

Abstract
Integration of IS1301 into an AT-rich inverted repeat located upstream of the ltx operon was previously shown to confer a hyperleukotoxic phenotype in Actinobacillus actinomycetemcomitans IS1 (T. He, T. Nishihara, D. R. Demuth, and I. Ishikawa, J. Periodontol. 70:1261-1268, 1999), but the mechanism leading to increased leukotoxin production was not determined. We show that an IS1 ltx promoter::lacZ reporter construct expresses 12-fold higher levels of β-galactosidase activity than a reporter containing the ltx promoter from A. actinomycetemcomitans 652, suggesting that IS1301 increases transcription of the ltx operon. Examination of the IS1301 sequence identified a potential outwardly directed promoter. However, site-specific mutagenesis of the −35 element of the putative promoter had no effect on the transcriptional activity of the IS1 reporter construct. Furthermore, reverse transcriptase PCR and real-time PCR experiments did not detect a transcript that was initiated within IS1301. These results suggest that increased expression of leukotoxin in strain IS1 does not arise from an outwardly directed IS1301 promoter. To determine how IS1301 alters transcriptional regulation of the ltx operon, cis-acting sequences that regulate leukotoxin expression were identified. The AT-rich sequence that resides downstream from the site of IS1301 insertion was shown to function as a positive cis-acting regulator of leukotoxin expression. This sequence resembles an UP element in its location, AT-rich content, and activity and is homologous to the consensus UP element sequence. In addition, a negative cis-acting sequence was identified upstream from the site of IS1301 insertion, and deletion of this region increased promoter activity by fourfold. Mobility shift experiments showed that this region bound to a protein(s) in extracts from A. actinomycetemcomitans 652. The specific sequences required for this interaction were localized to a 26-nucleotide segment of the ltx promoter that resides 17 bp upstream from the site of IS1301 insertion. Together, these results suggest that positive and negative cis-acting sequences regulate leukotoxin expression and that IS1301 may increase transcription of the ltx operon in A. actinomycetemcomitans IS1 by displacing a negative cis-acting regulator approximately 900 bp upstream from the basal elements of the ltx promoter.
Actinobacillus actinomycetemcomitans is a capnophilic gram-negative bacterium that is associated with various forms of early-onset periodontal diseases (48, 55, 57), endocarditis (2), and subcutaneous abscesses (38). Although the mechanisms by which A. actinomycetemcomitans causes disease are not well understood, the organism expresses a wide variety of potential virulence-associated factors that may contribute to pathogenesis. For example, the breakdown of the extracellular matrix and the induction of bone resorption may be facilitated by expression of collagenase (41), lipopolysaccharide(23, 32), and GroEL-like proteins (8). A. actinomycetemcomitans expresses various adherence factors (36), including fimbriae (18, 21, 42), and invades human epithelial cells (35, 50). The organism also produces several toxins which target various components of the immune system and may play a role in modulating the host response. An immunosuppressive protein which induces G2 arrest of human lymphocytes is encoded by the cdtB gene of a cytolethal distending toxin (cdt) operon (45, 46). A. actinomycetemcomitans also expresses a leukotoxin which is a member of the repeats in toxin (RTX) family of bacterial cytolysins (53) and kills cells of the lymphocytic and monomyelocytic lineages (22, 31, 33). Leukotoxin interacts with lymphocyte function-associated antigen 1 (32) and causes the rapid development of cation-selective membrane pores (20). Korostoff et al. have also shown that leukotoxin perturbs mitochondrial function, leading to apoptosis of HL-60 cells exposed to sublytic doses of toxin (26, 27).
The leukotoxin is expressed from an operon consisting of at least four genes, designated ltxC, ltxA, ltxB, and ltxD, of which the ltxA gene encodes the structural toxin (28, 30). The three remaining genes are required for the activation and secretion of the leukotoxin. The ltxC gene is analogous to hlyC of the Escherichia coli hemolysin operon, which is involved in the acylation of the hemolysin protein (19). Although it has not yet been demonstrated in A. actinomycetemcomitans, it is likely that ltxC carries out a similar function. The ltxB and ltxD genes function in the signal peptide-independent secretion of the leukotoxin (29) and are homologous to hylBD of the E. coli hemolysin operon. In addition, an open reading frame (orfA) that encodes a putative polypeptide of unknown function exists upstream from ltxCABD and is cotranscribed with the ltx operon. Leukotoxin expression varies greatly among A. actinomycetemcomitans strains, but most strains express relatively low levels of toxin. However, several hyperleukotoxic strains have been identified (4, 15, 25) and recent studies suggest that these organisms are associated with human periodontal diseases (5, 10, 12, 14). Hyperexpression of leukotoxin in A. actinomycetemcomitans correlates with the presence of specific genetic rearrangements, both insertions and deletions, in the region immediately upstream of the ltx operon (4, 15). For example, in A. actinomycetemcomitans JP2, a deletion of 530 bp encompassing parts of orfA and the orfA-ltxC intergenic region results in a 10-fold increase in toxin expression (4). He et al. also showed that integration of IS1301 upstream from orfA in A. actinomycetemcomitans IS1 increases toxin expression by eightfold (15). A spontaneous revertant of IS1 which lost IS1301 exhibited significantly reduced leukotoxic activity (15).
The mechanisms by which genetic rearrangements in the ltx operon influence toxin expression are not understood, in part because relatively little is known about the regulatory mechanisms that govern leukotoxin expression. In this report, we show that the acquisition of IS1301 results in increased transcription of the ltx operon but that IS1301 does not possess an outwardly directed promoter that transcribes ltx sequences. In the ltx promoter, we identified a positive cis-acting sequence resembling an UP element downstream from the site of IS1301 insertion and a negative cis-acting regulator that resides upstream from IS1301. Our results suggest that IS1301 may disrupt normal regulation of the ltx operon and increase leukotoxin expression by displacing a negative cis-acting sequence 900 bp upstream from the basal ltx promoter elements.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
A. actinomycetemcomitans 652 is a minimally leukotoxic strain and was previously described (4). A. actinomycetemcomitans IS1 is a hyperleukotoxic strain that contains an insertion sequence related to IS1301 of Neisseria meningitidis integrated upstream from the ltx operon (15). All A. actinomycetemcomitans strains were grown aerobically at 37°C in brain heart infusion medium (Difco) supplemented with 0.5% yeast extract and 0.4 mg of NaHCO3 per liter. Where necessary for the selection of recombinant organisms, medium was supplemented with ampicillin (50 μg per ml) and/or kanamycin (25 μg per ml). Electroporation of A. actinomycetemcomitans with the shuttle vector pYGK (3) was carried out as described by Sreenivasan et al. (51). E. coli strains were cultured in Luria-Bertani (LB) broth supplemented where necessary with ampicillin (50 μg per ml) and/or kanamycin (25 μg per ml).
Construction of ltx promoter::lacZ reporter plasmids.
Reporter plasmids containing the glyA-ltxC intergenic region from A. actinomycetemcomitans strains 652 (652lacZ) and IS1 (ISlacZ) were constructed by amplifying the appropriate promoter fragment by PCR by using genomic DNA as a template. The promoter fragment from strain 652 was amplified by using primers 1 and 2 (Table 1); the promoter from strain IS1 was generated by using primers 3 and 2. Amplification reactions were carried out by using the following conditions: 95°C for 1 min, 55°C for 2 min, and 72°C for 3 min for 30 cycles. The PCR products were purified by using the QIA-quick PCR purification kit (Qiagen) according to the instructions from the manufacturer and cloned into pGEM-TEasy (Pharmacia). Plasmids were used to transform competent E. coli DH5α, and recombinants were selected on LB agar containing 50 μg of ampicillin per ml. Plasmid was isolated from recombinant organisms and confirmed by restriction digestion. Promoter fragments were subsequently released from pGEM-TEasy by digestion with KpnI and BamHI and ligated into pYGKlacZ that had been cleaved with the same enzymes. Plasmid pYGK is an E. coli-A. actinomycetemcomitans shuttle vector that is maintained at a copy number of approximately three to four copies per cell (3). Transformation of E. coli DH5α and confirmation of the desired plasmids were as described above except that recombinants were selected on LB agar containing 25 μg of kanamycin per ml. Purified plasmid obtained from recombinant E. coli cultures was subsequently introduced into A. actinomycetemcomitans 652 by electroporation according to the method of Sreenivasan et al. (51).
