

Summary
Transcriptional activation of the interferon-β (IFN-β) gene requires assembly of an enhanceosome containing ATF-2/c-Jun, IRF-3/IRF-7, and NFκB. These factors bind cooperatively to the IFN-β enhancer and recruit coactivators and chromatin remodelling proteins to the IFN-β promoter. We describe here a crystal structure of the DNA-binding domains of IRF-3, IRF-7 and NFκB, bound to one half of the enhancer, and use a previously described structure of the remaining half to assemble a complete picture of enhanceosome architecture in the vicinity of the DNA. Association of eight proteins with the enhancer creates a continuous surface for recognizing a composite DNA binding element. Paucity of local protein-protein contacts suggests that cooperative occupancy of the enhancer comes from both binding-induced changes in DNA conformation and interactions with additional components such as CBP. Contacts with virtually every nucleotide pair account for the evolutionary invariance of the enhancer sequence.
Introduction
Transcriptional enhancers are cis-regulatory elements of eukaryotic gene expression, often located at considerable distances from the promoters they regulate (Carey and Smale, 2000). Activation and nuclear localization of specific sets of transcription factors in response to specific signals determine the occupancy of any particular enhancer, which can thus detect and integrate information from multiple signal transduction pathways (Ptashne, 2004). The multiple sites within enhancers create a combinatorial code, which directs the transcriptional machinery to specific promoters in response to a given signal. Enhancers vary greatly in length and complexity. At one end of the complexity gradient are enhancers with transcription-factor binding sites tightly clustered in a relatively compact genomic segment (e.g., the 55 base pairs (bp) of the enhancer for the interferon-® gene). Transcription from promoters they regulate appears to require cooperative formation of an “enhanceosome” – an assembly of distinct sets of proteins on the enhancer DNA (Thanos and Maniatis, 1995). At the other end of the gradient are “modular” enhancers, with rather loosely clustered binding sites, generally covering much longer stretches of the genome. Whereas enhanceosome assembly ensures that enhancers operate as a single unit, modular enhancers represent a more flexible form of information processing (Arnosti and Kulkarni, 2005).
The best-characterized compact enhancer in the human genome is the virus-inducible enhancer of the interferon-® (IFN-®) gene. Binding sites for the heterodimer ATF-2/c-Jun, interferon response factors IRF-3 and IRF-7, and NFκB (p50:RelA) are tightly clustered in a 55 bp stretch of DNA. The ”AT-hook” protein HMGA1 (formerly designated HMGI(Y)) also binds to this sequence and promotes enhanceosome assembly (Thanos et al., 1993), although the work reported here shows that it is unlikely to be present in the final assembly. The enhanceosome components bind to a nucleosome-free region of the IFN-® promoter, spanning the interval from −102 to −47 bp relative to the transcription start site. The IFN-® enhancer has been subdivided into four positive regulatory domains (PRDs). The structure described here shows that the enhancer forms essentially one composite binding element and that binding sites overlap, but the PRD designations still remain useful. IRF proteins bind to PRDI and III; NFκB to PRDII, and ATF-2/c-Jun to PRDIV (Figure 1). Each of these factors can interact with the co-activator, CREB-binding protein (CBP) or with its closely related paralog, p300. Assembly of all these factors into an “enhanceosome” is thought to provide stringent specificity and stability (Maniatis et al., 1998; Munshi et al., 1999). In vivo, the IFN-β enhancer is nucleosome-free. It is flanked by two nucleosomes, one of which masks the TATA box and the start site of transcription (Agalioti et al., 2000). Virus infection leads to the activation of ATF-2/c-Jun, IRF-3/7, and NFκB, and their binding to the nucleosome-free enhancer. Nucleosome acetylation and chromatin remodeling by the SWI/SNF complex reposition the nucleosome covering the TATA box and allow access by TBP (Lomvardas and Thanos, 2001).
Figure 1.
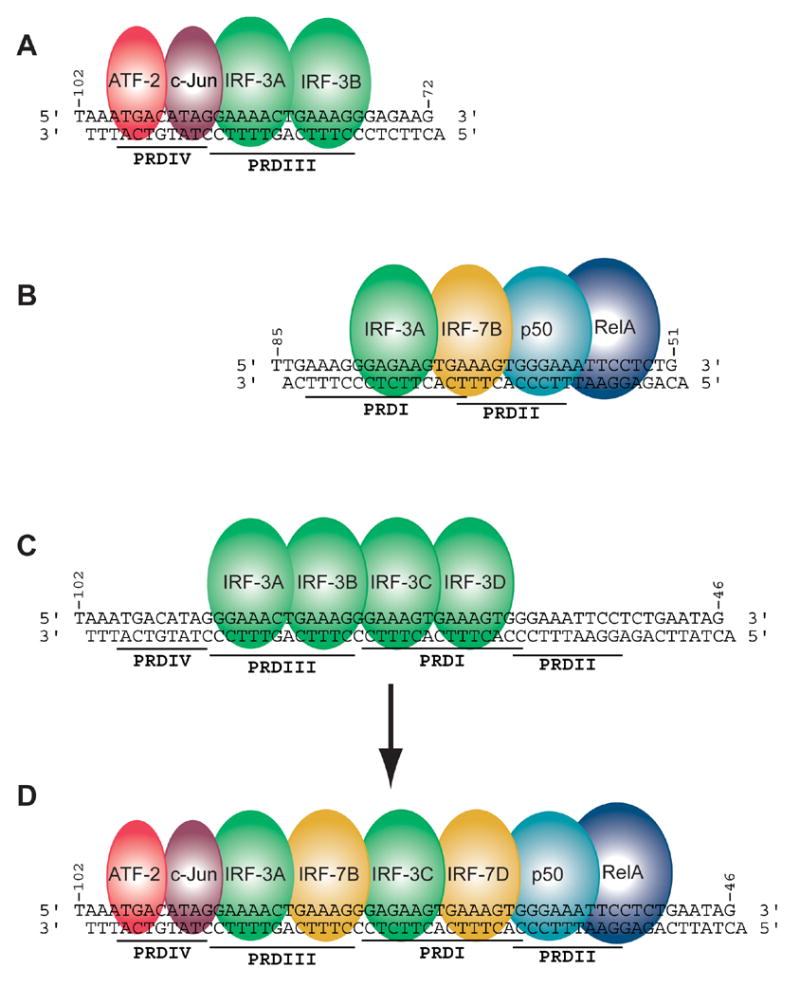
Strategy for assembling the complete INF-β enhanceosome structure. Three sets of overlapping crystal structures were determined. (A) ATF-2/c-Jun in complex with two IRF-3 molecules on the PRDIV and PRDIII regions of the enhancer (Panne et al., 2004). (B) NFκB in complex with IRF-7 and IRF-3 on PRDI-PRDII. (C) Four IRF-3 molecules in complex with the full length enhancer. (D) Fully assembled INF-β enhanceosome.
The IFN-® enhanceosome is an example of signal integration through assembly of a set of “generic” transcription factors, each of which works in conjunction with other factors at other enhancers. The individual factors do not activate IFN-® gene expression by themselves, and failure to mobilize any one of the factors abrogates IFN-® transcription entirely (Maniatis et al., 1998). Although multiple copies of individual binding sites can function as virus-inducible elements, these artificial enhancers can respond to stimuli other than virus infection ( Thanos and Maniatis, 1995b). The authentic enhanceosome is thus a coincidence detector. It responds to the coordinate activation of a specific set of transcription factors, which assemble on the IFN-® enhancer into a complex for recruiting RNA polymerase II.
The IRF family of transcription factors includes nine mammalian members, IRF-1 to IRF-9, as well as several viral homologs (Mamane et al., 1999). All IRFs are characterized by a well-conserved N-terminal DNA-binding domain of about 120 amino acids, which recognizes similar DNA sequences termed IRF-binding element/IFN-stimulated response element (ISRE), the consensus being 5′-AANNGAAA-3′ (Fujii et al., 1999). A challenge for structural studies of INF-β regulation has been uncertainty concerning the occupancy of the four IRF binding sites in the PRDIII-I region during different stages of viral infection and in different cell types, because techniques such as overexpression by transient transfection frequently mask in vivo specificity. Initial studies implicated IRF-1 in interferon-β transcription (Fujita et al., 1988; Thanos and Maniatis, 1995), but later gene inactivation studies showed that IRF-1 is not required for viral induction of interferon-β in most cell types, and that IRF-3 and IRF-7 are the relevant factors (Honda et al., 2005; Sato et al., 2000; Wathelet et al., 1998). IRF-3 is constitutively expressed, but levels of IRF-7 are increased through positive feedback by IFN-α/β stimulation (Marie et al., 1998; Sato et al., 1998). These properties suggested that IRF-7 has a role in later stages of virus infection and that the immediate early enhanceosome might contain only IRF-3 (Sato et al., 2000). We (Figure S1) and others (Escalante and Aggarwal, personal communication) have therefore attempted to crystallize the enhanceosome with IRF-3 bound to the PRDI-III region. It is now clear, however, that IRF-7 is constitutively expressed at high levels in plasmocytoid dendritic cells, the primary source of type I IFN in response to infection, and that it is essential for interferon-β expression (Honda et al., 2005). Thus, IRF-7 is likely to be a component even of the early enhanceosome.
