

Abstract
A Vibrio cholerae arcA mutant was constructed and used to examine the role of the global anaerobiosis response regulator ArcA in the expression of virulence factors in this important human pathogen. In V. cholerae, expression of the major virulence factors cholera toxin (CT) and toxin-coregulated pilus (TCP) is regulated by the transcriptional activator ToxT. toxT expression, in turn, is controlled by the transmembrane DNA binding proteins ToxR and TcpP. In the V. cholerae arcA mutant, although ToxR and TcpP were unaffected, Northern blot and reverse transcription-PCR analyses indicated that the expression of toxT was significantly decreased with concomitant reduction in the expression of CT and TCP. CT and TCP expression was completely restored in the V. cholerae arcA mutant strain by expressing a cloned toxT gene in the mutant. These results suggest that ArcA functions as a positive regulator of toxT expression under both aerobic and anaerobic conditions, although as expected, the effect was more pronounced during anaerobic growth. This was reflected in a reduction of virulence of the V. cholerae arcA mutant strain in the infant mouse cholera model.
Vibrio cholerae, a gram-negative noninvasive enteric bacterium, is the causative agent of the diarrheal disease cholera (22). Cholera remains a major cause of human mortality, not only in the developing countries, where it exists almost in endemic form, but also in certain developed countries, where it frequently assumes epidemic proportions. For successful infection of their human host, V. cholerae cells must colonize the intestine and produce copious amounts of cholera toxin (CT), a potent enterotoxin that causes the severe watery diarrhea characteristic of the disease. A toxin-coregulated pilus (TCP) coordinately expressed with CT greatly enhances colonization of the intestinal epithelium by the bacterium (16, 42). Additional factors include other potential toxins, accessory colonization factors, outer membrane proteins, hemolysins, and hemagglutinins, all of which may contribute to the virulence of this important human pathogen.
Expression of a subset of the virulence factors of V. cholerae is coordinately controlled by a regulatory cascade in which the inner membrane DNA binding proteins, ToxR and TcpP, control the expression of ToxT, a transcriptional activator that directly regulates the expression of several virulence genes, including those encoding CT and TcpA, the major subunit of TCP (9, 24). Expression of the virulence factors of this regulon, known, mainly for historical reasons, as the ToxR regulon, is coordinately regulated and strongly influenced by environmental signals. A number of parameters, including temperature, osmolarity, pH, amino acids, and bile, are known to modulate expression of the ToxR regulon, and these environmental signals exert their effects at different levels of the regulatory cascade (38).
The conditions that control virulence gene expression in pathogens also stimulate global regulatory changes in many genes. A feature of the formidable adaptation strategy of pathogens for optimal survival in their hosts is the integration of virulence gene expression with regulatory circuits that respond to environmental conditions, typically those resembling the in vivo sites of infection (11). In Salmonella, Shigella, Yersinia, Bordetella, and many other pathogens, general bacterial signal transduction and stress response regulatory circuits responding to osmolarity, iron concentration, inorganic-ion concentrations, nutrient availability, and many other cues are integrated into the virulence regulon, usually involving specific adaptor molecules (7, 13, 29, 41). Low oxygen concentration is a characteristic feature of the physiological sites of infection of many mammalian pathogens. Indeed, anaerobiosis has been shown to affect the production of virulence factors in several bacterial pathogens, and Salmonella mutants defective in anaerobic respiration are impaired in the ability to replicate in host cells (6). Bacterial adaptation to growth under oxygen deprivation involves several global regulatory systems, like FNR and ArcA/ArcB (2, 30, 34). The central regulators of each of these systems control the expression of a large number of genes whose products are mainly involved in adaptation to anaerobiosis. In addition, FNR functions as a regulator of virulence genes in several pathogens. Expression of the major outer membrane protein antigen in Neisseria gonorrhoeae and a phospholipase virulence factor in Yersinia enterocolitica have been reported to be dependent on FNR (17, 35). Although ArcA has been shown to control resistance to reactive oxygen and nitrogen species in Salmonella, it is not directly involved in virulence regulation (28). The present study describes experiments to examine the role of ArcA in the expression of the virulence factors of V. cholerae. The results obtained suggest that ArcA positively modulates the expression of toxT, the transcriptional activator of the genes encoding CT and TcpA, and increases V. cholerae virulence in an experimental animal model.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. The V. cholerae and E. coli strains were maintained at −70°C in Luria-Bertani (LB) medium containing 20% (vol/vol) glycerol. Ampicillin (100 μg ml−1) and streptomycin (150 μg ml−1) were used where appropriate. Tetracycline was used at a concentration of 15 μg ml−1 for Escherichia coli and 5 μg ml−1 for V. cholerae. The plasmid pBluescript KS was used as a cloning vector, and the suicide vector pGP704 was used for site-directed mutagenesis (31). All plasmids were maintained and amplified in E. coli DH5α except pGP704 and its derivatives, which were maintained in E. coli SM10 λ pir. Plasmids were introduced into E. coli cells by transformation and into V. cholerae by triparental mating using E. coli MM294(pRK 2013) as a donor of mobilization factors. V. cholerae cells were grown with aeration either in LB medium, pH 6.6, at 30°C or in LB medium, pH 8.6, at 37°C. For anaerobic growth, overnight cultures were diluted 1:100 in 3 ml of LB contained in 10-mm-diameter 4-cm-long screw-cap tubes filled to the brim and incubated without shaking.
TABLE 1.
