

Abstract
Ion channels exist in all cells and are enormously varied in structure, function and regulation. Some progress has been made in understanding the role that ion channels play in the control of blood pressure, but the discipline is still in its infancy. Ion channels provide many different targets for intervention in disorders of blood pressure and exciting advances have been made in this field. It is possible that new drugs, as well as antisense nucleotide technology or gene therapy directed towards ion channels, may form a new class of treatments for high and low blood pressure in the future.
Keywords: Bartter's syndrome, blood pressure, epithelium, ion channels, Liddle's syndrome, vascular tone
Introduction
Ion channels are found in all living cells and their normal function is vital for life. Ion channels are actively involved in determining the movement of ions into and out of cells, and so are crucial for the electrical activity of nerves, muscles and sensory organs and are central to homeostasis. The study of ion channels has advanced rapidly as the structure and function of many channels has been described, and the term ‘channelopathy’ has recently been coined to group the diseases caused by ion channel abnormalities. Ion channels are targets for the action of many drugs used to treat not only known channelopathies, but also multifactorial conditions such as hypertension. As ion channels come under close scrutiny as potential targets for new drugs, knowledge of this field will become increasingly important in Clinical Pharmacology. In part 1 of this article the structure and function of ion channels in general are reviewed. In part 2 evidence that ion channels are, or might be, involved in the pathogenesis of hypertension is considered and the place of drugs which alter ion channel activity in the treatment of high blood pressure is discussed. Reviews of other ‘channelopathies’ can be found elsewhere [1–3].
Part 1 – Structure and function of ion channels
Lipid membranes separate individual cells from each other and from extracellular fluid, and partition organelles within cells. These membranes are barriers to the indiscriminate movement of water and water-soluble substances such as ions, and allow cells (or organelles) to determine their internal environment. Ions diffuse only slowly across lipid membranes themselves; more rapid movement is achieved with the aid of transport proteins, which sit in and cross these membranes and either form ion channels or act as ion carriers. Ion channel proteins are assembled in the cell membrane to form pores with ‘watery centres’ that allow electrically charged, water-soluble ions to cross the hydrophobic lipid bilayer. Ions move through these channels passively by diffusion down electrical and chemical ionic gradients. The group of transport proteins that act as ion carriers are also known as ‘co’ or ‘counter transporters’, or ‘ion exchangers’. These proteins all transport ions by binding to the ions on one side of the membrane, then ‘carrying’ the ions by undergoing a conformational change which moves the ions to the other side of the membrane. Movement of ions by carrier proteins is either active, where carrier proteins (also known as pumps) use energy in the form of ATP to move ions against an energy gradient, or passive, where ions diffuse down existing energy gradients in combination with carrier proteins. This review focuses on ion channels, in particular those expressed in the cell surface membrane which, mediate communication between the cell and its external environment. Ion transport proteins which act as ion carriers, such as the sodium-lithium counter-transporter, are outside the scope of this review.
Structure of ion channels
Ion channels vary widely in structure but as a class have some features in common. Channels are usually assembled within the membrane from several subunits, which may be different proteins or multiple copies of a single protein. Each protein subunit is folded into a complex tertiary structure that causes it to cross the cell membrane at least twice. The regions of the subunits embedded in the membrane together form the ion-conducting pore. Other regions of the subunits are situated either in the cytoplasm or outside the cell and are important for the normal function and regulation of channels. Cytoplasmic (intracellular) regions of protein subunits are involved in the regulation of channel activity. For example epithelial sodium channels are inactivated by channel removal from the cell membrane, a process mediated by cytoplasmic carboxyl termini of the β and γ subunits [4]. By contrast voltage-gated cation channels may be inactivated when a cytoplasmic region of the channel protein plugs the inner mouth of the channel pore, preventing movement of ions through the channel [5]. Extracellular regions of channel subunits appear to bind external messengers and drugs and determine the identity of ions that can enter the channel from outside the cell. For example the extracellular loop of the epithelial sodium channel appears to be involved in binding of amiloride, a drug which blocks this sodium channel [6], while an extracellular loop of voltage-gated cation channels determines which particular cation (Na+, K+ or Ca++) that channel will actually transport [7].
Normal function of ion channels (Figure 1)
Figure 1.
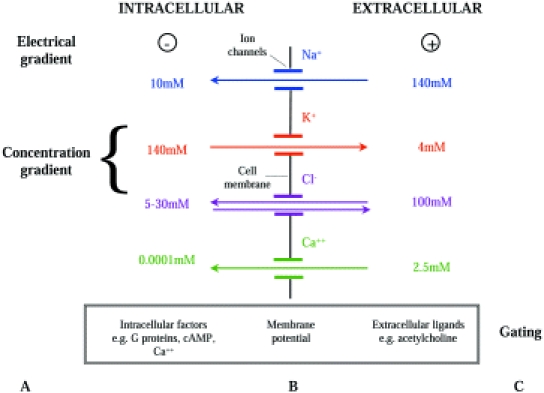
Factors which determine ion transport through ion channels in cell membranes. Movement of ions across the cell membrane is determined by: A – the electrical and concentration gradients for ions across the membrane, B – properties of ion channels in the cell membrane (channel selectivity, conductance, number and open probability) and C – factors which gate the ion channels, i.e. determine whether the channels are open or closed.
Ion channels permit the diffusion of ions across cell membranes. The nature and direction of ion movement across the cell membrane is determined by the ion selectivity of the open channels and the electrical and chemical energy gradients across the cell membrane. The magnitude of the flux of ions across the membrane is determined by the numbers of channels in the membrane, whether or not the channels are open (determined by ‘gating’), the fraction of time the channels are open (open probability) and ease of diffusion or flow of ions through the channels (conductance). Coordination of the activity of the many different channel types present in membranes of individual cells is essential if normal cell function is to be maintained.
Selectivity of ion channels
Ion channels are classified by the type of ion that can pass through the channel pore. Cation channels permit diffusion of positively charged ions and anion channels negatively charged ions. Channel types may also be selective for each of the major cations (sodium, potassium and calcium) and for the major anion (chloride). Under physiological conditions ions tend to move in only one direction through individual channel types, either into or out of the cell. As ions are charged, this direction of diffusion is determined both by the concentration gradient for that ion and the electrical potential across the cell membrane.
Driving forces for ion transport across the cell membrane
In the body ions generally diffuse down a chemical (or concentration) gradient from regions of high concentration to regions of low concentration. Hence movement of an ion across the cell membrane is determined by the relative concentrations of that ion inside and outside the cell. For example the extracellular concentration of Na+ is 140 mmol l−1 whereas the intracellular concentration of Na+ is 10 mmol l−1. Therefore if Na+ channels are open in the cell membrane, Na+ will diffuse into the cell. Conversely K+ concentrations are lower in extracellular fluid (4 mmol l−1) than in intracellular fluid (140 mmol l−1) so when potassium channels are open potassium will diffuse out of the cell. As they are electrically charged, ions also move in an electrical field with cations moving away from positive charge towards negative charge, and anions moving from negative to positive charge. The potential across the membrane may reinforce or sometimes oppose the movement of ions down their concentration gradient.
As the ions move, there is a tendency for the concentration gradient to be reduced and so the chemical driving forces promoting diffusion lessen and the electrical forces opposing diffusion increase. Movement ceases when electrical and chemical forces become exactly balanced, preventing further movement of that ion in either direction. The electrical force across the membrane at this point is known as the Nernst potential. The Nernst potential can be predicted for individual ions. Where only Na+ can diffuse across the cell membrane a typical value for membrane potential is + 70 mV. Where only K+, Ca++ or Cl– can diffuse, the membrane potential is typically −98 mV, + 150 mV or −30 mV, respectively (positive and negative signs denote the intracellular potential relative to a ground reference electrode).
