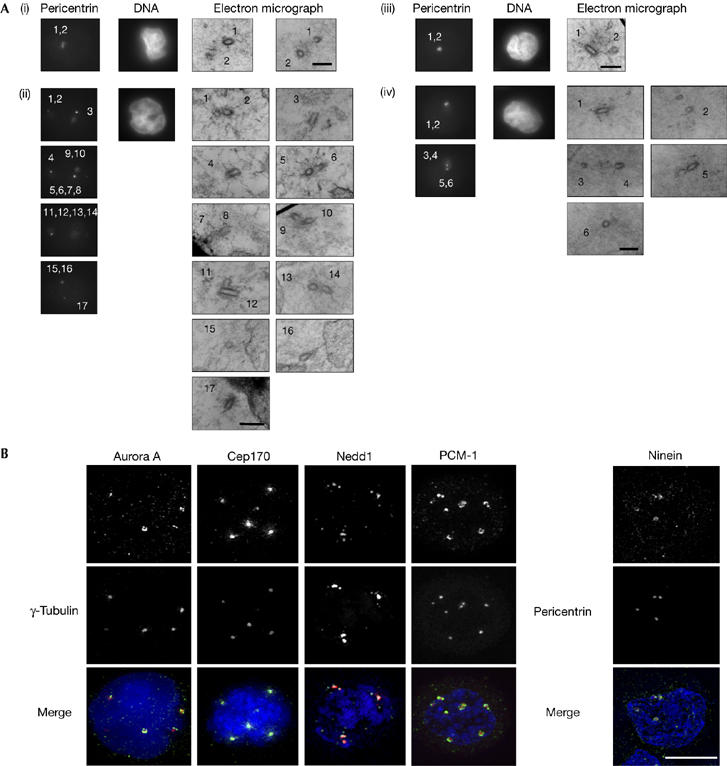
Microscopy analysis of amplified centrosomes in human cells. (A) Electron microscopy was carried out on human control (panels i,ii) or A-T (panels iii,iv) lymphoblastoid cells 48 h after 10 Gy irradiation. Cells were processed for immunofluorescence microscopy of pericentrin and staining of the DNA, as indicated. Cells were then flat-embedded and ultrathin serial sections were cut. Numbers on each pericentrin micrograph indicate the positions of the centrioles shown in the corresponding electron micrographs. Scale bars, 0.5 μm. (B) Localization of centrosomal proteins to amplified centrosomes in human lymphoblastoid cells. Cells were stained with antibodies to the indicated centrosomal or pericentrosomal proteins (green) and to γ-tubulin or pericentrin (red), then counterstained with DAPI (blue). Cells shown were fixed and stained 48 h after 10 Gy ionizing radiation treatment. Scale bar, 10 μm. DAPI, 4,6-diamidino-2-phenylindole.
To define what elements of the DNA damage response regulate centrosome amplification after irradiation of human cells, we examined lymphoblastoid cells with defects in ATM and ATR. The point mutation in ATR in Seckel syndrome cells causes a reduction in the protein level, but not its complete loss (O'Driscoll et al, 2003). Centrosome amplification after irradiation was observed in A-T cells by using electron microscopy (Fig 2Aiii,iv) and was quantitated by light microscopy with γ-tubulin as a marker (Fig 3B). DNA-damage-induced centrosome amplification was also seen in Seckel syndrome cells (Fig 3B). Normal localization of Aurora A, Cep170, Nedd1, ninein, PCM1 and pericentrin was observed in amplified centrosomes in A-T and Seckel syndrome cells (data not shown). This amplification was dose- and time-dependent in both lymphoblastoid cell lines, and similar actual numbers of centrosomes were seen in wild-type, A-T and Seckel syndrome cells (data not shown). These findings show that neither ATM nor ATR is essential for centrosome amplification.
Figure 3.
