

Abstract
Our long-term goal is to direct the evolution of novel protease variants. To this end we have engineered a new type of protease-activated reporter enzyme. Many protease-activated enzymes evolved in nature, but the introduction of novel regulatory mechanisms into normally unregulated enzymes poses a difficult design challenge. Random Elongation Mutagenesis [1] was used to fuse the p6 peptide, which is recognized and cleaved by HIV protease, and twelve random sequence amino acids to the C-termini of beta-glucuronidase (GUS) and alkaline phosphatase (AP). The resulting GUS- p6-(NNN)12 and AP-p6-(NNN)12 libraries were expressed in E. coli and screened for clones that were inactivated by the C-terminal extension (tail). The inactivated clones were co-expressed with HIV protease, and those that were re-activated were isolated. The AP and GUS activities of the most responsive clones were each >3.5-fold higher when co-expressed with HIV protease, and this activation is correlated with in vivo proteolysis. It should be possible to generalize this strategy to different reporter enzymes, different target proteases, and perhaps to other types of protein-modifying enzymes.
Keywords: Molecular switch, biosensor, reporter, random elongation mutagenesis, directed evolution
INTRODUCTION
The objective of this study is to develop high throughput protease assays for directed evolution studies, but the cleavage of most natural substrates is difficult to detect. We therefore sought to convert constitutively active reporter enzymes into protease-activated molecular switches. The design of reporters that are inactivated by proteases is relatively straightforward [2–9], but high throughput screens based on inactivation are more likely to produce false positives. Previous workers have designed clever molecular switches [10–16], but to our knowledge no one has described a general method to convert unregulated enzymes into zymogens.
Although many protease-activated enzymes exist in nature, the introduction of novel regulatory mechanisms into existing proteins remains a technical challenge to protein engineers. Many eukaryotic proteases are expressed in an inactive form until they are themselves proteolytically processed. The enzyme must initially fold into a particular catalytically inactive conformation and display its pro-peptide correctly. The pro-peptide acts as an auto-inhibitory domain by stabilizing the inactive conformation. Upon proteolytic removal of the pro-peptide, the activity of the protein is restored through a change in conformation. Such systems apparently evolve readily in nature, but are difficult to design by structure-based site-directed mutagenesis.
These post-translationally regulated enzymes are complex molecular machines, so it seems likely that they evolved from ancestors that were not post-translationally regulated. The regulatory mechanism could have evolved in two sequential steps (Fig. 1). First, recombination events, frame-shift mutations or point mutations in the stop codon extended one copy of the structural gene. A subset of these essentially random sequence extensions reduced the primary catalytic activity of the enzyme, either by occluding the active-site (intrasteric regulation) or stabilization of an inactive conformation (allosteric regulation). Second, the cell evolved novel mechanisms that blocked the intra-molecular interactions of the newly evolved auto-inhibitory domain, such as proteolysis, effector-binding or covalent modification. The capacity to regulate enzyme activities through post-translational modifications would have given the cell a selective advantage over its competitors. The evolution of the auto-inhibited proteins such as importin alpha, kinases, and phosphatases [17] might also have followed this three-step scheme.
Fig. (1).
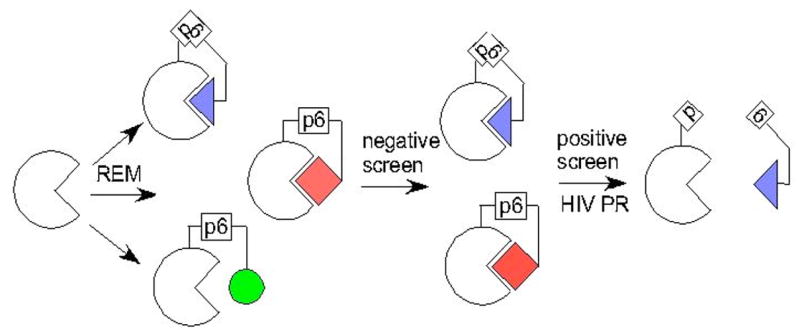
Evolution of novel auto-inhibitory domains. Ancient enzymes (represented as empty semi-circles with triangular active-sites) were probably constitutively active. Mutations and recombination events (Random Elongation Mutagenesis) fused random sequence peptides to the termini (represented as filled geometric shapes). Post-translational regulation evolved through selection against constitutively active variants (circle). Constitutively inactive variants (square, as HIV protease recognizes p6 peptide sequence only in bent conformation) were also disadvantageous.