All cells have a membrane potential at rest which is a product of the concentration gradient for ions across the cell membrane and the ion channels open in that cell membrane. In many cells the predominant channel type open at rest is the potassium channel, and the membrane potential is close to the Nernst potential for potassium (−70 to −100 mV).
Number of ion channels
Ion channel expression in the cell membrane is dynamic with channels or channel subunits moving into and out of the cell membrane [8]. For example epithelial sodium channels appear to be inserted into the cell membrane by exocytosis from membrane vesicles and removed from the cell membrane by targeted endocytosis [8]. The turn-over of epithelial sodium channels in the cell membrane appears to be rapid with a short half life of 1–3.6 h [9, 10]. Insertion or removal of channel proteins or protein subunits from the cell membrane is one mechanism used to regulate ion channel function. For example, aldosterone controls sodium absorption across rat distal colon by regulating the expression of sodium channel subunits in epithelial cell membranes [11]. When the rat is sodium replete and aldosterone levels are low, there is no sodium absorption through epithelial sodium channels and only one sodium channel subunit, the α subunit, can be found in colonic epithelial cells. If the rat becomes sodium depleted and aldosterone levels increase, β and γ sodium channel subunit production increases and these are assembled with the α subunit to form active epithelial sodium channels, stimulating sodium absorption [11].
Gating of channels and open probability
Ion channels may be open or closed and switching between these states is called ‘gating’. Channels are gated by electrical or chemical factors which cause small changes in the shape of the channel protein and open or close the channel pore. Voltage-gated channels are gated by changes in the cell membrane potential, while ligand-gated channels, such as nicotinic channels, are gated by molecules (in this case acetylcholine) which bind directly to the channel. Calcium-activated channels, cyclic nucleotide-gated channels and G protein-gated channels are all gated by interaction with their specific intracellular molecules.
The gating of channels forms one of the most important mechanisms for controlling channel function. Channel gating determines the proportion of the time the channel spends in the open state (open probability of the channel). The gating behaviour of the channel can in turn be fine tuned for further regulation of channel activity. For example, in addition to its effects on channel numbers, aldosterone also appears to enhance sodium absorption through epithelial sodium channels by increasing the open probability of channels already in the cell membrane [12]. How aldosterone does this is not clear, it has been suggested that it acts either by methylation of channel proteins, by alteration of intracellular pH or through a change in intracellular calcium concentration [13].
Conductance of channels
Single channel conductance describes the ease of movement of ions through an individual channel and is designated γ. Conductance is the opposite of resistance and can be defined by Ohms law as the ratio of i, the current through that channel to V, the potential difference (voltage) across the membrane γ = i/V. Membrane conductance (G) for all the channels of one type in the cell membrane is a product of the number of channels of that type (N), the open probability of the channels (Po) and the single channel conductance, G = N.Po.γ. Some drugs act to change ion transport across cell membranes by altering ion channel conductance. For example sodium cromoglycate and frusemide block chloride secretion through chloride channels in airway epithelia by reducing conductance of the chloride channels [14]. Chloride channels may play a role in determining airway calibre, and blockade of airway chloride channels by sodium cromoglycate and frusemide may be one mechanism by which these drugs prevent osmotically by induced bronchoconstriction [14].
Coordination of channel activity
The surface membrane of every cell contains many different channel types that vary depending on the type of cell. Some coordination of the activity of these channels (as well as of other ion transporters) is important for normal cell function, particularly where cell function is to transport ions across polarized cells, or to generate electrical signals in nonpolarized cells (Figure 2).
Figure 2.

Ion channel activity and the normal function of polarized and nonpolarized cells. Coordination of the activity of ion channels and other transporters is essential for normal cell function of both A – polarized and B – nonpolarized cells. A – In the collecting duct of the renal tubule, sodium is absorbed across epithelial cells by coordinated activity of apical Na+ channels and basolateral Na+-K+-ATPase pumps. The Na+-K+-ATPase pumps in the basolateral membrane of tubular cells use energy to pump Na+ out of the cell into the interstitium in exchange for K+. This sets up a concentration gradient for Na+ across the cell from lumen to interstitium. Na+ is then absorbed from the lumen down this concentration gradient through Na+ channels in the apical membrane of the cells. K+ is recycled out of the cell by apical and basolateral potassium channels. B – Coordinated activity of potassium and calcium channels is important for the control of tone in arteriolar smooth muscle. (a) Potassium efflux through potassium channels hyperpolarizes the cell membrane and triggers closure of voltage-gated calcium channels and a fall in intracellular calcium concentration. (b) Closure of potassium channels reduces potassium efflux, depolarizes the cell membrane and triggers opening of voltage-gated calcium channels with a rise in intracellular calcium. Intracellular calcium determines vascular tone by promoting actin–myosin interaction and smooth muscle contraction.
Polarized cells
Epithelial cells lining viscera have different ion transporters in their luminal (apical) membrane from those in the interstitial (basolateral) membrane. This may be achieved by targeted transport of ion channels to each specific cell membrane by microtubules in the cytoplasm [15]. The polarized distribution of ion transport proteins generates movement of ions across the cell. For example sodium absorption across the collecting duct in the renal tubule is achieved by the coordinated activity of apical Na+ channels and basolateral Na+-K+-ATPase pumps (Figure 2). The Na+-K+-ATPase pumps in the basolateral membrane of tubular cells use energy to pump sodium out of the cell into the interstitium in exchange for potassium. This sets up a concentration gradient for sodium across the cell from lumen to interstitium. Sodium then diffuses from the lumen down this concentration gradient through epithelial sodium channels in the apical membrane of epithelial cells. Absorption of sodium is controlled by factors which alter activity of either apical sodium channels or Na+-K+-ATPase pumps. For example aldosterone increases sodium absorption by stimulating activity of both these sodium transporters.
Non-polarized cells
In nonpolarized cells ion channels direct transport of ions into or out of the cell rather than across the cell. Coordination of ion transport activity allows the generation or transduction of electrical signals in nerve and muscle and the regulation of intracellular calcium in muscle cells which controls muscle contraction and smooth muscle tone.
Measurement of ion channel activity
Activity of ion channels can be studied in vitro using patch clamp electrophysiology (Figure 3). In this technique a micropipette with tip diameter of 1–2 µm is brought up against the cell membrane to be studied. Gentle suction is applied which forms a high resistance seal between the pipette and the membrane and isolates the ‘patch’ of membrane within the tip. This is known as an on-cell or cell-attached patch. Usually the patch is then pulled off the cell (forming an isolated inside-out patch) or the patch is ruptured to record from the whole cell. The pipette itself acts as an electrode as it is filled with an electrically conducting solution. A second, reference electrode is placed in the fluid bathing the cell or patch. Electrical properties of the patch or single cell can thus be measured and the effects of changes in chemical or electrical energy gradients as well as of channel agonists and antagonists can be studied.
Figure 3.
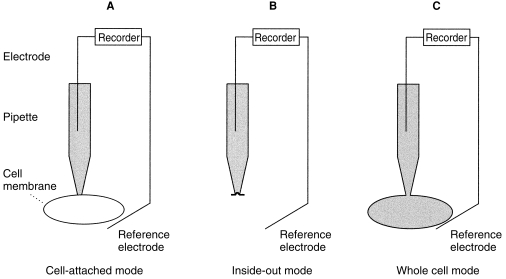
Patch clamp technique for the measurement of ion channel activity. A – Cell-attached mode. The micropipette used for patch-clamping is brought up to the cell membrane and gentle suction applied so that a ‘patch’ of membrane is isolated in the tip of the pipette by a high resistance seal. Using this technique the properties of single channels or groups of channels in the patch can be defined when intracellular contents are left intact. The effect of extracellular agents on channel activity can be studied by adding the agents to the pipette solution. B – Inside-out mode. Once the seal between pipette and membrane patch is established the patch is ‘torn’ away from the rest of the cell and the intracellular surface of the membrane is exposed. This makes it possible to control the intracellular environment artificially and to study channel activity after exposure of the cytoplasmic membrane surface to channel agonists and antagonists. C – Whole cell mode. A cell-attached patch is established, then extra suction is applied to suck out the patch of membrane, leaving the pipette in contact with the intracellular contents. Using this mode current and conductance, which are the sum of the properties of all the channels in the membrane, are measured. Properties of different channel types can be determined by examining the effect of extracellular or intracellular channel agonists and antagonists on membrane current and conductance.
