

Abstract
Amyloid β peptide (Aβ)-containing plaques are a hallmark of Alzheimer disease. Here, we show that the neurotoxic Aβ, a major plaque component, is a potent activator of the transcription factor NF-κB in primary neurons. This activation required reactive oxygen intermediates as messengers because an antioxidant prevented Aβ-induced NF-κB activation. Maximal activation of NF-κB was found with 0.1 μM Aβ-(1–40) and 0.1 μM Aβ-(25–35) fragments, making a role for NF-κB in neuroprotection feasible. Using an activity-specific mAb for the p65 NF-κB subunit, activation of NF-κB also was observed in neurons and astroglia of brain sections from Alzheimer disease patients. Activated NF-κB was restricted to cells in the close vicinity of early plaques. Our data suggest that the aberrant gene expression in diseased nervous tissue is at least in part due to Aβ-induced activation of NF-κB, a potent immediate–early transcriptional regulator of numerous proinflammatory genes.
Keywords: reactive oxygen intermediates, senile plaques, neuroprotection, H2O2, neuronal cell death
A characteristic of Alzheimer disease (AD) is the accumulation of a 40–43-amino acid peptide termed “amyloid β-peptide” (Aβ or βA4) (1, 2). This peptide is the major component of senile plaques found in AD and in Down syndrome. Aβ is a proteolytic fragment of the large amyloid precursor protein (APP) (3). Several animal models recently have underscored the importance of APP for neuronal function. A knockout mouse, in which a shorter exon 2-lacking form of APP is expressed, shows memory defects (4). Two APP-null mutation strategies recently have been developed (5, 6). APP-null mutation leads to impaired neuronal function and reactive gliosis (5). Overexpression of APP leads to the formation of senile plaques and synaptic loss (7). In addition, Aβ was found to be neurotoxic for primary neurons and clonal cell lines (8–11). The neurotoxic action of Aβ could be blocked by antioxidants (10) and relied on the production of reactive oxygen intermediates, e.g., hydrogen peroxide (11).
AD is accompanied by the expression of inflammatory cytokines and cell adhesion molecules, including interleukin 6 (IL-6), IL-1β, and intercellular adhesion molecule 1, in neurons and microglia (12). Several studies suggest that the neurodegenerative effects in AD can, at least in part, be attributed to the action of neurotoxic cytokines, such as IL-6 (13, 14). We have noticed that many of the genes newly induced in AD are under immediate–early transcriptional control of NF-κB, a factor present in neurons as well as microglia (15, 16). Many mostly pathogenic stimuli can activate NF-κB (17), leading to the expression of a cellular defense program. In this line, activated NF-κB is a positive regulator of genes, whose products mediate the acute phase response, lymphoproliferation, leukocyte adhesion, chemoattraction of macrophages, B and T cell activation, and antiviral response. Very recently, the neurotransmitter glutamate and its agonists kainate and N-methyl-d-aspartate were shown to cause activation of NF-κB in primary neurons (18, 19). Glutamate plays a role in both neurotransmission and neurodegeneration and has been shown to induce oxidative stress in neurons (20).
In nonstimulated cells, NF-κB is sequestered in the cytoplasm by inhibitory subunits called IκB proteins. Stimulation of cells by diverse inducers causes phosphorylation of IκB-α and subsequent degradation of IκB-α by the proteasome (21). Liberated NF-κB is transported in the nucleus, where it induces transcription of target genes, including IκB-α as an autoregulatory loop (21). Reactive oxygen intermediates (ROIs) can function as second messengers in NF-κB activation (22–25). Cytoplasmic NF-κB, containing p50 and p65 (RelA) DNA-binding subunits, was found in all regions of the brain (26) and in basically all cell types of the nervous system (16, 18, 27, 28). The p65 subunit is essential for survival (29).
Here we report that the neurotoxic peptide Aβ is a potent inducer of NF-κB in primary neurons and astrocytes. This activity of Aβ required the production of ROIs as messengers. Immunohistochemical analysis of brain sections from AD patients using a mAb with selectivity for the activated nuclear form of p65 revealed that NF-κB was activated in neurons and astrocytes. Cells with activated NF-κB were restricted to the close proximity of early plaque stages. We discuss the possibility that (i) Aβ-induced NF-κB activation contributes to the pathological changes observed in AD via the induction of proinflammatory and cytotoxic genes or, more likely, that (ii) Aβ-induced NF-κB activation is part of a cellular defense program.
MATERIALS AND METHODS
Primary Culture Aβ Treatment and Cell Survival Analysis.
