

Abstract
Oleamide is an endogenous fatty acid primary amide that possesses sleep-inducing properties in animals and has been shown to effect serotonergic systems and block gap junction communication in a structurally specific manner. Herein, the structural features of oleamide required for inhibition of the gap junction-mediated chemical and electrical transmission in rat glial cells are defined. The effective inhibitors fall into two classes of fatty acid primary amides of which oleamide and arachidonamide are the prototypical members. Of these two, oleamide constitutes the most effective, and its structural requirements for inhibition of the gap junction are well defined. It requires a chain length of 16–24 carbons of which 16–18 carbons appears optimal, a polarized terminal carbonyl group capable of accepting but not necessarily donating a hydrogen bond, a Δ9 cis double bond, and a hydrophobic methyl terminus. Within these constraints, a range of modifications are possible, many of which may be expected to improve in vivo properties. A select set of agents has been identified that serves both as oleamide agonists and as inhibitors of fatty acid amide hydrolase, which is responsible for the rapid inactivation of oleamide.
Oleamide (1) ![]() is an endogenous fatty acid primary amide shown to accumulate in the cerebrospinal fluid under conditions of sleep deprivation (1–3). In a structurally specific manner, it has been shown to induce physiological sleep in animals when administered by i.p. or i.v. injection (1). Consistent with its role as a prototypical member of a new class of biological signaling molecules, enzymatic regulation of the endogenous concentrations of oleamide has been described (1, 4–7) or proposed (8). Fatty acid amide hydrolase (FAAH, oleamide hydrolase) is an integral membrane protein that degrades 1 to oleic acid, and potent inhibitors (Ki = 13 μM–1 nM) of the enzyme have been detailed (4). The characterization and neuronal distribution of FAAH have been disclosed (5–7), and it was found to possess the capabilities of hydrolyzing a range of fatty acid amides including anandamide, which serves as an endogenous ligand for cannabinoid receptors (9–11). In contrast to anandamide, an appealing feature of the members of this new class of biological signaling agents is the primary amide, suggesting that their storage and release may be controlled in a manner analogous to that of short peptide hormones and messengers terminating in a primary amide (8).
is an endogenous fatty acid primary amide shown to accumulate in the cerebrospinal fluid under conditions of sleep deprivation (1–3). In a structurally specific manner, it has been shown to induce physiological sleep in animals when administered by i.p. or i.v. injection (1). Consistent with its role as a prototypical member of a new class of biological signaling molecules, enzymatic regulation of the endogenous concentrations of oleamide has been described (1, 4–7) or proposed (8). Fatty acid amide hydrolase (FAAH, oleamide hydrolase) is an integral membrane protein that degrades 1 to oleic acid, and potent inhibitors (Ki = 13 μM–1 nM) of the enzyme have been detailed (4). The characterization and neuronal distribution of FAAH have been disclosed (5–7), and it was found to possess the capabilities of hydrolyzing a range of fatty acid amides including anandamide, which serves as an endogenous ligand for cannabinoid receptors (9–11). In contrast to anandamide, an appealing feature of the members of this new class of biological signaling agents is the primary amide, suggesting that their storage and release may be controlled in a manner analogous to that of short peptide hormones and messengers terminating in a primary amide (8).
In addition to its sleep-inducing properties in animals, oleamide has been shown to effect serotonergic systems (7, 12–14) and to block gap junction communication (15) in a structurally specific manner. Although it was found to inhibit the gap junction-mediated chemical and electrical transmission in rat glial cells, it had no inhibitory effect on mechanically stimulated or glutamate-induced calcium wave propagation, thereby decoupling two previously indistinguishable glial cell communication pathways (15). Given the central role of intercellular chemical and electrical signaling in the central nervous system, the oleamide inactivation of glial cell gap junction channels may influence higher order neuronal function including sleep. Herein, we describe a study of the structural features of oleamide required for this inhibition of glial cell gap junction communication.
MATERIALS AND METHODS
Materials.
The agents examined were purchased (Sigma, Pfaltz & Bauer, and Aldrich), prepared as described (3, 4, 16–19), or synthesized following protocols previously detailed (3, 4, 16–18).
Methods.
Rat glial cells (20) were cultured in plastic tissue culture ware in Richter’s improved minimal essential medium (Irvine Scientific), supplemented with 10% fetal calf serum and 50 μg/ml gentamicin sulfate, and incubated in a humidified atmosphere of 95% air/5% CO2 at 37°C. The cells were passaged by trypsinization and used at passages 6–10 and experimented at ≈90% confluent. The tested compounds were dissolved in EtOH and added to the culture media (1 μL/ml medium) providing the indicated concentrations (100, 50, or 20 μM) from 1,000-fold higher stock solutions. Control cultures were treated with EtOH (1 μL/ml medium, final 0.1% EtOH concentration). Microinjection of fluorescent Lucifer yellow CH dye into cultured cells and quantitation of the dye transfer frequency to directly adjacent neighboring cells were performed as described (21). In brief, micropipettes (approximate tip diameter of 0.5 μm) were loaded with the dye solution (5% Lucifer yellow CH dye in aqueous 0.1 M LiCl) by backfilling. Cells were visualized by a Nickon Diaphot inverted phase contrast/epifluorescent microscope and impaled with dye-filled micropipettes by using an Eppendorf micromanipulator (model 5240). Impaled cells were loaded with dye by iontophoresis for 2–3 s at 20 nA. After 5 min, the transfer of dye to directly adjacent cells was determined by using epifluorescent illumination. For each treatment, compound was maintained in the culture dish for 4 h, and then 10 cells were microinjected in each of two dishes and the total numbers of dye-coupled and nondye-coupled adjacent neighboring cells were calculated and compared with control (0% inhibition) to determine the percentage of inhibition of dye-coupling.