Where cell-attached or inside-out patches are studied then properties (number, open probability, conductance, gating) of single channels or groups of channels in the patch can be defined. Where whole cell patch clamping is used, current and conductance across the cell membrane are measured which are the sum of the properties of all the channels in the membrane. Properties of different channel types can be determined by examining the effect of specific channel agonists and antagonists on membrane current and conductance, but individual channel properties cannot generally be studied using the whole cell technique.
The combination of molecular biology with patch clamp techniques has been used to study the genes encoding ion channels. Many ion channels have been identified and characterized following their expression in vitro in cells such as the Xenopus oocyte.
Part 2. Ion channels and the control of blood pressure
Many genetic, pathophysiological and pharmacological studies provide evidence that ion channel function is an important determinant of blood pressure levels. This section describes ion channel abnormalities which cause both high and low blood pressure and discusses how drugs that affect ion channels are used, or are being developed, for therapy (Table 1).
Table 1.
Ion channels and the control of blood pressure
| Ion channel | Location where channel influences blood pressure | Effect of increased channel activity on blood pressure | Drugs which open ion channel | Effect of decreased channel activity on blood pressure | Drugs which block ion channel |
|---|---|---|---|---|---|
| Epithelial sodium channel | Renal collecting duct | Increases blood pressure (Liddle's syndrome, T594M mutation?) | Decreases blood pressure (Pseudohypoaldosteronism) | Amiloride, triamterene | |
| Renal outer medullary potassium channel (ROM K) | Kidney (loop of Henle) | Low/normal blood pressure (Bartter's syndrome) | Glibenclamide | ||
| Cystic fibrosis transmembrane regulator (CFTR) | ?Renal tubules ??Sweat ducts | Some evidence for low blood pressure (Cystic fibrosis) | |||
| Cation channel | Vascular endothelial cells | ?Lowers blood pressure | LP-805 | ||
| Voltage-gated calcium channel | Vascular smooth muscle cells | Lowers blood pressure | Nifedipine Amlodipine Diltiazem Verapamil | ||
| ATP-sensitive potassium channel | Vascular smooth muscle cells | Lowers blood pressure | Nicorandil Cromokalim Pinacidil | Increases blood pressure | Glibenclamide |
Ion channels and volume control
The epithelial sodium channel
Epithelial sodium channels (ENaC) are present in the luminal (apical) membranes of epithelial cells in the distal nephron (distal tubule and collecting duct) and distal colon. Sodium entry through epithelial sodium channels is the rate limiting step for sodium absorption across these epithelia. Regulation of sodium channel function at these sites allows fine tuning of sodium balance in the body and is critical for blood pressure regulation. Evidence for the importance of the epithelial sodium channel in the control of blood pressure comes not least from the fact that both high and low blood pressure can be caused by epithelial sodium channel abnormalities.
Epithelial sodium channels are composed of α, β and γ subunits [16, 17]. The three subunits share similar primary sequences and are each inserted into the cell membrane in the same way. Each subunit starts with the amino terminus inside the cell, then crosses the membrane and forms a large extracellular loop, before crossing back through the cell membrane and ending with the carboxyl terminus inside the cytoplasm. The transmembrane domains form the sodium-conducting pore and the intracellular termini interact with intracellular molecules such as cytoskeletal proteins, G proteins and protein kinase C.
Liddle's syndrome
Liddle's syndrome is a rare inherited form of hypertension caused by mutations of the epithelial sodium channel which increase sodium reabsorption in the renal distal tubule [18]. Patients with Liddle's syndrome develop high blood pressure, often in their teenage years. This hypertension is associated with hypokalaemic alkalosis, as potassium and hydrogen excretion in the renal distal tubule are linked to sodium channel activity, and with suppression of the renin-aldosterone axis secondary to sodium retention [19]. Mutations which cause Liddle's syndrome affect the cytoplasmic tail of either the β or γ subunits of the epithelial sodium channel and invariably remove or alter a short amino acid sequence called the PY motif, so called as it consists of several proline (P) residues followed by a tyrosine residue (Y) [20, 21]. The PY motif is important for the removal of apical sodium channels from the cell membrane back into the cell. Disruption of the PY motif appears to impair this sodium channel endocytosis, so increasing the number of sodium channels in the cell membrane [22, 23]. Liddle's mutations may also increase the open probability of affected channels [23]. Increased numbers of functioning channels and increased open probability of epithelial sodium channels in the apical cell membrane permit continued and essentially uncontrolled sodium absorption which accounts for the sodium retention and hypertension of Liddle's syndrome.
Evidence for the importance of renal sodium absorption in Liddle's syndrome comes from a patient with Liddle's syndrome who developed end-stage renal failure and required a renal transplant [24]. Following the transplant of a kidney with ‘normal’ epithelial sodium channels the Liddle's patient had only mild high blood pressure with resolution of the hypokalaemia and alkalosis and normal activity of the renin-angiotensin-aldosterone axis. However increased renal sodium channel activity has not been demonstrated directly in Liddle's patients as sodium channels in the renal tubule are inaccessible to direct clinical measurement. Increased sodium absorption through sodium channels which have structural [25, 26] and functional [27, 28] similarities to renal sodium channels has been demonstrated in both lymphocytes [29] and the nasal epithelium [30] of patients with Liddle's syndrome.
Hypertension in Liddle's syndrome can be controlled by a low salt diet in combination with either amiloride or triamterene, drugs which block the epithelial sodium channels and prevent the excessive renal sodium reabsorption.
Pseudohypoaldosteronism
Pseudohypoaldosteronism type 1 (PHA 1) is a rare inherited condition in which patients have low blood pressure. In several affected families mutations that inactivate epithelial sodium channels have been identified. Mutations of all three sodium channel subunits have been implicated in the development of PHA 1 [31, 32]. As a result of some of these mutations, patients have gross truncation of the affected sodium channel subunit which may interfere with the assembly, or normal structure, of sodium channels. In other patients, mutations cause changes in specific amino acids near the first transmembrane domain of the β or γ subunit. These changes result in a partial loss of channel function, perhaps by altering single channel conductance or open probability. In these families absent or reduced activity of mutated epithelial sodium channels results in failure of sodium reabsorption in the distal nephron with salt wasting. Not only does blood pressure fall but also plasma renin activity and aldosterone concentrations are raised in response to the sodium deficiency, and hyperkalaemia and acidosis occur as potassium and hydrogen excretion are coupled to sodium reabsorption in the distal renal tubule.
Sodium channel polymorphisms and blood pressure variation
The observation that both high and low blood pressure could be caused by abnormalities of the epithelial sodium channel led to the speculation that variations in sodium channel activity could contribute to the ‘normal’ distribution of blood pressure levels seen in the general population. Mutation detection and association studies have been performed to investigate the role of epithelial sodium channel abnormalities in the development of high blood pressure. In most ethnic groups there is no evidence that sodium channel polymorphisms contribute to the development of hypertension. Sodium channel polymorphisms causing single amino acid substitutions in the C-terminus or exon 8 of the β subunit occur in 1% of white French adults, but none of these substitutions affect sodium channel activity when tested in the Xenopus oocyte expression system [33]. In Scandanavian subjects there is no association between hypertension and a marker for β and γ subunits [34].