Cerebellar granule cell cultures were prepared from 6–7-day-old Wistar rats (18). At this stage, α-p65 mAb immunoreactivity in cerebellar neurons is very low (18). Aβ-(25–35) (lots 506861 and 510178) and Aβ-(1–40) (lots 506773 and 510598) (Bachem) fragments were dissolved in water at 1 mg/ml directly before use or the solution was aliquoted and stored frozen at −20°C. Refreezing was avoided. One batch of Aβ-(1–40) tested for aggregation with Congo red staining and electron microscopy was obtained from Boehringer Ingelheim. A scrambled control peptide (sequence KSGNMLGIIAG; ref. 11) was synthesized and purified by HPLC. Substance P, bovine catalase, pyrrolidine dithiocarbamate (PDTC), and 3-amino-1,2,4-triazole were from Sigma. Inhibition of bovine catalase (specific activity 2200 units/mg protein) was done with a 5-fold molar excess of 3-amino-1,2,4-triazole for 1 h at ambient temperature followed by dialysis on Millipore filters. Cells were treated with the indicated concentrations of peptides in medium with serum for 45 min to 3 days. Thereafter, cells were fixed for 2 min in ethanol and for 5 min in 3.7% formaldehyde and immunostained. The sequence of the peptide used for antibody competition is described elsewhere (28). For analysis of neuronal survival in parallel to immunostaining, the fluorescent nuclear dye 6-diamidino-2-phenylindole (DAPI) was used. Nuclear chromatin morphology was analyzed with a × 40 objective. Nonviable neurons were recognized by nuclear condensation and/or fragmented chromatin. In phase contrast images, those neurons were irregularly shaped with shrunken cell body and/or dystrophic neurites. The number of viable and nonviable neurons was counted in three to five fixed fields per chamber.
Immunostaining Methods.
Isocortical tissue was obtained post mortem from patients with histopathologically confirmed AD. Cryostat sections (8 μm) were cut from frozen tissue using a Jung cryostat (Leica, Heidelberg) and mounted on gelatine-coated slides. Immunohistochemistry was performed essentially as previously described (15, 18, 28). For double labeling experiments, the brain sections were incubated with the two antibodies (diluted 1:50): an mAb against p65 (Boehringer Mannheim) and a polyclonal rabbit anti-Aβ antiserum (anti-β amyloid, Alzheimer; Boehringer Mannheim). Bound antibodies were detected with a biotinylated anti-mouse IgG/avidin Cy3 complex and an anti-rabbit IgG coupled with dichlorotriazinyl amino fluorescein. In addition, 50μm paraffin-embedded sections were cut from autopsy material obtained from the local Department of Neuropathology. Before immunohistochemistry, sections were treated for 10 min in a microwave oven in 0.1 M citrate buffer (pH 6.0). α-p65 mAb (1:50) was detected with a peroxidase-coupled secondary antibody using nickel-enhanced diaminobenzidine as substrate (Vectastain Elite, Vector Laboratories). Peptide preabsorbtion was done as described (18). Color slides for Figs. 1, 2, 3, 4 were digitized using a Nikon Coolscan connected to an Apple Macintosh computer. Color slides for Fig. 5 were digitized with a Linotype-Hell Topaz Scanner at high resolution. Mounting of figures was done using adobe photoshop software.
Figure 1.

Activation of NF-κB by Aβ in rat cerebellar granule cells. Primary neuronal cells were treated with 100 nM Aβ-(1–40) (d-f) and Aβ-(25–35) (g-i) for 45 min or left untreated (a-c). (a, d, g) Indirect immunofluorescence analysis of cell cultures for p65 NF-κB immunoreactivity using the activity-specific α-p65 mAb (28) and a Cy3-conjugated second antibody (see Materials and Methods). (b, e, h) DNA staining of nuclei in cell cultures using DAPI. (c, f, i) Phase contrast micrographs. N, neurons; A, astrocytes. (Bar = 50 μm.)
Figure 2.

Concentration dependence of Aβ-induced NF-κB activation in cerebellar granule cells. (A) Granule cells treated with Aβ-(1–40). (B) Granule cells treated with Aβ-(25–35). (Upper) Indirect immunofluorescence analysis of primary cell cultures for binding of α-p65 mAb. Cells were stimulated for 45 min with the indicated concentrations of Aβ-(1–40). Sections with two to four neurons are shown. Neurons were identified by DAPI staining and by their morphology upon phase contrast microscopy. Analysis of the immunofluorescence intensity of a larger area of the specimen in one optical plane. Histograms for each concentration are shown. The ordinate (linear scale) depicts relative amounts of pixels. The fluorescence intensities of the Cy3 chromophor are shown on a linear scale on the abscissa. Arrows indicate the maximal signals obtained at 100 nM Aβ peptides.
Figure 3.

The effect of the antioxidant PDTC and catalase on Aβ-induced NF-κB activation. Granule cells were incubated for 45 min with 100 nM Aβ alone [a and b, Aβ-(1–40); c and d, Aβ-(25–35)] or with Aβ and 100 μM PDTC, which was added to cell cultures 10 min before Aβ [e and f, Aβ-(1–40); g and h, Aβ-(25–35)], or with 100 nM Aβ plus 400 μg/ml catalase, which was added to cells 1 h before the Aβ peptide [i and j, Aβ-(1–40); k and l, Aβ-(25–35)]. Cell cultures were analyzed by indirect immunofluorescence for an increase of α-p65 mAb immunoreactivity (a, e, i, c, g, k) and by nuclear DAPI-staining (b, f, j, d, h, l).