Bacterial strains and plasmids used in the study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strain | ||
| V. cholerae O395 | Smr | Laboratory collection |
| V. cholerae O395d2 | O395 arcA::pGP704 Smr Apr | This study |
| E. coli SM10 | thi thr leu tonA lacY supE recA::RP4-2-Tc::Mu Km | 31 |
| E. coli MM294 | endA hsdR pro supF | J. J. Mekalanos |
| Plasmids | ||
| pBluescript KS | Cloning vector; Apr | Stratagene |
| pNS1 | pBluescript KS derivative with 1.2-kb fragment carrying V. cholerae arcA gene | This study |
| pMT5 | V. cholerae toxT cloned in plasmid pMMB66HE | 10 |
| pMT5-tet | pMT5 carrying tet gene from plasmid pBR322. | This study |
| pKEK162 | pUC118 carrying toxT gene | 36 |
| pKEK162-tet | pKEK162 carrying tet gene from plasmid pBR322 | This study |
Cloning of V. cholerae arcA gene and construction of arcA mutant.
A sequence from the V. cholerae genome sequence database (15) identified by a BLAST search as encoding an ArcA homolog provided the basis for the design of the oligonucleotide primers 5′-CTGACAAGCGAATACTCTCA-3′ and 5′-ACGCAAAGCAGTGATAAAA-3′, which were used to amplify the chromosomal arcA gene from V. cholerae strain O395. The 1.2-kb PCR fragment was cloned at the EcoRV site of the plasmid pBluescript KS to give the plasmid pNS1 (Table 1). An internal 100-bp fragment corresponding to nucleotides 70 to 173 from the 5′ end of the open reading frame (ORF) was amplified using appropriate primers. The fragment was cloned at the EcoRV site of the suicide vector pGP704(Apr) and transformed into a λ pir lysogen of E. coli SM10. Ampicillin-resistant transformants containing the recombinant plasmid were selected and conjugally transferred to V. cholerae strain O395 (Smr). Transconjugants resistant to both ampicillin and streptomycin were selected. Southern blot analysis was used to confirm that integration had occurred at an appropriate position within the chromosomal arcA gene.
RNA isolation and RT-PCR.
For isolation of RNA, cells were grown to the late logarithmic phase in LB medium (pH 6.6) at 30°C, conditions optimum for expression of the ToxR regulon (31). Total RNA was extracted and purified using guanidinium isothiocyanate (1). The RNA was treated with RNase-free DNase I (amplification grade; Gibco-BRL), and reverse transcription (RT)-PCR was performed using a single-tube RT-PCR kit (Gibco-BRL) in the presence of an RNase inhibitor (RNasin; Gibco-BRL). Two hundred nanograms of DNase-treated RNA was used in all reactions. Amplification was for 25 to 35 cycles (94°C for 30 s, 55°C for 30 s, and 72°C for 1 min, followed by a 7-min extension at 72°C) for ctxAB-, tcpA-, toxT-, toxR-, and tcpP-specific primers, and 25 cycles were used for 16S rRNA. Genomic DNA served as a positive control, and DNase-treated RNA that had not been reverse transcribed was used as a negative control. Primers for RT-PCR, corresponding to internal regions of the ORFs, were designed based on the V. cholerae genome sequence (15). Twenty-microliter aliquots removed at 25, 30, and 35 cycles of each PCR were electrophoresed on 1% agarose gels with ethidium bromide, and the gels were analyzed using a Gel Doc 1000 system (Bio-Rad Laboratories, Richmond, Calif.). The PCR products were normalized according to the amount of 16S rRNA detected in the same cDNA sample. Each set of experiments was repeated at least three times. Comparisons of relative intensities of PCR products obtained from wild-type V. cholerae and arcA mutant strains were made using the two-sample t test.
Northern blot analysis.
RNA samples (15 to 20 μg per well) were electrophoresed on 1% agarose- 2.1 M formaldehyde- mopholinepropanesulfonic acid gels, blotted onto nylon membranes using 20× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), and hybridized with labeled probes as described elsewhere (12).
GM1-ganglioside-dependent enzyme-linked immunosorbent assays for assay of CT.
CT production was assayed in culture supernatants by GM1-linked immunosorbent assays using polyclonal rabbit serum directed against purified CT. Anti-rabbit immunoglobulin G conjugated with horseradish peroxidase (Jackson Laboratory, Bar Harbor, Maine) was used as a secondary antibody. Dilutions of CT in known concentrations (Sigma) were used to estimate the amounts of CT in the samples, expressed as the amount of toxin per unit of optical density at 600 nm of the culture.
LD50 assays.
Fifty-percent lethal doses (LD50s) were determined in 5- to 7-day-old infant mice by oral challenge with various doses of viable bacteria. The mice were taken from their mothers at least 6 h before infection. The V. cholerae strains were grown in LB medium, pH 6.5, at 30°C to the late logarithmic phase and diluted as required in 0.15 M NaCl containing blue food coloring; 100 μl of various doses of inoculum was delivered orally to the infant mice. Four or more mice were used per dose, and survival was determined at 24 h. LD50s representing the means of three independent experiments were calculated.
RESULTS
Cloning of the arcA gene from V. cholerae and construction of an arcA mutant.
A search of the V. cholerae genome sequence using the BLAST algorithm revealed the presence of an ORF with 88.2% identity and 94.1% similarity to the E. coli arcA gene. The E. coli arcA gene is reported to be flanked by creD and yjjY, which encode a colicin E2 tolerance protein and a protein of unknown function, respectively (3). arcB, encoding the sensor component of the ArcA-ArcB two-component system, is located at an unlinked locus in the E. coli genome (19). Examination of the arcA locus in the publicly available V. cholerae strain N16961 genome sequence revealed that the arcB gene was located immediately downstream of arcA in the opposite orientation (15). A hypothetical ORF was present upstream of the arcA gene. Thus, the genomic organization of arcA in V. cholerae is different from that in E. coli. The arcA gene from V. cholerae strain O395 was PCR amplified as a 1.2-kb fragment and cloned into the plasmid pBluescript KS to give the plasmid pNS1 (Table 1). The nucleotide sequence of the cloned fragment was determined, and both the gene sequence and the genomic organization were found to be identical to that of strain N16961 in the V. cholerae genome sequence database.