We describe here the structure of a substantial part of the IFN-β enhanceosome, including the PRDI and PRDII regions in complex with the DNA-binding domains of NFκB, IRF-7 and IRF-3. Together with our previously published structure of the PRDIV-PRDIII half of the complex (Panne et al., 2004) and with a structure of the overlapping PRDIII-PRDI region also described here, we can construct a complete picture of the DNA-proximal enhanceosome architecture. Our structure shows IRF-3 bound to ISRE sites A and C and IRF-7 to sites B and D in the PRDIII-I region of the enhancer, consistent with the dependence of IFN-β transcription on both IRF family members. The composite model shows that association of eight proteins with enhancer DNA creates a continuous surface for the recognition of 50 base pairs. Contrary to some suggestions, the DNA is essentially straight. The various transcription-factor binding sites overlap, and local conformational changes in the DNA contribute to specificity and to transcription-factor positioning, as already seen in the PRDIV-PRDIII structure (Panne et al., 2004). The positive-regulatory domains of the IFN-β enhancer form a single, composite binding element. The unit of regulation for virus-activated transcription is thus the entire nucleotide sequence rather than individual PRD elements, consistent with strict conservation at almost every position of the enhancer in mammalian genomes.
Results
Strategy and component structures
We have previously reported a structure that contains the ATF-2/c-Jun heterodimeric bZIP segments and two IRF-3 DNA-binding domains, in complex with a DNA fragment containing base pairs −102 to −72 (PRDIV and III) (Panne et al., 2004). We now describe a crystal structure that contains an IRF-3 DNA-binding domain, an IRF-7 DNA-binding domain, and the Rel-homology regions of NFκB p65 (RelA) and p50, bound to DNA base pairs −85 to −51 (PRDI and PRDII) (Figure 2). Using a third crystal structure that overlaps the first two, with four IRF-3 DNA-binding domains bound to a 57 bp enhancer DNA, base pairs −102 to −46 (PRDIII and PRDI) (Figure S1), we have reconstructed the complete set of DNA-binding domains and enhancer DNA that determines the global organization of the enhanceosome. The strategy is illustrated schematically in Figure. 1. Before describing the new structures in detail, we summarize necessary information about the components from various other structures.
Figure 2.
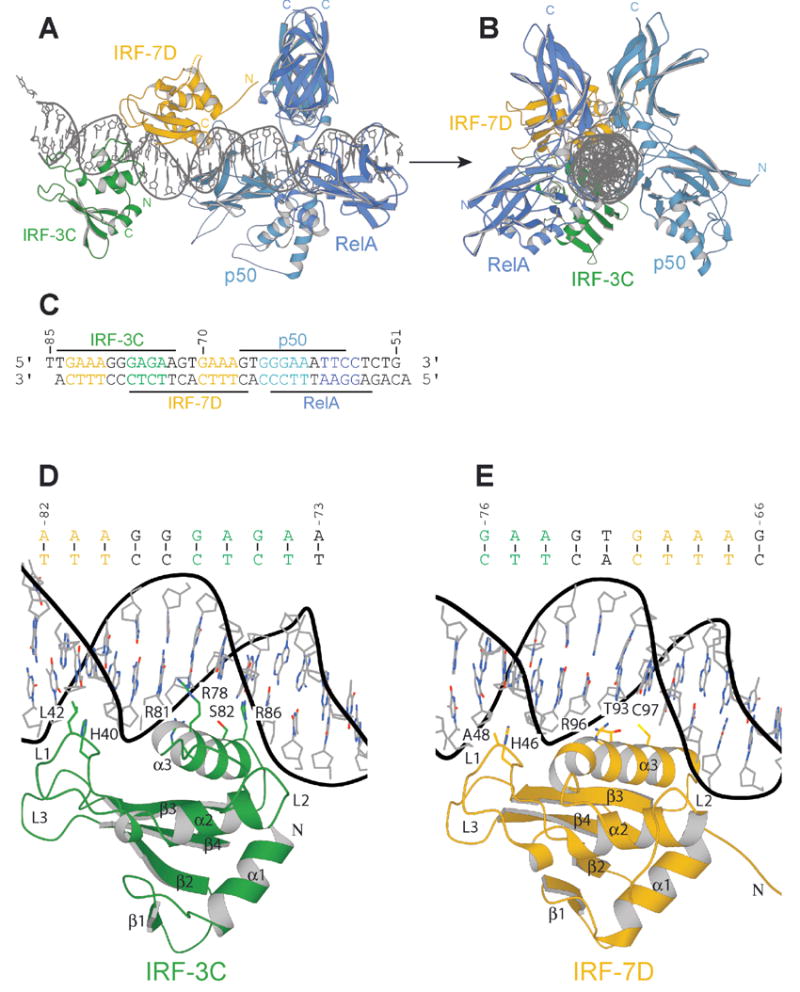
Structure of the NFκB:IRF-7:IRF-3:DNA complex. Ribbons representation of the complex viewed (A) normal to and (B) along the DNA helical axis. The Rel-homology region (RHR) of p50 is in light blue; that of RelA, in dark blue. IRF-7D is in yellow; IRF-3C, in green. The two DNA strands are in grey. (C) Sequence of the DNA duplex corresponding to the PRDI-II region of the IFN-β enhancer (−85 to −51 nucleotides from the start site of transcription). (D) Protein-DNA contacts of IRF-3C. (E) Protein-DNA contacts of IRF-7D.
IRF DNA-binding domains
All IRF proteins have an N-terminal DNA-binding domain of about 120 amino-acid residues, with a conserved α/® architecture comprising a four-stranded antiparallel ®-sheet (®1-®4), three helices (α1-α3), and three long loops (L1-L3) (Figure 2D, E) (Escalante et al., 2002a; Escalante et al., 1998; Fujii et al., 1999; Panne et al., 2004). Each DNA-binding domain contacts the DNA backbone over a 12 bp stretch. A set of protein-DNA contacts in the major and minor grooves determine specificity for the IRF recognition sequence 5′ AANNGAAA 3′. Conserved residues Asn79, Arg81 and Ser82 (IRF-3 numbering), all in α3, specify the downstream GAAA, and water-mediated contacts in the minor groove from His40 determine the preferred upstream AA (Figure 2D). Additional DNA contacts with non-conserved residues Leu42, Arg78, and Arg86 explain the more restricted binding specificity of IRF-3 (Panne et al., 2004). Leu42, in L1, inserts into the minor groove adjacent to His40 and contacts the base pair 5′ to the consensus site (5′ NAANNGAAA 3′), disfavoring a G:C or C:G at this position. (We have noted previously that this interference at least partly explains the binding register at site A (Panne et al., 2004)). Arg78 and Arg86 contact the regions flanking the core GAAA repeat; although these side chains are quite adaptable they favor a G:C or C:G on either side of the core (Escalante and Aggarwal, personal communication; Panne et al., 2004).
IRF-7 is essential for viral induction of IFN-® (Honda et al., 2005). We suggest, based on the specificities just described, that sites B and D are selective for IRF-7, and sites A and C, for IRF-3. First, site D is suboptimal for IRF-3 because of the potential interference between Leu42 and G75 in the wild-type sequence. IRF-7 has alanine instead of leucine at the corresponding position (Figure 2E), allowing it to tolerate the guanine N2. Second, sites A and C appear to prefer IRF-3, because they accommodate bidentate hydrogen bonding of Arg78 with two successive guanines (Escalante and Aggarwal, personal communication). IRF-7 has threonine at the corresponding position (Figure 2). Third, the methyl group of this threonine can form a van der Waals contact with the methyl group of a thymine just upstream of the consensus guanine, as in sites B and D. Specificity for T at this position in IRF-7 sites and for G in IRF-3 sites has also been detected in SELEX experiments (Lin et al., 2000a), as well as in preferential IRF-3 and IRF-7 sites involved in transcription of the interferon-α genes (Morin et al., 2002).