TABLE 1.
Oligonucleotide primers
| Primer | Sequencea |
|---|---|
| 1 | 5′-GCGGTACTAAACGTCTTCCGGTTTAC-3′ |
| 2 | 5′-GCGGATCCATATTAAATCTCCTTGT-3′ |
| 3 | 5′-GCGGTACCTTCTCAGTCTAATTTTTGCG-3′ |
| 4 | 5′-GACGGCCGGGGCCCAAAATTAAATAATTTTTTCTA TTGACTATT-3′ |
| 5 | 5′-GACGGCCGGGGCCCACATAAAATTATTAGTTTTTT GATAATAC-3′ |
| 6 | 5′-GAGGGCCCGATAATACTTTATCTACGCAATCC-3′ |
| 7 | 5′-GCGGTACCCAAAAAACTAATAATTTTATGAAAT-3′ |
| 8 | 5′-GCGGTACCCTAATAATTTTATGAAATTAAATAAT-3′ |
| 9 | 5′-GCGGTACCCTATTGACTATTAAAGAATCCGG-3′ |
| 10 | 5′-ATCTTACAGATCAAAACCTGATAAC-3′ |
| 11 | 5′-TTTGATAATACTTTATGTACGCAATCC-3′ |
| 12 | 5′-AGAAAAAATTATTTAATTTCATAAAATTAT-3′ |
| 13 | 5′-AACAAAGCGGTAATGAAAATTG-3′ |
| 14 | 5′-ATACGCAAAAATTAGACTGAG-3′ |
| 15 | 5′-TATCTACGCAATCCCGTATTG-3′ |
| 16 | 5′-TAGGTAATTTATCCGGTCAAAGG-3′ |
| 17 | 5′-TAGTTTTTTCGCCGAGAATG-3′ |
| 18 | 5′-TTTGTACTCGAAAGATCAACAGC-3′ |
| 19 | 5′-CTATTGACTATTAAAGAATC-3′ |
| 20 | 5′-TTTTAGGTTTAGGGCGATG-3′ |
| 21 | 5′-TTTATTCCCGTTTAGATGTTGG-3′ |
| 22 | 5′-GGTGATGATGGTGATGAGG-3′ |
| 23 | 5′-TAGGTTGATATTTGGTTGATATCCCTACTCGAAAG ATCAACAGCC-3′ |
| 24 | 5′-GGCTGTTGATCTTTCGAGTAGGGATATCAACCAAA TATCAACCTA-3′ |
Mutations introduced into primer sequences 22 and 23 are shown in bold italics.
Promoter constructs IR20 and IRL were synthesized in pUC19 by using PCR products derived from A. actinomycetemcomitans 652 genomic DNA. Construct IR20 contains 20 bp separating the two halves of the AT-rich inverted repeat (IR) sequence. The additional 20 nucleotides encode ApaI and EagI restriction sites which were included in the primer sequences used to generate the promoter fragments. The upstream promoter fragment was synthesized by using primers 1 and 5 (Table 1) and encompasses the sequences from the 3′ end of glyA up to and including the left segment of the IR. Amplification of the downstream promoter fragment utilized primers 2 and 4. The PCR products were cleaved initially at the KpnI and BamHI sites that were present in primers 1 and 2, respectively (Table 1), and subsequently digested with EagI. The resulting fragments were ligated into pUC19 to generate plasmid pIR5. Plasmid pIR5 was isolated from recombinant E. coli colonies, and the complete insert was released by digestion with KpnI and BamHI. The insert was subsequently transferred to pYGKlacZ and introduced into A. actinomycetemcomitans 652 as described above.
Construct IRL lacks the upstream portion of the AT-rich IR and was synthesized from two PCR products by using a strategy similar to that described above for clone IR20. The upstream promoter fragment was synthesized by using primers 1 and 6. The downstream fragment was identical to the fragment used as described above for the construction of IR20. Fragments were digested with the appropriate restriction enzymes, sequentially ligated into pUC19, and transferred into pYGKlacZ as described above.
Promoter fragments for the deletion constructs 191lacZ (primers 2 and 7), 197lacZ (primers 2 and 8), and 229lacZ (primers 2 and 9) were amplified by PCR from A. actinomycetemcomitans 652 genomic DNA by using the primer pairs indicated. Fragments were purified, cleaved with KpnI and BamHI, and ligated into pYGKlacZ as already described. All ltx promoter constructs were confirmed by nucleotide sequencing.
Determination of β-galactosidase activity.
Recombinant A. actinomycetemcomitans strains containing the lacZ reporter constructs were grown to mid-exponential phase and centrifuged to pellet bacterial cells. The cell pellets were washed and suspended in 0.1 M sodium phosphate, pH 7.5, at an optical density (OD) of 0.5 at 600 nm. β-galactosidase activity was determined by using o-nitrophenyl-β-galactoside (ONPG; Sigma Chemical Co.) as follows: 5 μl of cell suspension, 10 μl of 0.01% sodium dodecyl sulfate, and 25 μl of chloroform were added to 227 μl of 0.1 M sodium phosphate, pH 7.5. After incubation at 25°C for 10 min, 3 μl of Mg buffer (0.1 M MgCl2, 4.5 M β-mercaptoethanol) and 66 μl of ONPG (4 mg per ml in 0.1 M sodium phosphate, pH 7.5) were added. Samples were incubated for 10 min at 37°C. Reactions were terminated by the addition of 0.5 ml of 1 M Na2CO3, and the relative levels of β-galactosidase activity were determined by measuring the OD at 420 nm. All reactions were run in triplicate. Units of β-galactosidase activity were calculated according to the method of Miller (37).
Preparation of bacterial cell extracts.
For protein extraction, 5 ml of B-PER bacterial protein extraction reagent (Pierce) supplemented with a protease inhibitor cocktail (Sigma Chemical Co.) was added to frozen bacterial pellets. The cell pellets were thawed on ice and resuspended by pipetting until the suspension was homogeneous. The suspension was gently mixed at room temperature for 10 min, and lysates were centrifuged at 27,000 × g for 15 min at 4°C to remove cellular debris and insoluble proteins. Cleared extracts were dialyzed overnight at 4°C against a solution containing 20 mM HEPES (pH 7.9), 2 mM MgCl2, 1 mM dithiothreitol (DTT), 1 mM EDTA, and 20% glycerol supplemented with 1 ml of protease inhibitor cocktail per liter, followed by dialysis for 8 h against a solution containing 20 mM HEPES (pH 7.9), 40 mM KCl, 1 mM DTT, 1 mM EDTA, 0.2 mM MgCl2, and 20% glycerol. Cell extracts were stored at −70°C.
Phosphocellulose chromatography.