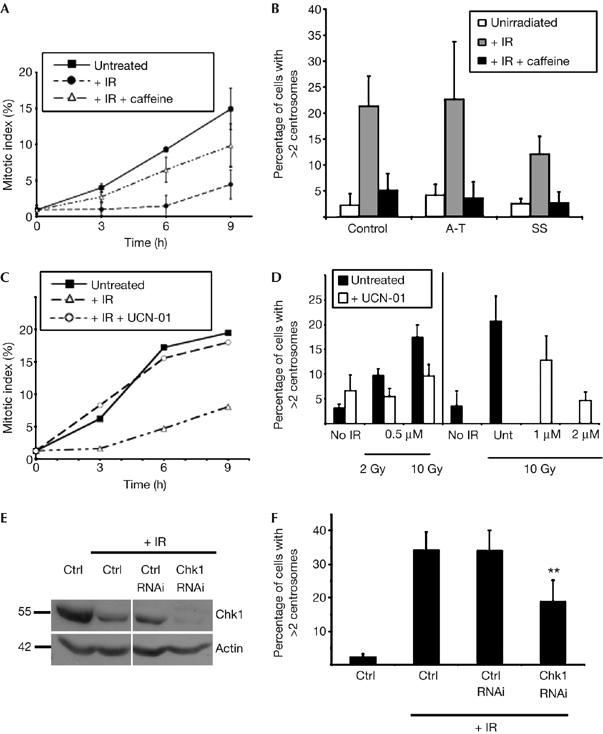
Dependence of DNA-damage-induced centrosome amplification in human cells on ATM/ATR and Chk1 kinase activity. (A) Repression of the G2–M checkpoint by caffeine treatment of human lymphoblastoid cells. Cumulative mitotic indices of human lymphoblastoid cells grown in the presence of colcemid for the time periods indicated after no treatment or after 10 Gy γ-irradiation (IR), with or without preincubation with 2 mM caffeine, as shown. A total of 200 cells were counted per time point in three separate experiments and data presented are the means±s.d. of these replicates. (B) Quantification of control, A-T and Seckel syndrome (SS) cells with multiple centrosomes before and 48 h after 10 Gy ionizing radiation in the presence or absence of caffeine. Centrosomes were counted by immunofluorescence microscopy of γ-tubulin. Data were obtained from at least 500 cells per experiment and histograms show the mean+s.d. of results from three separate blind experiments. (C) UCN-01-mediated abrogation of radiation-induced cell-cycle arrest. Cumulative mitotic indices of human lymphoblastoid cells grown in the presence of colcemid for the time points indicated after no treatment or after 2 Gy γ-irradiation, with or without preincubation with 0.5 μM UCN-01. A total of 200 cells were counted per time point. (D) UCN-01-mediated suppression of centrosome amplification 48 h after the indicated dose of γ-irradiation in the presence or absence of the indicated concentration of UCN-01. Centrosomes were counted by immunofluorescence microscopy of γ-tubulin. Data were obtained from at least 500 cells per experiment and histograms show the mean+s.d. of results from three separate blind experiments. (E) Immunoblot analysis of Chk1 repression in U2OS cells by RNAi at 72 h after transfection, the time at which centrosome counts were performed. Immunoblot for actin was used as a loading control. (F) Chk1 RNA interference-mediated suppression of centrosome amplification 48 h after 20 Gy irradiation of U2OS cells. Centrosomes were counted by immunofluorescence microscopy of γ-tubulin. Data were obtained from at least 500 cells per experiment. Asterisks indicate significant difference from irradiated controls (paired t-test, P<0.01). ATM, ataxia telangiectasia, mutated; ATR, ATM and Rad3-related; Chk1, checkpoint kinase 1; RNAi, RNA-mediated interference.
Next, we used pharmacological means to probe the pathways by which DNA damage causes centrosome amplification. Caffeine is an in vitro inhibitor of the ATM/ATR kinases (Sarkaria et al, 1999). As shown in Fig 3A, 2 mM caffeine treatment suppressed the G2-phase checkpoint normally observed in human lymphoblastoid cells after irradiation. Caffeine treatment resulted in near-normal numbers of centrosomes after irradiation of control, A-T and Seckel syndrome cells (Fig 3B). That a caffeine-sensitive activity allows centrosome amplification in both ATM- and ATR-deficient cells indicated that they might act in a complementary or redundant manner in driving centrosomal responses to DNA damage.
One response that is controlled by complementary activities of ATM and ATR is Chk1 activation. UCN-01 (7-hydroxystaurosporine) is a Chk1 inhibitor (Sarkaria et al, 1999; Graves et al, 2000). We tested whether disruption of Chk1 function by UCN-01 treatment affected centrosome amplification after ionizing radiation in human lymphoblastoid cells. At the minimum levels of UCN-01 required to inhibit the G2–M checkpoint (Fig 3C), we saw a reduction in the centrosome amplification caused by DNA damage (Fig 3D). At higher UCN-01 concentrations, we noted a complete repression of centrosome amplification (Fig 3D, right panel). Next, we used RNA-mediated interference to knock down the expression of Chk1. Owing to the difficulties we experienced in transfecting the lymphoblastoid cells, we carried out these experiments in U2OS cells. Cells were transfected with inhibitory RNA duplexes and irradiated 24 h after transfection and then analysed 72 h after transfection. As shown in Fig 3E,F, reduction of Chk1 caused a significant repression of centrosome amplification after DNA damage.