However people of African origin with high blood pressure have low plasma renin activity and salt-sensitive hypertension, similar to the picture seen in patients with Liddle's syndrome, which suggest that sodium retention and volume-expansion may underlie the high blood pressure. There is some evidence that sodium channel polymorphisms may contribute to the development of high blood pressure in African and Caribbean people. The T594M mutation of the β subunit C terminus has been shown to be associated with high blood pressure in a case-control study of black Londoners [35]. However this association was not seen in African Americans [36]. In patients with the T594M mutation there is disruption of a putative protein kinase C binding site. Protein kinase C mediates inhibition of sodium channel activity which reduces sodium absorption through sodium channels [37]. The T594M mutation therefore could increase sodium channel activity by interfering with mechanisms which reduce channel activity. Patch clamp studies in lymphocytes from individuals with the T594M mutation show that protein kinase C inhibition of sodium channel activity is indeed reduced in heterozygotes and abolished in homozygotes [38]. Physiological studies in people with the T594M mutation are currently ongoing. Further studies are required to determine whether sodium channel blocking drugs such as amiloride or triamterene have a role in the treatment of hypertension in people with the T594M mutation.
The ROMK potassium channel
A renal outer medullary potassium channel (ROMK) is found in the apical membrane of epithelial cells in the thick ascending limb (TAL) of the loop of Henle. The channel facilitates the reabsorption of up to 30% of filtered NaCl and as such is important in volume regulation (Figure 4) [39]. The role of ROMK channels is to recycle K+ from the tubular cells to the lumen. Na+ and Cl– are primarily absorbed across the thick ascending loop of Henle by a sodium(potassium)chloride cotransporter and so can only be absorbed in the presence of K+ [39]. Sodium absorption via paracellular routes is also driven by the lumen-positive potential difference created by K+ excretion through the ROMK channel [40].
Figure 4.
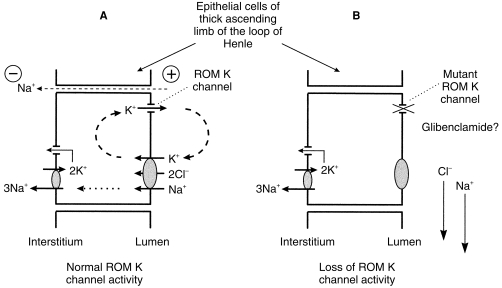
The Renal Outer Medullary K channel (ROM K) and blood pressure. A – Na+ and Cl– are reabsorbed in the thick ascending loop of Henle by the actions of an apical Na-K-2 Cl cotransporter, thus their absorption at this site is dependent on adequate supplies of luminal K+. The activity of apical ROMK channels is essential for Na+ and Cl– reabsorption as ROMK channels increase luminal K+ concentrations by recycling K+ from the tubular cells to the lumen. K+ secretion through ROMK channels also creates a lumen-positive potential difference which drives sodium absorption via paracellular routes. B – Where potassium secretion through ROMK channels is reduced or blocked by channel mutations (Bartter's syndrome) or drugs (e.g. Glibenclamide), luminal K+ concentrations fall which impairs Na-K-2 Cl cotransporter activity and paracellular Na+ absorption. Up to 30% of filtered NaCl is normally reabsorbed in the thick ascending limb of the loop of Henle and failure of this process results in salt-wasting, volume contraction and stimulation of the renin-angiotensin-aldosterone axis.
Mutations of ROMK channels have been identified as the underlying cause in some patients with Bartter's syndrome. Bartter's syndrome comprises an inherited group of renal tubular disorders in which low or normal blood pressure is associated with hypokalaemic alkalosis and diverse other clinical features [41]. ROMK mutations reduce potassium secretion through ROMK channels by interfering with phosphorylation, proteolytic processing or protein trafficking of the channel [42]. Failure of potassium secretion through ROMK channels reduces sodium absorption in the TAL, increases sodium delivery to the distal renal tubule and results in salt-wasting, volume contraction and stimulation of the renin-angiotensin-aldosterone axis (Figure 4) [43]. Increased distal sodium delivery interferes greatly with homeostasis of other cations. Sodium reabsorption through epithelial sodium channels is stimulated to compensate for sodium loss and this is coupled to increased potassium and hydrogen ion excretion causing hypokalaemic alkalosis. Distal calcium and magnesium reabsorption are also impaired and the resultant hypercalciuria often causes nephrocalcinosis.
Bartter's syndrome, in people homozygous for inactivating ROMK mutations, thus presents with a plethora of clinical problems, generally in the neonatal period. In particular at this age salt wasting causes severe dehydration, although subsequent compensatory mechanisms may correct blood pressure to normal levels. It is possible that people heterozygous for ROMK mutations are protected against the development of high blood pressure by relative sodium depletion.
Drugs which block ROMK channels could potentially reduce blood pressure. The sulphonylurea glibenclamide has been shown to block ROMK channels [44]. In micropuncture studies of rat renal tubules, glibenclamide infusion increased delivery of sodium to the early distal tubule and caused natriuresis with diuresis but without altered potassium excretion [45]. These effects are consistent with blockade of ROMK channels in the loop of Henle. Glibenclamide is also natriuretic in humans [46] but despite this effect it has not been shown to lower blood pressure in vivo [47]. Glibenclamide probably lacks antihypertensive actions in patients because it also blocks potassium channels in vascular smooth muscle which would be predicted to raise blood pressure (see below). Potassium channel blocking agents with actions on renal but not vascular potassium channels might have a future role in the treatment of high blood pressure.
The CFTR chloride channel
The cystic fibrosis transmembrane regulator (CFTR) is an epithelial chloride channel which also acts as a regulator of the function of other ion channels. Mutations which inactivate the CFTR chloride channel cause cystic fibrosis, a lethal autosomal recessive condition characterized by lung and pancreatic disease. The role of CFTR in determining blood pressure is speculative. Indirect evidence for a role for CFTR in blood pressure regulation comes from a study which found significantly lower blood pressure in a group of young adults with cystic fibrosis than in age-and sex-matched controls [48]. In most tissues which express CFTR, including airway epithelia, the function of CFTR is to secrete chloride ions. However CFTR is important for chloride reabsorption in the sweat ducts [49] and probably in the proximal renal tubules [50]. Loss of CFTR activity could therefore lead to chloride (and hence sodium and water) loss in sweat and urine with volume depletion. It is possible therefore that heterozygotes with CFTR mutation could be protected against the development of hypertension by this mechanism, although there are no published studies which address this question.
Ion channels and vascular tone
Blood pressure is determined by cardiac output and peripheral arteriolar resistance. Peripheral resistance in turn is determined mainly by vascular tone generated by the contraction of vascular smooth muscle. Ion channels have many crucial roles in the regulation of vascular tone. In vascular endothelial cells ion channel activity is important for controlling the release of vasoactive mediators which in turn modulate vascular smooth muscle tone. In smooth muscle cells the coordinated action of ion channels is one of the mechanisms which control intracellular calcium and hence the degree of contraction of the cell. Vascular ion channels are important targets for drugs to treat pathological states of high or low blood pressure.
Endothelial ion channels
Physiology
The endothelium is integral to the regulation of vascular tone. Its predominant influence is to cause relaxation of vascular smooth muscle by the production of vasodilator mediators, including nitric oxide and prostacyclin, but it also produces vasoconstrictor substances such as endothelin. The mechanisms which regulate the production and release of these mediators are not fully understood. However they appear to be calcium-dependent and determined by intracellular calcium which in turn is influenced by orchestrated ion channel activity in the cell membrane [51]. Vasoactive substances, such as bradykinin or histamine, and mechanical stresses which influence vascular tone, act at least in part through an effect on ion channel function. They act directly to stimulate opening of calcium and cation channels in the cell membrane, which increases calcium entry into the cell. The stimuli also act indirectly to activate K+ channels in the cell membrane and so increase K+ efflux. This change causes hyperpolarization of the cell membrane and hence increases the driving force for Ca2+ entry into the cell. Increased intracellular calcium increases production and release of vasodilator mediators which reduce vascular tone.