Cellulose phosphate ion exchanger P11 (Whatman) was prepared according to the manufacturer's instructions, and chromatography was carried out essentially as described by Samuels et al. (44). Prior to loading of the crude cell extract, the column was washed with three column volumes of a solution containing 20 mM HEPES (pH 7.9), 1 mM DTT, 1 mM EDTA, 0.1 M KCl, 0.2 mg of bovine serum albumin/ml, and 20% glycerol and subsequently with three column volumes of buffer A (20 mM HEPES [pH 7.9], 1 mM DTT, 1 mM EDTA, 40 mM KCl, 20% glycerol). Cell extracts were loaded onto the column, and nonspecifically bound material was removed by washing three times with buffer A. Bound protein was eluted with 1 M KCl. Fractions containing eluted protein were identified by ODs at 280 nm, pooled, and dialyzed against a solution containing 20 mM HEPES (pH 7.9), 1 mM DTT, 1 mM EDTA, 0.1 M KCl, and 20% glycerol. Phosphocellulose-fractionated extracts were stored at −70°C.
Analysis of protein interaction with the ltx promoter.
Prior to use of protein extracts in gel shift assays, the suspensions were treated to remove biotin and/or biotin-containing proteins by incubation with magnetic beads derivatized with streptavidin (streptavidin MagneSphere paramagnetic particles; Promega). The streptavidin beads were washed three times before use with a solution containing 0.1 M NaCl, 10 mM HEPES (pH 7.9), and 5% glycerol. Bacterial cell extracts (100 μl) were incubated with the streptavidin beads (25 μl) for 15 min at 25°C, and subsequently, the beads were removed by using a magnetic separator. Routinely, crude bacterial cell extracts were extracted three times with streptavidin beads; the phosphocellulose-fractionated extracts were treated with a single application of streptavidin beads.
Promoter fragments B101, B101IR, B180, and B154 used in the mobility shift assays were amplified by PCR by using genomic DNA from A. actinomycetemcomitans 652 as the template. Primers used to produce the promoter fragments were as follows (Table 1): B101, primers 10 and 11; B101IR, primers 10 and 12; B180, primers 13 and 15; and B154, primers 13 and 14. To synthesize biotin-labeled probes, the upstream primer used in each reaction was modified with biotin at its 5′ end. These primers were obtained from BioSynthesis, Inc. (Lewisville, Tex.). Promoter fragments B101IRL and B101IR20 were amplified by PCR with the primers used to produce B101IR. The templates for these reactions were plasmids pIRL and pIR20, respectively. Therefore, probes B101IRL and B101IR20 contain the modifications of the AT-rich IR sequence described above.
Analysis of protein-promoter interactions.
Routinely, gel mobility shift reactions were carried out in a reaction volume of 10 μl containing 2 ng of biotinylated probe, 0 to 10 ng of unlabeled probe as a competitive inhibitor, 1 μg of poly(dI-dC), 2.5 μl of protein extract, 15 mM HEPES (pH 7.9), 24 mM KCl, 100 mM NaCl, 1 mM MgCl2, 1 mM DTT, 0.25 mM EDTA, and 5% glycerol. Mixtures for reactions that were carried out with unfractionated protein extracts contained 7.5 μg of protein and 1 μg of poly(dI-dC). Mixtures for reactions using phosphocellulose-fractionated protein extracts contained 0.3 μg of protein and 10 ng of poly(dI-dC). In all reactions, the biotinylated promoter probes were incubated with the protein extract at room temperature for 30 to 60 min. Reaction mixtures were loaded onto a 5% polyacrylamide gel and electrophoresed at 100 V in 1× Tris-borate-EDTA. The labeled probes and probe-protein complexes were subsequently transferred onto a HyBond N+ nylon membrane (Amersham Pharmacia Biotech) by using a semidry blotter (Bio-Rad, Inc.) at 12 to 15 V for 30 min. After the transfer was completed, the DNA was cross-linked to the membrane by exposure to UV light (1,200 μJ for 30 s) by using a UV Stratalinker 1800 (Stratagene). Detection of the labeled probes was accomplished by using a streptavidin-horseradish peroxidase conjugate and the BM chemiluminescence blotting substrate kit (Roche Diagnostics, Inc.) according to the manufacturer's protocol. Reaction products were visualized by exposure to Kodak X-Omat-AR X-ray film for 5 to 30 min.
To quantitate the formation of the probe-protein complex, digital images of each exposed film were analyzed by using Corel Photopaint. We first determined the background for each lane by dividing the total number of pixels (PTOT) that were present in a defined area of each lane (midway between the bands representing the protein-probe complex and the labeled probe itself) by the average grayscale level (IAVE) for these pixels. Grayscale level was expressed on a scale of 1 to 255 where black was 1 and white was 255. The resulting background level for each lane was then subtracted from the result of a similar analysis of the region encompassing the protein-probe complex band. Percent inhibition of complex formation by unlabeled probe at a given concentration, x, was calculated by using the following equation:
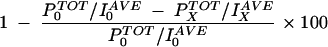 |
where P0TOT and I0AVE are the total number of pixels and average grayscale intensity, respectively, of the protein-probe band in the absence of unlabeled probe and PxTOT and IxAVE are the total number pixels and average grayscale intensity of the protein-probe band in the presence of unlabeled probe at concentration x.
Site-specific mutagenesis of the ltx promoter.
Site-specific mutagenesis was carried out by using the Quikchange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocols. The plasmid template for mutagenesis reactions was pISlacZ. Reactions utilized oligonucleotide primers 23 and 24. Recombinant clones containing the mutations were confirmed by nucleotide sequencing. Confirmed clones were introduced into A. actinomycetemcomitans 652 and analyzed for lacZ expression as described above.
RT-PCR and real-time PCR with A. actinomycetemcomitans RNA.
Real-time PCRs were performed by using Ready-To-Go reverse transcriptase PCR (RT-PCR) beads (Amersham Biosciences). A reaction volume of 50 μl was used to perform two reactions with 25 μl each. The RT step was carried out according to the manufacturer's instructions. Each reaction mixture contained 7.5 pmol of the appropriate antisense primer and 50 ng of total bacterial RNA. After completion of the cDNA synthesis, 7.5 pmol of the appropriate sense primer and 0.5× SYBR-Green dye (Roche Applied Science) were added. Amplification was carried out by using the Smart Cycler system (Cepheid). The amplification conditions for the real-time PCR were as follows: denaturation at 95°C for 270 s for a single cycle, followed by 45 cycles of denaturation at 95°C for 30 s, annealing at 56 or 58°C for 30 s, and elongation at 72°C for 60 s. The threshold cycle for each real-time PCR was determined from a second derivative plot of total fluorescence as a function of cycle number by using the software package supplied with the Smart Cycler system. The primers used for real-time PCRs were as follows: primer 16 (Table 1) anneals 15 nucleotides downstream from the transcriptional start site of the orfA promoter (Porf), and primers 18 and 19 anneal 95 and 42 nucleotides upstream from the Porf transcriptional start site, respectively. Each of these primers was used in conjunction with reverse primer 17, which anneals in the orfA open reading frame, or with reverse primer 20, which anneals to the 3′ end of orfA. The annealing temperature for these reactions was 56°C. Control primers 21 and 22 anneal to the cytolethal distending toxin gene B (cdtB) of A. actinomycetemcomitans. The annealing temperature in these reactions was 58°C. All reactions were carried out at least two times with consistent results.
RT-PCRs were carried out by using Ready-To-Go RT-PCR beads (Amersham Biosciences) according to the manufacturer's instructions. Each reaction mixture contained 100 ng of total RNA and 15 pmol of the appropriate primers. cDNA synthesis was carried out at 42°C for 30 min, followed by a single cycle at 95°C for 5 min. Amplification conditions were as follows: 30 cycles of denaturation at 94°C for 60 s, annealing at 55°C for 60 s, and elongation at 72°C for 90 s. A. actinomycetemcomitans total RNA was isolated from mid-exponential-phase aerobic cultures by using TRIzol reagent (Invitrogen Life Technologies) according to the manufacturer's instructions. Contaminating genomic DNA was removed from the RNA preparations by digestion with RQ RNase-free DNase I (Promega).