Alternation of the sites for IRF-3 and IRF-7 is also optimal for concomitant binding by IRF dimers. IRF-3 is activated by phosphorylation of C-terminal residues, leading to dimerization, nuclear translocation, and interaction with CBP/p300 (Lin et al., 1998). Sites A and C are on the same face of a DNA duplex, and the C-termini of the DNA-binding domains at these sites project in the same direction, about 40Å apart. Binding of an IRF dimer at sites A and C (or B and D) would allow attack from one side of the duplex and a simple geometry for the overall complex. The C-termini of the domains positioned at sites A and B project in opposite directions, about 60Å apart; binding of a dimer at sites A and B (or C and D) would require wrapping of an extended hinge around the enhancer. The N-terminal DNA binding domain and dimerization domains of IRF-3 are linked through a flexible segment of ~70 amino-acid residues (Qin et al., 2005), long enough to accommodate either arrangement, but the alternating characteristics of the binding site sequences clearly suggests that IRF-3 binds preferentially to sites A and C and IRF-7, to sites B and D.
NFκB
The Rel homology region (RHR) of the NFκB heterodimer p50/RelA has been crystallized on a number of different DNA sites (Berkowitz et al., 2002; Chen et al., 1998a; Chen-Park et al., 2002; Escalante et al., 2002b). In all these structures, the p50 subunit binds at the 5′ end and the RelA subunit at the 3′ end of the κB site (Figure 2). The main determinant of this orientation appears to be a contact of His64 of p50 with the upstream guanine (5′-GGGAAATTCC-3′; in the description below, we number the base-pairs in this sequence 1–10); the homologous residue in RelA is Ala, which does not confer specificity. Therefore, the p50 half-site is generally described as 5 base-pairs in length (5′-GGGAAATTCC-3′); the RelA half site, as 4 base-pairs in length (5′-GGGAAATTCC-3′) (Chen and Ghosh, 1999). The approximate dyad passes through the central base-pair, assigned to neither half site (Figures 2 and 3).
Figure 3.
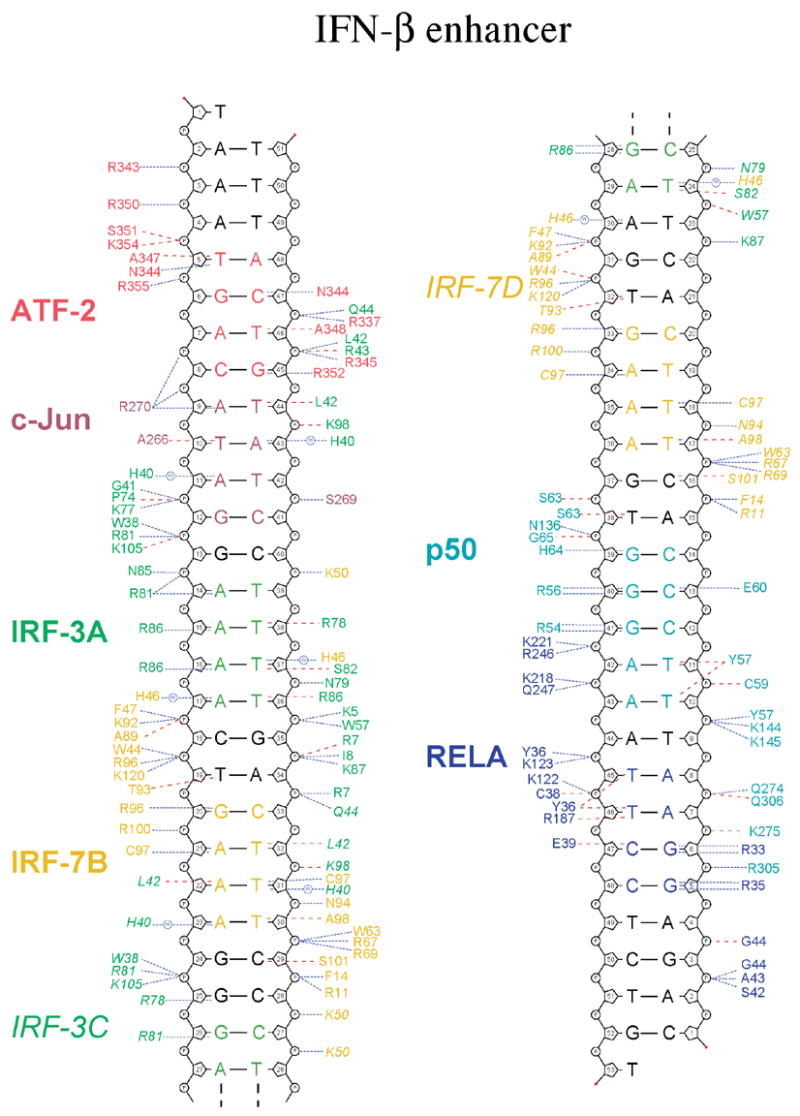
Protein-DNA contacts. Schematic diagram of protein-DNA contacts generated using NUCPLOT (Luscombe et al., 1997). Residues from ATF-2 are in red, c-Jun, in dark red, IRF-3A and C, in green, and IRF-7B and D, in yellow. The RHR of p50 is in light blue; that of RelA, in dark blue. The core binding sites for each protein are indicated in the corresponding colors. Blue lines indicate hydrogen bonds; red lines, van der Waals contacts.
Structure of an NFκB:IRF-7:IRF-3:DNA complex at PRDI-II
Design of a fusion construct
IRF-7 has an insertion of 9 amino-acid residues between helices α2 and α3 (Figure S3). With IRF-7 bound to site D, this insertion projects towards loop L1 of p50. We considered that this loop might be involved in direct protein-protein contacts with p50, but NFκB binds full-length enhancer DNA together with the DNA-binding domains of IRF-3 and/or IRF-7 without apparent cooperativity (Figure S2). Efforts to crystallize such complexes, either on full-length enhancer or on a fragment containing sites C and D plus PRDII yielded only crystals with NFκB, IRF-3, or IRF-7 bound alone to the DNA duplexes. To test whether the specific order with IRF-3 bound to site C and IRF-7 bound to site D might be critical for cooperative assembly, we designed polypeptide linkers between IRF-3 and IRF-7 to stabilize the relative order of the domains. We have shown previously that adjacent IRFs can be linked in this way, without affecting binding or structure (Panne et al., 2004). Neither an IRF-3:IRF-3 or an IRF-3:IRF-7 dimer showed cooperative binding with NFκB, however (Figure S2). From modeled structures based on IRF:DNA and NFκB:DNA complexes, we noticed that the N- and C-termini of the domains are oriented favorably for construction of the fusion protein RelA(RHR):IRF-7(DBD):IRF-3(DBD) and of an extension of the same protein with an additional IRF-7(DBD):IRF-3(DBD). (The colon (:) indicates a linker sequence of varying length.) These fusion proteins were co-expressed with the p50 RHR to generate an NFκB heterodimer with attached IRF domains. The purpose of the fusion construct was to fix the relative order of individual domains on the enhancer (IRF-3 on site C, IRF-7 on site D and NFκB on PRDII) and to stabilize the assembly. Analysis of binding is shown in Figure S2. We experimented with linkers of two lengths (see Experimental Procedures); the shorter linkers allowed crystallization of the heterodimer of p50 with RelA(RHR):IRF-7(DBD):IRF-3(DBD) bound to a 35 base-pair DNA duplex spanning PRDI and PRDII.
Structure determination
The complex crystallized in space group P212121 (a=95.5Å, b=116.4Å, c=134.8Å) and gave measurable diffraction to dmin=2.8Å. We determined the structure by molecular replacement and refined it to Rwork=24.5%, Rfree=28.6% (Table 1). The asymmetric unit contains one complex; the 5′ end of the DNA stacks against RelA of a symmetry-related molecule, with the first two nucleotides of the coding strand displaced from the duplex (Figure 2A, B). The 3′ end does not have any crystal-packing interactions. Thus, end-to-end DNA packing, as frequently found in crystals of DNA:protein complexes, does not constrain the DNA conformation. The refined model contains residues 19-291 of RelA, 37-350 of p50, 8-128 of IRF-7, 9-111 of IRF-3, and DNA nucleotide pairs spanning the region from −85 to −51 of the enhancer (Figure 2). The designed linkers connecting the individual domains in the fusion protein are disordered, as anticipated. The distances spanned are ~26 Å and ~27 Å for the two linkers, much less than the extended length of a 15-residue polypeptide chain. The single-chain strategy is further validated by the following observations. First, the conformations of individual domains of the fusion protein are identical to those crystallized independently, in the context of other DNA substrates and crystal lattices. Second, the DNA binding interfaces are just as expected for the individual constituents (see below). Contacts between the various DNA-binding domains and the DNA are summarized in Figure 3.
Table 1.