To confirm the role of Chk1, we turned to the Chk1−/− DT40 line (Zachos et al, 2003). Chk1-deficient cells showed only background levels of centrosome amplification after irradiation (Fig 4A). When Chk1 was constitutively expressed in these cells, DNA damage again induced centrosome abnormalities (Fig 4A,B). Notably, the expression of a kinase-dead Chk1 mutant (Asp130Ala) did not restore DNA-damage-induced centrosome amplification (Fig 4A) and also expression of a Chk1 mutant in which serine 345, a PI3K kinase target site, is replaced by alanine (Ser345Ala). Fig 4B confirms the expression of the Chk1 transgenes. The Asp130Ala mutant consistently shows an altered electrophoretic mobility, but repeated sequencing of the expression construct has confirmed that the Asp130Ala alteration is the only mutation. The G2–M checkpoint, which is abrogated by Chk1 deficiency (Zachos et al, 2003), was restored by expression of wild-type but not by kinase-dead or Ser345Ala Chk1 (Fig 4C,D).
Figure 4.
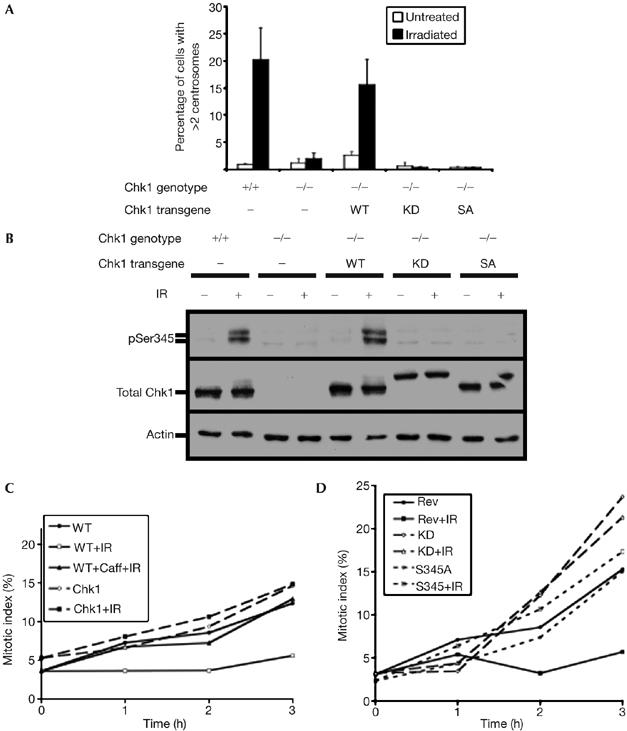
Genetic analysis of the requirement of Chk1 activity for DNA-damage-induced centrosome amplification. (A) Centrosome amplification 12 h after 10 Gy irradiation in Chk1−/− DT40 cells and in Chk1−/− cells expressing wild-type (WT), kinase-dead (KD) or Ser345Ala (SA) mutant Chk1 transgenes. Centrosomes were counted by immunofluorescence microscopy of γ-tubulin spots. Data were obtained from at least 500 cells per experiment and histograms show the mean+s.d. of results from three separate blind experiments. (B) Immunoblot analysis of Chk1 levels in cells used for the experiments shown in (A). Antibodies recognizing Chk1 protein, Chk1 pSer345 and actin were hybridized to total cell extract from cells of the indicated genotype before or 2 h after 10 Gy γ-irradiation. (C,D) Cumulative mitotic indices over time of colcemid-blocked WT, Chk1−/−Chk1+ (Rev), Chk1−/−Chk1D130A+ (KD) or Chk1−/−Chk1S345A+ (SA) cells after no treatment or after 2 Gy γ-irradiation (IR), with or without preincubation with 2 mM caffeine (Caff). A total of 200 cells were counted per time point. Chk1, checkpoint kinase 1.
To test our model of G2-phase centrosome amplification after DNA damage, we carried out fluorescence-activated cell sorting analysis of human and chicken cells after ionizing radiation. We saw no evidence of mitotic failure (polyploid cells) as an alternative explanation for centrosome amplification (Fig 5A). As we also saw Chk1-dependent centrosome amplification occurring after DNA topoisomerase II inhibition (data not shown); we conclude that Chk1-controlled centrosome amplification is a general response to DNA damage in tumour cells and we present the current models (shown in Fig 5B) for how multiple centrosomes might arise. It remains to be defined whether the principal role of Chk1 is the imposition of a cell-cycle delay or whether Chk1 signals directly to the centrosome duplication machinery. Candidate pathways include Cdc25 regulation of Cdk2. The centrosomal localization of Chk1 (Kramer et al, 2004) also indicates a potential role at the centrosome.