A site-directed insertion mutation in the V. cholerae arcA gene was constructed by chromosomal integration of the mobilizable suicide plasmid pGP704 containing an internal 100-bp fragment of the cloned arcA gene, thereby disrupting the ORF. Cointegrate Smr Apr transconjugants containing no plasmid were selected, and Southern blot analysis was used to confirm that the mobilized plasmid had integrated at the appropriate site in the arcA gene. The V. cholerae arcA mutant was designated strain O395d2. Even though the convergent orientation of the downstream arcB gene makes it unlikely that arcA insertion mutation could be polar, plasmid pNS1 containing the entire arcA structural gene was introduced into the V. cholerae arcA mutant strain to test if the cloned arcA gene could complement the phenotypes of strain O395d2 described below.
Characterization of the V. cholerae arcA mutant.
The V. cholerae arcA mutant strain O395d2 produced the same smooth colony morphology as the parental strain, O395, although the mutant colonies were smaller than those of the wild type. Since the arcA gene product is a central regulator of multiple genes involved in anaerobic respiration (18), the growth of V. cholerae strain O395d2 was examined under aerobic and anaerobic conditions and compared to that of the parental strain, O395. No significant difference was observed between the growth curves of the mutant and wild-type strains grown in LB or minimal medium under aerobic or anaerobic conditions. It has recently been reported that arcA mutation has no effect on the anaerobic growth of Salmonella enterica serovar Enteritidis (28).
Since mutations in the E. coli arcA gene result in sensitivity to the dye toluidine blue (18), the V. cholerae arcA mutant strain O395d2 was examined for toluidine blue sensitivity. The MIC of toluidine blue decreased from 2 μg ml−1 for the wild-type V. cholerae strain O395 to 0.15 to 0.2 μg ml−1 for the arcA mutant strain O395d2. Thus, similar to E. coli, mutation in arcA renders V. cholerae cells ∼10 times more sensitive to toluidine blue.
CT production is reduced in the V. cholerae arcA mutant.
To examine if ArcA has any role in the virulence of V. cholerae, production of CT, the major virulence factor of the organism, was examined in the arcA mutant strain O395d2. When grown in LB medium (pH 6.6) at 30°C, conditions optimum for CT production (31), although the parental strain produced ∼10 μg of CT per ml of culture supernatant per unit of optical density at 600 nm, only ∼1 μg could be detected in culture supernatants of the arcA mutant strain. Thus, CT production in strain O395d2 was reduced by ∼90% compared to that in the parental strain, O395, when the strains were grown under conditions optimum for CT production (Fig. 1). When grown under nonpermissive conditions (LB medium [pH 8.6]; 37°C), only ∼20% of the amount of CT produced under permissive conditions was detected in the wild-type strain, O395. Under these conditions, a further decrease in CT was observed in the arcA mutant strain O395d2, suggesting that ArcA contributes to the residual CT production under nonpermissive conditions (Fig. 1).
FIG. 1.
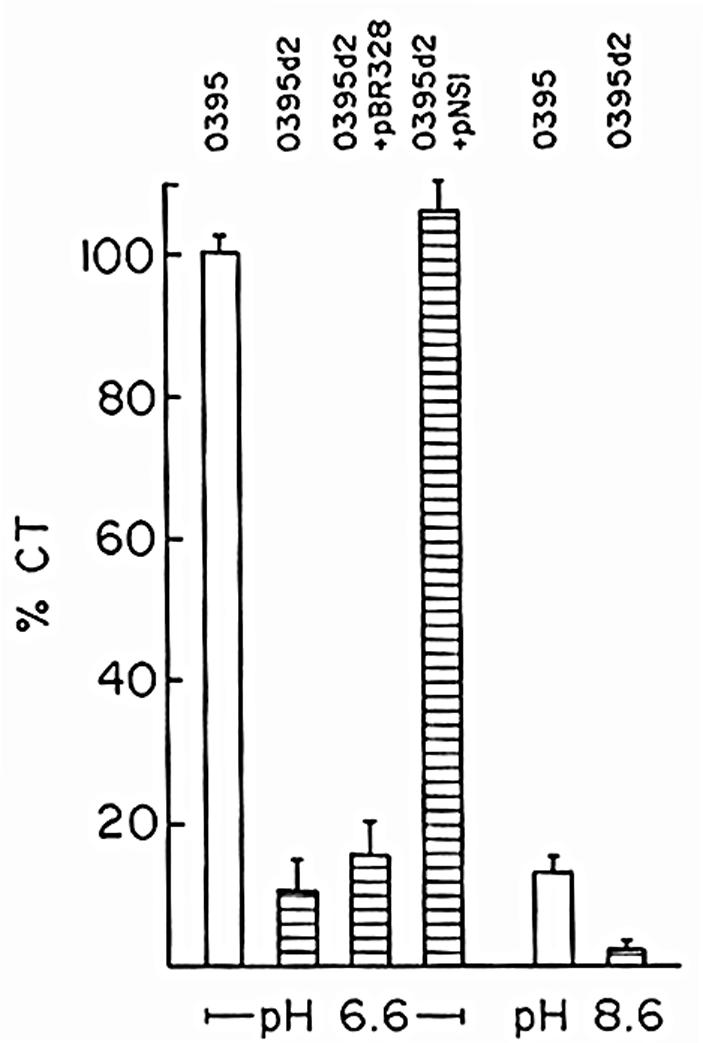
CT production in V. cholerae wild-type and arcA mutant strains. V. cholerae O395 and strain O395d2 containing the plasmid pBR328 or pNS1 were grown in LB medium, pH 6.6 or 8.6, to the stationary phase. CT was measured in culture supernatants corresponding to 109 CFU and expressed as percentages of the amount obtained in culture supernatants of strain O395 grown in LB medium, pH 6.6, at 30°C. The data represent the averages of five independent experiments. The error bars indicate the standard errors of the mean.