RESULTS
IS1301 increases transcription of the ltx operon.
A clinical isolate of A. actinomycetemcomitans (designated strain IS1) was previously shown to possess an insertion sequence related to IS1301 of N. meningitidis in an AT-rich IR situated upstream from the ltx operon (15) (Fig. 1A). Since the nucleotide sequence flanking IS1301 in the ltx promoter of A. actinomycetemcomitans IS1 is >99% identical to the promoter sequence of the minimally leukotoxic A. actinomycetemcomitans strain 652 (15), we hypothesized that IS1301 increases transcription of the ltx operon by introducing a new outwardly directed promoter and/or by inactivating a negative cis-acting sequence (e.g., the AT-rich IR) that may be involved in the normal regulation of toxin expression. To test this hypothesis, we first constructed ltx promoter::lacZ reporter plasmids containing the intact ltx promoter regions (i.e., glyA-ltxC intergenic region shown in Fig. 1A) from A. actinomycetemcomitans IS1 and A. actinomycetemcomitans 652. As shown in Fig. 1B, ISlacZ containing the promoter from strain IS1 directed approximately 12-fold higher levels of β-galactosidase activity than the ltx promoter from strain 652 (clone 652lacZ). Consistent with this finding, real-time PCRs showed that the level of ltx mRNA was approximately eightfold higher in A. actinomycetemcomitans IS1 than in strain 652 (Fig. 1C). These results suggest that the hyperleukotoxic phenotype of A. actinomycetemcomitans IS1 arises from increased transcription of the ltx operon.
FIG. 1.

IS1301 increases transcription of the ltx operon. (A) Schematic representation of the A. actinomycetemcomitans 652 and A. actinomycetemcomitans IS1 leukotoxin operons and upstream sequences. ltxA encodes the toxin polypeptide, and ltxC, ltxB, and ltxD encode toxin-activating and secretion proteins. orfA represents an open reading frame encoding a hypothetical protein of unknown function that is cotranscribed with ltxCABD. The upstream gene, glyA, encodes serine hydroxymethyltransferase and is not a part of the leukotoxin transcriptional unit. The locations of an AT-rich imperfect IR and −10 and −35 promoter elements of the orfA promoter (Porf) are shown. In A. actinomycetemcomitans IS1, a copy of IS1301 is integrated into the IR sequence. IS1301 carries a single gene, trnA, encoding a transposase (15). The arrows represent the annealing sites for oligonucleotide primers 16 to 20 used for RT-PCR and real-time PCRs. (B) Transcriptional activity of ltx promoter::lacZ reporter constructs. The glyA-ltxC intergenic regions of A. actinomycetemcomitans 652 and A. actinomycetemcomitans IS1 were fused to lacZ to generate constructs 652lacZ and ISlacZ, respectively. With the exception of the IS1301 sequence, the nucleotide sequences of the IS1 and 652 promoter fragments are >99% identical. For each construct, β-galactosidase activity is shown on the right and was measured from cell extracts obtained from the recombinant strains by using ONPG as the substrate. (C) Real-time PCR results obtained by using total RNA from A. actinomycetemcomitans strains 652 and IS1. Reactions were carried out as described in Materials and Methods by using the primer pairs indicated. The cytolethal distending toxin B gene (cdtB) is transcribed independently of the ltx operon and was used in a control reaction in these experiments.
IS1301 does not introduce an outwardly directed promoter.
The introduction of an outwardly directed fusion promoter is a common mechanism whereby IS elements increase the expression of genes that are located immediately downstream from the site of IS insertion (6, 17, 34, 40, 47, 52, 54). Examination of the nucleotide sequence of IS1301 identified a putative outwardly directed −35 sequence situated 18 bp upstream from a sequence with similarity to the E. coli consensus −10 element (Fig. 2A), suggesting that a potential fusion promoter may exist in A. actinomycetemcomitans IS1. To determine whether this promoter is functional, a lacZ reporter was constructed that contained mutations in the −35 sequence of the putative promoter. As shown in Fig. 2B, mutation of the putative −35 sequence (from TTGTAC to CCCTAC) had no effect on the expression of lacZ. To eliminate the possibility that other IS1301 sequences function to promote transcription of the ltx operon, RT-PCRs were carried out by using either forward primer 16 (Fig. 1A) that anneals 15 nucleotides downstream from the transcriptional initiation site of Porf or forward primer 18 or 19, which anneals 95 or 42 nucleotides upstream of the Porf initiation site, respectively (Fig. 1A). As shown in Fig. 2C, the expected products were obtained from amplification reactions using primer 16 with downstream primer 17 or 20. In contrast, no amplification products were detected in reactions using primers that annealed upstream from the Porf transcriptional initiation site (i.e., primers 18 and 19). Furthermore, real-time PCRs using the same primer pairs generated results that were consistent with those of RT-PCR (data not shown). Together, our results suggest that the hyperleukotoxic phenotype of A. actinomycetemcomitans IS1 does not arise from increased transcription of the ltx operon by an outwardly directed IS1301 promoter.
FIG. 2.

IS1301 does not possess an outwardly directed promoter. (A) The nucleotide sequence from the stop codon of trnA to the transcriptional initiation site of Porf is shown. The bold italic sequence is the terminal repeat of IS1301. The terminal repeat contains a putative −35 element (TTGTAC; underlined), indicated by the larger font, that is 18 nucleotides upstream from a potential −10 sequence (AATAAT; underlined). The −35 and −10 promoter elements of Porf are also labeled and underlined. The nucleotides that were substituted by site-specific mutagenesis are shown above the sequence. (B) Activities of the IS1 promoter::lacZ reporter construct (IS1) and the construct carrying site-specific mutations (−35M) were determined as described in Materials and Methods. The activity of the unaltered IS1 reporter was normalized to 1.0. (C) Results of RT-PCR with total IS1 RNA. The annealing sites for the primers used in the RT-PCR for each lane are shown in Fig. 1A. Lane 1, primers 16 and 17; lane 2, primers 16 and 20; lane 3, primers 19 and 17; lane 4, primers 18 and 17; lane 5, no-RT control with primers 16 and 17; lane 6, 100-bp DNA size standard ladder.
Identification of cis-acting regulators of ltx operon expression.
To determine whether IS1301 disrupts the normal transcriptional regulation of the ltx operon, cis-acting sequences that control leukotoxin expression in A. actinomycetemcomitans 652 were identified. We first examined the possibility that the AT-rich IR region represents a negative regulator of leukotoxin expression that is inactivated by integration of the IS element. This was accomplished by constructing variants of the A. actinomycetemcomitans 652 ltx promoter that contained insertions or deletions within the IR region. We surmised that altering or deleting the IR in the promoter of strain 652 may also result in increased leukotoxin expression. However, as shown in Fig. 3, deletion of the upstream segment of IR (clone IRL) or of the entire AT-rich IR region (clone 229lacZ) resulted in a two- to threefold decrease in β-galactosidase activity relative to that of the control construct containing the intact 652 ltx promoter (652lacZ). In addition, an insertion of 20 random nucleotides between the two opposing segments of the AT-rich IR (clone IR20) resulted in a similar reduction of lacZ expression. Clone YGKlacZ, which contains lacZ without an A. actinomycetemcomitans promoter, expressed background levels of β-galactosidase activity similar to those of untransformed A. actinomycetemcomitans controls, indicating that no spurious vector-derived promoter sequences contributed to the β-galactosidase activity of the reporter constructs described above. These results suggest that the AT-rich IR sequence functions as a positive cis-acting regulatory element. Indeed, the nucleotide sequence, the location, and the activity of the AT-rich IR region resemble those of an UP element. As shown in Fig. 3B, this region of the A. actinomycetemcomitans ltx promoter exhibits significant similarity to the consensus UP element sequence (9).