Figure 4.

Colocalization of α-p65 mAb and anti-Aβ immunoreactivities in senile plaques from brains of AD patients. In double immunofluorescence experiments, cryosections from patients with AD were immunostained with polyclonal anti-Aβ (b) and monoclonal anti-p65 (a) antibodies. Bound antibodies were detected by species-specific second antibodies conjugated to either Cy3 [red fluorescence, anti-p65 (a)] or dichlorotriazinyl amino fluorescein [green fluorescence, anti-Aβ (b)]. (Bar = 10 μm.)
Figure 5.
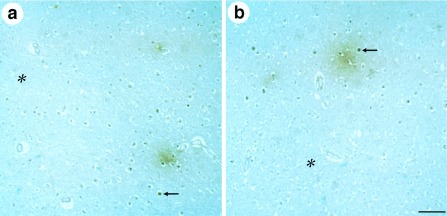
Immunohistochemical analysis of plaques in cortical sections of AD brains for α-p65 mAb immunoreactivity. Plaques are visualized by immunoreactivity (brown color). Examples of α-p65 immunoreactive cell nuclei (arrows) and surrounding plaques are depicted. Different examples for primitive plaque types are shown. Regions distant from plaques have reduced α-p65 immunoreactivity (asterisks). (Bar = 50 μm.)
RESULTS
Nanomolar Concentrations of Aβ Peptides Activate Transcription Factor NF-κB in Primary Neurons.
We investigated by an immunocytochemical approach whether Aβ could activate transcription factor NF-κB in primary cultures of cerebellar granule cells. A recently developed mAb (α-p65 mAb; ref. 28) was used that selectively stains the activated nuclear form of NF-κB and allows monitoring of NF-κB activation at the single cell level. The antibody recognizes an epitope on the DNA-binding p65 subunit, which is shielded by IκB in the inactive cytoplasmic form of NF-κB (28). Because the p65 gene is not rapidly inducible (30, 31), the appearance of nuclear p65 immunoreactivity results from the release of IκB during posttranslational activation.
An untreated control culture of cerebellar granule cells showed weak α-p65 mAb immunoreactivity, which did not overlap with the nuclei. Each image in Fig. 1, a-c shows a number of neurons (small nuclei) and an astrocyte (large nucleus). Treatment with 100 nM of Aβ peptides [Aβ-(1–40) and Aβ-(25–35)] for 45 min induced a strong increase in p65 activity in both neurons and astrocytes (Fig. 1 d and g). α-p65 mAb immunoreactivity now colocalized with the nuclear DAPI staining (compare Fig. 1 Top and Middle). This shows that Aβ can activate NF-κB in both neurons and astrocytes. Aβ-(1–40) activated NF-κB in the same concentration range as Aβ-(25–35), (see below) but substance P failed to activate NF-κB at concentrations up to 15 μM (data not shown). The concentration profile of the NF-κB activation induced by Aβ-(1–40) (Fig. 2A) and Aβ-(25–35) (Fig. 2B) was measured. Activation was quantified using histograms of full microscopic views, depicted on a linear scale with the relative amount of pixels shown on the ordinate and the fluorescence intensity shown on the abscissa (Fig. 2). We found that 100 nM reproducibly gave the strongest activation with both Aβ-(1–40) and Aβ-(25–35) resulting in a peak at the highest fluorescence intensity (Fig. 2 A and B, arrows). At a concentration of 1 μM Aβ-(1–40), p65 was still activated (Fig. 2A) in contrast to neurons treated with 1 μM Aβ-(25–35). Treatment with higher concentrations of peptides resulted in an induction of an α-p65 immunoreactivity, which no longer colocalized with the nuclear DNA staining but was concentrated in perinuclear aggregates. This was much more pronounced in cells treated with Aβ-(25–35) (Fig. 2B).
Next, we tested whether the observed NF-κB activation by the Aβ peptides was specific. A peptide of the same amino acid composition as Aβ-(25–35) but with a randomized sequence (11) was tested at a concentration of 100 nM. The scrambled peptide failed to detectably activate NF-κB (data not shown). To assess the specificity of the α-p65 mAb, the antibody was preincubated with the p65 peptide used to raise the mAb (28). Peptide-preabsorbed α-p65 mAb did not stain granule cells treated with Aβ (data not shown).
Activation of NF-κB by Aβ Is Time-Dependent.