Figure 5.
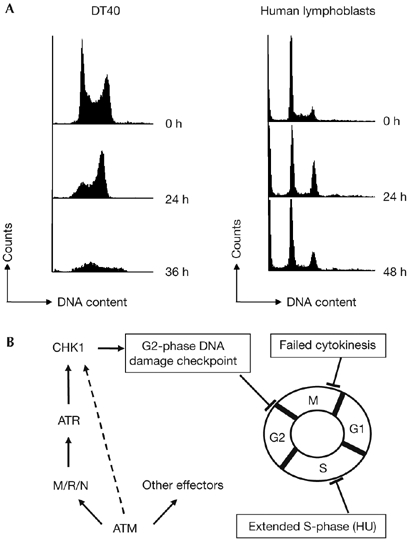
Cell-cycle analysis after irradiation. (A) FACS plots of DT40 and lymphoblast cells at the indicated time points after 10 Gy irradiation. (B) Current models for centrosome amplification incorporating the current data on pathways controlling DNA-damage-induced centrosome amplification. FACS, fluorescence-activated cell sorting.
Methods
Cloning. Site-directed mutagenesis using a Quikchange kit (Stratagene, La Jolla, CA, USA) was used to replace the sequence encoding Ser 345 or Asp 130 with the sequence for alanine residues in pcDNA3.1zeo-Chk1 (encoding chicken Chk1; Zachos et al, 2003) to generate pcDNA3.1zeo-Chk1S345A or pcDNA3.1zeo-Chk1KD, respectively. Constructs were verified by DNA sequencing.
Cell culture and analysis. DT40 cell culture was as described by Dodson et al (2004). Chk1−/− DT40 cells were transfected with linearized Chk1 expression constructs and overexpressing clones selected as described by Zachos et al (2003). Human lymphoblastoid cells GM07521 (apparently normal), GM01525 (A-T) and GM18367 (Seckel syndrome) were obtained from Coriell Cell Repositories and U2OS cells were from the ATCC (Middlesex, UK). UCN-01 was provided by the Division of Cancer Treatment and Diagnosis, National Cancer Institute (Bethesda, MD, USA), and was dissolved in dimethyl sulphoxide at 1 mg/ml. Ionizing radiation experiments were carried out using a 137Cs source at 23.5 Gy/min (Mainance Engineering, Hampshire, UK). Microscopy analysis of mitotic indices and flow cytometry were carried out as described by Dodson et al (2004).
RNA-mediated interference. An siGENOME™SMART pool of RNA duplexes inhibitory to Chk1 and an siCONTROL non-targeting short interfering RNA (siRNA) pool (Dharmacon, Lafayette, CO, USA) were transfected into U2OS cells by using lipofectamine (Invitrogen, Carlsbad, CA, USA). A 300 pmol portion of siRNA was complexed with 12 μl lipofectamine in serum-free medium and added to cells in a 6 ml final volume.
Microscopy. Monoclonal antibodies 3G11 (Dammermann & Merdes, 2002) recognizing pericentrin and 35C1 (Abcam, Cambridge, UK) against Aurora A were used at dilutions of 1:250 and 1:1,000, respectively. Polyclonal antibodies to Cep170 were used at 1:1,000 after methanol fixation (Guarguaglini et al, 2005) and antisera to Nedd1, ninein and PCM1 were used as described by Dammermann & Merdes (2002) and Haren et al (2006). Immunofluorescence microscopy of pericentrin before electron microscopy and of phospho-H2AX was carried out after fixation with 4% paraformaldehyde and permeabilization with 0.15% Triton X-100, both in cytoskeleton buffer (137 mM NaCl, 5 mM KCl, 1.1 mM Na2HPO4, 0.4 mM KH2PO4, 2 mM MgCl2, 2 mM EGTA, 5 mM PIPES, 5.5 mM glucose). Further preparation and electron microscopy were carried out as described by Dodson et al (2004). Otherwise, immunofluorescence microscopy of pericentrin was carried out as described by Dodson et al (2004). Antibodies, cell fixation and staining and light microscopy using an Olympus BX51 microscope were as described previously (Dodson et al, 2004).