Our plan was to create protease-activated reporters by recapitulating this hypothetical evolutionary process in vitro. We were inspired by a Random Elongation Mutagenesis (REM) study that showed that proteins are stabilized by selected peptide fusions [1]. We reasoned that some C-terminal peptide fusions could decrease activity, either by intrasteric or allosteric regulation, and that these novel auto-inhibitory activities could in turn be alleviated by proteolytic removal. Here we describe the REM of two dissimilar reporter genes and the identification of protease-activated variants.
MATERIALS AND METHODS
Materials
Escherichia coli strain InvaF’ was from Invitrogen (Carlsbad, CA); E. coli E15 (phoA8(del), garB10, fhuA22, ompF627(T2R), fadL701(T2R), relA1, pit-10, spoT1, rrnB-2, mcrB1, creC510) was obtained from the E. coli Genetic Stock Center, and converted into E15 (DE3) with the Lambda DE3 Lysogenation Kit (Novagen, Madison, WI). The lacI expression vector pREP4 was from Qiagen (Chatsworth, CA), pBAD myc his A from Invitrogen, and pSL1180 from Amersham/GE Healthcare. pCDF-Duet and pET28a(+) were from Novagen (Madison, WI). DNA sequencing kits were from Perkin-Elmer/Applied Biosystems (Foster City, CA). DNA purification columns were purchased from Qiagen (Chatsworth, CA). The Butterfly nitrocellulose membranes were from Schleicher and Schuell (Keene, NH). The histochemical GUS substrate, 5-bromo-4-chloro-3-indolyl-beta-D-glucuronide (X-gluc), was from Gold Biotechnology (St. Louis, MO); the histochemical AP substrate, 5-bromo-4-chloro-3-indolyl-phosphate (BCIP), was from Sigma Chemicals (St. Louis, MO). All DNA modifying enzymes (restriction enzymes, taq polymerase, T4 DNA ligase) were from New England Biolabs (Beverly, MA). Oligonucleotides were synthesized by IDT (Coralville, IA). Primary antibodies were from Abcam (Cambridge, MA) and secondary antibodies were from Rockland Immunochemicals (Gilbertsville, PA).
Plasmids and Cloning
The constitutive E. coli GUS expression vector, Plac*-6his-gusA-pET20, was previously described [18]. We subcloned the p6 (VSFNFPQITL), HA (YPYDVPDYA) and (NNN)12 in two consecutive whole plasmid PCR reactions [19]. First, we used primers HA-pET30 and N36-3’pETout2 to amplify Plac*-6his-gusA-pET20. The PCR product was purified, self-ligated and electroporated into InvαF’/pREP4. We purified the resulting Plac*-6his-gusA-HA-(NNN)12- pET20 plasmid library and PCR amplified it using primers p6-3’pET and SalI-HA. Again the PCR product was purified, self-ligated and electroporated into InvαF’/pREP4. The transformants were propagated in LB-ampicillin/kanamycin, and the Plac*-6his-gusA-p6-HA-(NNN)12-pET20 plasmid library was purified.
The inducible HIV protease expression vectors, p1+IQ (lacZ−) and PBAD-HIV PR-pACYC184 were constructed as follows. The expression vector p1+IQ[5] was obtained from the American Type Culture Collection. The lacZ gene was excised from this vector using Bam HI; the remaining DNA was purified, self-ligated and used to transform E. coli strain InvαF’. The HIV protease gene was subcloned from p1+IQ (lacZ−) into pBAD myc his A (Invitrogen) [20] using restriction enzymes Eco RI and Hind III. The araC-PBAD-HIV PR expression cassette was subcloned from HIV PR-pBAD plasmid into pACYC184 (New England Biolabs) using Sph I and Sal I.