Complementation of the V. cholerae arcA mutant strain O395d2 with plasmid pNS1 carrying the full-length arcA gene could restore CT to wild-type levels, indicating that the mutation in arcA was indeed responsible for reduced CT production in strain O395d2 (Fig. 1).
Transcriptional repression of ctxAB and tcpA genes in the V. cholerae arcA mutant.
To examine if the inhibition of CT production in the V. cholerae arcA mutant strain O395d2 was at the level of transcription, RNA was isolated from parent and mutant strains and Northern blot analysis was carried out using a 1.9-kb HindIII-XbaI fragment of the plasmid pCVD15 (21), carrying the ctxAB gene, as a probe. The amount of ctxAB-specific mRNA in strain O395d2 was considerably less than that in strain O395 (Fig. 2A). Hybridization to 16S rRNA was used as a control for equal loading (Fig. 2C). The amounts of ctxAB-specific mRNA in strains O395 and O395d2 were also estimated by RT-PCR. 16S rRNA production was used as an internal control. Analysis of the results obtained indicated a statistically significant (P = 0.001) difference in the relative intensities of PCR products corresponding to ctxAB mRNA obtained from wild-type and arcA mutant strains. After normalization according to the amounts of 16S rRNA present in each RNA population, the amount of ctxAB-specific mRNA in V. cholerae O395 was about fourfold higher than that in strain O395d2 (Fig. 3A, group I, lanes a and b, and B).
FIG. 2.
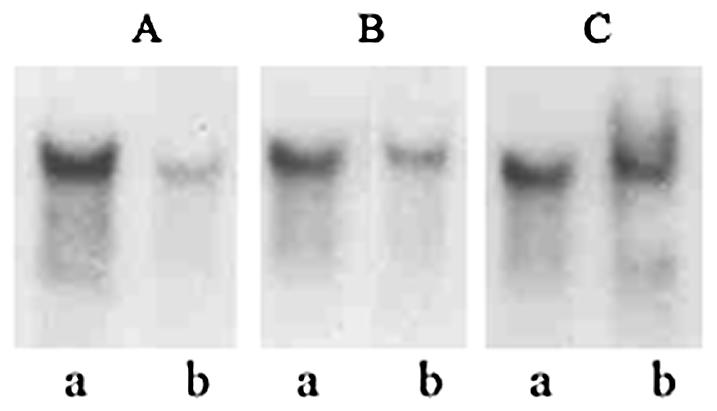
Northern blot analysis. V. cholerae O395 (lanes a) and O395d2 (lanes b) were grown in LB medium (pH 6.6) at 30°C; RNAs were isolated, and Northern blots were prepared and probed with [32P]dCTP-labeled fragments of the ctxAB (A), tcpA (B), or 16S rRNA (C) gene. Fifteen micrograms of RNA was loaded per well.
FIG. 3.

Expression of ctxAB and tcpA in V. cholerae wild-type and arcA mutant strains. (A) RT-PCR was performed with RNAs isolated from strains O395 (lanes a), O395d2 (lanes b), and O395d2 carrying the plasmid pNS1 (lanes c) for estimation of ctxAB mRNA (I), tcpA mRNA (II), and 16S rRNA (III). DNase-treated RNA samples that had not been reverse transcribed were used as negative controls (IV). Lane M, 100-bp DNA ladder. (B) Densitometric analysis of the ctxAB- and tcpA-specific RT-PCR products from strains O395 (open bars), O395d2 (horizontally hatched bars), and O395d2 carrying plasmid pNS1(diagonally hatched bars) after normalization according to the amount of RT-PCR product corresponding to 16S rRNA detected in the same cDNA sample. The results represent the averages of three independent experiments. The error bars indicate standard errors of the mean.
Since ctxAB gene expression is coordinately regulated with expression of the tcpA gene (42), transcription of tcpA was also examined in the V. cholerae arcA mutant. Both Northern blot analysis (Fig. 2B, lanes a and b) and RT-PCR (Fig. 3A, group II, lanes a and b, and B) indicated that the amount of tcpA-specific transcript was reduced two- to threefold in the arcA mutant strain O395d2 compared to that in the parental strain, O395. The difference was statistically significant (P = 0.001). Thus, a mutation in the arcA gene represses the expression of the two major virulence genes of the ToxR regulon, though the repression was more pronounced for ctxAB. Complementation of the arcA mutant strain O395d2 with the plasmid pNS1 carrying the full-length V. cholerae arcA gene could restore CT production to wild-type levels (Fig. 1). RT-PCR analysis indicated that expression of both ctxAB and tcpA increased to wild-type levels in V. cholerae O395d2 carrying the plasmid pNS1 (Fig. 3). These results suggest that repression of ctxAB and tcpA expression in the arcA mutant was indeed attributable to disruption of the arcA coding sequence and was not due to an unrecognized secondary mutation.
toxT expression is repressed in the V. cholerae arcA mutant.