Data collection and refinement statistics
| NFκB:IRF-7:IRF-3:DNA | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 95.49, 116.37, 134.77 |
| Resolution (Å) | 2.8 |
| Rsym | 7.1 (60.0) * |
| I / σI | 37.6 (4.3) * |
| Completeness (%) | 99 (100) * |
| Redundancy | 14.1 (13.5)* |
| Refinement | |
| Resolution (Å) | 2.8 |
| No. reflections | 37592 |
| Rwork/Rfree | 24.5/28.4 |
| No. atoms | 7915 |
| Protein | 6462 |
| DNA | 1428 |
| Water | 25 |
| Average B-factors (Å2) over all residues in each chain | |
| Protein | 57.46, 66.92, 52.53, 107.511 |
| DNA | 59.59, 49.462 |
| Water | 36.08 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.3 |
Highest resolution shell is shown in parenthesis.
RelA, p50, IRF-7D, IRF-3C
Coding strand, Template strand
IRF-3 and IRF-7
Site C has one important deviation from the consensus ISRE: the consensus GAAA is interrupted by a guanine 5′TGAAAGGGAGAA3′. There are two possible binding registers for IRF-3. In one, α3 would lie over AGAA, preserving the canonical two base-pair inter-site spacing with the downstream IRF-7; in the other, α3 lies over GAGA, preserving a two base-pair inter-site spacing with an upstream IRF-7 (not present in our crystals). We observe the latter configuration, which is also present in a structure of four IRF-3 DNA-binding domains bound to PRDI-III (Escalante and Aggarwal, personal communication). We can identify several structural reasons for this selectivity. On the AGAA site, a minor-groove –NH2 from the first nucleotide of the G triplet (G79) would repel His40. Binding at GAGA not only avoids this clash, but also allows Arg78 to have the regularly observed bidentate major-groove interaction with two consecutive guanines (G78 and G77) (Figure 2D; also Escalante and Aggarwal, personal communication). Although Arg86 is disordered in our density map, it could interact with the non-consensus G in site C. In the context of the enhanceosome, the selection of this binding site permits a hydrogen bond to the main chain carbonyl of Leu42 from Arg7 of the upstream IRF-3A, as observed in a four-site IRF-3:DNA structure described in the supplement (Figure S1 and Table S1). IRF-7 does not have Arg at the corresponding position in its structure, so that the additional base-pair between sites B and D does not sacrifice this potential contact (Figure S3).
The principal difference between the IRF-7 DNA-binding domain and those of IRF-1, IRF-2, IRF-3, and IRF-4, for which structures have been determined previously (Escalante et al., 2002a; Escalante et al., 1998; Fujii et al., 1999; Panne et al., 2004), is in the three loops, L1-L3 (Figure S2). L2, between helices α2 and α3, has a 9-residue insertion, which is in part unstructured, reflecting a series of Gly and Pro residues. The DNA recognition helix, α3, has an N-terminal extension of 5 residues extending away from the DNA, so that it contains 21 instead of the usual 16 residues (Figure 2E). Modeling of IRF-1 onto the PRDI site proximal to NFκB showed that loop L2 of either IRF would sterically overlap with loop L1 of p50 (Escalante et al., 2002b). The insertion in IRF-7 leads to a rearranged L2, which together with a shift in L1 of p50 (see below) avoids steric interference, allowing IRF-7 and NFκB to co-occupy the enhancer.
Conserved interactions with GAAA in the major groove of site D include hydrogen bonds between Arg96 (IRF-7 numbering) and the initial G and a non-polar contact between Ala98 and the methyl group of the thymine paired with the third A. The interactions of Cys97 with the two central base pairs resemble those of its homolog, Ser82, in IRF-3 (Figures 2E and 3). The water-mediated, minor-groove contacts of His46 with two upstream adenines (5′ AANNGAAA 3′) are as described above for IRF-3, but Ala 48, which replaces Leu 42 of IRF-3, allows the protein to accommodate an upstream G:C base pair. Also as described above, the van der Waals contact between Thr93 and the C5-methyl group of T -71 strengthens the IRF-7 preference for sites B and D.
NFκB
As in other NFκB-containing structures, including the initially studied p50 homodimer:DNA complexes (Ghosh et al., 1995; Muller et al., 1995), homologous residues (two arginines and a glutamate) in both p50 and RelA recognize the core guanine bases in each half-site. These contacts, and others summarized in Figure 3, position the two RHR-N domains with respect to the DNA duplex. We have compared our structure of the p50:RelA heterodimer with others determined on a variety of DNA sites. These structures differ among each other in the relative orientations of the RHR-N and RHR-C domains, which are flexibly hinged, and in the DNA conformations to which they accommodate. The dimeric RHR-C domains provide a convenient reference frame, as their contact is essentially invariant. Our structure differs from that of a p50:RelA heterodimer bound with 12 bp of precisely the same DNA sequence (PRDII) (Berkowitz et al., 2002; Escalante et al., 2002b) by as much as either of them differs from other published structures (Figure S4 and (Berkowitz et al., 2002)). That is, we can find in the comparison no clear correlation between DNA sequence and NFκB conformation. The DNA in our enhanceosome complex is longer than the fragments used in earlier work with NFκB alone, and there are indeed additional phosphate backbone contacts made by S63, G65, and N136 of p50 and by S42, A43, and G44 of RelA that are not seen in the complex on a 12 bp site. We believe that the comparison of our structure with the earlier PRDII complex (Berkowitz et al., 2002; Escalante et al., 2002b) calls into question the notion, that subtle differences in κB-site sequences can have reliable “allosteric” effects on the conformation of bound NFκB (Chen-Park et al., 2002).
An interaction not present in earlier structures is the contact between loops L1 of p50 and L2 of IRF-7 on site D (Figure 4S). The former shifts by about 5 Å toward IRF-7, with respect to its position in the p50:RelA:PRDII complex, but the interaction between the two loops is still tenuous (Figure S4). The only evident contact is between the side chain of Asn75 in p50 and the main-chain carbonyl of Gly68 in IRF-7. Loop L1 in p50 is relatively flexible, and this single contact is unlikely to have any propagated conformational effects on the rest of NFκB.
Figure 4.
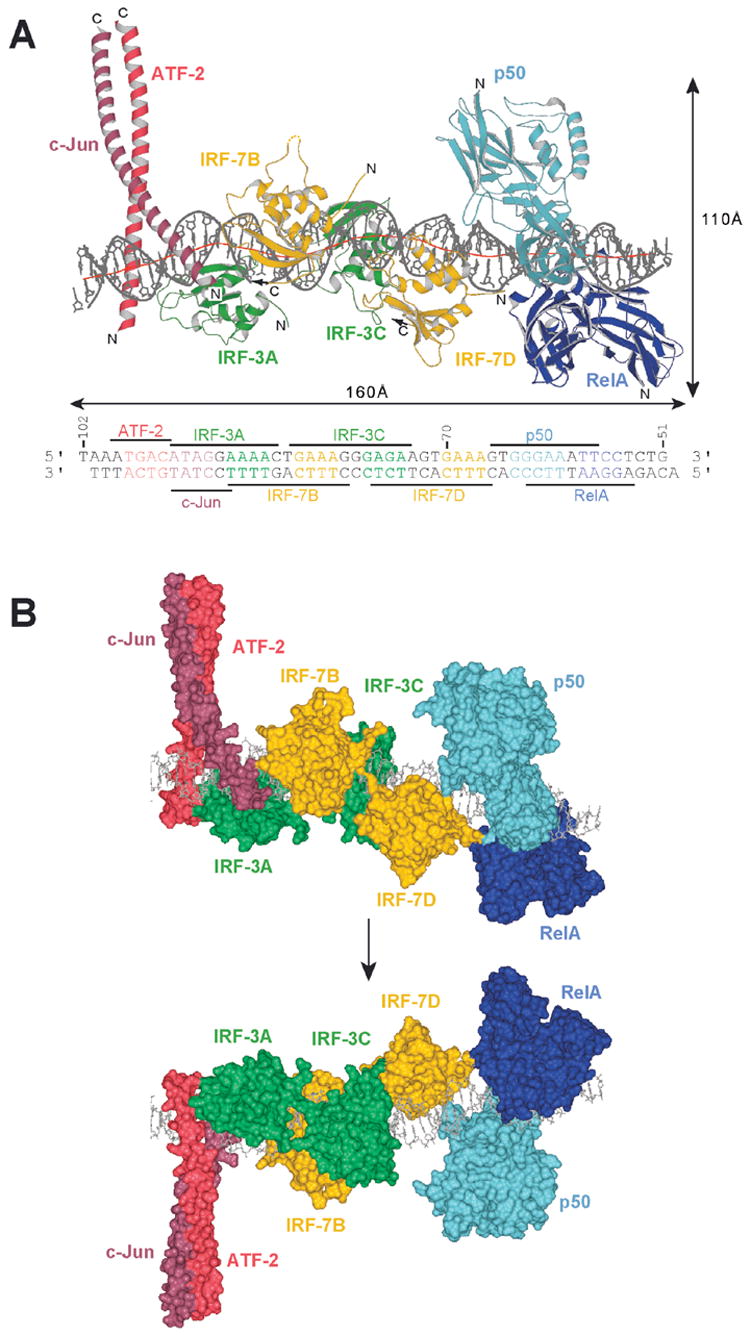
Overall structure of the interferon-β enhanceosome: (A) Side view of the complex, showing bending of the DNA around the four IRF domains. The red line shows the local DNA helical axis as calculated with the program Curves (Lavery and Sklenar, 1988). The RHR of p50 is in light blue; that of RelA, in dark blue. IRF-7B and D are in yellow; IRF-3A and C, in green. ATF-2 is in light red; c-Jun, in dark red. The two DNA strands are in grey. (B) Molecular surface representation of the interferon-β enhanceosome showing that the eight polypetides form a composite surface for DNA recognition. The two views, related by a 180° rotation as suggested by the arrow, are from opposite sides of the complex.