The PBAD-HIV PR-pCDF expression vectors were constructed in two stages. First, we made the PBAD-pCDF expression vector by subcloning the araC repressor and PBAD promoter from pBAD myc his A into pSL1180 using Sph I and Nco I; the PBAD promoter was then subcloned from PBAD-pSL1180 into pCDF Duet using Nco I and Xba I. Second, the HIV protease gene in p1+IQ was PCR amplified using primers 5'NdeI-HIVpr and pUC-600, subcloned into pET28a+ using Nde I and Hind III and sequenced to confirm its wild-type identity. The subcloning fused DNA encoding a hexa-histidine tag to its 5' end. The inactivating D25N mutation was introduced into 6his-HIV PR-pET28 by whole circle PCR using primers HIVPR-D25N-62 and HIVPR-62out. The mutant gene was also sequenced to confirm its identity. The wild-type and D25N variants of the 6his-HIV PR gene were subcloned from their respective 6his-HIV PR- pET28 plasmids into PBAD-pCDF using Nco I and Xho I.
The phoA gene was amplified from the chromosome of E. coli using the primers, 5’-phoA and 3’phoA (Table 1), and cloned into pET28 using NdeI and HindIII. Whole plasmid PCR was used to insert the p6 peptide and 36 degenerate nucleotides (NNN)12 just upstream of the alkaline phosphatase (phoA) stop codon. The 3'spacer-p6-phoA and N36-3'-pET-out primers were used to amplify the entire phoA-pET28a+ plasmid. The PCR product was purified, self-ligated and electroporated into E. coli InvαF. The resulting phoA-p6-(NNN)12-pET28 plasmid library was purified and transformed into E15(DE3) for the high throughput screen.
Table 1.
Sequences of Primers Used in this Study
| Primer Name | Sequence (5’ to 3’) | Purpose |
|---|---|---|
| HA-pET30 | ccccgctagcataatccggcacatcatacgg
atacgcaagcttgtcgacggagctcgaa |
Constructing Plac*-6his-gusA-p6-HA-(NNN)12-pET20 |
| N36-3’pETout2 | ccccgctagc (nnn)12 tagcggccgca
ccaccaccactgagatccggct |
Constructing Plac*-6his-gusA-p6-HA-(NNN)12-pET20 |
| p6-3’pET | ggcgtcgactatccgtatgatgtgccggattatgct | Plac*-6his-gusA-p6-HA-(NNN)12-pET20 library |
| SalI-HA | ggcgtcgactatccgtatgatgtgccggattatgct | Plac*-6his-gusA-p6-HA-(NNN)12-pET20 library |
| 5’NdeI-HIVpr | ctaccattaccagttggtctggtgtca | Cloning of HIV PR |
| pUC-600 | gaaaacgttcttcggggcgaa | Cloning of HIV PR |
| HIVPR-D25N-62 | gaagctctattaaatacaggagcagatg | Cloning of D25N HIV PR |
| HIVPR-62out | ctttagttgcccccctatctttattgtg | Cloning of D25N HIV PR |
| 5’phoA | gggtctagacatatgaaacaaagcactattgcactggcac | Amplification of phoA from E. coli chromosome |
| 3'phoA | cccaagcttgagatctttcagccccagagcggctttcatgg | Amplification of phoA from E. coli chromosome |
| 3'p6-spacer-phoA | cgctagcgtgatctgcgggaagttgaagctcacggtcgcat
catccgggaagctctcgagtgcggccgcaagcttgaga |
phoA-p6-(NNN)12-pET28 library construction |
| N36-3'pETout | (nnn)12 tgagatccggctgctaacaaagcccga | phoA-p6-(NNN)12-pET28 library construction |
| 3’pET3 | gcatgccgtaaagcactaaatcggaacc | Sequencing of gusA and phoA tails |
| gusA-1440 | tatgtccaaagcggcgatttggaaacg | Sequencing of gusA tail |
| phoA 1325 | acataccggcagtcagttgcgtattgc | Sequencing of phoA tail |
DNA Sequencing
The most responsive clones, gusA (clone 461) and phoA (clone 1), were sequenced using the primers 3’pET for both 3’ends and gusA1440 and phoA-1325 for the 5’ ends, respectively. We employed the ABI BigDye 3.1 protocol, and sequencing reaction products were separated and analyzed at the Emory/Georgia Tech FAME Center. To obtain the phoA-1325 sequence, we employed several techniques including sequencing of the PCR product, altering the PCR conditions and outsourcing the job to utilizing SEQWRIGHT (Houston, TX), a company which specialized in the sequencing of difficult templates, especially GC-rich templates.