FIG. 3.

(A) Identification of cis-acting sequences that influence ltx operon expression in A. actinomycetemcomitans 652. The control construct, 652lacZ, contains the entire glyA-ltxC intergenic region fused to lacZ. Constructs IRL and IR20 were derived from 652lacZ and contain 20 random nucleotides separating the two segments of the IR and a deletion of the upstream segment of the IR, respectively. Deletion construct 229lacZ lacks the AT-rich IR and all additional sequences that reside upstream from the −35 element of Porf; construct 191lacZ contains the AT-rich sequence but lacks all other upstream sequences; construct 197lacZ contains only the portion of IR that resides downstream from the site of IS1301 insertion (see the text). The negative control, YGKlacZ, contains only a promoterless lacZ in the shuttle vector pYGK. For each construct, β-galactosidase activity is shown on the right and was measured from cell extracts obtained from the recombinant strains by using ONPG as the substrate. (B) The AT-rich IR sequence resembles an UP element. The upper line represents the consensus UP element sequence (9), where W is A or T and N is any nucleotide; the lower sequence is the AT-rich IR element of the ltx promoter from A. actinomycetemcomitans 652 (Aa652). Homologous residues are indicated by colons; nucleotides that are analogous to the nonspecific residues in the consensus are indicated with X.
To identify potential regulatory sequences that reside upstream from the site of IS1301 insertion, reporter constructs that contained progressive deletions within the glyA-orfA intergenic region were constructed. The control construct 652lacZ contains the entire glyA-orfA intergenic region (Fig. 1A) comprising 262 bp upstream from the transcriptional initiation site of Porf. Deletion constructs 191lacZ, 197lacZ, and 229lacZ possess 79, 71, and 39 nucleotides upstream from the Porf initiation site, respectively. As shown in Fig. 3, deletion of nucleotides upstream from residue −79 resulted in a fourfold increase in β-galactosidase activity, suggesting that a negative cis-acting sequence resides upstream from the AT-rich IR region in strain 652. Deletion of an additional 8 nucleotides (−79 to −71), which removes all of the nucleotide sequence that exists upstream from the site of IS1301 insertion (at nucleotide −71) in A. actinomycetemcomitans IS1, does not alter lacZ expression. This suggests that nucleotides −71 to −79 are not necessary for activation of transcription by the AT-rich IR. Together, these results suggest that two cis-acting sequences regulate transcription of the ltx operon in A. actinomycetemcomitans 652: a negative cis-acting sequence that resides upstream from the site of IS1301 integration and the AT-rich IR (nucleotides −40 to −71), which functions as a positive cis-acting regulator of leukotoxin expression.
Interaction of protein factors with the A. actinomycetemcomitans 652 promoter.
To determine whether the IR or the upstream negative cis-acting region interacts with putative trans-acting proteins, oligonucleotide probes comprising nucleotides −77 to −166 (B101) and nucleotides −39 to −166 (B101IR) were reacted with a protein extract from A. actinomycetemcomitans 652 and analyzed by gel electrophoresis. These probes differ only in the presence of the IR sequence in the B101IR probe. A schematic representation of the glyA-orfA intergenic region and the probes used for mobility shift experiments is shown in Fig. 4A. Representative results from the mobility shift experiments using probes B101 and B101IR are shown in Fig. 4B and C, respectively. The mobility of each probe was retarded after incubation with the crude A. actinomycetemcomitans protein extract. Identical shifted probe bands were observed when the labeled probes were incubated with phosphocellulose chromatography-enriched protein extracts (data not shown). Interestingly, the addition of unlabeled B101 to these reaction mixtures as a competitive inhibitor (1.0, 2.5, and 5.0 M equivalents) inhibited the interaction of the protein factor(s) with both B101 and B101IR in a dose-dependent manner. This suggests that the upstream sequence, and not the IR itself, interacts with the polypeptide(s) in the cell extract. To further confirm that the mobility shift observed with probe B101IR (Fig. 4C) was not dependent upon the presence of the IR sequence, mobility shift experiments were carried out by using two additional probes that contained disrupted IR regions. Oligonucleotide probes B101IRL and B101IR20 were amplified by PCR from the plasmid constructs pIRL and pIR20 (Fig. 3A) and correspond to probe B101IR except that they contain a deletion and an insertion in the IR sequences, respectively. As shown in Fig. 4D, the mobility of all three probes (B101IR, B101IRL, and B101IR20) was retarded after incubation with the A. actinomycetemcomitans protein extract, suggesting that the interaction of the protein factor with these probes is independent of the IR. Together, these results suggest that the interaction of the putative trans-acting protein occurs only at the upstream cis-acting region.
FIG. 4.


(A) Schematic representation of the glyA-orfA intergenic region showing the probes used for mobility shift experiments. Probe B101IR contains the entire AT-rich IR sequence and extends from nucleotides −39 to −166, where +1 is the transcriptional initiation site of Porf. Probe B101 lacks the IR region and extends from −77 to −166. Probe B180 extends from −87 to −218, and probe B154 extends from −111 to −218. (B and C) For mobility shift experiments, 2 ng of biotinylated ltx promoter fragments B101 (B) and B101IR (C) was incubated with 7.5 μg of a protein extract derived from A. actinomycetemcomitans 652 and electrophoresed in 5% polyacrylamide gels. Competition experiments were carried out as described above in the presence of 2 to 10 ng of unlabeled B101 probe. Lanes 1, labeled probe; lanes 2, labeled probe and protein extract; lanes 3, labeled probe, extract, and 2 ng of unlabeled B101; lanes 4, labeled probe, extract, and 5 ng of unlabeled B101; lanes 5, labeled probe, extract, and 10 ng of unlabeled B101. (D) DNA probes B101IRL and B101IR20 correspond to B101IR but contain altered IR sequences. These probes were amplified from the recombinant plasmid constructs IRL and IR20 (Fig. 3). Biotinylated probes B101IR (lanes 1 to 4), B101IRL (lanes 5 to 8), and B101IR20 (lanes 9 to 12) were incubated with 7.5 μg of A. actinomycetemcomitans protein extract in the absence and presence of unlabeled B101IR probe (2 and 5 ng) and electrophoresed in 5% polyacrylamide gels. Lane 1, labeled B101IR; lane 2, B101IR and extract; lane 3, B101IR, extract, and 2 ng of unlabeled B101IR; lane 4, B101, extract, and 5 ng of unlabeled B101IR; lane 5, labeled B101IRL alone; lane 6, B101IRL and extract; lane 7, B101IRL, extract, and 2 ng of unlabeled B101IR; lane 8, B101IRL, extract, and 5 ng of unlabeled B101IR; lane 9, labeled B101IR20 alone; lane 10, B101IR20 and extract; lane 11, B101IR20, extract, and 2 ng of unlabeled B101IR; lane 12, B101IR20, extract, and 5 ng of unlabeled B101IR. (E) Probes B180 and B154 lack 10 and 36 bp, respectively, from the 3′ end of probe B101. Biotinylated B180 and B154 were incubated with 30 μg of A. actinomycetemcomitans protein extract and electrophoresed in 5% polyacrylamide gels. Lane 1, labeled B154; lane 2, B154 and extract; lane 3, labeled B180; lane 4, B180 and extract.
To more precisely localize the sequences necessary for interacting with the A. actinomycetemcomitans protein extract, mobility shift experiments were carried out by using labeled probes B180 and B154, encompassing nucleotides −87 to −218 and −111 to −218, respectively (Fig. 4). As shown in Fig. 4E, no shifted probe band was observed when B154 was incubated with the protein extract. In contrast, the mobility of probe B180 was retarded, suggesting that the sequences between nucleotides −87 and −111 are essential for interaction with the trans-acting polypeptide.