RESULTS
Fatty Acid Primary Amides.
The first series examined were the primary amides (1, 3, 22–24) of the naturally occurring fatty acids and related synthetic analogs (Table 1) 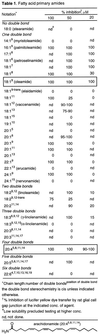 . In these studies, the number, position, and stereochemistry of the double bond(s) and the length of the agent were assessed. From these studies, which were assisted with the inclusion of the primary amides of nonnaturally occurring fatty acids, a clear depiction of the structural requirements of the endogenous agent emerged. The effective inhibitors of the gap junction-mediated dye transfer fall into two classes of which oleamide and arachidonamide (9–11, 25–32) are the prototypical members.
. In these studies, the number, position, and stereochemistry of the double bond(s) and the length of the agent were assessed. From these studies, which were assisted with the inclusion of the primary amides of nonnaturally occurring fatty acids, a clear depiction of the structural requirements of the endogenous agent emerged. The effective inhibitors of the gap junction-mediated dye transfer fall into two classes of which oleamide and arachidonamide (9–11, 25–32) are the prototypical members.
For agents related to oleamide that contain one double bond, the presence, position, and stereochemistry of the olefin as well as the chain length were found to have a defined relationship. The monounsaturated fatty acid primary amides of 16–24 carbons containing a Δ9 cis double bond were all found to be effective inhibitors of the gap junction-mediated dye transfer. Removal of the Δ9 double bond (18:0) or its replacement with a trans double bond (18:19-trans) resulted in no observable inhibition at the concentrations examined. Shortening the chain length to 14 carbons (14:19) resulted in the loss of activity, and lengthening the chain slightly diminished the inhibitory effectiveness. No further substantial trend in the length was observed with the longer fatty acid primary amides provided they possessed a Δ9 cis double bond: 14:19 ≪ 16:19 = 18:19 > 20:19 = 22:19 = 24:19. The position of the cis double bond had a well defined effect on the potency. A Δ9 cis olefin exhibited the strongest inhibitory effect, and the potency typically declined as its position was moved in either direction. This effect was apparent in the comparisons made in the 18:1 series and was especially clear in the 20:1 series where the potency declined as the distance from the Δ9 position increased. Thus, although 18:18 was less effective while 18:17 and 18:16 were as effective as oleamide, the potency decline as the distance from the Δ9 position increased was more apparent in the 20:1 series. Although this location in oleamide is central to its structure and potentially represents a relationship with either the carboxamide or methyl terminus, the potent activity of palmitoleamide (16:19) suggested that it is the positional relationship with the amide that is important. This relationship was confirmed in the 20:1, 22:1, and 24:1 series where inhibitory activity was observed with the synthetic Δ9 olefins rather than the naturally occurring isomers bearing a cis double bond 9 carbons from the methyl terminus: 20:19 = 20:111, 22:19 > 22:113, and 24:19 > 24:115. In this context, it is interesting that erucamide (22:113), which has been detected in human cerebrospinal fluid (1), is not an inhibitor of the gap junction and does not induce physiological sleep in rats.
With the polyunsaturated fatty acid primary amides, those containing two double bonds exhibited modest activity. Typically, this activity was considerably diminished relative to oleamide except for linoleamide (18:29,12), which also possesses the cis Δ9 double bond. Even with linoleamide, the inhibition potency was lower than oleamide, indicating that the additional Δ12 cis double bond was not beneficial. The fatty acid primary amides containing three double bonds were even less active with essentially all members being ineffective at 50 μM, including α-linolenamide (20:39,12,15), which incorporates the Δ9 cis double bond of oleamide, and 20:38,11,14, which incorporates an alternative Δ8 or Δ11 cis double bond. The exception to this generalization is γ-linolenamide (18:36,9,12), which proved to be an effective but less potent inhibitor. Similarly, arachidonamide containing four cis double bonds was an effective inhibitor of the gap junction-mediated dye transfer although it was not quite as potent as oleamide. This effect also was observed with a fatty acid primary amide containing five but not six cis double bonds.
These results are consistent with two classes of fatty acid primary amides that inhibit the gap junction-mediated dye transfer. The prototypical member of the more potent class is oleamide, and the second, slightly less effective, class contains arachidonamide.
The Carboxamide.