Western Blotting
Cultures containing gusA (clone 461) and phoA (clone 1) were expressed as described for the enzyme assays. One milliliter of culture was centrifuged for 5 minutes on a tabletop microfuge at 13,000rpm to collect the cells. The supernatant was discarded and the cells were resuspended in 100uLs 1X sodium dodecyl sulphate (SDS) buffer. Samples were then boiled and spun again at 13,000rpm. Ten microliters of each sample were loaded onto a 6% polyacrylamide protein gel and the proteins were transferred onto a nitrocellulose membrane. After transfer, the membrane was blocked for one hour using 4% non-fat milk dissolved in phosphate buffered saline (PBS). The membrane was then probed with a primary antibody for one hour. A polyclonal rabbit anti-_-glucuronidase antibody diluted 1:7,000 and a monoclonal mouse anti-HA antibody diluted 1:10,000 was used for the engineered gusA and a monoclonal mouse anti-alkaline phosphatase antibody diluted 1:10,000 was used for the engineered phoA. The membrane was then washed 4X with PBS containing 0.1% Tween-20 and probed with goat anti-rabbit or goat anti-mouse IRD labeled antibodies diluted 1:10,000. After washing again 4X with PBS containing 0.1% Tween-20, the immobilized antibody-antigen complexes were visualized and quantified on a LI-COR Odyssey Infrared imager.
RESULTS
The technical endpoint of this study is to attach cleavable auto-inhibitory domains to two E. coli-derived reporter enzymes, GUS and AP. These reporters are good starting points because they are commonly used, well characterized and different enough from each other to demonstrate that our approach is not restricted to any particular protein fold. It would be difficult to predict the exact amino acid sequence of a cleavable auto-inhibitory peptide, so random mutagenesis and screening were employed. Different vectors and E. coli strains were used for the over-expression of gusA and phoA, respectively, but the overall strategy was identical. Random sequences were inserted at the C-termini of both genes by an inverse PCR method called “whole plasmid PCR”[21], and screened for clones that were active only when co-expressed with Human Immunodeficiency Virus protease (HIV PR). This protease is a good model because it is well characterized, can be expressed in E. coli [5], and is very relevant to human disease.
A constitutive GUS expression vector, Plac*-6his-gusA- pET20 [18] was used as an ancestral (starting) plasmid. DNA encoding three consecutive peptides was inserted just upstream of the 3' stop codon in sequential whole plasmid PCRs [19] (Fig. 4 a ). The first peptide was p6 (VSFNFPQITL), which when displayed in the correct orientation is efficiently cleaved by HIV PR [5]. The second was the HA epitope (YPYDVPDYA), which could be used to monitor protein expression. The third was degenerate (NNN)12 sequence encoding twelve random amino acids.
Fig. (4).

Sequencing of the engineered reporter proteins (A) p6-HA-(NNN)12 fused to beta-glucuronidase (B) A chromatogram showing high quality sequencing halting just upstream of the random nucleotides fused to AP.
E. coli InvαF' cells were co-transformed with the Plac*- 6his-gusA -p6-HA-(NNN)12-pET20 library and the inducible HIV protease expression vector, PBAD-HIV PR-pACYC184. The transformed cells were propagated on LB agar plates supplemented with 50 micrograms/mL ampicillin and 34 micrograms/mL chloramphenicol and 80 micrograms/mL X-gluc. The GUS variants were expressed under these conditions; HIV PR was not. We examined ~20,000 colonies and picked ~400 that exhibited diminished GUS activity (lighter blue) relative to the parental control colonies. The latter were re-streaked on LB-amp X-gluc plates and identical plates supplemented with 0.2% L-arabinose, which induced the expression of HIV protease. Thirteen clones exhibited greater GUS activity on the plates supplemented with 0.2% L-arabinose than on the plates lacking this inducer, and one of these (gusA -p6-HA-(NNN)12 clone 461) reproducibly exhibited the most pronounced HIV protease-dependent activation.