Endothelial ion channels and blood pressure
Normal function of endothelial ion channels thus appears to be important in the control of vascular tone and dysfunction of these ion channels could contribute to alterations in blood pressure. However the detailed role of endothelial ion channels in blood pressure regulation is complex and not fully understood. As yet no specific abnormalities of endothelial ion channel structure or function have been identified as a cause of blood pressure abnormalities. However hypertension has been shown to induce secondary changes in endothelial ion channel function in rat models of hypertension [52]. The altered haemodynamic forces of hypertension stimulate increased density of stretch-activated potassium channels and pressure-activated calcium channels in endothelial cell membranes. Upregulation of stretch-activated potassium channels would lead to hyperpolarization of the cell membrane and an increased driving force for calcium entry into the cell through the increased numbers of pressure-activated calcium channels [52]. The response to mechanical stresses thus would be increased production of vasodilator mediators and this in turn may represent an important adaptive mechanism for vessel wall protection in hypertension.
Agents which alter endothelial ion channel activity could potentially be developed for use as drugs to raise or lower blood pressure. The experimental antihypertensive drug LP-805 activates calcium influx into endothelial cells via a cation channel. It may be that it lowers blood pressure by increasing the calcium available to stimulate release of vasodilator mediators such as nitric oxide [53].
Ion channels in vascular smooth muscle
Physiology
Hyperpolarization of the cell membrane has opposite effects on intracellular calcium levels in endothelial and smooth muscle cells. In endothelial cells hyperpolarization of the cell membrane results in increased calcium influx and a rise in intracellular calcium levels. In vascular smooth muscle cells however, hyperpolarization of the cell membrane actually inhibits calcium entry into the cell and causes a fall in intracellular calcium. Hyperpolarization has different effects in these two cell types because calcium entry pathways in endothelial and smooth muscle cell membranes are gated differently. Calcium enters endothelial cells through receptor-mediated, leak and stretch-activated pathways [54], hence hyperpolarization promotes calcium entry and causes an increase in intracellular calcium by creating a favourable electrical gradient for calcium influx through these pathways. In vascular smooth muscle cells however, calcium entry is primarily through L-type (long-lasting), voltage-gated calcium channels [55]. Hyperpolarization of smooth muscle cell membranes results in closure of these voltage-gated calcium channels and hence a fall in intracellular calcium concentrations.
The membrane potential of vascular smooth muscle cells thus is an important determinant of intracellular calcium which in turn controls the degree of contraction of smooth muscle cells, vascular tone and blood pressure. Activity of ion channels, particularly potassium channels, controls the membrane potential [56]. When potassium channels are open, the electrochemical gradient for potassium drives potassium out of the cell and the cell membrane potential becomes more negative. This hyperpolarization of the cell membrane triggers closure of voltage-gated calcium channels, a fall in intracellular calcium and hence vasodilation. Closure of potassium channels reduces potassium efflux from the cell and results in depolarization of the cell membrane, calcium channel opening, an increase in intracellular calcium and vasoconstriction.
Smooth muscle in resistance vessels exists in a state of partial contraction maintained by an intracellular calcium level which appears to be achieved by coordination of ion channel activity [57]. A variety of potassium channel types gated by calcium, voltage or ATP, maintain the cell membrane potential at a level close to that at which calcium channels are activated. Calcium channel activation triggers membrane depolarization and calcium entry that is limited by the opening of calcium-sensitive and other potassium channels which hyperpolarize the cell membrane and close the calcium channels once more. The degree of smooth muscle contraction which is maintained by these mechanisms is probably important in the long term regulation of arterial blood pressure. More detailed reviews of the structure, function and regulation of ion channels in vascular smooth muscle can be found elsewhere [56, 58].
Vascular smooth muscle ion channels and blood pressure
Hypertension
In theory increased calcium channel activity, or reduced potassium channel activity, should cause an increase in blood pressure by increasing vascular tone. Increased L-type Ca++ current density (whole cell current normalized to cell surface area) has been demonstrated in smooth muscle cells from large arteries from the spontaneously hypertensive rat and may contribute to the development of high blood pressure in these animals [59]. However potassium channel activity appears to be increased rather than decreased in animal models of hypertension. Calcium-activated potassium current density is increased in aortas from hypertensive rats compared with controls [60] and single-channel Ca++ activated K+ current is increased in arterial smooth muscle cells from rats with genetic or renal hypertension [61]. ATP-sensitive potassium channels in vascular smooth muscle cells from spontaneously hypertensive rats have impaired sensitivity to the opening actions of levocromakalim, which might imply that opening of these channels is already increased in hypertension62. It is possible that this increased activity of vascular K+ channels may be a compensatory mechanism which opposes the increase in vascular tone in hypertension. Indeed these changes in potassium channel function revert to normal if the hypertension is reversed with treatment [62, 63]
Drug treatment for hypertension
Calcium channel blocking drugs and potassium channel opening drugs can, by acting directly or indirectly, close voltage-gated calcium channels, reduce calcium flux into smooth muscle cells and hence relax vascular smooth muscle (Figure 5).
Figure 5.
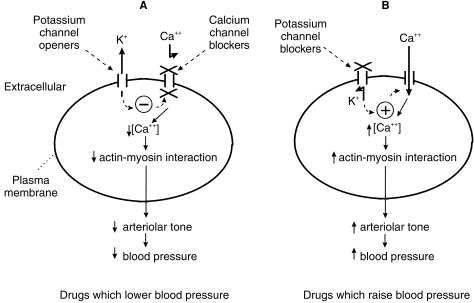
Pharmacological agents which modify blood pressure through actions on vascular smooth muscle cell membrane ion channels.A – Drugs which lower blood pressure. Potassium channel openers (e.g. pinacidil) open ATP-sensitive K+ channels and increase diffusion of K+ out of the cell. K+ efflux hyperpolarizes the cell membrane which triggers closure of voltage-gated calcium channels and a fall in intracellular Ca++ concentration. Ca++ channel blockers (e.g. Nifedipine, diltiazem, verapamil) block voltage-gated Ca++ channels directly, thus also reducing intracellular Ca++. A fall in intracellular Ca++ reduces the interaction between actin and myosin, lowers arteriolar smooth muscle tone and hence causes a fall in blood pressure. B – Drugs which raise blood pressure. Drugs which block ATP-sensitive K+ channels (e.g. glibenclamide) reduce K+ efflux from the cell which results in depolarization of the cell membrane and triggers opening of voltage-gated Ca++ channels. Ca++ influx through these channels increases intracellular Ca++ which in turn promotes actin–myosin interactions and an increase in arteriolar tone. K+ channel blocking agents may have a role in the treatment of hypotension in septic or haemorrhagic shock.
Ca++ - channel blockers
The blood pressure-lowering effect of drugs which block L-type calcium channels (e.g. nifedipine, verapamil) has been recognized for over 30 years. More recently other types of voltage-gated calcium channel have been identified, which are also potential targets in the treatment of hypertension. T-type (transient) calcium channels have been identified in vascular smooth muscle cells [64] and differ from L-type calcium channels as they are gated at more negative membrane potentials and, once open, stay open for a shorter period of time. T-type calcium channels appear to play a role in the control of blood pressure [65] and are also involved in the promotion of growth and proliferation of vascular smooth muscle cells [66]. Mibefradil is the first of a new class of calcium channel blocking drugs which blocks both T and L-type Ca++ channels but has selectivity for the T-type channel [67]. Mibefradil lowers blood pressure [68] and has an antiproliferative effect on vascular smooth muscle in vitro and in animal studies [66]. It has recently been withdrawn from the market because of its propensity to interact with other drugs [69]. However newer, less troublesome T-type Ca2+ channel blockers could have potential both in the treatment of hypertension and in the prevention or reversal of hypertension-induced cardiovascular remodelling that may contribute to the morbidity and mortality of hypertension [70].