Model for the complete enhanceosome
The structure just described of NFκB:IRF-7:IRF-3 on PRDI-II and the structure of ATF-2/c-Jun/IRF-3 on PRDIV-III together cover the entire IFN-β enhanceosome. Because these structures overlap in the region spanning nucleotides −72 to −85 of the enhancer, we could in principle reconstruct the entire DNA-proximal assembly from these two structures alone. We have chosen to use as an additional guide a structure containing four IRF-3 DNA binding domains that cover PRDIII-PRDI – that is, all four IRF sites. That structure, which contained a single base-pair deletion in PRDI, is described in the supplementary data (Figure S1, Table S1). A related structure containing the wild-type PRDI has been determined by another group (Escalante and Aggarwal, personal communication), and the two are in excellent agreement. The details of how the structure of NFκB:IRF-7:IRF-3 on PRDI-II was overlapped with that of ATF-2/c-Jun/IRF-3 on PRDIV-III (Panne et al., 2004) are provided in the section Experimental Procedures. Excellent spatial superpositions at the ends of the overlapped structures validate our approach to “glue” together the PRDIV-PRDIII and PRDI-PRDII structures at the single interface between them (Figure 7S). In the model shown in Figure 4, we have replaced IRF-3 at site B (from the structure of (Panne et al., 2004)) with IRF-7, using criteria described in the section Experimental Procedures. The fully assembled enhanceosome has a length of ~160 Å. Binding of the eight proteins to DNA buries 13900 Å2 or 72% of the solvent accessible surface area of the enhancer DNA. We base the description below on the concatenated structure, although the analysis of local DNA conformation is of course based on one or the other of the individual coordinate sets.
Protein interactions
A striking characteristic of the complex is the paucity of specific protein interactions, despite the close packing of the various transcription-factor DNA-binding domains. We observed this property in analyzing the ATF-2/c-Jun/IRF-3/DNA complex, and it continues to be true all along the length of the enhancer, as noted at various points in the structure description above. Successive proteins do contact each other, but with relatively tenuous side-chain interactions. Nonetheless, EMSA assays show that the IRF proteins bind cooperatively, with no detectable, single-occupancy intermediate, provided that the DNA substrate has at least two binding sites in a tandem orientation (Escalante and Aggarwal, personal communication; Falvo et al., 2000; Fujii et al., 1999; Panne et al., 2004). We therefore extend the concept proposed earlier, that some degree of cooperativity can arise through DNA conformability in the absence of strong protein contacts, when binding sites overlap (as they do here) and the required or imposed DNA conformations at the overlapping sites are complementary (Panne et al., 2004).
Enhancer DNA conformation
The conformation of the 57 bp enhancer DNA exhibits local variations about a straight, B-form structure. The axis traces a gently sinusoidal curve, with a net overall bend of ~13–15° and no sharp kinks (Figure 4A). Structures of DNA binding domains from IRF-1, IRF-2, IRF-3, and IRF-4, complexed with DNA, show that these domains stabilize a characteristic DNA conformation, in which the DNA duplex bends gently around the IRF recognition helix (α3) (Escalante et al., 2002a; Escalante et al., 1998; Fujii et al., 1999; Panne et al., 2004). This DNA conformation is present in both the IRF-3 and IRF-7 sites and accounts for the sinusoidal path of the helix axis. IRF binding sites A, B and C are separated by 6 bp (just over half a turn of the DNA helix), and bends at these sites are therefore of opposite phase. Site D is separated by 7 bp from site C, and because of this extra base pair, the bends at sites C and D do not cancel each other, leaving the enhancer with a ~13–15° bend centered on the A-tract in site D (Figure 4A). Similar conclusions follow from the structure of 4 IRF-3 domains on PRDIII-PRDI (Escalante and Aggarwal, personal communication). Thus, the net bend is largely a consequence of the relative positions of the IRF domains, as the ATF-2/c-Jun and NFκB sites in the complex are relatively straight, consistent with measurements of bending in solution (Falvo et al., 1995). The compression associated with bending around each of the IRF domains leads to a periodic opening and closing of the major and minor grooves along the enhancer (Figure S5). The closely spaced arrangement of DNA-binding domains along the enhancer would not allow major bends, even in response to constraints introduced by links between upstream and downstream elements outside the 57 bp segment in our crystals (“DNA looping”), as neighboring proteins would then collide (Figure 4B). The relatively undistorted conformation of the enhancer rules out earlier suggestions that the DNA might wrap around a transcription-factor core and contradicts models that postulate long-range DNA bending as a critical part of protein-DNA recognition at these sites (Munshi et al., 1999).
HMGA1a binding
The HMGA1a protein is thought to “orchestrate” assembly and disassembly of the IFN-® enhanceosome, perhaps through modification of DNA conformation and modulation of protein-protein interactions (Yie et al., 1999). The structure shows that an assembled enhanceosome cannot accommodate HMGA1a, which binds in the DNA minor groove through so-called “AT-hook” contacts from one or more of three related, flexibly linked segments in the short (~110-residue) protein. There are four potential HMGA1a sites in the IFN-® enhancer: the A:T-rich region around −60 in PRDII, a site 10 bp downstream around position −48, and two sites flanking PRDIV around −100 and −88. HMGA1a variants form complexes with enhancer DNA, as detected by EMSA, but do not form stable ternary complexes in the presence of the transcription factors. That is, complexes formed in the presence of HMGA1a do not display further gel retardation when compared to complexes formed in the absence of HMGA1a, and there is no electron density for HMGA1a in maps from co-crystallization efforts (Berkowitz et al., 2002; Panne et al., 2004). One of the HMGA1a sites in PRDIV, the minor groove of the AT-rich sequence at −88, is blocked by loop L1 of IRF-7B (Panne et al., 2004), ruling out two concomitant interactions at that end of the enhancer. At the other end, superposition of the NMR structure of the second and third DNA-binding segments of HMGA1a bound to PRDII (Huth et al., 1997) onto the NFκB-bound PRDII site in our structure shows that binding of NFκB and HMGA1a require very different minor-groove widths. Thus, steric occlusion at PRDIV and conformational incompatibility at PRDII lead us to conclude that HMGA1a is not part of the completed IFN-β enhanceosome.
Discussion
The virus-induced human IFN-β enhancer is arguably the most thoroughly characterized transcriptional regulatory element in any higher eukaryotic genome. The structure reported here allows the assembly of a model of the full enhancer bound to the DNA-binding domains of all the relevant transcription factors. It shows in molecular detail the interactions of ATF-2/c-Jun, IRF-3, IRF-7 and NFκB with the enhancer DNA.
The nucleotide sequence of the IFN-® enhancer is nearly invariant over roughly 100 million years of evolution, unlike the sequence of the gene (Figure S6). Thus, the precise organization of the assembled transcription factors has had strong and continuing selective advantage. Moreover, mutational analyses have shown that virtually every nucleotide in the enhancer DNA sequence matters for some aspect of the response to viral infection (Du and Maniatis, 1992; Goodbourn and Maniatis, 1988; Thanos and Maniatis, 1992). The enhanceosome structure accounts for this conservation by showing that the transcription factors form a composite surface for recognition of the entire sequence and that adjacent trascription-factor binding sites overlap (Figures 3 and 4). For example, IRF-3 and IRF-7 specify additional bases around the core IRF binding site 5′ AANNGAAA 3′ through non-conserved amino-acid residues such as Leu42, Arg78 and Arg86 in IRF-3 and Thr93 in IRF-7. These additional DNA contacts explain a number of observed sequence preferences (Lin et al., 2000a; Morin et al., 2002). They also account for the requirement of IRF-7 in the early IFN-β response to viral infection (Honda et al., 2005), by showing how loop L2 of IRF-7 at site D avoids interference with NFκB and how DNA sequence just outside the cores of sites B and D leads to a preference for IRF-7 over other family members.