To quantify the HIV protease-dependence, InvαF’ cells were co-transformed with the selected (clone 461) Plac*-6his-gusA-p6-HA-(NNN)12-pET20, and either PBAD-WT HIV PR-pCDF or PBAD-D25N HIV PR-pCDF. The latter vectors produce the wild-type or a catalytically inactive (but correctly folded) variant of HIV protease [22], but are otherwise identical. The transformed cells were propagated to mid-log phase in liquid LB medium supplemented with 50 micrograms/mL ampicillin and 50 micrograms/mL spectinomycin. Gene expression was induced for three hours with 0.5 mM IPTG and 0.2% L-arabinose. We added 50 microliters of each culture (in triplicate) to 250 microliters 2 mM para-nitrophenyl-beta-D-glucuronide in 50 mM Tris, pH7.6. The chromogenic substrate is able to permeate the intact cells and react with the GUS [21]. The formation of the para-nitrophenol product was followed in a microplate spectrophotometer (405 nm) for 30 minutes (Fig. 2a). The cells expressing the gusA-p6-HA-(NNN)12 and wild-type HIV protease exhibited >3.5-fold more GUS activity than isogenic cells expressing the D25N protease mutant (Fig. 2c). These results show that the GUS activity of gusA-p6-HA-(NNN)12 clone 461 is dependent upon the catalytic activity of HIV protease rather than some other indirect mechanism.
Fig. (2).
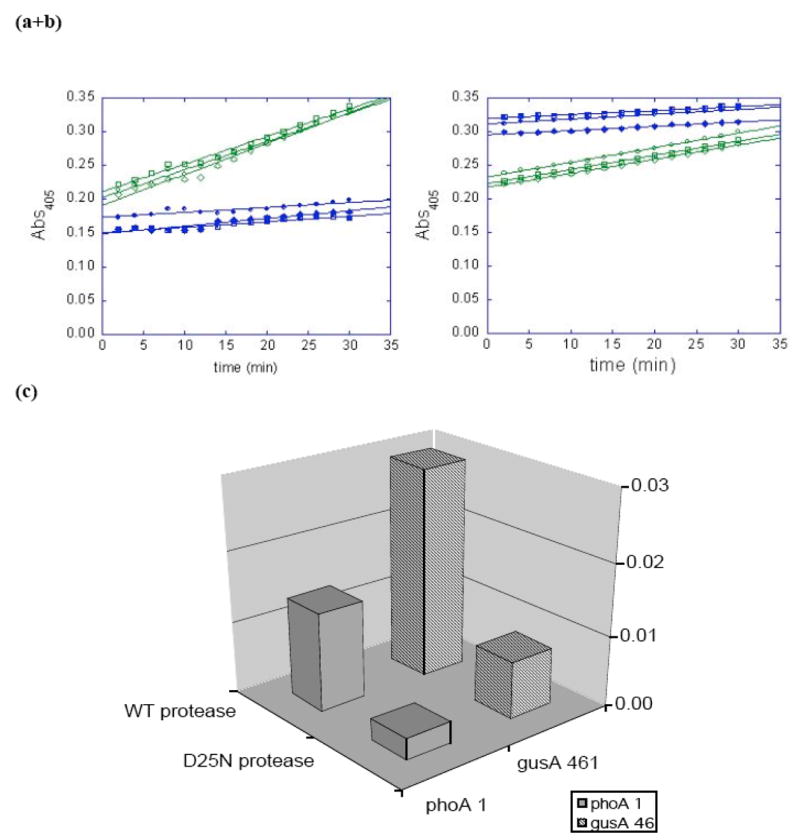
HIV protease activates beta-glucuronidase and alkaline phosphatase variants. E. coli cells were propagated and induced to co-express a reporter (selected beta-glucuronidase (A) or alkaline phosphatase (B)) variant AND HIV protease (wild-type = □, △, ○; catalytically inactive D25N = ■, ▲,●). The intact cells were reacted with pNP-glucuronide (A) or pNP-phosphate (B), which were apparently cell-permeable; the formation of the pNP product was followed in a microplate spectrophotometer at 405 nm. The rates of production formation were calculated from the slopes of the lines in plots 2A and B (C).