Expression of the ctxAB and tcpA genes is coordinately regulated by the transcriptional activator ToxT (9). Since expression of both ctxAB and tcpA was reduced in the V. cholerae arcA mutant, toxT expression was examined in the mutant strain O395d2. Strains O395 and O395d2 were grown to mid-logarithmic phase under inducing conditions, RNAs were isolated, and toxT-specific transcripts were estimated by RT-PCR. toxT expression was found to be fivefold lower in strain O395d2 than in the parental strain, O395 (Fig. 4A). Thus, ArcA stimulates the expression of toxT in V. cholerae. Introduction of the full-length arcA gene in the plasmid pNS1 into the mutant O395d2 restores the toxT transcripts to wild-type levels (Fig. 4A, lanes c to e).
FIG. 4.

Effects of arcA mutation on toxT, toxR, and tcpP gene expression. (A) RT-PCR was performed for 30 (lanes a and b) and 35 (lanes c, d, and e) cycles with RNA isolated from V. cholerae strains O395 (lanes a and c), O395d2 (lanes b and d), and O395d2 carrying plasmid pNS1 (lane e) for estimation of toxT mRNA. (B) RT-PCR was performed for 25 cycles with RNA isolated from strain O395 grown under aerobic (lane a) or anaerobic (lane b) conditions and for 35 cycles with RNA from aerobically (lane c) or anaerobically (lane d) grown strain O395d2 for estimation of toxT mRNA. (C) RT-PCR was performed for 25 (lanes a and b) and 30 (lanes c and d) cycles with RNAs isolated from strains O395 (lanes a and c) and O395d2 (lanes b and d) for estimation of tcpP mRNA. (D) RT-PCR was performed for 30 cycles with RNAs isolated from strains O395 (lane a) and O395d2 (lane b) for estimation of toxR mRNA.
To examine if the reduction in CT production in the V. cholerae arcA mutant was indeed due to repression of toxT expression, plasmids pMT5-tet and pKEK162-tet containing the V. cholerae toxT gene (Table 1) (10, 36) were introduced into the arcA mutant strain O395d2, as well as the wild-type strain O395, and CT was examined in these cells under optimum ToxR-inducing conditions. CT production was restored to wild-type levels in strain O395d2 bearing the plasmid pMT5-tet or pKEK162-tet, although the plasmids had almost no effect on CT production in the wild-type strain (Fig. 5). These results indicated that ArcA does not directly affect expression of ctxAB or tcpA but most likely acts upstream of ToxT.
FIG. 5.

Complementation of CT production in the V. cholerae arcA mutant by ToxT. Strains O395 and O395d2 with or without the plasmid pMT5-tet or the plasmid pKEK162-tet were grown to stationary phase in LB medium (pH 6.6) at 30°C, and CT was measured in the culture supernatants corresponding to 109 CFU and expressed as percentages of the amount obtained in culture supernatants of strain O395 grown in LB (pH 6.6) at 30°C. The error bars indicate standard errors of the mean.
Since the proportion of active phosphorylated ArcA is known to increase during anaerobic growth of an organism (18, 20), the expression of toxT was also examined in anaerobically grown V. cholerae cells. V. cholerae strain O395 was grown under aerobic and anaerobic conditions to a density corresponding to 0.5 A600 units, RNA was extracted, and the amount of toxT-specific mRNA was estimated by RT-PCR. A fourfold increase in toxT mRNA was observed in the anerobically grown V. cholerae cells compared to that in cells grown with aeration (Fig. 4B, lanes a and b). In strain O395d2, the level of toxT mRNA was not appreciably altered between aerobic and anaerobic growth conditions (P = 0.2) (Fig. 4B, lanes c and d), indicating that the low levels of toxT mRNA produced in the arcA mutant were, as expected, not dependent on the oxygen concentration.
ArcA has no effect on toxR and tcpP expression.
Expression of toxT requires the transmembrane DNA binding proteins ToxR and TcpP, which act synergistically to activate transcription from the toxT promoter (8, 14). Since toxT expression was reduced in the V. cholerae arcA mutant, expression of ToxR and TcpP was examined in this strain. Expression of both tcpP and toxR in the V. cholerae arcA mutant strain O395d2 was found to be comparable to that in the parental strain, O395 (Fig. 4C and D). ToxR, in addition to its role in the virulence cascade, regulates the production of OmpU and OmpT, the major outer membrane porins of V. cholerae. While OmpU is positively regulated by ToxR, OmpT expression is apparently under the negative regulation of ToxR (31). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis of outer membrane proteins indicated that the OmpU-OmpT profile in the V. cholerae arcA mutant was similar to that in the wild-type strain O395 (data not shown).
LD50 assays in infant mouse cholera model.
In view of the fact that mutation in the arcA gene of V. cholerae reduces expression of the major virulence genes, the effect of the arcA mutation on the virulence of V. cholerae was examined in vivo using the infant mouse cholera model. Five-day-old mice were infected with V. cholerae strain O395, the arcA mutant strain O395d2, and strain O395d2 carrying the plasmid pNS1, and the LD50s were determined. The LD50 for strain O395d2 was >3 × 108, while that for strain O395 was <2 × 106. Thus, the V. cholerae arcA mutant was ∼100 times less virulent than the parental wild-type strain, O395, in the infant mouse cholera model. When strain O395d2 was complemented with the plasmid pNS1, the LD50 of the strain decreased to <2.5 × 106, which was comparable to that of the parental strain.