A hallmark of combinatorial transcriptional control is synergy, mediated largely in this case by enhanceosome formation (Merika and Thanos, 2001; Struhl, 2001). Synergy implies strong cooperativity at some level of assembly, such as direct interactions between adjacently bound transcription factors. When c-Fos, c-Jun, and NFAT bind the ARRE2 site of the IL-2 enhancer, contacts between adjacent proteins do impart both cooperativity and specificity: an extended network of polar interactions, which includes all three proteins and the DNA backbone, establishes a preferred orientation for the Fos:Jun heterodimer on its binding site and a particular conformation for the two-domain RHR of NFAT (Chen et al., 1998b). Extended contacts between transcription factors are noticeably absent in the IFN-β enhanceosome, however. Despite the density with which the eight bound proteins are packed along the essentially straight segment of enhancer DNA, the structure and the binding measurements reported here (Figure S2) and in our previous paper (Panne et al., 2004) show that the relatively tenuous local protein interfaces between abutting DNA-binding domains impart very little cooperativity. For example, the L2 loop of IRF-7 has an insertion of 9 amino-acid residues (with respect to IRF-3) between helicesα2 and α3 (Figure S3). Although this loop extends towards p50, IRF-7 does not bind cooperatively with NFκB to the enhancer (Figure S2), and the structure shows that the glycine- and proline-rich L2 loop is largely disordered and moves out of the way to accommodate loop L1 of p50 without making extensive contacts (Figure S4).
What interactions can give rise to cooperativity of transcription factor association with the IFN-β enhancer in the absence of strong contacts between adjacent proteins? In principle, cooperative binding can arise through nucleotide sequence dependent structural changes in the DNA that allow formation of complementary DNA conformations for adjacently bound transcription factors (Escalante et al., 2002a; Klemm and Pabo, 1996; Panne et al., 2004). This conformational complementarity appears to be the case for ATF-2/c-Jun and all four IRFs (but not for NFκB, which has a site that does not overlap that of its neighbor). We have shown that cooperative binding of ATF-2/c-Jun and IRF-3 depends on the inherent asymmetry of the ATF-2/c-Jun binding site and that modifying it into a consensus AP-1 recognition element eliminates the cooperativity (Panne et al., 2004). That is, the ATF-2/c-Jun site is actually a composite element that accommodates not just ATF-2/c-Jun but also part of the adjacent IRF-3. Similarly, all four IRF binding sites are composite elements, and the structures show a remarkably precise sequence organization to accommodate a specific array of IRFs.
Local complementarity of DNA conformation at overlapping sites cannot, however, account for the strong in vivo synergy of IFN-β gene regulation, as binding analyzed by the EMSA experiments in Figure S2 would than have shown a more striking cooperative character. Previous work has shown that interactions beyond the DNA-binding domains provide additional driving force for cooperative assembly. Have we failed to visualize important pairwise interactions between the transcription factors? Except for the dimerization domains of IRF-3 and IRF-7, which not only hold the dimers together but also bind the co-activator CBP/p300 (Qin et al., 2005), essentially all of the regions of the various DNA-bound proteins known to have well-defined, folded structure are included in the structures and in our binding measurements. That is, the remaining parts of ATF-2, c-Jun, and NFκB are probably flexibly extended, and various segments are known to interact with specific co-activators or co-repressors or to serve as signals for nuclear localization or for degradation. These extended regions are unlikely to form specific contacts with each other or with the IRFs. The absence of pairwise interactions in vitro, using purified full-length activators further supports this contention (D.Panne; unpublished observation).
The high mobility group protein HMGA1a has also been implicated in cooperative enhanceosome assembly (Thanos et al, 1993). Unlike the stably folded, “architectural” HMG proteins such as LEF-1, HMG-1, and SRY, which alter DNA conformation and create a platform for association of transcriptional activators, HMGA1a merely requires an accessible, A:T-rich minor groove. The enhanceosome structure shows that the mapped HMGA1a binding sites are not accessible and that HMGA1a is unlikely to be part of the final assembly. Enhanceosome assembly is asynchronous (Munshi et al., 2001). HMGA1a could therefore act as a molecular chaperone during different stages of the assembly process and then dissociate from the final complex – a mode of action also proposed for HMG-1 in certain cases (Thomas, 2001).
Multivalent interactions of the co-activators, CBP and p300, with all the assembled transcription factors participate in activating transcription directed by the IFN-β enhancer (Merika et al., 1998; Wathelet et al., 1998). CBP and p300 are large, extended, flexible molecules, with a series of domains, some widely spaced, that bind segments of the activation regions of various transcription factors. The IRF-binding domain (IBiD) near the C-terminus of CBP interacts with IRF-3 (Lin et al., 2001; Qin et al., 2005); the KIX domain, near the N-terminus, with RelA and c-Jun (Bannister et al., 1995); the CH2 domain, between KIX and IBiD, with ATF-2 (Kawasaki et al., 1998).
In transient transfection experiments with IFN-β reporter genes, insertion of an integral DNA turn (10 base pairs) between the PRDI-PRDII and the PRDIV-PRDIII domains of the IFN-β enhancer does not compromise activation; insertion of a half-integral turn of the helix (5 bp) between the sites essentially disables the enhancer (Thanos and Maniatis, 1995). These experiments reveal the importance of the position of transcription factors on the face of the DNA helix in the assembly of the preinitiation complex, and they illustrate the adaptability of CBP and p300 in spanning variable intervals between DNA-bound transcription factors. They do not, however, reflect all the biological specificity that has led to the evolutionary invariance of the enhancer sequence. In particular, the transfection experiments are unlikely to reflect the subtleties of enhanceosome assembly and enhancer function of the endogenous gene in the context of chromatin. For example, the level of induction of the endogenous gene is orders of magnitude higher than observed with the transfected reporter (T.M. upublished data), and thus the effects of insertions on cooperative binding might not be observed at all in the transfection experiments.
The IFN-β enhanceosome is a precise and specific assembly of “generic” transcription factors that participate in many other regulatory complexes as well. Faithful coincidence detection requires that a functional response should occur only when the right set of transcription factors is on the enhancer and only when all those factors are indeed present. The structure shows that this combinatorial specificity is encoded not just in the various binding sites but also in their overlap and in their positions with respect to each other. That is, precision of the assembly contributes directly to its specificity (e.g., to the requirement for IRF-3 and IRF-7), even in the absence of extended protein-protein interfaces. The strict evolutionary conservation of the IFN-β enhancer sequence correlates with its organizational precision, and we suggest that other strictly conserved enhancer sequences – for example, the 300 bp IL-2 enhancer/promoter – may have similar structural characteristics. These characteristics also imply that non-consensus binding-site sequences can have critical functional importance, a property that will need to be included in computional algorithms for detecting transcription-factor sites in genome sequences.
The IFN-β enhanceosome structure further shows that cooperativity of assembly probably resides at a level of interaction not represented by contacts between neighboring DNA-binding domains but probably at the level of co-activators. The flexibility of CBP/p300 allows it to serve as a signal integrator not only for enhanceosomes of tightly defined geometry, but also for “modular” enhancers with more variably spaced binding sites. One of the best-studied modular elements, the even-skipped stripe-2 enhancer of Drosophila ( Small et al., 1991), shows local evolutionary conservation over segments longer than a single transcription-factor binding site, even when the larger-scale organization of the enhancer is clearly variable. Thus, conserved sub-elements may have a precise, enhanceosome-like molecular architecture within a generally more flexible complete enhancer (Ludwig et al., 2000; Ludwig et al., 2005). A generic adaptor (CBP/p300) would then pass on to the Pol II machinery a summary of tightly regulated signals from several specifically arrayed sets of generic activators.
Experimental Procedures
Protein constructs and structure determination
To obtain a defined complex for crystallization, we created a single-chain construct linking residues 19-291 of RelA, 8-128 of IRF-7 and 4-113 of IRF-3, as well as a variant construct with an additional IRF-7/IRF-3 pair at its C-terminus. The domains were joined by flexible linkers, L1-L4. In previous work, we joined two IRF-3 DNA-binding domains in tandem with a 26-residue flexible linker (Panne et al., 2004) and used this covalently linked dimer, which bound the enhancer more tightly than a pair of unlinked domains, for crystallization (Panne et al., 2004). In that work, we also crystallized the components in the absence of the covalent linker and found no structural differences. Thus the linker allowed stabilization of the assembly without introducing structural constraints. Moreover, the structure reported here of four IRF-3 DNA-binding domains bound to the INF-β enhancer was obtained using no linker between individual IRF-3 domains (Figure S1), and it agrees very well with the linked complexes in regions of overlap (see Results). We are therefore confident that the linkers do not perturb the structures of the DNA:protein complexes.