DISCUSSION
A. actinomycetemcomitans strains vary greatly in the production of leukotoxin (4, 24). Most strains resemble the minimally leukotoxic A. actinomycetemcomitans 652 and express relatively low levels of toxin. However, several groups have shown that severe forms of early-onset human periodontal diseases are associated with organisms exhibiting a hyperleukotoxic phenotype (5, 10, 12-14, 56). These strains express 8- to 10-fold higher levels of leukotoxin than A. actinomycetemcomitans 652 (4, 15, 25) The increased expression of leukotoxin by these organisms correlates with the presence of specific genetic rearrangements, both insertions and deletions, in the glyA-orfA intergenic region situated immediately upstream of the ltx operon (4, 15). However, the regulation of leukotoxin expression in A. actinomycetemcomitans is not well understood and it is not known how these rearrangements lead to increased expression of the toxin genes. Our results show that insertion of IS1301 into an AT-rich IR in the glyA-orfA intergenic region of A. actinomycetemcomitans results in a hyperleukotoxic phenotype. The acquisition of the IS element leads to an increase in the transcription of the ltx operon and results in higher steady-state levels of ltx mRNA and toxin protein.
One mechanism whereby IS elements increase the expression of distal genes is by the introduction of an outwardly directed fusion promoter (6, 17, 34, 40, 47, 52, 54). Examination of the IS1301 sequence showed that it contains a sequence (TTGTAC) that resembles the consensus E. coli −35 promoter element in its downstream terminal repeat. Furthermore, in A. actinomycetemcomitans IS1, this putative −35 element resides 18 nucleotides upstream from the sequence AATAAT, which resembles the consensus −10 element (TATAAT). Thus, insertion of IS1301 may form a σ70-like promoter upstream from the ltx operon. However, site-specific mutagenesis of this putative −35 sequence did not influence the transcriptional activity of the IS1 ltx promoter. In addition, RT-PCRs did not detect significant levels of ltx transcripts that are initiated upstream from the transcriptional initiation site of the orfA promoter (Porf). Thus, transcription of the ltx operon in strain IS1 does not appear to be driven by an IS1301 promoter. Therefore, the induction of ltx operon transcription must occur by an alternative mechanism such as the disruption by IS1301 of the normal transcriptional regulation of the operon.
Two cis-acting sequences that influence the expression of the ltx operon in A. actinomycetemcomitans 652 were identified in the ltx promoter region upstream from orfA (Porf). One of these sequences is an AT-rich imperfect IR that is located immediately upstream from the −35 sequence in Porf. Deletion or alteration of this sequence led to decreased transcriptional activity of Porf, suggesting that the AT-rich IR functions as a positive regulator of leukotoxin expression. Indeed, the structural and functional characteristics of this sequence resemble those of an UP element. UP elements are positive cis-acting AT-rich sequences that are usually located immediately upstream from the −35 promoter element and stimulate transcription by interacting with the β-subunit of RNA polymerase (7, 9, 43). The AT-rich IR is homologous to the consensus UP element sequence (9) in 19 of 22 positions. Functional experiments are currently under way to determine whether the β-subunit of A. actinomycetemcomitans RNA polymerase interacts with the AT-rich IR in Porf. The second cis-acting regulatory region identified in our studies resides upstream from the putative UP element in A. actinomycetemcomitans 652. Deletion of this region resulted in a fourfold increase in the transcriptional activity of Porf, indicating that this sequence functions to down regulate leukotoxin expression. Furthermore, mobility shift experiments showed that this region interacts with a protein factor(s) in extracts from A. actinomycetemcomitans 652 and that nucleotides −87 to −111 (Fig. 4) are required for this interaction to occur. The nature of this polypeptide and the mechanism by which it regulates leukotoxin expression are not known; studies to isolate and identify the putative trans-acting regulator are ongoing. Interestingly, the interaction of the protein factor with B101IR was inhibited more effectively with unlabeled B101 (Fig. 4, compare panels B and C). The reason for this is not clear. However, AT-rich sequences are known to promote stable bends in DNA (39) and it is possible that the AT-rich IR forms a secondary structure that sterically inhibits protein interactions with the upstream sequence in probe B101IR.
The positions of the two cis-acting regulators relative to the site of IS1301 insertion suggest a possible mechanism that may contribute to the hyperleukotoxic phenotype exhibited by A. actinomycetemcomitans IS1. In strain IS1, IS1301 is integrated into the upstream portion of the AT-rich IR, at nucleotide −71. The surrounding sequence, ACTAA, closely resembles the reported target integration sequence (AYTAG, where Y represents pyrimidine) for IS1301 in N. meningitidis strains (16). Thus, the acquisition of IS1301 displaces 6 nucleotides of the AT-rich IR (residues −71 to −77) and the putative negative cis-acting sequence (nucleotides −87 to −111) approximately 900 bp upstream from the basal promoter elements of Porf. Our results show that deletion of nucleotides −71 to −79 (clone 197lacZ) did not affect the transcriptional activity of Porf reporter constructs. In contrast, deleting the portion of the AT-rich sequence that resides downstream from the site of IS1301 insertion (clone 229lacZ) significantly reduced transcriptional activity. Thus, the acquisition of IS1301 has little effect on transcriptional stimulation of the ltx operon mediated by the putative UP element. Our results suggest that, in the absence of an IS1301 promoter, the hyperleukotoxic phenotype arises at least in part from the displacement of the negative cis-acting element by IS1301. The uncoupling of negative regulation may therefore allow the ltx operon of strain IS1 to be predominantly controlled by the positive cis-acting element, leading to increased overall expression of leukotoxin. It is also possible that IS1301 possesses additional positive cis-acting regions that stimulate the transcriptional activity of Porf.
The insertion and excision of IS1301 in siaA of N. meningitidis have been reported to function as a reversible genetic switch that controls the biosynthesis of sialic acid (11). This switch regulates capsule expression and lipo-oligosaccharide sialylation which in turn controls the bacterium's adherence to and invasiveness in mucosal epithelial cells (11). Reversible IS-mediated regulation of gene expression has also been reported in Pseudomonas aeruginosa PAO (49) and Pseudomonas atlantica (1). The excision and reinsertion of IS elements at defined sites in the genome suggest that the transposase exhibits a high degree of specificity in recognizing the specific target sequence. Indeed, IS1301 integrates at a defined target sequence (see above) and also requires additional structural features, e.g., extended palindromic symmetry, stem-loop formation, and the presence of AT-rich tracts (16). All of these features are conserved at the site of IS1301 integration into A. actinomycetemcomitans IS1. While the frequency of excision and reinsertion of IS1301 upstream from the ltx operon in A. actinomycetemcomitans is not currently known, it is tempting to speculate that a similar reversible genetic switch may regulate the expression of leukotoxin. The resulting flux in leukotoxin expression may therefore influence virulence of A. actinomycetemcomitans and contribute to the periodic nature of periodontal disease.
Acknowledgments
This work was supported by Public Health Service grant DE-10729 from the National Institute of Dental and Craniofacial Research.