A study of the requirements at the carboxylate terminus proved revealing in that it was surprisingly tolerant (Table 2) 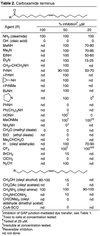 . Oleic acid was found to be inactive at the concentrations examined, and oleyl alcohol, methyl or ethyl oleate, and oleyl acetate exhibited progressively reduced effectiveness. Oleic acid (33, 34), arachidonic acid (35, 36), and α-linolenic acid (37) have been reported to inhibit the gap junction in rat cardiac myocytes, striatal astrocytes, and rat liver epithelial cells, respectively. The limited studies of their structural requirements for gap junction inhibition including the chain length, degree and site of unsaturation, and the stereochemistry of the double bond(s) roughly follow those detailed herein although fewer fatty acids have been examined in these efforts (33–39). The comparisons detailed in Table 2 suggest that the most effective derivatives of the fatty acids in rat glial cells are the primary amides. However, a wide range of secondary or tertiary amides were found to be potent inhibitors of the gap junction-mediated dye transfer. Thus, the hydrogen bond donor capability of the primary amides is not essential to the expression of their properties. Many of these were equipotent with oleamide, and the series examined exhibited a clear trend favoring the smaller secondary or tertiary amide substituents. This trend is evident upon comparing the effects of R = NH2 ≥ MeNH > Et2N > c-(CH2)4N > PhNH. Although the secondary amides typically were more potent than the corresponding tertiary amides, the comparable potency of the N-methyl and N,N-dimethyl amide (MeNH = Me2N) is notable. This potency is consistent with a requirement for small amide substituents, and the most potent of the agents examined was the O-methyl,N-methyl hydroxamide (R = MeONMe). Although the esters of oleic acid were inactive at 50–100 μM, oleyl aldehyde proved to be surprisingly effective while its corresponding dimethyl acetal was inactive. Oleyl aldehyde was found to be nearly equipotent with oleamide, and the related agent R = CF3 possessing a strongly electrophilic and polarized carbonyl was significantly more potent than oleamide. In contrast, related but less electrophilic ketones (R = BrCH2 > ClCH2) were substantially less effective. Thus, a smooth trend favoring the more polarized or electrophilic carbonyls was observed. The behavior of the former two agents (R = H, CF3) is especially significant because they are also potent inhibitors (Ki = 190 and 1 nM, respectively) of fatty acid amide hydrolase (4). Such agents possess dual activities serving both as effective agonists of oleamide and as potent inhibitors of the enzyme responsible for its degradation. Both exhibit a reversible effect on the gap junction inhibition and removal of the media containing the agents and replacement with fresh media restored full gap junction function. Although such inhibitors may exert their effects by inhibiting FAAH and potentiating the effect of endogenous oleamide, both the very low levels of FAAH and endogenous oleamide in rat glial cells are not sufficient to account for the gap junction inhibition effect.
. Oleic acid was found to be inactive at the concentrations examined, and oleyl alcohol, methyl or ethyl oleate, and oleyl acetate exhibited progressively reduced effectiveness. Oleic acid (33, 34), arachidonic acid (35, 36), and α-linolenic acid (37) have been reported to inhibit the gap junction in rat cardiac myocytes, striatal astrocytes, and rat liver epithelial cells, respectively. The limited studies of their structural requirements for gap junction inhibition including the chain length, degree and site of unsaturation, and the stereochemistry of the double bond(s) roughly follow those detailed herein although fewer fatty acids have been examined in these efforts (33–39). The comparisons detailed in Table 2 suggest that the most effective derivatives of the fatty acids in rat glial cells are the primary amides. However, a wide range of secondary or tertiary amides were found to be potent inhibitors of the gap junction-mediated dye transfer. Thus, the hydrogen bond donor capability of the primary amides is not essential to the expression of their properties. Many of these were equipotent with oleamide, and the series examined exhibited a clear trend favoring the smaller secondary or tertiary amide substituents. This trend is evident upon comparing the effects of R = NH2 ≥ MeNH > Et2N > c-(CH2)4N > PhNH. Although the secondary amides typically were more potent than the corresponding tertiary amides, the comparable potency of the N-methyl and N,N-dimethyl amide (MeNH = Me2N) is notable. This potency is consistent with a requirement for small amide substituents, and the most potent of the agents examined was the O-methyl,N-methyl hydroxamide (R = MeONMe). Although the esters of oleic acid were inactive at 50–100 μM, oleyl aldehyde proved to be surprisingly effective while its corresponding dimethyl acetal was inactive. Oleyl aldehyde was found to be nearly equipotent with oleamide, and the related agent R = CF3 possessing a strongly electrophilic and polarized carbonyl was significantly more potent than oleamide. In contrast, related but less electrophilic ketones (R = BrCH2 > ClCH2) were substantially less effective. Thus, a smooth trend favoring the more polarized or electrophilic carbonyls was observed. The behavior of the former two agents (R = H, CF3) is especially significant because they are also potent inhibitors (Ki = 190 and 1 nM, respectively) of fatty acid amide hydrolase (4). Such agents possess dual activities serving both as effective agonists of oleamide and as potent inhibitors of the enzyme responsible for its degradation. Both exhibit a reversible effect on the gap junction inhibition and removal of the media containing the agents and replacement with fresh media restored full gap junction function. Although such inhibitors may exert their effects by inhibiting FAAH and potentiating the effect of endogenous oleamide, both the very low levels of FAAH and endogenous oleamide in rat glial cells are not sufficient to account for the gap junction inhibition effect.