Potassium channel openers
A large number of molecules open potassium channels. These can be classified according to their actions into 1) agents which open ATP-sensitive K+ channels, e.g. cromokalim, pinacidil, 2) agents which activate guanylate cyclase in addition to opening ATP-sensitive K+ channels, e.g. nicorandil, 3) agents which open calcium-dependent K+ channels, e.g. dehydro-saponin 1 [71]. Drugs which open ATP-sensitive K+ channels act as arteriolar vasodilators, increasing forearm blood flow [72, 73] and lowering blood pressure [74]. Pinacidil is available in several countries for the treatment of high blood pressure and, while it does lower blood pressure, its use as monotherapy is frequently associated with side-effects such as peripheral oedema, tachycardia, palpitations and headache [75, 76]. Nicorandil acts as both an arteriolar and venous dilator, probably as a consequence of its dual mode of action, and increases coronary blood flow [77]. Its principle use is in the prevention and treatment of angina. Potassium channel openers are not currently licensed for the treatment of hypertension in the United Kingdom.
Hypotension
Work done in animal models suggests that potassium channels play a role in the development of the pathologically low blood pressure of shock. Glibenclamide, which blocks ATP-sensitive K+ channels, caused an increase in blood pressure in experimental endotoxin-induced shock [78, 79] and in haemorrhagic shock [80, 81]. It thus appears that ATP-sensitive K+ channels are activated in ‘shock’ and cause vasodilation and hypotension probably by hyperpolarization of vascular smooth muscle cell membranes (Figure 5). Glibenclamide also reduces the mortality rate in animals with haemorrhagic shock, suggesting that drugs which block K+ channels may have an important therapeutic role in the management of shock [80].
Acknowledgments
The author thanks Professor Joe Collier for his considerable help and encouragement and his perceptive comments during the preparation of the manuscript and Sahantha De Silva for background research.
References
- 1.Ackerman MJ, Clapham DE. Ion channels – Basic science and clinical disease. N Engl J Med. 1997;336:1575–1586. doi: 10.1056/NEJM199705293362207. [DOI] [PubMed] [Google Scholar]
- 2.Chaudhuri A, Behan P. Channelopathies in neurological disorders. CNS. 1998;1:12–15. [Google Scholar]
- 3.Schwiebert EM, Benos DJ, Fuller CM. Cystic fibrosis: a multiple exocrinopathy caused by dysfunctions in a multifunctional transport protein. Am J Med. 1998;104:576–590. doi: 10.1016/s0002-9343(98)00119-3. [DOI] [PubMed] [Google Scholar]
- 4.Goulet CC, Volk KA, Adams CM, Prince LS, Stokes JB, Snyder PM. Inhibition of the epithelial Na+ channel by interaction of Nedd4 with a PY motif deleted in Liddle's syndrome. J Biol Chem. 1998;273:30012–30017. doi: 10.1074/jbc.273.45.30012. [DOI] [PubMed] [Google Scholar]
- 5.Catterall WA. Structure and function of voltage-gated ion channels. Ann Rev Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- 6.Ismailov IL, Kieber-emmons T, Lin C, et al. Identification of an amiloride binding domain within the alpha-subunit of the epithelial Na+ channel. Biol Chem. 1997;272:21075-21083. doi: 10.1074/jbc.272.34.21075. [DOI] [PubMed] [Google Scholar]
- 7.Kukuljan M, Labarca P, Latorre R. Molecular determinants of ion conductance and inactivation in K+ channels. Am J Physiol. 1995;268:C535–C556. doi: 10.1152/ajpcell.1995.268.3.C535. [DOI] [PubMed] [Google Scholar]
- 8.Hamm-alvarez SF, Sheetz MP. Microtubule-dependent vesicle transport: modulation of channel and transporter activity in liver and kidney. Physiol Rev. 1998;78:1109–1129. doi: 10.1152/physrev.1998.78.4.1109. [DOI] [PubMed] [Google Scholar]
- 9.Staub O, Gautschi I, Ishikawa T, et al. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997;16:6325–6336. doi: 10.1093/emboj/16.21.6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimkets RA, Lifton RP, Canessa CM. The activity of the epithelial sodium channel is mediated by clathrin mediated endocytosis. J Biol Chem. 1997;272:25537–25541. doi: 10.1074/jbc.272.41.25537. [DOI] [PubMed] [Google Scholar]
- 11.Asher C, Wald H, Rossier BC, Garty H. Aldosterone-induced increase in the abundance of Na+ channel subunits. Am J Physiol. 1996;271:C605–C611. doi: 10.1152/ajpcell.1996.271.2.C605. [DOI] [PubMed] [Google Scholar]
- 12.Kemendy AE, Kleyman TR, Eaton DC. Aldosterone alters the open probability of amiloride-blockable sodium channels in A6 epithelia. Am J Physiol. 1992;263:C825–C837. doi: 10.1152/ajpcell.1992.263.4.C825. [DOI] [PubMed] [Google Scholar]
- 13.Garty H, Palmer LG. Epithelial sodium channels: Function, structure and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 14.Alton EWFW, Kingsleigh-smith DJ, Munkonge FM, et al. Asthma prophylaxis agents alter the function of an airway epithelial chloride channel. Am J Respir Cell Mol Biol. 1996;14:380–387. doi: 10.1165/ajrcmb.14.4.8600943. [DOI] [PubMed] [Google Scholar]
- 15.Lafont F, Burkhardt JK, Simons K. Involvement of microtubule motors in basolateral and apical transport in kidney cells. Nature. 1994;372:801–803. doi: 10.1038/372801a0. [DOI] [PubMed] [Google Scholar]
- 16.Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. 1993;361:467–470. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- 17.Canessa CM, Schild L, Buell G, et al. Amiloride-sensitive epithelial Na+ channel is made up of three homologous subunits. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 18.Palmer BF, Alpern RJ. Liddle's syndrome. Am J Med. 1998;104:301–309. doi: 10.1016/s0002-9343(98)00018-7. [DOI] [PubMed] [Google Scholar]
- 19.Liddle GW, Bledsoe T, Coppage WS. A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans Assoc Am Phys. 1963;76:199–213. [Google Scholar]
- 20.Shimkets RA, Warnock DG, Bositis CM, et al. Liddle's syndrome: heritable human hypertension caused by mutations in the β subunit of the epithelial sodium channel. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 21.Hansson JH, Nelson-williams C, Suzuki H, et al. Hypertension caused by a truncated epithelial sodium channel γ subunit: genetic heterogeneity of Liddle syndrome. Nature Genet. 1995;11:76–82. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- 22.Snyder PM, Price MP, McDonald FJ, et al. Mechanism by which Liddle's syndrome mutations increase activity of a human epithelial Na+ channel. Cell. 1995;83:969–978. doi: 10.1016/0092-8674(95)90212-0. [DOI] [PubMed] [Google Scholar]