Aβ peptides were applied for several days on cerebellar granule cells. NF-κB activation was measured at different time points (data not shown). NF-κB activation shows an inverted U-shaped concentration dependence for Aβ peptides. Maximal activation was observed at a narrow concentration range of 0.1 μM Aβ-(25–35) and 0.1 μM Aβ-(1–40) up to 1 μM Aβ-(1–40) after 1 h, and up to 24 h, of peptide exposure. No activation of NF-κB could be detected at higher concentrations even after an exposure of up to 3 days. Nevertheless, neurotoxicity of both Aβ peptides (10 μM) was evident after 1 day of exposure and increased to more than 80% nonvital cells after 3 days. The neurotoxicity of the Aβ peptides was even more pronounced when the treatment was done without serum.
Of interest, after 1 day of exposure with 0.1 μM Aβ-(1–40) an up to 30% increase in viability of granule cells was observed. This correlates very well with the maximal activation of NF-κB at this neuroprotective dose of 0.1 μM Aβ-(1–40) and is in a time frame to allow de novo gene expression. As observed with activation by other stimuli, e.g., tumor necrosis factor (TNF) or phorbol esters, NF-κB activation is a transient process, which is inhibited through de novo expression of the NF-κB-target gene IκB as part of an autoregulatory loop (21). To test the mechanisms involved in the repression of NF-κB activation at high concentrations of Aβ, we tested several concentrations of H2O2 for NF-κB activation (data not shown). Surprisingly, here also an inverted U-shaped concentration curve was observed, and with H2O2 amounts higher than 40 μM, a perinuclear staining was evident. One explanation for this unusual localization of activated p65 might be an oxidative modification of p65 interfering with nuclear transport. Taken together, these data point to a role of NF-κB in neuroprotection rather than in neurodegeneration.
Activation of NF-κB by Aβ Is Dependent on ROIs.
Aβ peptide solutions are capable of generating ROIs (11, 32), and ROIs were shown to be critical for the neurotoxic action of Aβ, as was evident from the protective effect of antioxidants, such as vitamin E or exogenously applied catalase (10, 11). Because NF-κB is activated by H2O2 in some T cell lines, in HeLa cells (23, 24), and in cerebellar granule cells, we tested whether ROIs, in particular H2O2, play a role in Aβ-induced NF-κB activation. Cultures of granule cells were stimulated with Aβ-(1–40) (Fig. 3, a-d, Upper) and Aβ-(25–35) (Fig. 3, a-d, Lower) alone, in combination with the antioxidant PDTC (Fig. 3, e-h), an inhibitor of cellular H2O2 production in response to okadaic acid stimulation (33), and in combination with catalase (Fig. 3, i-l). PDTC (100 μM) almost completely blocked the appearance of Aβ-activated p65 in nuclei (Fig. 3, e and g). Only a weak perinuclear p65 staining was evident.
TNF-induced NF-κB activation can be suppressed by stable overexpression of catalase or exogenously applied enzyme (25), suggesting that H2O2 is a critical ROI factor in TNF signaling. Because catalase also prevents the cytotoxic effects of Aβ (11), we wondered whether NF-κB activation by Aβ also was suppressed by the H2O2-degrading enzyme. Primary granule neurons were preincubated for 1 h with 400 μg/ml catalase before the addition of 100 nM Aβ peptides [Fig. 3, i-l: Aβ-(1–40) Upper; Aβ-(25–35) Lower]. As shown in Fig. 3, i-l, exogenous catalase effectively suppressed the appearance of Aβ-activated NF-κB in nuclei. In comparison to PDTC, inhibition with catalase is much less efficient. For inhibition of TNF-mediated NF-κB activation, a prolonged pretreatment (30 h) with exogenous catalase was necessary (25). Here, only a 1-h pretreatment with catalase was needed to clearly suppress NF-κB activity. We tested also catalytically inactive catalase. Therefore we preincubated catalase with the specific inhibitor 3-amino-1,2,4-triazole (25). The treated catalase was tested for inhibition of Aβ-(1–40)- and Aβ-(25–35)-mediated NF-κB activations. Surprisingly, the inactivated catalase acted also as an inhibitor of NF-κB activation (data not shown). One possible explanation might be an interference of catalase with Aβ aggregation.
NF-κB Immunoreactivity Is Present in Senile Plaques.
We investigated whether NF-κB is activated in the vicinity of Aβ-containing plaques in the brains of AD patients. The first series of experiments were performed with cryosections from brains of patients with AD stained by an indirect immunofluorescence technique because of this technique’s suitability for double labelings. A total of 10 sections originating from the limbic cortex and hippocampus of four AD brains were analyzed. Bindings of polyclonal anti-Aβ antibodies and of a monoclonal α-p65 antibody were visualized by respective, specific secondary antibodies. In Fig. 4a, red fluorescence (Cy3) corresponds to activated p65 and green fluorescence (dichlorotriazinyl amino fluorescein) corresponds to anti-Aβ immunoreactivity (Fig. 4b). An example of a senile plaque, as detected by an anti-Aβ antiserum, is shown in Fig. 4b, and the same structure is stained with α-p65 in Fig. 4a. Out of 300 anti-Aβ immunoreactive plaques inspected, ≈65% were found to also be strongly positive for α-p65 immunoreactivity. No staining was evident when the mAb was preabsorbed with the corresponding p65 peptide epitope (data not shown).