Thus, a range of effective although sterically undemanding substitutions can be made for the carboxamide. To date, the effective inhibitors possess a polarized carbonyl at the C terminus potentially capable of accepting but not necessarily donating a hydrogen bond.
Oleyl Ethanolamide, Anandamide, and Related Structures.
An important subset of modified carboxamides is the ethanolamide derivatives (40–43). In our earlier study, anandamide as well as oleamide were shown to selectively inhibit gap junction communication (dye transfer and electrical coupling) without affecting calcium wave transmission although oleamide was more potent (15). This effect in glial cells is distinct from the report that anandamide and oleyl ethanolamide not only inhibit gap junction-mediated dye transfer but also block the propagation of astrocyte calcium waves generated either by mechanical stimulation or local glutamate application (32). Consequently, the corresponding ethanolamide of oleic acid (3) and the bis-(ethanol)amides of oleic and arachidonic acid were examined (Table 3) 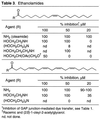 . Oleamide proved more potent than oleyl ethanolamide and discernibly more potent than anandamide at 20 μM although both were effective inhibitors at 50 μM. Although the effective properties of the oleyl ethanolamide (R = HOCH2CH2NH) may suggest that it represents an important member of another class of active endogenous agents, it is less potent than oleamide, N-ethyl oleamide, which lacks the hydroxyl group, as well as N-propyl oleamide, which contains a methyl group in place of the hydroxyl group (Table 2). In addition, oleyl propanolamide exhibited comparable properties. Thus, the hydroxyl group of oleyl ethanolamide does not appear to contribute to its gap junction inhibition properties and actually may diminish them. Similar observations were made in the comparison of arachidonyl amide (R = NH2) and anandamide (R = HOCH2CH2NH), where the latter was less potent. Comparable observations have been made in the examination of anandamide analogs (R = CH3CH2CH2NH > HOCH2CH2NH) with their binding to the central nervous system cannabinoid receptor (25–31). In contrast, the bis-(ethanol)amides were ineffective at inhibiting the gap junction at 50 μM, which is consistent with their behavior as bulky tertiary amides that exhibit lower activity.
. Oleamide proved more potent than oleyl ethanolamide and discernibly more potent than anandamide at 20 μM although both were effective inhibitors at 50 μM. Although the effective properties of the oleyl ethanolamide (R = HOCH2CH2NH) may suggest that it represents an important member of another class of active endogenous agents, it is less potent than oleamide, N-ethyl oleamide, which lacks the hydroxyl group, as well as N-propyl oleamide, which contains a methyl group in place of the hydroxyl group (Table 2). In addition, oleyl propanolamide exhibited comparable properties. Thus, the hydroxyl group of oleyl ethanolamide does not appear to contribute to its gap junction inhibition properties and actually may diminish them. Similar observations were made in the comparison of arachidonyl amide (R = NH2) and anandamide (R = HOCH2CH2NH), where the latter was less potent. Comparable observations have been made in the examination of anandamide analogs (R = CH3CH2CH2NH > HOCH2CH2NH) with their binding to the central nervous system cannabinoid receptor (25–31). In contrast, the bis-(ethanol)amides were ineffective at inhibiting the gap junction at 50 μM, which is consistent with their behavior as bulky tertiary amides that exhibit lower activity.
Recent studies have identified 2-arachidonyl glycerol as a potential endogenous cannabinoid receptor agonist (44), and gap junction inhibition by diacylglycerols including 1-oleoyl-2-acetylglycerol in Chinese hamster V79 cells has been disclosed (38). Consequently, both racemic and (2S)-1-oleoyl-2-acetylglycerol were evaluated as potential inhibitors. Consistent with the behavior of simple oleic acid esters (Table 2), both were not inhibitors of rat glial gap junction-mediated dye transfer.
Methyl Terminus.
From the studies of the fatty acid primary amides, the length of the fatty acid chain did not reduce substantially the gap junction inhibition properties provided it incorporated the key Δ9 cis double bond: 18:19 > 20:19 = 22:19 = 24.19. This result indicates that the methyl terminus can accommodate extensions by a alkyl chain without losing the agonist properties. Similarly, capping the methyl terminus with a methyl ether (R = CH2OCH3) did not affect the gap junction inhibition. In contrast, the introduction of polar functionality at this site eliminated the inhibitory properties. Thus, the symmetrical 18:19 dicarboxamide as well as the terminal alcohol and carboxylic acid exhibited no gap junction inhibition at 50 μM (Table 4) 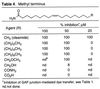 .