- 23.Firsov D, Schild L, Gautschi I, Merillat AM, Schneeberger E, Rossier BC. Cell surface expression of the epithelial sodium channel and a mutant causing Liddle's syndrome: a quantitative approach. PNAS. 1996;93:15370–15375. doi: 10.1073/pnas.93.26.15370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Botero-velez M, Curtis JJ, Warnock DG. Liddle's syndrome revisited – A disorder of sodium reabsorption in the distal tubule. N Engl J Med. 1994;330:178–181. doi: 10.1056/NEJM199401203300305. [DOI] [PubMed] [Google Scholar]
- 25.McDonald FJ, Price MP, Snyder PM, Welsh MJ. Cloning and expression of the beta- and gamma-subunits of the human epithelial sodium channel. Am J Physiol. 1995;268:C1157–1163. doi: 10.1152/ajpcell.1995.268.5.C1157. [DOI] [PubMed] [Google Scholar]
- 26.Burch LH, Talbot CR, Knowles MR, Canessa CM, Rossier BC, Boucher RC. Relative expression of the human epithelial Na+ channel subunits in normal and cystic fibrosis airways Am. J Physiol. 1995;269:C511–C518. doi: 10.1152/ajpcell.1995.269.2.C511. [DOI] [PubMed] [Google Scholar]
- 27.Chinet TC, Fullton JM, Yankaskas JR, Boucher RC, Stutts MJ. Sodium-permeable channels in the apical membrane of human nasal epithelial cells. Am J Physiol. 1993;265:C1050–C1060. doi: 10.1152/ajpcell.1993.265.4.C1050. [DOI] [PubMed] [Google Scholar]
- 28.Eaton DC, Becchetti A, Ma H, Ling BN. Renal sodium channels: Regulation and single channel properties. Kidney Int. 1995;48:941–949. doi: 10.1038/ki.1995.375. [DOI] [PubMed] [Google Scholar]
- 29.Bubien JK, Ismailov IL, Berdiev BK, et al. Liddle's disease: abnormal regulation of amiloride-sensitive Na+ channels by β subunit mutation. Am J Physiol. 1996;270:C208–C213. doi: 10.1152/ajpcell.1996.270.1.C208. [DOI] [PubMed] [Google Scholar]
- 30.Baker EH, Jeunemaitre X, Portal AJ, et al. Abnormalities of nasal potential difference measurement in Liddle's Syndrome. J Clin Invest. 1998;102:10–14. doi: 10.1172/JCI1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strautnieks SS, Thompson RJ, Gardiner RM, Chung E. A novel splice-site mutation in the γ subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nature Genet. 1996;13:248–250. doi: 10.1038/ng0696-248. [DOI] [PubMed] [Google Scholar]
- 32.Chang SS, Grunder S, Hanukoglu A, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nature Genet. 1996;12:248–253. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 33.Persu A, Barbry P, Bassilana F, et al. Genetic analysis of the beta subunit of the epithelial Na+ channel in essential hypertension. Hypertension. 1998;32:129–137. doi: 10.1161/01.hyp.32.1.129. [DOI] [PubMed] [Google Scholar]
- 34.Melander O, Orho M, Fagerudd J, et al. Mutations and variants of the epithelial sodium channel gene in Liddle's syndrome and primary hypertension. Hypertension. 1998;31:1118–1124. doi: 10.1161/01.hyp.31.5.1118. [DOI] [PubMed] [Google Scholar]
- 35.Baker EH, Dong YB, Sagnella GA, et al. Association of hypertension with T594M mutation in β subunit of epithelial sodium channels in black people resident in London. Lancet. 1998;351:1388–1392. doi: 10.1016/s0140-6736(97)07306-6. [DOI] [PubMed] [Google Scholar]
- 36.Su YR, Rutkowski MP, Klanke CA, et al. A novel variant of the β-subunit of the amiloride-sensitive sodium channel in African Americans. J Am Soc Nephrol. 1996;7:2543–2549. doi: 10.1681/ASN.V7122543. [DOI] [PubMed] [Google Scholar]
- 37.Shimkets RA, Lifton R, Canessa CM. In vivo phosphorylation of the epithelial sodium channel. Proc Natl Acad Sci U S A. 1998;95:3301–3305. doi: 10.1073/pnas.95.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui Y, Su YR, Rutkowski M, Reif M, Menon AG, Pun RYK. Loss of protein kinase C inhibition in the beta-T594M variant of the amiloride-sensitive Na+ channel. Proc Natl Acad Sci U S A. 1997;94:9962–9966. doi: 10.1073/pnas.94.18.9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greger R. Ion transport mechanisms in thick ascending limb of Henle's loop of mammalian nephron. Physiol Rev. 1985;65:760–797. doi: 10.1152/physrev.1985.65.3.760. [DOI] [PubMed] [Google Scholar]
- 40.Hebert SC, Andreoli TE. Ionic conductance pathways in the mouse medullary thick ascending limb of Henle. The paracellular pathway and electrogenic Cl– absorption. J Gen Physiol. 1986;87:567–590. doi: 10.1085/jgp.87.4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon DB, Karet FE, Rodriguez-soriano J, et al. Genetic heterogeneity of Bartter's syndrome revealed by mutations in the K+ channel, RomK. Nat Genet. 1996;14:152–156. doi: 10.1038/ng1096-152. [DOI] [PubMed] [Google Scholar]
- 42.Schwalbe RA, Bianchi L, Accili EA, Brown AM. Functional consequences of RomK mutants linked to antenatal Bartter's syndrome and implications for treatment. Hum Mol Genet. 1998;7:975–980. doi: 10.1093/hmg/7.6.975. [DOI] [PubMed] [Google Scholar]
- 43.Guay-woodford LM. Bartter Syndrome: Unravelling the pathophysiologic enigma. Am J Med. 1998;105:151–161. doi: 10.1016/s0002-9343(98)00196-x. [DOI] [PubMed] [Google Scholar]
- 44.McNicholas CM, Guggino WB, Schwiebert EM, Hebert SC, Giebisch G, Egan ME. Sensitivity of a renal K+ channel (ROMK2) to the inhibitory sulfonylurea compound glibenclamide is enhanced by coexpression with the ATP-binding cassette transporter cyctic fibrosis transmembrane regulator. Proc Natl Acad Sci USA. 1996;93:8083–8088. doi: 10.1073/pnas.93.15.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bailey MA, Walter SJ. Renal effects of glibenclamide: a micropuncture study. J Pharmacol Exp Ther. 1998;285:464–467. [PubMed] [Google Scholar]
- 46.Rado JP, Borbely L, Szende L, Fischer J, Tako J. Investigation of the diuretic effect of glibenclamide in healthy subjects and in patients with pituitary and nephrogenic diabetes insipidus. Horm Metab Res. 1974;6:289–292. doi: 10.1055/s-0028-1093850. [DOI] [PubMed] [Google Scholar]
- 47.Williams S, Abbott D, Morfis L, Manwaring P, Diamond T, Howes LG. Effects of glibenclamide on blood pressure and cardiovascular responsiveness in non-insulin dependent diabetes mellitus. J Hypertens. 1998;16:705–711. doi: 10.1097/00004872-199816050-00019. [DOI] [PubMed] [Google Scholar]
- 48.Lieberman J, Rodbard S. Low blood pressure in young adults with cystic fibrosis: an effect of chronic salt loss in sweat? Ann Intern Med. 1975;82:806–808. doi: 10.7326/0003-4819-82-6-806. [DOI] [PubMed] [Google Scholar]
- 49.Quinton PM. Missing Cl conductance in cystic fibrosis. Am J Physiol. 1986;251:C649–C652. doi: 10.1152/ajpcell.1986.251.4.C649. [DOI] [PubMed] [Google Scholar]
- 50.Schweibert EM, Lopes AG, Guggino WB. Chloride channels along the nephron. In: Guggino WB, editor. Chloride Channels. San Diego: Academic Press; 1994. pp. 265–315. [Google Scholar]
- 51.Nilius B, Viana F, Droogmans G. Ion channels in vascular endothelium. Annu Rev Physiol. 1997;59:145–170. doi: 10.1146/annurev.physiol.59.1.145. [DOI] [PubMed] [Google Scholar]