Early Plaque Stages Are Surrounded by Neurons with Activated NF-κB.
Using paraffin-embedded sections of AD brains and peroxidase-conjugated anti-mouse-IgG, distinct plaque stages in the isocortex of AD patients were analyzed for α-p65 mAb immunoreactivity. Although this procedure is of lower sensitivity compared with immunofluorescence staining methods, as evident from a lower density of NF-κB-positive plaques, it has the advantages that lipofuscin granules are not detected and that different plaque stages can be identified. We have analyzed diffuse, primitive, and classical plaques in a total of 30 samples from the cerebral cortex of four patients with AD. A total of 100 plaques of each sample was inspected. The majority of plaques was significantly labeled with α-p65 mAb (see Fig. 4) whereby the intensity of labeling correlated with the plaque stage. The strongest staining was observed in primitive plaques, and diffuse and classical plaques showed only weak p65 staining (data not shown). In primitive plaque stages, α-p65 mAb immunostaining clearly labeled plaques (Fig. 5, brown color). Two examples of regions with primitive plaques are shown at low power view in Fig. 5 and are consistent with the results from indirect immunofluorescence labeling (see Fig. 4). A number of cells surrounding the plaque (Fig. 5, arrows), among them pyramidal neurons (identified by their triangular shape at high power magnification; data not shown), showed a strong nuclear α-p65 mAb immunoreactivity (dark brown) indicating both the presence of activated transcription factor NF-κB and an inducing signal derived from the nearby primitive plaque. In brain regions free of senile plaques, very few anti-p65 immunoreactive cells were observed (Fig. 5, asterisks). This observation in post mortem tissue was in contrast to freshly prepared rodent brains, which have a relatively high density of α-p65 mAb-positive neurons (15, 28) and may reflect ceased neuronal activity. Taken together, our data suggest that predominantly early plaque stages can emit a signal activating NF-κB in closely adjacent neurons and astroglia of the cerebral cortex of patients with AD.
DISCUSSION
The Mechanism of NF-κB Activation by Aβ.
In this study, we show, in primary neuronal cultures, that the transcription factor NF-κB can be potently activated by nanomolar concentrations of both neurotoxic peptides Aβ-(1–40) and Aβ-(25–35). Cerebellar granule cells were used as a convenient well characterized model of cultured neurons. Similar activation of NF-κB was detected in post mortem sections from cortex and hippocampus of patients with AD, suggesting that this response was not limited to cerebellar neurons. The Aβ-(25–35) peptide has been shown to induce oxidative stress in primary neurons and to activate a κB-dependent reporter gene construct in transiently transfected B12 neuroblastoma cells (11). Here, we directly observed NF-κB activation at a single cell level in both cultured primary neurons and astrocytes and provide evidence that this also may occur in a pathological in vivo situation.
It has been reported that the neurotoxic action of Aβ relies on H2O2 production (11). Here we show that NF-κB activation by Aβ was prevented by PDTC, a compound shown to prevent cellular H2O2 production and the de novo phosphorylation and subsequent degradation of IκB-α in response to TNF and IL-1 (34, 35). We also found that the Aβ-mediated NF-κB activation could be blocked by exogenous catalase. This might be a result of a catalase catalyzed degradation of H2O2. But, surprisingly, catalase treated with the specific inhibitor 3-amino-1,2,4-triazole also blocked NF-κB activation. A nonenzymatic mechanism for catalase in inhibiting Aβ-mediated NF-κB activation also is supported by the short time of preincubation (1 h), which is sufficient for blocking, whereas TNF-mediated NF-κB activation is only blocked after a prolonged pretreatment (30 h) with exogenous catalase (25). Taken together, these data suggest a structural interaction of catalase with Aβ peptides, independent of enzymatic activity. The consequence might be an interference with Aβ aggregation and/or receptor binding.
Aβ has been reported to generate ROIs in the absence of cells (32), so we tested exogenous H2O2 for NF-κB activation. In HeLa and Jurkat T cell clones, 50–250 μM of exogenously applied H2O2 was required to activate NF-κB (23, 24). Corresponding to the inverted U-shaped activation curve of Aβ, 10–40 μM of H2O2 was efficient in activating NF-κB in cerebellar granule cells. Perinuclear aggregates of p65 could be detected in cells exposed to high amounts of Aβ or H2O2. One possible explanation for this unphysiological location is an accumulation of covalently modified NF-κB due to increased oxidative stress. Future studies with purified proteins will further address this question.
Recently, it was shown that Aβ interacts with tachykinin receptors (36). However, substance P, one of the natural ligands of tachykinin receptors, failed to activate NF-κB in primary cerebellar granule cells, suggesting that tachykinin receptors were not involved in NF-κB activation by Aβ.