.
Putative Precursors.
The potential that oleamide may be stored as a N-oleoyl glycinamide derivative whose formation from oleoyl-CoA is catalyzed by acyl-CoA:glycine N-acyltransferase and released upon α-hydroxylation of glycine by peptidylglycine α-amidating monooxygenase or a related enzyme led us to examine a set of N-oleoyl glycine derivatives even before its potential was demonstrated experimentally (8). Both N-oleoyl glycine as well as N-oleoyl glycinamide were ineffective as inhibitors of gap junction-mediated dye transfer (Table 5)  . Thus, a putative storage precursor to oleamide as well as N-oleoyl glycinamide, which is representative of a peptide or protein terminating with an oleoyl glycinamide, failed to inhibit the gap junction consistent with their role as inactive precursors (45). N-Oleoyl glycine ethyl ester was an effective, albeit less potent, inhibitor and likely represents simply another allowable N-substitution on the terminal carboxamide (cf. Table 2). In contrast, N-oleoyl sarcosine as well as its ethyl ester were found to be effective inhibitors. Initially surprising, this observation suggests that the intramolecular hydrogen-bonded rotamer of the free acid or the deprotonated carboxylate, which would be observed with N-oleoyl glycine but less favored with N-oleoyl sarcosine, ties up the amide carbonyl disrupting its intermolecular hydrogen bond acceptor capabilities that may be required for gap junction inhibition.
. Thus, a putative storage precursor to oleamide as well as N-oleoyl glycinamide, which is representative of a peptide or protein terminating with an oleoyl glycinamide, failed to inhibit the gap junction consistent with their role as inactive precursors (45). N-Oleoyl glycine ethyl ester was an effective, albeit less potent, inhibitor and likely represents simply another allowable N-substitution on the terminal carboxamide (cf. Table 2). In contrast, N-oleoyl sarcosine as well as its ethyl ester were found to be effective inhibitors. Initially surprising, this observation suggests that the intramolecular hydrogen-bonded rotamer of the free acid or the deprotonated carboxylate, which would be observed with N-oleoyl glycine but less favored with N-oleoyl sarcosine, ties up the amide carbonyl disrupting its intermolecular hydrogen bond acceptor capabilities that may be required for gap junction inhibition.
Modifications in the Olefin.
A series of agents (2–10) was examined to define the role of the olefin and to provide insights into a conformational dependence on activity (Table 6) 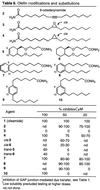 . Thus, whereas the saturated agent octadecanamide and the trans-9-octadecenamide were ineffective (cf. Table 1), 9-octadecynamide (2) was a potent inhibitor, being only slightly less active than oleamide. Its conformational characteristics, which more closely resemble those of the inactive 18:0 and 18:19-trans than oleamide, suggest that the appropriate presentation of a π-system in addition to the conformation of the cis olefin may be important. Consistent with this, the epoxide 3 was inactive and the cyclopropane 4 was less active although both may closely mimic the conformational characteristics of oleamide. Although there are a number of plausible explanations for this, 4 may benefit from the partial π characteristics of the cyclopropane. Alternatively, the incorporation of the epoxide oxygen in 3 may alter the hydrophobic character of the agent and diminish gap junction inhibition. Consistent with either explanation, both cis- and trans-5 and -6 proved to be weak inhibitors (cis > trans) of the gap junction-mediated dye transfer, and 5, which possesses the cis Δ9 double of oleamide, was more potent. The removal of the double bond reduced but did not eliminate the inhibition properties, suggesting an important role for both the π-character of the double bond as well as its imposed conformational preference for a hairpin conformation. Consistent with this, incorporation of a benzene ring into the structure provided the agents 7–10 that inhibit the gap junction-mediated dye transfer. The most potent in the series was 8, being nearly equipotent with oleamide, and it most closely mimics the hairpin conformation of oleamide. This potency was followed by that of 7, which represents an extended version of oleamide. Both 9 and 10 were less effective, and both mimic alternatives to the hairpin conformation of oleamide.
. Thus, whereas the saturated agent octadecanamide and the trans-9-octadecenamide were ineffective (cf. Table 1), 9-octadecynamide (2) was a potent inhibitor, being only slightly less active than oleamide. Its conformational characteristics, which more closely resemble those of the inactive 18:0 and 18:19-trans than oleamide, suggest that the appropriate presentation of a π-system in addition to the conformation of the cis olefin may be important. Consistent with this, the epoxide 3 was inactive and the cyclopropane 4 was less active although both may closely mimic the conformational characteristics of oleamide. Although there are a number of plausible explanations for this, 4 may benefit from the partial π characteristics of the cyclopropane. Alternatively, the incorporation of the epoxide oxygen in 3 may alter the hydrophobic character of the agent and diminish gap junction inhibition. Consistent with either explanation, both cis- and trans-5 and -6 proved to be weak inhibitors (cis > trans) of the gap junction-mediated dye transfer, and 5, which possesses the cis Δ9 double of oleamide, was more potent. The removal of the double bond reduced but did not eliminate the inhibition properties, suggesting an important role for both the π-character of the double bond as well as its imposed conformational preference for a hairpin conformation. Consistent with this, incorporation of a benzene ring into the structure provided the agents 7–10 that inhibit the gap junction-mediated dye transfer. The most potent in the series was 8, being nearly equipotent with oleamide, and it most closely mimics the hairpin conformation of oleamide. This potency was followed by that of 7, which represents an extended version of oleamide. Both 9 and 10 were less effective, and both mimic alternatives to the hairpin conformation of oleamide.