DISCUSSION
The major virulence genes of V. cholerae are carried within two horizontally acquired mobile genetic elements, CTXφ and VPIφ, which are the genomes of filamentous bacteriophages (23, 44). Similar to many pathogens that have acquired virulence genes on accessory genetic elements by horizontal transfer, in V. cholerae, several genes from the ancestral genome that have functions in normal bacterial physiology have been coopted to control the expression of the virulence genes of the pathogenicity islands. Virulence gene regulators that are not exclusively associated with the pathogenicity island and that probably participate in additional regulatory networks include the transmembrane proteins ToxR and ToxS (8), the cytoplasmic proteins AphA and AphB (25, 39), the heat shock proteins DnaK and σ32 (4, 33), and VarA, a member of the two-component family of response regulators (45), all of which activate the expression of virulence factors in V. cholerae. In addition, the expression of the virulence factors is negatively regulated by the cyclic AMP-cyclic AMP receptor protein complex (37) and the histone-like nucleoid-associated protein H-NS (32). In this study, we present evidence that another protein with global effects, ArcA, which controls the expression of a large number of anoxia-responsive genes, exerts a positive regulatory effect on the ToxR virulence regulon of V. cholerae. The V. cholerae arcA gene was cloned, and an internal fragment was used to construct a site-directed insertion mutation in the arcA gene of the wild-type strain, O395. The V. cholerae arcA mutant exhibited a dye-sensitive phenotype characteristic of E. coli arcA mutants. Furthermore, mutation in the arcA gene had significant effects on the expression of the major virulence genes of V. cholerae. Expression of both ctxAB and tcpA was reduced, and a significant decrease in the expression of the toxT gene was observed (Fig. 2 to 4). Since these defects could be complemented by the cloned arcA gene, the phenotypes were directly attributable to ArcA. Moreover, expression of toxT from an inducible promoter completely restored CT to wild-type levels in the V. cholerae arcA mutant strain (Fig. 5). This indicates that ArcA most likely acts upstream of toxT expression. Since neither the ToxR nor the TcpP protein, which act synergistically to activate toxT expression, was affected in the V. cholerae arcA mutant, it may be assumed that ArcA controls toxT expression.
ArcA-ArcB is a two-component signal transduction system which facilitates adaptation to anoxia (30). In response to the redox state of the cell, the ArcB kinase phosphorylates ArcA, and the concentration of phosphorylated ArcA (ArcA-P) is predicted to increase progressively during the transition from aerobic to anaerobic growth and to reach peak levels in anoxic cells (18, 20, 26). The Arc system appears to be active over a broad range of intracellular redox conditions, and significant levels of ArcA-P have been detected even in vigorously aerated cells (20). Indeed it has recently been demonstrated that ArcA is important for resistance to reactive nitrogen and oxygen intermediates in S. enterica serovar Enteritidis, even under aerobic conditions (28). The ArcA-P present during aerobic growth of V. cholerae may be responsible for the ∼5-fold-higher toxT expression observed in aerobically grown wild-type cells than in the arcA mutant (Fig. 4A). If ArcA-P is indeed a transcriptional activator of the toxT gene, a further increase in toxT transcription would be expected in cells grown anaerobically, in view of the fact that the ArcA-P concentration in the oxygen-deprived cells should be higher than that in cells grown under oxygen-rich conditions. Indeed, RT-PCR analysis with RNA isolated from wild-type V. cholerae cells grown under aerobic or anaerobic conditions indicated that toxT expression was ∼4-fold higher in the anaerobically grown cells (Fig. 4B, lanes a and b). In the arcA mutant strain O395d2, however, since only ArcA-independent expression of toxT occurred, no difference was observed between toxT expression levels in cells grown under aerobic and anaerobic conditions (Fig. 4B, lanes c and d). Taken together, these results indicate that ArcA positively influences the expression of the toxT gene in V. cholerae and that the effect is more pronounced under anaerobic conditions. The intestinal environment is assumed to be anaerobic, and indeed, we have found significant induction of genes known to be upregulated by anaerobiosis (melR) during intraintestinal growth of V. cholerae using the infant mouse cholera model (unpublished observation). It was therefore interesting to examine the virulence of the V. cholerae arcA mutant strain in vivo. The results obtained suggest that the V. cholerae arcA mutant is moderately attenuated for virulence: the LD50 of the mutant is ∼100-fold higher than that of the wild-type strain. This result was not unexpected in view of the fact that toxT expression, though reduced, was not abolished in the arcA mutant (Fig. 4).
Since ArcAB controls the expression of many anoxia-induced genes, it is difficult to explain why no growth impairment of the V. cholerae arcA mutant was observed under anaerobic conditions. The complete genome sequence of V. cholerae (15) reveals that the organism has several global regulatory systems known to respond to molecular oxygen, of which FNR and ArcAB are thought to be of particular importance in relation to cellular adaptation to anaerobic growth conditions. In E. coli, there is partial overlap between the FNR- and ArcA-regulated genes, so that many genes required for anaerobic growth may be controlled by either FNR or ArcA (2, 18, 34). In V. cholerae, since ArcA is not required for anaerobic growth, it is attractive to hypothesize that the overlap between the FNR and ArcAB regulons is more extensive, so that genes essential for anaerobic growth are not solely controlled by any one regulator. Such a strategy may be expected to contribute to the evolutionary success of a pathogen that has to survive under anaerobic conditions within the host body. It may be mentioned in this context that ArcA is not essential for anaerobic growth of S. enterica (28).