Activated NF-κB in Brains of Patients with AD.
In this study, we provide immunohistochemical evidence that Aβ may activate NF-κB in brains from patients with AD. It must be emphasized that post mortem material was analyzed, with an autolysis time up to 12 h. During this time, the IκB-shielded epitope for α-p65 mAb could become exposed on p65 because of enhanced proteolysis. However, a major argument against such an artificial NF-κB activation is that very few cells in cryosections and paraffin-embedded sections were immunoreactive, except for those in the close vicinity of certain plaque stages (see Fig. 5).
Two types of α-p65 mAb immunoreactivity were observed in AD brains. First, early plaque stages were directly stained, in particular primitive plaques, which showed a strong reactivity in their center. The same staining pattern was reported for the inflammatory cytokine IL-6 (14), whose gene is known to be inducibly controlled by NF-κB (12). Likewise, IL-1 and intercellular adhesion molecule 1, which are both encoded by target genes for NF-κB (12), were found in plaques (37, 38). It has been suggested that the center of classical plaques contains an activated microglial cell, producing inflammatory cytokines such as IL-6 and IL-1 (39) that could be produced as a consequence of activated NF-κB. Recently, the APP gene was identified as a NF-κB target gene (40), explaining the up-regulation of this gene by IL-1. In addition, Aβ-(25–35) can stimulate the expression of IL-1 but not of IL-6 (41). The whole system consists of several feedback loops, like IL-1 and APP, which stimulate their own synthesis directly or indirectly via microglia activation (39). In this system, NF-κB seems to function as a central regulator. NF-κB is present in synapses (19, 26), so the p65 immunoreactivity found within plaques may alternatively (or in addition) originate from disintegrated synapses (42). The absence of nuclear DAPI staining from the plaque center (data not shown) would support this model.
Second, α-p65 mAb immunostaining was seen in neurons and astroglia surrounding primitive plaques. These discrete nuclear stainings indicate the exposure of cells to a strong NF-κB-activating signal originating from the plaque. Aβ is the major plaque component, so Aβ is a likely candidate. The reduced p65 immunostaining in the vicinity of classical plaques could come from the progressive loss of neurons by apoptotic cell death (43).
In the context of a neurodegenerative disease, our study makes a link among one causative agent (Aβ), production of secondary ROI signals, activation of NF-κB, an ROI-inducible transcription factor, and expression of a number of inflammatory genes known to be regulated by NF-κB. Below we discuss the possibility that the activation of NF-κB could initiate a neurodegenerative or neuroprotective gene expression program, depending on the cellular context.
A Possible Role for NF-κB in Neurodegeneration.
Do such toxic compounds as ROIs really depend on a transcription factor to exert their neurotoxic effects? High ROI doses may not need transcriptional events; however, a chronic exposure to low ROI doses may depend on a genetic program to exert a neurotoxic effect. Upon oxidative stress, NF-κB may induce the expression of gene products with a direct or indirect neurotoxic activity. Such indirect neurotoxic effects might well be the consequence of NF-κB activation in glial cells. On the other hand, candidates for neuronal NF-κB target genes with a direct neurodegenerative effect are the inducible NO synthase (44) and IL-6 (45). Novel major histocompatibility complex class I expression in stressed neurons could have an indirect neurodegenerative effect by targeting the cells for immunosurveillance (46). Of interest, stimulation of N-methyl-d-aspartate and non-N-methyl-d-aspartate receptors on cerebellar granule neurons leads to a rapid activation of NF-κB and a subsequent surface expression of major histocompatibility complex class I molecules (18).
A Possible Role for NF-κB in Neuroprotection.
Depending on the genetic program of a cell, NF-κB may be able to activate expression of genes with neuroprotective function, e.g., in neurons, whereas neurodegenerative functions are directed in glia. This could explain why both TNF α and β can induce neuroprotection against Aβ-induced neuronal death when applied 24 h before Aβ (47). Recently, we found that nerve growth factor can activate NF-κB via the p75 neurotrophin receptor (48). Of interest, this receptor is abundantly expressed on cholinergic neurons and is necessary for sprouting after lesions (49).
A further possibility is that short term stimuli of NF-κB lead to neuroprotection whereas chronic stimuli, e.g., long term incubation with Aβ peptides, cause NF-κB inhibition and subsequent neurodegeneration. A link of NF-κB activation and neuroprotection is supported by the following observations: (i) The strongest NF-κB activation is only found at extremely low concentrations of Aβ (maximal at 0.1 μM); (ii) this inverted U-shaped activation curve is found with both Aβ-(25–35) and Aβ-(1–40); (iii) for Aβ-(1–40), a concentration of 0.1 μM is neuroprotective in cerebellar granule cell paradigm used here; and (iv) in vivo only, early plaque types are surrounded by neurons with activated NF-κB.