Coding sequences for the RelA-IRF-7-IRF-3 fusion proteins used here were cloned with a C-terminal hexahistidine-tag into the pET Duet vector (Novagen), along with the RHR (residues 37-350) of p50 as the second encoded chain. Linker design was initially based on the one used previously (Panne et al., 2004). These constructs failed to yield crystals, however, and we then further adjusted the linker sequences. The most important change was to reduce the linker length in L2 and L4 (linking IRF-7 to IRF-3) from 54 to 16 amino acids. We used the restriction enzyme SapI for cleavage at the fusion junctions, allowing us to design and clone the linker sequences without introducing additional, unwanted amino acids. The NFκB:IRF-7:IRF-3 construct with the shorter linker sequences yielded the crystals reported here. Expression, purification, crystallization and data collection were performed as described in the Supplement.
The structure was determined by molecular replacement using MOLREP (Vagin and Teplyakov, 1997). As search models, we used the structures of NFκB bound to DNA from the PRDII site of the INF-β enhancer (1LE5; Berkowitz et al., 2002) and of the IRF-3:DNA complex (1T2K; Panne et al., 2004). After locating the NFκB:DNA complex, we fixed its position and orientation and searched for the two IRF:DNA complexes. The additional DNA was built by placing the phosphates of nucleotide pairs visually into density and restraining their positions during initial refinement. The planarity of the base-pairs and the sugar pucker were restrained to conform to standard B-DNA. The dihedral torsion angles of α helices were restrained to those of an α-helical conformation. In later refinement cycles, these conformational restraints were removed. Iterative model building was performed using the programs O and Coot (Emsley and Cowtan, 2004; Jones et al., 1991); refinement calculations used CNS 1.2 (Brunger et al., 1998). The DNA helical parameters were analyzed using 3DNA (Lu and Olson, 2003), and the global helical axis was plotted using Curves (Lavery and Sklenar, 1988).
Model for the complete enhanceosome
The model in Figure 4 was obtained by superposing the overlapping parts of two structures, those of ATF-2/c-Jun/IRF-3/DNA (1T2K) and NFκB:IRF-7:IRF-3:DNA (2O6I), with the structure of four IRF-3 DNA-binding domains bound to the full length enhancer (2O6G) as a guide. Cα residues 5-110 from IRF-3B (chain B, 1T2K) were superposed, using the program O, with those of IRF-3B (chain F, 2O6G). These two chains superpose with a root mean square deviation (rmsd) of 0.54 Å. Cα residues 9-110 from IRF-3C (chain D, 2O6I) were superposed with those of IRF-3C (chain G, 2O6G). These two chains superpose with a rmsd of 0.98 Å. The two structures contain overlapping DNA nucleotides (region −85 to −72 of the enhancer), one set of which was removed in the final assembly. The excellent superposition in this region can be seen in Figure S7 (rmsd 1.2 Å). Finally, to complete the model, Cα residues 6-65 and 90-127 of a copy of IRF-7D (chain C, 1O6I) were superposed with residues 6-65 and 72-109 of IRF-3 (chain B, 1T2K). These two chains superpose with a rmsd of 0.98 Å as shown in Figure S7.
Supplementary Material
Figure S1: Structure of the IRF-3:DNA complex. (A) Ribbons representation of the complex containing four copies of IRF-3 bound to a sequence-optimized enhancer. (B) Sequence of the wild-type and the optimized enhancer. The positions of the four Positive Regulatory Domains (PRDI-IV) are shown below the enhancer sequence. The two sites containing a nucleotide change A89G and the deletion G75 are indicated by arrows. (C) Alignment of the four IRF binding sites of the wild-type and mutant sequence.
Figure S2: Electrophoretic mobility shift assays.
(A) Comparison of NFκB and a IRF-3 fusion protein (IRF-3)2binding to the 57mer DNA duplex used for crystallization. The DNA contains the mutant interferon-β enhancer sequence as shown in Figure S1. In lanes 1–4, 10 μM NFκB heterodimer was preincubated with 20 μM substrate DNA for 10 minutes before addition of increasing concentrations of (IRF-3)2. In lanes 5–8, (IRF-3)2 was mixed directly with 20 μM substrate DNA. (IRF-3)2 concentrations were 6 μM (lanes 1, 5), 12 μM (lanes 2, 6), 24 μM (lanes 3, 7), 48 μM (lane 4, 8). Note the relative homogeneous species (lane 4) containing NFκB and two (IRF-3)2 molecules. (B) NFκB binding in the presence of either the IRF-3 fusion protein (IRF-3)2 (lanes 1–4) or the IRF-7/IRF-3 fusion protein (IRF-7-3)2 (lanes 5–8) to the 35mer DNA duplex used for crystallization (Figure 2). In lanes 1–4, 15 μM (IRF-7-3)2; and in lanes 5–8, 15 μM (IRF-3)2 was preincubated with 20 μM substrate DNA for 10 minutes before addition of increasing concentrations of NFκB 4.8 μM (lanes 1, 5), 9.6 μM (lanes 2, 6), 20 μM (lanes 3, 7), 39 μM (lanes 4, 8). Note the absence of cooperative binding of NFκB with either (IRF-3)2 or (IRF-7-3)2. (C) Electrophoretic mobility shift assay of the five part fusion protein NFκB:IRF-7:IRF-3:IRF-7:IRF-3 binding to a 60mer DNA duplex. The following amounts of fusion protein 1.25 μM (lane 1), 2.5 μM (lane 2), 5 μM (lanes 3), 10 μM (lane 4) were incubated for 10 minutes with 10 μM substrate DNA and then deposited on the gel.
Figure S3: Sequence alignment of the DNA-binding domains of human IRF proteins. Helices are marked with boxes; β-sheets, with arrows and loops with lines above the sequences. Residues that are conserved in most IRF proteins are shown in red; chemically conserved residues, in blue. Residues involved in recognition of the core AANNGAAA sequence of the binding site are marked with an asterisk (*) above the sequence. Residues involved in additional direct DNA contacts in IRF-3 and IRF-7 are marked with #. Arg7, involved in contacts between IRF-3A and IRF-3C, is indicated by +.
Figure S4: Interface between loop L1 of p50 and loop L2 of IRF-7D. For comparison of IRF loop conformations, IRF-3 (green) has been superposed on IRF-7D (yellow). The two domains superpose with a root mean square deviation (rmsd) of 0.98Å. For comparison of p50 loop comparisons, the dimerization domains (RHR-C domains) of published NFκB:DNA complexes can be superposed on the RHR-C domains in our structure. These superpositions reveal rigid body displacements of the p50 and RelA RHR-N domains with respect to the RHR-C dimer module, similar to shifts described previously (Berkowitz et al., 2002; Escalante et al., 2002b). Thus, the RHR-C domains of NFκB bound to the PRDII site (1LE5) superpose on those in our structure with a rmsd of 0.65Å, and in this superposition, the RHR-N domains of p50 in the two complexes are displaced from each other by ~5.6Å (shown in red), and those of RelA, by ~6.5Å. For comparison, the RHR-C domains of NFκB bound to the Ig-κB site (1VKX) superpose on those in our structure with a rmsd of 0.81Å, and in this superposition, the RHR-N domains of p50 in the two complexes are displaced from each other by ~4.2Å, and those of RelA, by ~1.0Å. Similarly, the RHR-C domains of NFκB bound to the HIV-κB site (1LEI) superpose on those in our structure with a rmsd of 0.65Å, and in this superposition, the RHR-N domains of p50 are displaced from each other by ~2.8Å, and those of RelA, by ~5.0Å. In short, complexes crystallized on identical DNA binding sites (the present structure and 1LE5) exhibit conformational differences that are as large as those crystallized on different DNA sites (the present structure and 1VKX or 1LEI). The superposed p50 subunit of the p50:RelA:PRDII complex (1LE5) is shown in red, and IRF-3, in green. Loop L2 of IRF-7D avoids overlap with loop L1 of p50, which has a different conformation than it does in the 1LE5 structure, shifted by about 5.6Å towards IRF-7D, as indicated by the arrows. The absence of a predicted clash between loop L2 of the superposed IRF-3 and L1 of p50 is due to a difference between the DNA conformations in the structure of NFκB bound to PRDII in isolation and in that of the enhanceosome complex.