Western blot analysis showed a correlation between cleavage of the p6 site in vivo and increased GUS activity (Fig. 3a). Antibodies specific to GUS and to the C-terminal HA tag, each associated with secondary antibodies conjugated to unique dyes (one that absorbs infrared light at 700nm and one that absorbs at 800nm), were used to observe cleavage of the p6 peptide in the same gel. The first panel in Fig. 3a shows a blot with the polyclonal anti-GUS antibodies. The band corresponding to GUS (~72kDa) is shown; there are many non-specific bands illuminated with this antibody which were eliminated for clarity. A doublet can be seen in the lane where gusA 461 is coexpressed with WT HIV PR, indicating cleavage of the roughly 3kDa “tail”. The second panel shows a blot with the anti-HA antibody and, though equal volumes of cells were loaded, there is clearly a decrease in the intensity of this band when coexpressed with WT HIV PR. Since the p6 region is present upstream of the HA sequence in the tail region, cleavage of p6 would eliminate the HA epitope from the tail. Finally, the last panel of this figure is a merge of the two antibodies. The doublet can again be seen in the lane with WT HIV PR, only now it is clear that the top band corresponds to the unaltered engineered protein, containing the HA tag and the bottom band represent the cleaved GUS. This result, combined with the activity assays, shows that cleavage at the p6 site correlates with increased GUS activity.
Fig. (3).
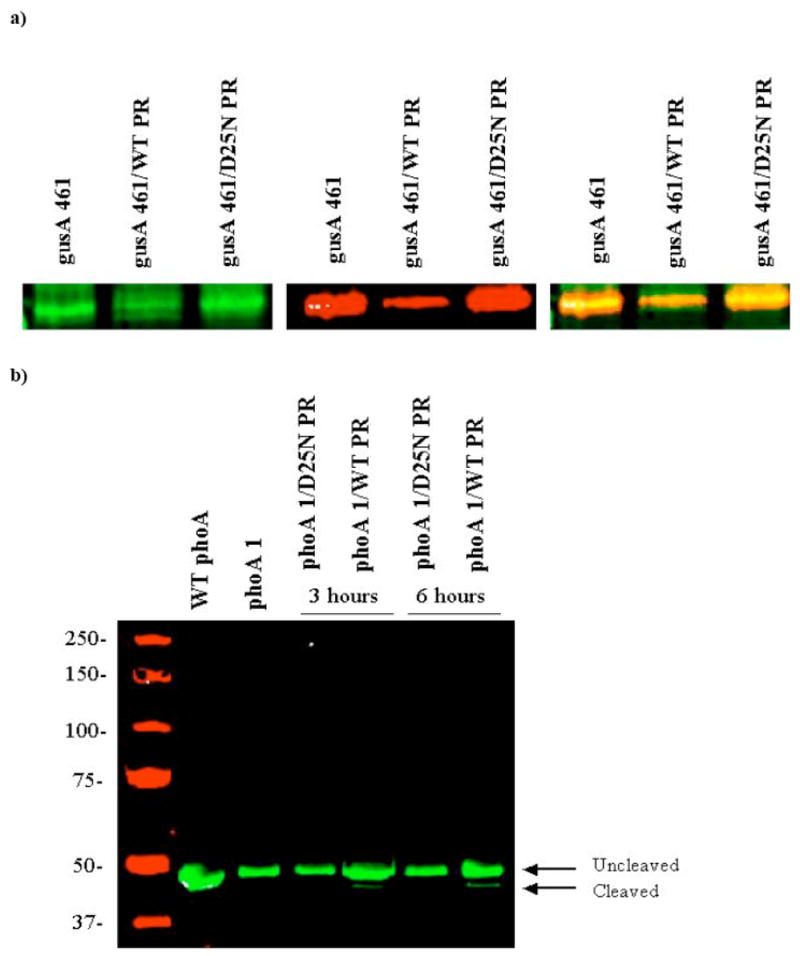
Western blots showing cleavage of the engineered reporter proteins. (A) A western blot was performed with anti-GUS (green) antibodies and anti-HA (red) antibodies. The left panel is a blot probed for GUS alone, the middle is probed for HA alone and the right-most panel shows a merge of the two images. The arrow and white box represents the expected size of GUS. (B) A western blot was performed with anti-AP antibodies. The blot shows a clear cleavage product when the engineered AP is coexpressed with WT HIV PR.