Modifications in the Linking Chain and Enzyme Inhibitors: Dual Activities.
In the examination of the fatty acid primary amides, it was established that increasing or shortening the distance between the carboxamide and cis double bond generally diminished the gap junction inhibition. Additional modifications in the 7 carbon chain linking the olefin and carboxamide also were examined (Table 7) 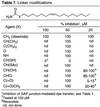 . The first set of agents contain modifications that slow the rate of degradation by enzymatic hydrolysis (1, 4, 12–14, 46). Both α-methyl oleamide and α,α-dimethyl oleamide were potent inhibitors of the gap junction, and both have been shown to be subject to slower FAAH hydrolysis than oleamide (12–14). In addition, the agents containing a terminal primary carbamate (X = O) or urethane (X = NH) were also potent inhibitors. The latter is especially interesting because the corresponding urethane of oleyl amine, which is two carbons longer in length and whose double bond resides not 9 but 11 carbons from the terminal carbonyl, is essentially inactive (Table 2). Similarly, α-hydroxy oleamide (X = CHOH), α-chlorooleamide (X = CHCl), and α-acetylthiooleamide (X = CHSAc) were effective inhibitors, although the free thiol (X = CHSH) was ineffective. This latter agent suffers from disulfide formation that may account for its ineffectiveness. The last two agents in Table 7 constitute analogs that are effective (Ki = 16 and 17 nM, respectively) inhibitors of FAAH (4). Both serve as oleamide agonists and as inhibitors (4, 47) of FAAH responsible for its degradation. For both, the gap junction inhibition was reversible and restored to full activity after suspension of the glial cells in fresh media.
. The first set of agents contain modifications that slow the rate of degradation by enzymatic hydrolysis (1, 4, 12–14, 46). Both α-methyl oleamide and α,α-dimethyl oleamide were potent inhibitors of the gap junction, and both have been shown to be subject to slower FAAH hydrolysis than oleamide (12–14). In addition, the agents containing a terminal primary carbamate (X = O) or urethane (X = NH) were also potent inhibitors. The latter is especially interesting because the corresponding urethane of oleyl amine, which is two carbons longer in length and whose double bond resides not 9 but 11 carbons from the terminal carbonyl, is essentially inactive (Table 2). Similarly, α-hydroxy oleamide (X = CHOH), α-chlorooleamide (X = CHCl), and α-acetylthiooleamide (X = CHSAc) were effective inhibitors, although the free thiol (X = CHSH) was ineffective. This latter agent suffers from disulfide formation that may account for its ineffectiveness. The last two agents in Table 7 constitute analogs that are effective (Ki = 16 and 17 nM, respectively) inhibitors of FAAH (4). Both serve as oleamide agonists and as inhibitors (4, 47) of FAAH responsible for its degradation. For both, the gap junction inhibition was reversible and restored to full activity after suspension of the glial cells in fresh media.
DISCUSSION
The disclosure that oleamide inhibits the gap junction-mediated intercellular chemical and electrical transmission without effecting calcium wave propagation in rat glial cells (15) suggests that this may constitute an important site of action for this new class of biologically active lipids. The effective inhibitors of the gap junction fall into two classes of fatty acid primary amides of which oleamide and arachidonamide are the prototypical members. Of these, oleamide constitutes the most effective and its structural requirements for inhibition of the gap junction are well defined. The inhibition requires a chain length of 16–24 carbons of which 16–18 appears optimal, a polarized terminal carbonyl capable of accepting but not necessarily donating a hydrogen bond, a Δ9 cis double bond, and a hydrophobic methyl terminus. Within these constraints, a range of modifications are acceptable, many of which may be used to enhance in vivo properties. In addition to those that would slow or avoid inactivation by FAAH hydrolysis (5), a set of especially interesting agents have been identified that serve both as oleamide agonists and as FAAH inhibitors (4).
The inhibition of the gap junction by oleamide is consistent with a number of mechanisms (48). Perturbations in the bulk membrane fluidity or the membrane–protein interface that would effect the conformation of the membrane-bound proteins as well as direct interaction with the gap junction proteins have been discussed (15, 48). Similarly, both progressive closure and ultimately collapse of the gap junction as well as gated closure of the gap junction have been considered. The compounds disclosed herein and the defined structural features of oleamide responsible for the properties may help distinguish among the mechanisms as well as provide new neuromodulatory agents applicable to sleep or mood disorders, analgesia, and disorders associated with higher neuronal function.