Bacteria possess numerous two-component signal transduction systems which facilitate adaptation to environmental conditions (40). In pathogens, virulence genes have coopted specific bacterial signal transduction systems that respond to environmental cues characteristic of the host system to ensure optimal virulence gene expression at the proper time during infection (11). However, the roles of two-component systems in the regulation of pathogenicity in V. cholerae have not been studied in detail. The vieSAB operon, with similarity to sensor-regulator systems, was expressed during in vivo growth of V. cholerae (27) and is required for CT production in vitro (43). Signature-tagged mutagenesis to screen for mutations affecting colonization in the infant mouse cholera model identified two phosphotransferases that are involved in modulating the expression of the ToxR regulon (5). More recently, varA, a member of the gacA family of two-component response regulators, has been shown to affect the ToxR regulon (45). The environmental cues responsible for the activation of these systems have not been elucidated. The Arc system is the first well-defined two-component system shown to modulate virulence expression in V. cholerae, although it is still not clear whether ArcA directly interacts with the toxT promoter. It is becoming increasingly clear that a complex interplay between global stress response regulators encoded within the ancestral genome and virulence genes encoded within pathogenicity islands imposes complex but subtle regulations at multiple levels of the virulence cascade in V. cholerae.
Acknowledgments
We are grateful to J. Das for generous advice and encouragement before his untimely death and to all members of the Biophysics Division for cooperation and helpful discussions during the study. We thank I. Guha Thakurta and P. Majumdar for excellent technical support. We are grateful to J. J. Mekalanos, Harvard Medical School, Boston, Mass.; V. J. DiRita, University of Michigan Medical School, Ann Arbor, Mich.; and K. E. Klose, University of Texas Health Science Center, San Antonio, Tex., for generous gifts of plasmids and strains.
This work was supported by research grant 61/2/2000-BMS from the Indian Council of Medical Research, Government of India. N.S. is grateful to the Indian Council of Medical Research for a research fellowship.
Editor: A. D. O'Brien
REFERENCES
- 1.Ausbel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.). 1989. Current protocols in molecular biology. John Wiley and Sons, New York, N.Y.
- 2.Bauer, C. E., S. Elsen, and T. H. Bird. 1999. Mechanisms for redox control of gene expression. Annu. Rev. Microbiol. 53:495-523. [DOI] [PubMed] [Google Scholar]
- 3.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. Complete genome sequence of Escherichia coli K-12. Science 277:1432-1434. [DOI] [PubMed] [Google Scholar]
- 4.Chakraborty, S., N. Sengupta, and R. Chowdhury. 1999. Role of DnaK in in vitro and in vivo expression of virulence factors of Vibrio cholerae. Infect. Immun. 67:1025-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiang, S. L., and J. J. Mekalanos. 1998. Use of signature-tagged transposon mutagenesis to identify Vibrio cholerae genes critical for colonization. Mol. Microbiol. 27:797-806. [DOI] [PubMed] [Google Scholar]
- 6.Contreras, I., C. S. Toro, G. Troncoso, and G. C. Mora. 1997. Salmonella typhi mutants defective in anaerobic respiration are impaired in their ability to replicate within epithelial cells. Microbiology 143:2665-2672. [DOI] [PubMed] [Google Scholar]
- 7.Cotter, P. A., and V. J. DiRita. 2000. Bacterial virulence gene regulation: an evolutionary perspective. Annu. Rev. Microbiol. 54:519-565. [DOI] [PubMed] [Google Scholar]
- 8.DiRita, V. J., and J. J. Mekalanos. 1991. Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell 64:29-37. [DOI] [PubMed] [Google Scholar]
- 9.DiRita, V. J. 1992. Co-ordinate regulation of virulence genes by ToxR in Vibrio cholerae. Mol. Microbiol. 6:451-458. [DOI] [PubMed] [Google Scholar]
- 10.DiRita, V. J., M. Neely, R. K. Taylor, and P. M. Bruss. 1996. Differential expression of the ToxR regulon in classical and E1 Tor biotypes of Vibrio cholerae is due to biotype-specific control over toxT expression. Proc. Natl. Acad. Sci. USA 23:7991-7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guiney, D. G. 1997. Regulation of virulence gene expression by the host environment. J. Clin. Investig. 99:565-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta, S., and R. Chowdhury. 1997. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect. Immun. 65:1131-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hale, T. L. 1991. Genetic basis of virulence in Shigella species. Microbiol. Rev. 55:206-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hase, C. C., and J. J. Mekalanos. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 95:730-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heidelberg, J. F., J. A. Eisen, W. C. Nelson, R. A. Clayton, M. L. Gwinn, R. J. Dodson, D. H. Haft, E. K. Hickey, J. D. Peterson, L. Umayam, S. R. Gill, K. E. Nelson, T. D. Read, H. Tettelin, D. Richardson, M. D. Ermolaeva, J. Vamathevan, S. Bass, H. Qin, I. Dragoi, P. Sellers, L. McDonald, T. Utterback, R. D. Fleishmann, W. C. Nierman, O. White, S. L. Salzberg, O. H. Smith, R. R. Colwell, J. J. Mekalanos, J. C. Venter, and C. M. Fraser. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrington, D. A., R. H. Hall, G. Losonsky, J. J. Mekalanos, R. K. Taylor, and M. M. Levine. 1988. Toxin, toxin co-regulated pili and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J. Exp. Med. 168:1487-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Householder, T. C., W. A. Belli, S. Lissenden, J. A. Cole, and V. L. Clark. 1999. cis and trans-acting elements involved in regulation of aniA, the gene encoding the major anaerobically induced outer membrane protein in Neisseria gonorrhoeae. J. Bacteriol. 