An alkaline phosphatase (AP) based HIV protease sensor was also created to demonstrate that the approach can be generalized to other reporter enzymes. Whole plasmid PCR was used to create a phoA-p6-(NNN)12-pET28 library. The library was electroporated into E. coli E15 (DE3) cells, which lack their chromosomal phoA genes. The transformed cells were propagated on LB-kanamycin agar plates. The resulting colonies were adsorbed to nitrocellulose membranes and transferred colony-side up to LB-kanamycin plates supplemented with 0.5 mM IPTG and 0.08 mg/mL (BCIP). We screened ~3000 colonies and identified six hypomorphs (less AP activity than wild-type control colonies). These were re-streaked onto LB-kanamycin plates, and filter-lifted onto LB-kanamycin, 0.5 mM IPTG, 80 micrograms/mL BCIP agar plates. The colonies derived from one of the six isolates (clone 1) consistently turned blue very slowly (~12 hours at 37 degrees, compared to 30 minutes for the wild-type phoA control), and this single clone was selected for further characterization.
E. coli E15 (DE3) cells were co-transformed with the selected (clone 1) phoA -p6-(NNN)12-pET28 plasmid and either PBAD-WT HIV PR-pCDF or PBAD-D25N HIV PR- pCDF. The transformed cells were propagated in LB-kanamycin/spectinomycin media to mid-log phase, and the proteins were induced with 0.5 mM IPTG and 0.2% L-arabinose for three hours. The cells were reacted with para-nitrophenyl-phosphate, and the formation of the pNP product was followed in a spectrophotometer for 30 minutes (Fig. 2b). The cells expressing the AP-p6-(NNN)12 and wild-type HIV protease exhibited almost 5-fold more activity than isogenic cells expressing the catalytically inactive D25N protease (Fig. 2c). These results demonstrate that the AP activity of clone 1 (like that of GUS clone 461) is dependent upon protease activity.
Western blot analysis was again used to monitor proteolysis of the engineered AP in vivo. E. coli cultures expressing AP clone 1 and either the WT or D25N HIV protease were lysed, and the protein was separated by SDS-PAGE and probed with a monoclonal anti-AP antibody (Fig. 3b). The engineered protein should run at about 50.5kDa and cleavage at the p6 site cuts off about 2kDa, leaving a product of 48.5kDa. A cleavage product can be seen in the lanes expressing the engineered protein along with the WT HIV PR of the expected size. The lower molecular weight of AP makes the cleavage product even easier to visualize than that of the GUS variant. These results therefore indicate a correlation between HIV PR activity, cleavage of the AP protein at the expected site and increased enzyme activity.
The gusA -p6-HA-(NNN)12 (clone 461) and p h o A-p6- (NNN)12 (clone 1) genes were sequenced. The gusA variant included the expected p6 and HA sequences, followed by PIVLCVGVYIYL (Fig. 4a). These peptides (including spacers) add a total of 38 hydrophobic amino acids to the C-terminus of GUS. We attempted to sequence the phoA-p6- (NNN)12 gene, but found that the otherwise high quality sequence stopped precisely where the (NNN)12 sequence was expected to start (Fig. 4b). This result is reproducible for both strands of DNA, even when it is encoded in a small PCR fragment. It suggests to us that the (NNN)12 sequence has significant secondary structure at the 65 degree temperature of the sequencing reaction.
DISCUSSION
The whole cell assays and Western blot experiments establish a correlation between HIV protease expression, proteolysis at inserted p6 sites and increased reporter enzyme activity. It is not clear, however, how the selected C-terminal peptides inhibit the activity of the reporter enzyme prior to proteolysis. The peptide extensions might occlude the active-site, as shown in Fig. 1. Alternatively, they might destabilize the entire fusion protein, or in the case of alkaline phosphatase interfere with proper secretion. We have purified HIV protease, and are working to purify the GUS-p6-HA-(NNN)12 (clone 461) and AP-p6-(NNN)12 proteins in order to investigate these mechanisms in vitro.
We have shown that Random Elongation Mutagenesis and high throughput screening can be used to produce protease-dependent enzyme activities. This approach could likely be extended to other reporters, other proteases and possibly other protein-modifying activities. Ultimately, it could lead to the fabrication of E. coli strains that are dependent upon non-native protease activities for survival. Such strains would enable ultra-high throughput genetic selections for the directed evolution of interesting protease variants, or for the identification of novel protease inhibitors.
Acknowledgments
IM constructed PBAD-HIV PR-pCDF and the Plac*-6his-gusA-HA-(NNN)12-pET20 library; he isolated GUS (clone 461) and fused the gene (and the selected peptide) to lacZ. This work was supported by the NIH/NIAID (1 R21AI054602-01).