- 52.Hoyer J, Kohler R, Haase W, Distler A. Up-regulation of pressure activated Ca2+ -permeable cation channel in intact vascular endothelium of hypertensive rats. Proc Natl Acad Sci USA. 1996;93:11253–11258. doi: 10.1073/pnas.93.20.11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inazu M, Zhang H, Daniel EE. LP-805, a releaser of endothelium-derived nitric oxide, activates an endothelial calcium permeable non-specific cation channel. Life Sci. 1993;53:315–320. doi: 10.1016/0024-3205(93)90626-e. [DOI] [PubMed] [Google Scholar]
- 54.Adams DJ, Barakeh J, Laskey R, Van Breeman C. Ion channels and regulation of intracellular calcium in vascular endothelial cells. FASEB J. 1989;3:2389–2400. doi: 10.1096/fasebj.3.12.2477294. [DOI] [PubMed] [Google Scholar]
- 55.Rubart M, Patlak JB, Nelson MT. Ca2+ currents in cerebral artery smooth muscle cells of rat at physiological Ca2+ concentrations. J Gen Physiol. 1996;107:459–472. doi: 10.1085/jgp.107.4.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clapp LH, Tinker A. Potassium channels in the vasculature. Curr Opin Nephrol Hypertens. 1998;7:91–98. doi: 10.1097/00041552-199801000-00015. [DOI] [PubMed] [Google Scholar]
- 57.Orallo F. Regulation of cytosolic calcium levels in vascular smooth muscle. Pharmacol Ther. 1996;69:153–171. doi: 10.1016/0163-7258(95)02042-x. [DOI] [PubMed] [Google Scholar]
- 58.Gollasch M, Nelson MT. Voltage-dependent Ca2+ channels in arterial smooth muscle cells. Kidney Blood Press Res. 1997;20:355–371. doi: 10.1159/000174250. [DOI] [PubMed] [Google Scholar]
- 59.Martens JR, Gelband CH. Ion channels in vascular smooth muscle: alterations in essential hypertension. Proc Soc Exp Biol Med. 1998;218:192–203. doi: 10.3181/00379727-218-44286. [DOI] [PubMed] [Google Scholar]
- 60.Rusch NJ, De Lucena RG, Wooldridge TA, England SK, Cowley Aw., Jr A Ca2+ dependent K+ current is enhanced in arterial membranes of hypertensive rats. Hypertension. 1992;19:301–307. doi: 10.1161/01.hyp.19.4.301. [DOI] [PubMed] [Google Scholar]
- 61.England SK, Wooldridge TA, Stekiel WJ, Rusch NJ. Enhanced single-channel K+ current in arterial membranes from genetically hypertensive rats. Am J Physiol. 1993;264:H1337–H1345. doi: 10.1152/ajpheart.1993.264.5.H1337. [DOI] [PubMed] [Google Scholar]
- 62.Ohya Y, Setoguchi M, Fujii K, Nagao T, Abe I, Fujishima M. Impaired action of levocromakalim on ATP-sensitive K+ channels in mesenteric artery cells from spontaneously hypertensive rats. Hypertension. 1996;27:1234–1239. doi: 10.1161/01.hyp.27.6.1234. [DOI] [PubMed] [Google Scholar]
- 63.Rusch NJ, Runnells AM. Remission of high blood pressure reverses arterial potassium channel alterations. Hypertension. 1994;23:941–945. doi: 10.1161/01.hyp.23.6.941. [DOI] [PubMed] [Google Scholar]
- 64.Benham CD, Tsien RW. Calcium-permeable channels in vascular smooth muscle: Voltage-activated, receptor operated, and leak channels. Soc Gen Physiol Ser. 1987;42:45–64. [PubMed] [Google Scholar]
- 65.Glasser SP. The relevance of T-type calcium antagonists: a profile of mibefradil. J Clin Pharmacol. 1998;38:659–669. doi: 10.1002/j.1552-4604.1998.tb04804.x. [DOI] [PubMed] [Google Scholar]
- 66.Schmitt R, Clozel J-P, Iberg N, Buhler FR. Mibefradil prevents neointima formation after vascular injury in rats. Arterioscler Thromb Vasc Biol. 1995;15:1161–1165. doi: 10.1161/01.atv.15.8.1161. [DOI] [PubMed] [Google Scholar]
- 67.Mishra SK, Hermsmeyer K. Selective inhibition of T-type Ca2+ channels by Ro 40–5967. Circ Res. 1994;75:144–148. doi: 10.1161/01.res.75.1.144. [DOI] [PubMed] [Google Scholar]
- 68.Oparil S. Mibefradil, a T-channel-selective calcium antagonist: clinical trials in hypertension. Am J Hypertens. 1998;11:88–94. doi: 10.1016/s0895-7061(98)00005-3. S S. [DOI] [PubMed] [Google Scholar]
- 69.Po AL, Zhang WY. What lessons can be learnt from withdrawal of mibefradil from the market? Lancet. 1998;351:1829–1830. doi: 10.1016/s0140-6736(05)78800-0. [DOI] [PubMed] [Google Scholar]
- 70.Giles TD. Hypertension and pathologic cardiovascular remodeling: a potential therapeutic role for T-type calcium antagonists. Clin Ther. 1997;19(Suppl A):27–38. doi: 10.1016/s0149-2918(97)80035-5. [DOI] [PubMed] [Google Scholar]
- 71.Edwards G, Weston AH. Pharmacology of the potassium channel openers. Cardiovasc Drugs Ther. 1995;9:185–193. doi: 10.1007/BF00878465. [DOI] [PubMed] [Google Scholar]
- 72.Webb DJ, Benjamin N, Vallance P. The potassium channel opening drug cromakalim produces arterioselective vasodilation in the upper limbs of healthy volunteers. Br J Clin Pharmacol. 1989;27:757–761. doi: 10.1111/j.1365-2125.1989.tb03437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Carlsen JE, Kardel T, Lund JO, McNair A, Trap-jensen J. Acute haemodynamic effects of pinacidil and hydralazine in essential hypertension. Clin Pharmacol Ther. 1985;37:253–259. doi: 10.1038/clpt.1985.36. [DOI] [PubMed] [Google Scholar]
- 74.Singer DR, Markandu ND, Miller MA, Sugden AL, MacGregor GA. Potassium channel stimulation in normal subjects and in patients with essential hypertension: an acute study with cromakalim (BRL 34915) J Hypertens. 1989;7:S294–S295. doi: 10.1097/00004872-198900076-00143. [DOI] [PubMed] [Google Scholar]
- 75.Carlsen JE, Jensen HA, Rehling M, Lund JO, Trap-jensen J. Long-term haemodynamic effects of pinacidil and hydralazine in arterial hypertension. Drugs. 1988;36(Suppl 7):55–63. doi: 10.2165/00003495-198800367-00010. [DOI] [PubMed] [Google Scholar]
- 76.Goldberg MR. Clinical pharmacology of pinacidil. A prototype for drugs that affect potassium channels. J Cardiovasc Pharmacol. 1988;12(Suppl 2):41–47. doi: 10.1097/00005344-198812002-00008. [DOI] [PubMed] [Google Scholar]
- 77.Goldschmidt M, Landzberg BR, Frishman WH. Nicorandil: a potassium channel opening drug for treatment of ischemic heart disease. J Clin Pharmacol. 1996;36:559–572. doi: 10.1002/j.1552-4604.1996.tb04219.x. [DOI] [PubMed] [Google Scholar]
- 78.Landry DW, Oliver JA. The ATP-sensitive K+ channel mediates hypotension in endotoxaemia and hypoxic lactic acidosis in dog. J Clin Invest. 1992;89:2071–2074. doi: 10.1172/JCI115820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vanelli G, Hussain SNA, Aguggini G. Glibenclamide, a blocker of ATP-sensitive potassium channels, reverses endotoxin-induced hypotension in pig. Exp Physiol. 1995;80:167–170. doi: 10.1113/expphysiol.1995.sp003832. [DOI] [PubMed] [Google Scholar]
- 80.Szabo C, Salzman AL. Inhibition of ATP-activated potassium channels exerts pressor effects and improves survival in a rat model of severe haemorrhagic shock. Shock. 1996;5:391–394. doi: 10.1097/00024382-199606000-00001. [DOI] [PubMed] [Google Scholar]
- 81.Salzman AL, Vromen A, Denenberg A, Szabo C. KATP-channel inhibition improves hemodynamics and cellular energetics in hemorrhagic shock. Am J Physiol. 1997;41:H688–H694. doi: 10.1152/ajpheart.1997.272.2.H688. [DOI] [PubMed] [Google Scholar]