181:531-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iuchi, S., and E. C. C. Lin. 1988. arcA (dye), a global regulatory gene in Escherichia coli mediating repression of enzymes in aerobic pathways. Proc. Natl. Acad. Sci. USA 85:1888-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iuchi, S., D. C. Cameron, and E. C. C. Lin. 1989. A second global regulator gene (arcB) mediating repression of enzymes in aerobic pathways of Escherichia coli. J. Bacteriol. 171:868-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iuchi, S., and E. C. C. Lin. 1992. Mutational analysis of signal transduction by ArcB: a membrane sensor protein for anaerobic expression of operons involved in the central aerobic pathways in Escherichia coli. J. Bacteriol. 174:3972-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaper, J. B., H. Lockman, M. M. Baldini, and M. M. Levine. 1984. A recombinant live oral cholera vaccine. Bio/Technology 2:345-349. [Google Scholar]
- 22.Kaper, J. B., J. G. Morris, and M. M. Levine. 1995. Cholera. Clin. Microbiol. Rev. 8:48-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karaolis, D. K., S. Somara., D. R. Maneval, Jr., J. A. Johnson, and J. B. Kaper. 1999. A bacteriophage encoding a pathogenicity island, a type-IV pilus and a phage receptor in cholera bacteria. Nature 399:375-379. [DOI] [PubMed] [Google Scholar]
- 24.Klose, K. E. 2001. Regulation of virulence in Vibrio cholerae. Int. J. Med. Microbiol. 291:81-88. [DOI] [PubMed] [Google Scholar]
- 25.Kovacikova, G., and K. Skorupski. 1999. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J. Bacteriol. 181:4250-4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwon, O., D. Georgellis, and E. C. C. Lin. 2000. Phosphorelay as the sole physiological route of signal transmission by the arc two-component system of Escherichia coli. J. Bacteriol. 182:3858-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee, S. H., M. J. Angelichio, J. J. Mekalanos, and A. Camilli. 1998. Nucleotide sequence and spatiotemporal expression of the Vibrio cholerae vieSAB genes during infection. J. Bacteriol. 180:2298-2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu, S., P. B. Killoran, F. C. Fang, and L. W. Riley. 2002. The global regulator ArcA controls resistance to reactive nitrogen and oxygen intermediates in Salmonella enterica serovar Enteritidis. Infect. Immun. 70:451-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lucas, R. L., and C. A. Lee. 2000. Unravelling the mysteries of virulence gene regulation in Salmonella typhimurium. Mol. Microbiol. 36:1024-1033. [DOI] [PubMed] [Google Scholar]
- 30.Lynch, A. S., and E. C. Lin. 1996. Responses to molecular oxygen, p. 1526-1538. In F. C. Neidhardt et al. (ed.), Escherichia coli andSalmonella typhimurium: cellular and molecular biology, vol. 1. ASM Press, Washington D.C.
- 31.Miller, V. L., and J. J. Mekalanos. 1988. A novel suicide vector and its use in the construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol. 170:2575-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nye, M. B., J. D. Pfau, K. Skorupski, and R. K. Taylor. 2000. Vibrio cholerae H-NS silences virulence gene expression at multiple steps in the ToxR regulatory cascade. J. Bacteriol. 182:4295-4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parsot, C., and J. J. Mekalanos. 1990. Expression of ToxR, the transcriptional activator of virulence factors in Vibrio cholerae, is modulated by the heat shock response. Proc. Natl. Acad. Sci. USA 87:9898-9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sawers, G. 1999. The aerobic/anaerobic interface. Curr. Opin. Microbiol. 2:181-187. [DOI] [PubMed] [Google Scholar]
- 35.Schmiel, D. H., G. M. Young, and V. L. Miller. 2000. The Yersinia enterocolitica phospholipase gene yplA is part of the flagellar regulon. J. Bacteriol. 182:2314-2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schuhmacher, D. A., and K. K. Klose. 1999. Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J. Bacteriol. 181:1508-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skorupski, K., and R. K. Taylor. 1997. Cyclic CMP and its receptor protein negatively regulate the coordinate expression of cholera toxin and toxin coregulated pilus in Vibrio cholerae. Proc. Natl. Acad. Sci. USA 94:265-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skorupski, K., and R. K. Taylor. 1997. Control of ToxR virulence regulon in Vibrio cholerae by environmental stimuli. Mol. Microbiol. 25:1003-1009. [DOI] [PubMed] [Google Scholar]
- 39.Skorupski, K., and R. K. Taylor. 1999. A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol. Microbiol. 31:763-771. [DOI] [PubMed] [Google Scholar]
- 40.Stock, J. B., A. M. Stock, and J. M. Mottonen. 1990. Sgnal transduction in bacteria. Nature 344:395-400. [DOI] [PubMed] [Google Scholar]
- 41.Straley, S. C., and R. D. Perry. 1995. Environmental modulation of gene expression and pathogenesis in Yersinia. Trends Microbiol. 3:310-317. [DOI] [PubMed] [Google Scholar]
- 42.Taylor, R. K., V. L. Miller, D. B. Furlong, and J. J. Mekalanos. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. USA 84:2833-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tischler, A. D., S. H. Lee, and A. Camilli. 2002. The Vibrio cholerae vieSAB locus encodes a pathway contributing to cholera toxin production. J. Bacteriol. 184:4104-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waldor, M. K., and J. J. Mekalanos. 1996. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272:1910-1914. [DOI] [PubMed] [Google Scholar]
- 45.Wong, S. M., P. A. Carroll, L. G. Rahme, F. M. Ausubel, and S. B. Calderwood. 1998. Modulation of expression of the ToxR regulon in Vibrio cholerae by a member of the two-component family of response regulators. Infect. Immun. 66:5854-5861. [DOI] [PMC free article] [PubMed] [Google Scholar]