References
- 1.Matsuura T, Miyai K, Trakulnaleamsai S, Yomo T, Shima Y, Miki S, Yamamoto K, Urabe I. Nat Biotechnol. 1999;17:58–61. doi: 10.1038/5232. [DOI] [PubMed] [Google Scholar]
- 2.Sices HJ, Kristie TM. Proc Natl Acad Sci U S A. 1998;95:2828–2833. doi: 10.1073/pnas.95.6.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kupiec JJ, Hazebrouck S, Leste-Lasserre T, Sonigo P. J Biol Chem. 1996;271:18465–18470. doi: 10.1074/jbc.271.31.18465. [DOI] [PubMed] [Google Scholar]
- 4.Dautin N, Karimova G, Ullmann A, Ladant D. J Bacteriol. 2000;182:7060–7066. doi: 10.1128/jb.182.24.7060-7066.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baum EZ, Bebernitz GA, Gluzman Y. Proc Natl Acad Sci U S A. 1990;87:10023–10027. doi: 10.1073/pnas.87.24.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Block TM, Grafstrom RH. Antimicrob Agents Chemother. 1990;34:2337–2341. doi: 10.1128/aac.34.12.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balint RF, Plooy I. Biotechnology (N Y) 1995;13:507–510. doi: 10.1038/nbt0595-507. [DOI] [PubMed] [Google Scholar]
- 8.Deo SK, Lewis JC, Daunert S. Anal Biochem. 2000;281:87–94. doi: 10.1006/abio.2000.4539. [DOI] [PubMed] [Google Scholar]
- 9.Murray MG, Hung W, Sadowski I, Das Mahapatra B. Gene. 1993;134:123–128. doi: 10.1016/0378-1119(93)90185-6. [DOI] [PubMed] [Google Scholar]
- 10.Buskirk AR, Liu DR. Chem Biol. 2005;12:151–161. doi: 10.1016/j.chembiol.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 11.Doi N, Yanagawa H. FEBS Lett. 1999;453:305–307. doi: 10.1016/s0014-5793(99)00732-2. [DOI] [PubMed] [Google Scholar]
- 12.Dueber JE, Yeh BJ, Chak K, Lim WA. Science. 2003;301:1904–1908. doi: 10.1126/science.1085945. [DOI] [PubMed] [Google Scholar]
- 13.Guntas G, Mitchell SF, Ostermeier M. Chem Biol. 2004;11:1483–1487. doi: 10.1016/j.chembiol.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 14.Radley TL, Markowska AI, Bettinger BT, Ha JH, Loh SNJ. Mol Biol. 2003;332:529–536. doi: 10.1016/s0022-2836(03)00925-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsien RY. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 16.Tucker CL, Fields S. Nat Biotechnol. 2001;19:1042–1046. doi: 10.1038/nbt1101-1042. [DOI] [PubMed] [Google Scholar]
- 17.Kobe B, Heierhorst J, Kemp BE. Intrasteric regulation of protein kinases. Adv Second Messenger Phosphoprotein Res. 1997;31:29–40. doi: 10.1016/s1040-7952(97)80006-7. [DOI] [PubMed] [Google Scholar]
- 18.Matsumura I, Olsen MJ, Ellington AD. Biotechniques. 2001;30:474–476. doi: 10.2144/01303bm01. [DOI] [PubMed] [Google Scholar]
- 19.Geddie ML, Rowe LA, Alexander OB, Matsumura I. Methods Enzymol. 2004;388:134–145. doi: 10.1016/S0076-6879(04)88012-1. [DOI] [PubMed] [Google Scholar]
- 20.Vickrey JF, Logsdon BC, Proteasa G, Palmer S, Winters MA, Merigan TC, Kovari LC. Protein Expr Purif. 2003;28:165–172. doi: 10.1016/s1046-5928(02)00650-2. [DOI] [PubMed] [Google Scholar]
- 21.Rowe LA, Geddie ML, Alexander OB, Matsumura I. J Mol Biol. 2003;332:851–860. doi: 10.1016/s0022-2836(03)00972-0. [DOI] [PubMed] [Google Scholar]
- 22.Babe LM, Rose J, Craik CS. Proc Natl Acad Sci U S A. 1995;92:10069–10073. doi: 10.1073/pnas.92.22.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]