Acknowledgments
We gratefully acknowledge the Skaggs Institute for Chemical Biology, the National Institutes of Health (CA42056, D.L.B.), and the National Science Foundation (predoctoral fellowship to B.F.C.) for financial support. We thank Dr. J. E. Trosko for providing the rat glial cells and Dr. H. J. Waller for assembling the iontophoresis injector used for this study.
ABBREVIATION
- FAAH
fatty acid amide hydrolase
References
- 1.Cravatt B F, Prospero-Garcia O, Siuzdak G, Gilula N B, Henriksen S J, Boger D L, Lerner R A. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- 2.Lerner R A, Siuzdak G, Prospero-Garcia O, Henriksen S J, Boger D L, Cravatt B F. Proc Natl Acad Sci USA. 1994;91:9505–9508. doi: 10.1073/pnas.91.20.9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cravatt B F, Lerner R A, Boger D L. J Am Chem Soc. 1996;118:580–590. [Google Scholar]
- 4.Patterson J E, Ollmann I R, Cravatt B F, Boger D L, Wong C-H, Lerner R A. J Am Chem Soc. 1996;118:5938–5945. [Google Scholar]
- 5.Cravatt B F, Giang D K, Mayfield S P, Boger D L, Lerner R A, Gilula N B. Nature (London) 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 6.Giang D K, Cravatt B F. Proc Natl Acad Sci USA. 1997;94:2238–2242. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas E A, Cravatt B F, Danielson P E, Gilula N B, Sutcliffe G. J Neurosci Res. 1997;50:1047–1052. doi: 10.1002/(SICI)1097-4547(19971215)50:6<1047::AID-JNR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 8.Merkler D J, Merkler K A, Stern W, Fleming F F. Arch Biochem Biophys. 1996;330:430–434. doi: 10.1006/abbi.1996.0272. [DOI] [PubMed] [Google Scholar]
- 9.Devane W A, Hanus L, Breuer A, Pertwee R G, Stevenson L A, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 10.Johnson D E, Heald S L, Dally R D, Janis R A. Prostaglandins Leukotrienes Essent Fatty Acids. 1993;48:429–437. doi: 10.1016/0952-3278(93)90048-2. [DOI] [PubMed] [Google Scholar]
- 11.Di Marzo V, Fontana A. Prostaglandins Leukotrienes Essent Fatty Acids. 1995;53:1–11. doi: 10.1016/0952-3278(95)90077-2. [DOI] [PubMed] [Google Scholar]
- 12.Huidobro-Toro J P, Harris R A. Proc Natl Acad Sci USA. 1996;93:8078–8082. doi: 10.1073/pnas.93.15.8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas E A, Carson M J, Neal M J, Sutcliff J G. Proc Natl Acad Sci USA. 1997;94:14115–14119. doi: 10.1073/pnas.94.25.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boger D L, Patterson J E, Jin Q. Proc Natl Acad Sci USA. 1998;95:4102–4107. doi: 10.1073/pnas.95.8.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan X, Cravatt B F, Ehring G R, Hall J E, Boger D L, Lerner R A, Gilula N B. J Cell Biol. 1997;139:1785–1792. doi: 10.1083/jcb.139.7.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roe E T, Scanlan J T, Swern D. J Am Chem Soc. 1949;71:2215–2218. [Google Scholar]
- 17.Roe E T, Miles T D, Swern D. J Am Chem Soc. 1952;74:3442–3443. [Google Scholar]
- 18.Simmons H E, Smith R D. J Am Chem Soc. 1959;81:4256–4264. [Google Scholar]
- 19.Patterson J E. Ph.D. thesis. La Jolla, CA: The Scripps Research Institute; 1997. [Google Scholar]
- 20.Suter S, Trosko J E, El-Fouly M H, Lockwood L R, Koestner A. Fundam Appl Toxicol. 1987;9:785–794. doi: 10.1016/0272-0590(87)90185-0. [DOI] [PubMed] [Google Scholar]
- 21.Guan X J, Bonney W J, Ruch R J. Toxicol Appl Pharmacol. 1995;130:79–86. doi: 10.1006/taap.1995.1011. [DOI] [PubMed] [Google Scholar]
- 22.Arafat E S, Trimble J W, Andersen R N, Dass C, Desiderio D M. Life Sci. 1989;45:1679–1687. doi: 10.1016/0024-3205(89)90278-6. [DOI] [PubMed] [Google Scholar]
- 23.Wakamatsu K, Masaki T, Itoh F, Kondo K, Katsuichi S. Biochem Biophys Res Commun. 1990;168:423–429. doi: 10.1016/0006-291x(90)92338-z. [DOI] [PubMed] [Google Scholar]