Editor: V. J. DiRita
REFERENCES
- 1.Bartlett, D. H., and M. Silverman. 1989. Nucleotide sequence of IS492, a novel insertion sequence causing variation in extracellular polysaccharide production in the marine bacterium Pseudomonas atlantica. J. Bacteriol. 171:1763-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Block, P. J., A. C. Fox, C. Yoran, and A. J. Kaltman. 1973. Actinobacillus actinomycetemcomitans endocarditis: report of a case and review of the literature. Am. J. Med. Sci. 276:387-392. [DOI] [PubMed] [Google Scholar]
- 3.Brogan, J. M., E. T. Lally, and D. R. Demuth. 1996. Construction of pYGK, an Actinobacillus actinomycetemcomitans/Escherichia coli shuttle vector. Gene 169:141-142. [DOI] [PubMed] [Google Scholar]
- 4.Brogan, J. M., E. T. Lally, K. Poulsen, M. Kilian, and D. R. Demuth. 1994. Regulation of Actinobacillus actinomycetemcomitans expression: analysis of the promoter regions of leukotoxic and minimally leukotoxic strains. Infect. Immun. 62:501-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bueno, L. C., M. P. Mayer, and J. M. DiRienzo. 1998. Relationship between conversion of localized juvenile periodontitis-susceptible children from health to disease and Actinobacillus actinomycetemcomitans leukotoxin promoter structure. J. Periodontol. 69:998-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeShazer, D., G. E. Wood, and R. L. Friedman. 1994. Molecular characterization of catalase from Bordetella pertussis: identification of the katA promoter in an upstream insertion sequence. Mol. Microbiol. 14:123-130. [DOI] [PubMed] [Google Scholar]
- 7.Estrem, S. T., W. Ross, T. Gaal, Z. W. S. Chen, W. Niu, R. H. Ebright, and R. L. Gourse. 1999. Bacterial promoter architecture: subsite structure of UP elements and interactions with the carboxy-terminal domain of the RNA polymerase β-subunit. Genes Dev. 13:2134-2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gouhlen, F., A. Hafezi, V.-J. Uitto, D. Hinode, R. Nakamura, D. Grenier, and D. Mayrand. 1998. Subcellular localization and cytotoxic activity of the GroEL-like protein isolated from Actinobacillus actinomycetemcomitans. Infect. Immun. 66:5307-5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gourse, R. L., W. Ross, and T. Gaal. 2000. UPs and downs in bacterial transcription initiation: the role of the alpha subunit of RNA polymerase in promoter recognition. Mol. Microbiol. 37:687-695. [DOI] [PubMed] [Google Scholar]
- 10.Guthmiller, J. M., E. T. Lally, and J. Korostoff. 2001. Beyond the specific plaque hypothesis: are highly leukotoxic strains of Actinobacillus actinomycetemcomitans a paradigm for periodontal pathogenesis? Crit. Rev. Oral Biol. Med. 12:116-124. [DOI] [PubMed] [Google Scholar]
- 11.Hammerschmidt, S., R. Hilse, J. P. M. van Putten, R. Gerardy-Schahn, A. Unkmeir, and M. Frosch. 1996. Modulation of cell surface sialic acid expression in Neisseria meningitidis via a transposable genetic element. EMBO J. 15:192-198. [PMC free article] [PubMed] [Google Scholar]
- 12.Haraszthy, V. I., G. Hariharan, E. M. Tinoco, J. R. Cortelli, E. T. Lally, E. Davis, and J. J. Zambon. 2000. Evidence for the role of highly leukotoxic Actinobacillus actinomycetemcomitans in the pathogenesis of localized juvenile and other forms of early-onset periodontitis. J. Periodontol. 71:912-922. [DOI] [PubMed] [Google Scholar]
- 13.Haubek, D., J. M. DiRienzo, E. M. B. Tinoco, and M. Kilian. 1997. Racial tropism of a highly toxic clone of Actinobacillus actinomycetemcomitans associated with juvenile periodontitis. J. Clin. Microbiol. 35:3037-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haubek, D., O. K. Ennibi, K. Poulsen, S. Poulesen, N. Benzarti, and M. Kilian. 2001. Early-onset periodontitis in Morocco is associated with the highly leukotoxic clone of Actinobacillus actinomycetemcomitans. J. Dent. Res. 80:1580-1583. [DOI] [PubMed] [Google Scholar]
- 15.He, T., T. Nishihara, D. R. Demuth, and I. Ishikawa. 1999. A novel insertion sequence increases the expression of leukotoxicity in Actinobacillus actinomycetemcomitans clinical isolates. J. Periodontol. 70:1261-1268. [DOI] [PubMed] [Google Scholar]
- 16.Hilse, R., S. Hammerschmidt, W. Bautsch, and M. Frosch. 1996. Site-specific insertion of IS1301 and distribution in Neisseria meningitidis strains. J. Bacteriol. 178:2527-2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hubner, A., and W. Hendrickson. 1997. A fusion promoter created by a new insertion sequence, IS1490, activates transcription of 2,4,5-trichlorophenoxyacetic acid catabolic genes in Burkholderia cepacia AC100. J. Bacteriol. 179:2717-2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inouye, T., H. Ohta, S. Kokeguchi, K. Fukui, and K. Kato. 1990. Colonial variation and fimbriation of Actinobacillus actinomycetemcomitans. FEMS Microbiol. Lett. 69:13-17. [DOI] [PubMed] [Google Scholar]
- 19.Issartel, J. P., V. Koronokis, and C. Hughes. 1991. Activation of Escherichia coli prohemolysin to the mature toxin by acyl carrier protein dependent fatty acylation. Nature (London) 351:759-761. [DOI] [PubMed] [Google Scholar]
- 20.Iwase, M., E. T. Laly, P. Berthold, H. M. Korchak, and N. S. Taichman. 1990. Effects of cations and osmotic protectants on cytolytic activity of Actinobacillus actinomycetemcomitans leukotoxin. Infect. Immun. 58:1782-1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kachlany, S. C., P. J. Planet, R. DeSalle, D. H. Fine, and D. H. Figurski. 2001. Genes for tight adherence of Actinobacillus actinomycetemcomitans: from plaque to plague to pond scum. Trends Microbiol. 9:429-437. [DOI] [PubMed] [Google Scholar]
- 22.Karakelian, D., J. D. Lear, E. T. Lally, and J. C. Tanaka. 1998. Characterization of Actinobacillus actinomycetemcomitans leukotoxin pore formation in HL60 cells. Biochim. Biophys. Acta 1406:175-187. [DOI] [PubMed] [Google Scholar]
- 23.Kato, T., K. Honma, A. Yamanaka, T. Miura, and K. Okuda. 2000. Heterogeneity in the immune response to serotype b LPS of Actinobacillus actinomycetemcomitans in inbred strains of mice. FEMS Immunol. Med. Microbiol. 28:67-70. [DOI] [PubMed] [Google Scholar]
- 24.Kolodrubetz, D., L. H. Phillips, P. J. Ezzo, and E. Kraig. 1995. Directed genomic integration in Actinobacillus actinomycetemcomitans: generation of defined leukotoxin-negative mutans. Infect. Immun. 63:2780-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolodrubetz, D., J. Spitznagel, Jr., B. Wang, L. H. Phillips, C. Jacobs, and E. Kraig. 1996. cis elements and trans factors are both important in strain-specific regulation of the leukotoxin gene in Actinobacillus actinomycetemcomitans. Infect. Immun. 64:3451-3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korostoff, J., J. F. Wang, I. Kieba, M. Miller, B. J. Shenker, and E. T. Lally. 1998. Actinobacillus actinomycetemcomitans leukotoxin induces apoptosis in HL-60 cells. Infect. Immun. 66:4474-4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korostoff, J., N. Yamaguchi, M. Miller, I. Kieba, and E. T. Lally. 2000. Perturbation of mitochondrial structure and function plays a central role in Actinobacillus actinomycetemcomitans leukotoxin-induced apoptosis. Microb. Pathog. 29:267-278. [DOI] [PubMed] [Google Scholar]