Taken together, these data underscore a role for NF-κB as an immediate sensor for increased levels of ROIs in neurons. In response to increased ROI levels, NF-κB could direct the expression of a cellular defense program, as shown for the immune system. Under chronic stimulation and/or stimulation with high amounts of Aβ, this sensing system seems to be overloaded, with the consequence that there will be no increase in cellular buffering mechanisms for ROIs, and this might result in further exacerbation of the neurodegenerative process.
Transcription factor NF-κB is activated by Aβ, ROIs, and inflammatory cytokines in various cell types of the brain and induces a number of genes with relevance for AD. Hence, the factor may play an important role in the development of the disease and define a novel drug target for slowing down or arresting the progression of AD.
We thank A. Schulze–Specking for superb technical assistance. Dr. Hubertus Stockinger (Boehringer Mannheim) is gratefully acknowledged for providing α-p65 mAb. Dr. Franz–Josef Schneider (Boehringer Ingelheim) is thanked for providing Aβ-(1–40). This work was supported by grants from the Bundesministerium für Bildung, Wissenschaft, Forschung, und Technologie (Schwerpunkt Autoimmunität), Volkswagen–Stiftung, and Sonderforschungsbereich 505 of the Deutsche Forschungsgemeinschaft.
ABBREVIATIONS
- AD
Alzheimer disease
- APP
amyloid precursor protein
- Aβ
amyloid β-peptide
- IL
interleukin
- PDTC
pyrrolidine dithiocarbamate
- ROI
reactive oxygen intermediate
- TNF
tumor necrosis factor α
- DAPI
6-diamidino-2 phenylindole
References
- 1.Glenner G G, Wong C W. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 2.Masters C L, Simms G, Weinmann N A, Multhaup G, McDonald B L, Beyreuther K. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang J, Lemaire H G, Unterbeck A, Salbaum M, Masters C L, Grzeschik K H, Multhaup G, Beyreuther K, Müller-Hill B. Nature (London) 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 4.Müller U, Cristina N, Li Z W, Wolfer D P, Lipp H P, Rulicke T, Brandner S, Aguzzi A, Weissmann C. Cell. 1994;79:755–765. doi: 10.1016/0092-8674(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 5.Zheng H, Jiang M, Trumbauer M E, Sirinahsinghji D J S, Hopkins R, et al. Cell. 1995;81:525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
- 6.Li Z W, Stark G, Gotz J, Rulicke T, Muller U, Weissmann C. Proc Natl Acad Sci USA. 1996;93:6158–6162. doi: 10.1073/pnas.93.12.6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Games D, Adams D, Alessandrini R, Barbour R, Blackwell C, et al. Nature (London) 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 8.Koh J, Yang L L, Cotman C W. Brain Res. 1990;533:315–320. doi: 10.1016/0006-8993(90)91355-k. [DOI] [PubMed] [Google Scholar]
- 9.Yankner B A, Duffy L K, Kirschner D A. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 10.Behl C, Davis J, Cole G M, Schubert D. Biochem Biophys Res Commun. 1992;186:944–950. doi: 10.1016/0006-291x(92)90837-b. [DOI] [PubMed] [Google Scholar]
- 11.Behl C, Davis J B, Lesley R, Schubert D. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 12.Aisen P S, Davis K L. Am J Psychiatry. 1994;151:1105–1113. doi: 10.1176/ajp.151.8.1105. [DOI] [PubMed] [Google Scholar]
- 13.Vandenabeele P A, Fiers W. Immunol Today. 1991;12:217–219. doi: 10.1016/0167-5699(91)90032-O. [DOI] [PubMed] [Google Scholar]
- 14.Strauss S, Bauer J, Ganter U, Jonas U, Berger M, Volk B. Lab Invest. 1992;66:223–230. [PubMed] [Google Scholar]
- 15.Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle P A. Mol Cell Biol. 1994;14:3981–3992. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaltschmidt C, Kaltschmidt B, Lannes-Vieira J, Kreutzberg G W, Wekerle H, Baeuerle P A, Gehrmann J. J Neuroimmunol. 1994;55:99–106. doi: 10.1016/0165-5728(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 17.Baeuerle P A, Henkel T. Annu Rev Immunol. 1994;12:141–179. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 18.Kaltschmidt C, Kaltschmidt B, Baeuerle P A. Proc Natl Acad Sci USA. 1995;92:9618–9622. doi: 10.1073/pnas.92.21.9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerrini L, Blasi F, Denis-Donini S. Proc Natl Acad USA. 1995;92:9077–9081. doi: 10.1073/pnas.92.20.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coyle T, Puttfarcken P. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 21.Verma I M, Stevenson J K, Schwarz E M, Van Antwerp D, Miyamoto S. Genes Dev. 1996;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- 22.Staal F J T, Roederer M, Herzenberg L A. Proc Natl Acad USA. 1990;87:9943–9947. doi: 10.1073/pnas.87.24.9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreck R, Rieber P, Baeuerle P A. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer M, Schreck R, Baeuerle P A. EMBO J. 1993;12:2005–2015. doi: 10.1002/j.1460-2075.1993.tb05850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt K N, Amstadt P, Cerutti P, Baeuerle P A. Chem Biol. 1995;2:13–22. doi: 10.1016/1074-5521(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 26.Kaltschmidt C, Kaltschmidt B, Baeuerle P A. Mech Dev. 1993;43:135–147. doi: 10.1016/0925-4773(93)90031-r. [DOI] [PubMed] [Google Scholar]
- 27.Sparacio S M, Zhang Y, Vilcek J, Benveniste E N. J Neuroimmunol. 1992;39:231–242. doi: 10.1016/0165-5728(92)90257-l. [DOI] [PubMed] [Google Scholar]
- 28.Kaltschmidt C, Kaltschmidt B, Henkel T, Stockinger H, Baeuerle P A. Biol Chem Hoppe-Seyler. 1995;376:9–16. doi: 10.1515/bchm3.1995.376.1.9. [DOI] [PubMed] [Google Scholar]
- 29.Beg A A, Sha W C, Bronson R T, Gosh S, Baltimore D. Nature (London) 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 30.Ueberla K, Lu Y, Chung E, Haseltine W A. J Acquired Immune Defic Syndr. 1993;6:227–230. [PubMed] [Google Scholar]
- 31.Liou H C, Sha W C, Scott M L, Baltimore D. Mol Cell Biol. 1994;14:5349–5359. doi: 10.1128/mcb.14.8.5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hensley K, Carney J M, Mattson M P, Aksenova M, Harris M, Wu J F, Floyd R A, Butterfield D A. Proc Natl Acad Sci USA. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmidt K N, Traenckner B-M, Meier B, Baeuerle P A. J Biol Chem. 1995;270:27136–27142. doi: 10.1074/jbc.270.45.27136. [DOI] [PubMed] [Google Scholar]
- 34.Henkel T, Machleidt T, Alkalay I, Krönke M, Ben-Neriah Y, Baeuerle P A. Nature (London) 1993;365:182–185. doi: 10.1038/365182a0. [DOI] [PubMed] [Google Scholar]
- 35.Sun S C, Ganchi P A, Ballard D W, Greene W C. Science. 1993;259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 36.Kimura H, Schubert D. Proc Natl Acad Sci USA. 1993;90:7508–7512. doi: 10.1073/pnas.90.16.7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verbeek M M, Otte-Holler I, Westphal J R, Wesseling P, Ruiter D J, Waal R M. Am J Pathol. 1994;144:104–116. [PMC free article] [PubMed] [Google Scholar]
- 38.Griffin W S, Sheng J G, Roberts G W, Mrak R E. J Neuropathol Exp Neurol. 1995;54:276–281. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 39.Mrak R E, Sheng J G, Griffin W S. Hum Pathol. 1995;26:816–823. doi: 10.1016/0046-8177(95)90001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grilli M, Ribola M, Alberici A, Valerio A, Memo M, PierFranco S. J Biol Chem. 1995;270:26774–26777. doi: 10.1074/jbc.270.45.26774. [DOI] [PubMed] [Google Scholar]
- 41.Del B R, Angeretti N, Lucca E, De S M, Forloni G. Neurosci Lett. 1995;188:70–74. doi: 10.1016/0304-3940(95)11384-9. [DOI] [PubMed] [Google Scholar]
- 42.Schubert W, Prior R, Weidemann A, Dircksen H, Multhaup G, Masters C L, Beyreuther K. Brain Res. 1994;563:184–194. doi: 10.1016/0006-8993(91)91532-6. [DOI] [PubMed] [Google Scholar]
- 43.Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski H M. Acta Neuropathol. 1995;89:35–41. doi: 10.1007/BF00294257. [DOI] [PubMed] [Google Scholar]
- 44.Minc-Golomb D, Tsarfaty I, Schwartz J P. Br J Pharmacol. 1994;112:720–722. doi: 10.1111/j.1476-5381.1994.tb13136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campbell I L, Abraham C R, Masliah E, Kemper P, Inglis J D, Oldstone M B, Mucke L. Proc Natl Acad Sci USA. 1993;90:10061–10065. doi: 10.1073/pnas.90.21.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neumann H, Cavalié A, Jenne D E, Wekerle H. Science. 1995;269:549–552. doi: 10.1126/science.7624779. [DOI] [PubMed] [Google Scholar]
- 47.Barger S W, Hörster D, Furukawa K, Goodman Y, Krieglstein J, Mattson M P. Proc Natl Acad Sci USA. 1995;92:9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carter B D, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Böhm-Matthaei R, Baeuerle P A, Barde Y A. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- 49.Lucidiphillipi C A, Clary D O, Reichardt L F, Gage F H. Neuron. 1996;16:653–663. doi: 10.1016/s0896-6273(00)80084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]