Immunoblot analysis. Cell extracts were analysed by western blotting as described previously (Zachos et al, 2003). We used monoclonal antibodies G4 against Chk1 (Santa Cruz, Santa Cruz, CA, USA) and AC-40 against actin (Sigma-Aldrich, Dublin, Ireland) and polyclonal antibodies against Chk1 phosphorylated at Ser 345 (New England Biolabs, Ipswich, MA, USA) or, for analysis of the RNAi experiment, polyclonal anti-actin (A2066) and monoclonal anti-Chk1 DCS310 (both Sigma).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary Fig S1
Acknowledgments
We thank G. Guarguaglini for antibodies. This research was partly supported by a Marie Curie European Reintegration Grant in the 6th European Community Research and Technological Development Framework Programme. E.B. is the recipient of a Health Research Board Postdoctoral Fellowship. Work in C.G.M.'s lab is supported by an Investigator award from Science Foundation Ireland.
References
- Balczon R, Bao L, Zimmer WE, Brown K, Zinkowski RP, Brinkley BR (1995) Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol 130: 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Brinkley BR (2001) Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol 11: 18–21 [DOI] [PubMed] [Google Scholar]
- Dammermann A, Merdes A (2002) Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J Cell Biol 59: 255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson H, Bourke E, Jeffers LJ, Vagnarelli P, Sonoda E, Takeda S, Earnshaw WC, Merdes A, Morrison C (2004) Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J 23: 3864–3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF (1996) Abnormal centrosome amplification in the absence of p53. Science 271: 1744–1747 [DOI] [PubMed] [Google Scholar]
- Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O'Connor PM, Piwnica-Worms H (2000) The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem 275: 5600–5605 [DOI] [PubMed] [Google Scholar]
- Griffin CS, Simpson PJ, Wilson CR, Thacker J (2000) Mammalian recombination-repair genes XRCC2 and XRCC3 promote correct chromosome segregation. Nat Cell Biol 2: 757–761 [DOI] [PubMed] [Google Scholar]
- Guarguaglini G, Duncan PI, Stierhof YD, Holmstrom T, Duensing S, Nigg EA (2005) The forkhead-associated domain protein Cep170 interacts with Polo-like kinase 1 and serves as a marker for mature centrioles. Mol Biol Cell 16: 1095–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiducci C, Cerone MA, Bacchetti S (2001) Expression of mutant telomerase in immortal telomerase-negative human cells results in cell cycle deregulation, nuclear and chromosomal abnormalities and rapid loss of viability. Oncogene 20: 714–725 [DOI] [PubMed] [Google Scholar]
- Haren L, Remy MH, Bazin I, Callebaut I, Wright M, Merdes A (2006) NEDD1-dependent recruitment of the γ-tubulin ring complex to the centrosome is necessary for centriole duplication and spindle assembly. J Cell Biol 172: 505–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hut HM, Lemstra W, Blaauw EH, van Cappellen GW, Kampinga HH, Sibon OC (2003) Centrosomes split in the presence of impaired DNA integrity during mitosis. Mol Biol Cell 14: 1993–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP (2006) ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 8: 37–45 [DOI] [PubMed] [Google Scholar]
- Keryer G, Ris H, Borisy GG (1984) Centriole distribution during tripolar mitosis in Chinese hamster ovary cells. J Cell Biol 98: 2222–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg EA, Bartek J, Lukas J (2004) Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat Cell Biol 6: 884–891 [DOI] [PubMed] [Google Scholar]
- Lingle WL, Barrett SL, Negron VC, D'Assoro AB, Boeneman K, Liu W, Whitehead CM, Reynolds C, Salisbury JL (2002) Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci USA 99: 1978–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q et al. (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA (1999) Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol 1: 88–93 [DOI] [PubMed] [Google Scholar]
- Meraldi P, Honda R, Nigg EA (2002) Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J 21: 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA (2002) Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer 2: 815–825 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT (1999) Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res 59: 4375–4382 [PubMed] [Google Scholar]
- Sato C, Kuriyama R, Nishizawa K (1983) Microtubule-organizing centers abnormal in number, structure, and nucleating activity in X-irradiated mammalian cells. J Cell Biol 96: 776–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Mizumoto K, Nakamura M, Tanaka M (2000) Radiation-induced centrosome overduplication and multiple mitotic spindles in human tumor cells. Exp Cell Res 255: 321–326 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 25: 3553–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M (2000) Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev 14: 1439–1447 [PMC free article] [PubMed] [Google Scholar]
- Wong C, Stearns T (2003) Centrosome number is controlled by a centrosome-intrinsic block to reduplication. Nat Cell Biol 5: 539–544 [DOI] [PubMed] [Google Scholar]
- Zachos G, Rainey MD, Gillespie DA (2003) Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J 22: 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos G, Rainey MD, Gillespie DA (2005) Chk1-dependent S–M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol Cell Biol 25: 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Watkins JL, Piwnica-Worms H (2002) Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA 99: 14795–14800 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Fig S1