- 28.Kraig, E., T. Dailey, and D. Kolodrubetz. 1990. Nucleotide sequence of the leukotoxin gene from Actinobacillus actinomycetemcomitans: homology to the alpha-hemolysin/leukotoxin gene family. Infect. Immun. 58:920-929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lally, E. T., E. E. Golub, I. R. Kieba, N. S. Taichman, S. Decker, P. Berthold, C. W. Gibson, D. R. Demuth, and J. C. Rosenbloom. 1990. Structure and function of the B and D genes of the Actinobacillus actinomycetemcomitans leukotoxin complex. Microb. Pathog. 11:111-121. [DOI] [PubMed] [Google Scholar]
- 30.Lally, E. T., E. E. Golub, I. R. Kieba, N. S. Taichman, J. Rosenbloom, J. C. Rosenbloom, C. W. Gibson, and D. R. Demuth. 1989. Analysis of the Actinobacillus actinomycetemcomitans leukotoxin gene: delineation of unique features and comparison to homologous toxins. J. Biol. Chem. 274:15451-15456. [PubMed] [Google Scholar]
- 31.Lally, E. T., R. B. Hill, I. R. Kieba, and J. Korostoff. 1999. The interaction between RTX toxins and target cells. Trends Microbiol. 7:356-361. [DOI] [PubMed] [Google Scholar]
- 32.Lally, E. T., I. R. Kieba, A. Sato, C. L. Green, J. Rosenbloom, J. Korostoff, J. F. Wang, B. S. Shenker, S. Ortlepp, M. K. Robinson, and P. C. Billings. 1997. RTX toxins recognize a β2 integrin on the surface of human target cells. J. Biol. Chem. 272:30463-30469. [DOI] [PubMed] [Google Scholar]
- 33.Lear, J. D., D. Karakelian, U. Furblur, E. T. Lally, and J. C. Tanaka. 2000. Conformational studies of Actinobacillus actinomycetemcomitans leukotoxin: partial denaturation enhances toxicity. Biochim. Biophys. Acta 1476:350-362. [DOI] [PubMed] [Google Scholar]
- 34.Lopez, P., F. L. de Felipe, and C. Magni. 1996. Transcriptional activation of the citrase permease P gene of Lactococcus lactis by an insertion sequence-like element in plasmid pCIT264. Mol. Gen. Genet. 250:428-436. [DOI] [PubMed] [Google Scholar]
- 35.Meyer, D. H., J. E. Lippman, and P. M. Fives-Taylor. 1994. Invasion of epithelial cells by Actinobacillus actinomycetemcomitans: a dynamic multistep process. Infect. Immun. 64:2988-2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer, D. H., and P. M. Fives-Taylor. 1994. Characteristics of adherence of Actinobacillus actinomycetemcomitans to epithelial cells. Infect. Immun. 62:928-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller, J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 38.Page, M. I., and E. O. King. 1966. Infection due to Actinobacillus actinomycetemcomitans and Haemophilus aphrophilus. N. Engl. J. Med. 275:181-188. [DOI] [PubMed] [Google Scholar]
- 39.Perez-Martin, J., F. Rojo, and V. de Lorenzo. 1994. Promoters responsive to DNA bending: a common theme in prokaryotic gene expression. Microbiol. Rev. 58:268-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Podglagen, I., J. Breuil, and E. Collatz. 1994. Insertion of a novel DNA sequence, IS1186, upstream of the silent carbapenemase gene cfiA promotes expression of carbapenem resistance in clinical isolates of Bacteroides fragilis. Mol. Microbiol. 12:105-114. [DOI] [PubMed] [Google Scholar]
- 41.Robertson, P. B., M. Lantz, T. Marchuka, K. S. Kornman, C. I. Trummel, and S. C. Holt. 1982. Collagenolytic activity associated with Bacteroides species and Actinobacillus actinomycetemcomitans. J. Periodontal Res. 17:175-283. [DOI] [PubMed] [Google Scholar]
- 42.Rosan, B., J. Slots, R. J. Lamont, M. A. Listgarten, and G. M. Nelson. 1988. Actinobacillus actinomycetemcomitans fimbriae. Oral Microbiol. Immunol. 3:58-63. [DOI] [PubMed] [Google Scholar]
- 43.Ross, W., K. K. Gosink, J. Salomon, K. Igarashi, C. Zou, A. Ishihama, K. Severinov, and R. L. Gourse. 1993. A third recognition element in bacterial promoters: DNA binding by the alpha subunit of RNA polymerase. Science 262:1407-1413. [DOI] [PubMed] [Google Scholar]
- 44.Samuels, M., A. Fire, and P. A. Sharp. 1982. Separation and characterization of factors mediating accurate transcription by RNA polymerase II. J. Biol. Chem. 257:14419-14427. [PubMed] [Google Scholar]
- 45.Shenker, B. J., T. McKay, S. Datar, M. Miller, R. Chowden, and D. R. Demuth. 1999. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing G2 arrest in human T cells. J. Immunol. 162:4773-4780. [PubMed] [Google Scholar]
- 46.Shenker, B. S., R. H. Hoffmaster, T. McKay, and D. R. Demuth. 2000. Expression of cytolethal distending toxin (cdt) operon in Actinobacillus actinomycetemcomitans: evidence that the CdtB protein is responsible for G2 arrest of the cell cycle in human T cells. J. Immunol. 165:2612-2618. [DOI] [PubMed] [Google Scholar]
- 47.Simpson, A. E., R. A. Skurry, and N. Firth. 2000. An IS257-derived promoter directs transcription of a tetA(K) tetracycline resistance gene in the Staphylococcus aureus chromosomal mec region. J. Bacteriol. 182:3345-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slots, J., H. S. Reynolds, and R. J. Genco. 1980. Actinobacillus actinomycetemcomitans in human periodontal disease: a cross sectional microbiological investigation. Infect. Immun. 29:1013-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sokol, P. A., M. Z. Juan, D. G. Storey, and P. Thirukkumaran. 1994. Genetic rearrangement associated with in vivo mucoid conversion of Pseudomonas aeruginosa PAO is due to insertion elements. J. Bacteriol. 176:553-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sreenivasan, P. K., D. H. Meyer, and P. M. Fives-Taylor. 1993. Requirements for invasion of epithelial cells by Actinobacillus actinomycetemcomitans. Infect. Immun. 61:1239-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sreenivasan, P. K., D. J. LeBlanc, L. N. Lee, and P. Fives-Taylor. 1991. Transformation of Actinobacillus actinomycetemcomitans by electroporation, utilizing constructed shuttle plasmids. Infect. Immun. 59:4621-4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teras, R., R. Horak, and M. Kivisaar. 2000. Transcription from fusion promoters generated during transposition of transposon Tn4652 is positively affected by integration host factor in Pseudomonas putida. J. Bacteriol. 182:589-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Welch, R. A. 1991. Pore forming cytolysins of Gram negative bacteria. Mol. Microbiol. 5:521-528. [DOI] [PubMed] [Google Scholar]
- 54.Wood, M. S., A. Byrne, and T. G. Lessie. 1991. IS406 and IS407, two gene activating insertion sequences for Pseudomonas cepacia. Gene 105:101-105. [DOI] [PubMed] [Google Scholar]
- 55.Zambon, J. J. 1985. Actinobacillus actinomycetemcomitans in human periodontal disease. J. Clin. Periodontol. 12:1-20. [DOI] [PubMed] [Google Scholar]
- 56.Zambon, J. J., V. Haraszthy, G. Hariharan, E. T. Lally, and D. R. Demuth. 1996. The microbiology of early onset periodontitis: association of highly toxic A. actinomycetemcomitans strains with localized juvenile periodontitis. J. Periodontol. 67:282-290. [DOI] [PubMed] [Google Scholar]
- 57.Zambon, J. J., J. Slots, and R. J. Genco. 1983. Serology of oral Actinobacillus actinomycetemcomitans and serotype distribution in human periodontal disease. Infect. Immun. 41:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]