- 24.Jain M K, Ghomashchi F, Yu B-Z, Bayburt T, Murphy D, Houck D, Brownell J, Reid J C, Solowiej J E, Wong S-M, et al. J Med Chem. 1992;35:3584–3586. doi: 10.1021/jm00097a018. [DOI] [PubMed] [Google Scholar]
- 25.Pinto J C, Potie F, Rice K C, Boring D, Johnson M R, Evans D M, Wilken G H, Cantrell C H, Howlett A C. Mol Pharmacol. 1994;46:516–522. [PubMed] [Google Scholar]
- 26.Felder C C, Briley E M, Axelrod J, Simpson J T, Mackie K, Devane W A. Proc Natl Acad Sci USA. 1993;90:7656–7660. doi: 10.1073/pnas.90.16.7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abadji V, Lin S, Taha G, Griffin G, Stevenson L A, Pertwee R G, Makriyannis A. J Med Chem. 1994;37:1889–1893. doi: 10.1021/jm00038a020. [DOI] [PubMed] [Google Scholar]
- 28.Adams I B, Ryan W, Singer M, Thomas B F, Compton D R, Razdan R K, Martin B R. J Pharmacol Exp Ther. 1995;273:1172–1181. [PubMed] [Google Scholar]
- 29.Adams I B, Ryan W, Singer M, Razdan R K, Compton D R, Martin B R. Life Sci. 1995;56:2041–2048. doi: 10.1016/0024-3205(95)00187-b. [DOI] [PubMed] [Google Scholar]
- 30.Priller J, Briley E M, Mansouri J, Devane W A, Mackie K, Felder C C. Mol Pharmacol. 1995;48:288–292. [PubMed] [Google Scholar]
- 31.Khanolkar A D, Abadji V, Lin S, Hill W A G, Taha G, Abouzid K, Meng Z, Fan P, Makriyannis A. J Med Chem. 1996;39:4515–4519. doi: 10.1021/jm960152y. [DOI] [PubMed] [Google Scholar]
- 32.Venance L, Piomelli D, Glowinski J, Glaume C. Nature (London) 1995;376:590–594. doi: 10.1038/376590a0. [DOI] [PubMed] [Google Scholar]
- 33.Hirschi K K, Minnich B N, Moore L K, Burt J M. Am J Physiol. 1993;265:C1517–C1526. doi: 10.1152/ajpcell.1993.265.6.C1517. [DOI] [PubMed] [Google Scholar]
- 34.Burt J M, Massey K D, Minnich B N. Am J Physiol. 1991;260:C439–C448. doi: 10.1152/ajpcell.1991.260.3.C439. [DOI] [PubMed] [Google Scholar]
- 35.Giaume C, Marin P, Cordier J, Glowinski J, Premont J. Proc Natl Acad Sci USA. 1991;88:5577–5581. doi: 10.1073/pnas.88.13.5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massey K D, Minnich B N, Burt J M. Am J Physiol. 1992;263:C494–C501. doi: 10.1152/ajpcell.1992.263.2.C494. [DOI] [PubMed] [Google Scholar]
- 37.Hasler C M, Bennink M R, Trosko J E. Am J Physiol. 1991;261:C161–C168. doi: 10.1152/ajpcell.1991.261.1.C161. [DOI] [PubMed] [Google Scholar]
- 38.Aylsworth C F, Trosko J E, Welsch C W. Cancer Res. 1986;46:4527–4533. [PubMed] [Google Scholar]
- 39.Spray D C, Burt J M. Am J Physiol. 1990;258:C195–C205. doi: 10.1152/ajpcell.1990.258.2.C195. [DOI] [PubMed] [Google Scholar]
- 40.Bachur N R, Masek K, Melmon K L, Udenfriend S. J Biol Chem. 1965;240:1019–1024. [PubMed] [Google Scholar]
- 41.Ramachandran C K, Murray D K, Nelson D H. Biochem Arch. 1992;8:369–377. [Google Scholar]
- 42.Schmid H H O, Schmid P C, Natarajan V. Prog Lipid Res. 1990;29:1–43. doi: 10.1016/0163-7827(90)90004-5. [DOI] [PubMed] [Google Scholar]
- 43.Hanus L, Gopher A, Almog S, Mechoulam R. J Med Chem. 1993;36:3032–3034. doi: 10.1021/jm00072a026. [DOI] [PubMed] [Google Scholar]
- 44.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski N E, Schatz A R, Gopher A, Almog S, Martin B R, Compton D R, et al. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 45.Matsumoto M, Park J, Sugano K, Yamada T. Am J Physiol. 1987;252:G315–G319. doi: 10.1152/ajpgi.1987.252.3.G315. [DOI] [PubMed] [Google Scholar]
- 46.Maurelli S, Bisogno T, De Petrocellis L, Di Luccia A, Marino G, Di Marzo V. FEBS Lett. 1995;377:82–86. doi: 10.1016/0014-5793(95)01311-3. [DOI] [PubMed] [Google Scholar]
- 47.Koutek B, Prestwich G D, Howlett A C, Chin S A, Salehani D, Akhavan N, Deutsch D G. J Biol Chem. 1994;269:22937–22940. [PubMed] [Google Scholar]
- 48.Lerner R A. Proc Natl Acad Sci USA. 1997;94:13375–13377. doi: 10.1073/pnas.94.25.13375. [DOI] [PMC free article] [PubMed] [Google Scholar]