Figure S5: DNA conformation in the enhanceosome: Plots of the variations in widths of the major and minor grooves in the fully assembled enhanceosome.
Figure S6: Sequence alignment of the INF-β enhancer from different species. Conserved residues are shown in red. The core binding sites for each protein are boxed.
Figure 7S: Superposition of the NFκB:IRF-7:IRF-3:PRDI-II structure on that of ATF-2:c-Jun:2(IRF-3):PRDIV-III (1T2K). The two structures contain overlapping DNA nucleotides, one set of which was removed in the final assembly shown in Figure 4 and in the upper part of this figure. The relevant region of overlap between the two structures is boxed. In the lower part of this figure, nucleotides and phosphate backbone from the NFκB:IRF-7:IRF-3:DNA structure are shown in grey. Nucleotides from the ATF-2:c-Jun:2(IRF-3):DNA structure are shown in brown, and the phosphate backbone, in blue. There is excellent superposition of the overlapping nucleotides. The final enhanceosome model shown in Figure 4 contains the IRF-7D chain from the NFκB:IRF-7:IRF-3:DNA structure superposed onto (and substituted for) the IRF-3B chain of the ATF-2:c-Jun:2(IRF-3):DNA structure. These two chains superpose with a rmsd of 0.98 Å. IRF-3B is shown in green; the superposed IRF-7 is shown in yellow (IRF-7B).
Acknowledgments
We thank Michael Carey and Ernest Fraenkel for comments on the manuscript. Data were collected at beamline 8.2.1 at the Advanced Light Source, Lawrence Berkeley Laboratory, and at BioCARS beamline 14C at the Advanced Photon Source, Argonne National Laboratory. SCH is an Investigator in the Howard Hughes Medical Institute. Coordinates and structure factors have been deposited in the RSCB Protein Data Bank with accession codes 2O6G for the IRF-3/DNA structure and 2O6I for the NFκB:IRF-7:IRF-3:DNA structure. TM was supported by grant R01AI020642 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- Arnosti DN, Kulkarni MM. Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem. 2005;94:890–898. doi: 10.1002/jcb.20352. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Oehler T, Wilhelm D, Angel P, Kouzarides T. Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene. 1995;11:2509–2514. [PubMed] [Google Scholar]
- Berkowitz B, Huang DB, Chen-Park FE, Sigler PB, Ghosh G. The x-ray crystal structure of the NF-kappa B p50. p65 heterodimer bound to the interferon beta -kappa B site. J Biol Chem. 2002;277:24694–24700. doi: 10.1074/jbc.M200006200. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Carey M, Smale ST. Transcriptional Regulation in Eukaryotes 2000 [Google Scholar]
- Chen FE, Ghosh G. Regulation of DNA binding by Rel/NF-kappaB transcription factors: structural views. Oncogene. 1999;18:6845–6852. doi: 10.1038/sj.onc.1203224. [DOI] [PubMed] [Google Scholar]
- Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998a;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- Chen FE, Kempiak S, Huang DB, Phelps C, Ghosh G. Construction, expression, purification and functional analysis of recombinant NFkappaB p50/p65 heterodimer. Protein Eng. 1999;12:423–428. doi: 10.1093/protein/12.5.423. [DOI] [PubMed] [Google Scholar]
- Chen L, Glover JN, Hogan PG, Rao A, Harrison SC. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998b;392:42–48. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- Chen-Park FE, Huang DB, Noro B, Thanos D, Ghosh G. The kappa B DNA sequence from the HIV long terminal repeat functions as an allosteric regulator of HIV transcription. J Biol Chem. 2002;277:24701–24708. doi: 10.1074/jbc.M200007200. [DOI] [PubMed] [Google Scholar]
- Du W, Maniatis T. An ATF/CREB binding site is required for virus induction of the human interferon-β gene. Proc Natl Acad Sci U S A. 1992;89:2150–2154. doi: 10.1073/pnas.89.6.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Escalante CR, Brass AL, Pongubala JM, Shatova E, Shen L, Singh H, Aggarwal AK. Crystal structure of PU. 1/IRF-4/DNA ternary complex. Mol Cell. 2002a;10:1097–1105. doi: 10.1016/s1097-2765(02)00703-7. [DOI] [PubMed] [Google Scholar]
- Escalante CR, Shen L, Thanos D, Aggarwal AK. Structure of NF-kappaB p50/p65 heterodimer bound to the PRDII DNA element from the interferon-beta promoter. Structure. 2002b;10:383–391. doi: 10.1016/s0969-2126(02)00723-2. [DOI] [PubMed] [Google Scholar]
- Escalante CR, Yie J, Thanos D, Aggarwal AK. Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature. 1998;391:103–106. doi: 10.1038/34224. [DOI] [PubMed] [Google Scholar]
- Falvo JV, Parekh BS, Lin CH, Fraenkel E, Maniatis T. Assembly of a functional beta interferon enhanceosome is dependent on ATF-2/c-Jun heterodimer orientation. Mol Cell Biol. 2000;20:4814–4825. doi: 10.1128/mcb.20.13.4814-4825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falvo JV, Thanos D, Maniatis T. Reversal of intrinsic DNA bends in the IFN beta gene enhancer by transcription factors and the architectural protein HMG I(Y) Cell. 1995;83:1101–1111. doi: 10.1016/0092-8674(95)90137-x. [DOI] [PubMed] [Google Scholar]
- Fujii Y, Shimizu T, Kusumoto M, Kyogoku Y, Taniguchi T, Hakoshima T. Crystal structure of an IRF-DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J. 1999;18:5028–5041. doi: 10.1093/emboj/18.18.5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Sakakibara J, Sudo Y, Miyamoto M, Kimura Y, Taniguchi T. Evidence for a nuclear factor(s), IRF-1, mediating induction and silencing properties to human IFN-beta gene regulatory elements. EMBO J. 1988;7:3397–3405. doi: 10.1002/j.1460-2075.1988.tb03213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh G, van Duyne G, Ghosh S, Sigler PB. Structure of NF-kappa B p50 homodimer bound to a kappa B site. Nature. 1995;373:303–310. doi: 10.1038/373303a0. [DOI] [PubMed] [Google Scholar]
- Goodbourn S, Maniatis T. Overlapping positive and negative regulatory domains of the human beta- interferon gene. Proc Natl Acad Sci U S A. 1988;85:1447–1451. doi: 10.1073/pnas.85.5.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- Gupta S, Campbell D, Derijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Huth JR, Bewley CA, Nissen MS, Evans JN, Reeves R, Gronenborn AM, Clore GM. The solution structure of an HMG-I(Y)-DNA complex defines a new architectural minor groove binding motif. Nat Struct Biol. 1997;4:657–665. doi: 10.1038/nsb0897-657. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Song J, Eckner R, Ugai H, Chiu R, Taira K, Shi Y, Jones N, Yokoyama KK. p300 and ATF-2 are components of the DRF complex, which regulates retinoic acid- and E1A-mediated transcription of the c-jun gene in F9 cells. Genes Dev. 1998;12:233–245. doi: 10.1101/gad.12.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm JD, Pabo CO. Oct-1 POU domain-DNA interactions: cooperative binding of isolated subdomains and effects of covalent linkage. Genes Dev. 1996;10:27–36. doi: 10.1101/gad.10.1.27. [DOI] [PubMed] [Google Scholar]
- Lavery R, Sklenar H. The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J Biomol Struct Dyn. 1988;6:63–91. doi: 10.1080/07391102.1988.10506483. [DOI] [PubMed] [Google Scholar]
- Lin CH, Hare BJ, Wagner G, Harrison SC, Maniatis T, Fraenkel E. A small domain of CBP/p300 binds diverse proteins: solution structure and functional studies. Mol Cell. 2001;8:581–590. doi: 10.1016/s1097-2765(01)00333-1. [DOI] [PubMed] [Google Scholar]
- Lin R, Genin P, Mamane Y, Hiscott J. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol Cell Biol. 2000a;20:6342–6353. doi: 10.1128/mcb.20.17.6342-6353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomvardas S, Thanos D. Nucleosome sliding via TBP DNA binding in vivo. Cell. 2001;106:685–696. doi: 10.1016/s0092-8674(01)00490-1. [DOI] [PubMed] [Google Scholar]
- Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig MZ, Bergman C, Patel NH, Kreitman M. Evidence for stabilizing selection in a eukaryotic enhancer element. Nature. 2000;403:564–567. doi: 10.1038/35000615. [DOI] [PubMed] [Google Scholar]
- Ludwig MZ, Palsson A, Alekseeva E, Bergman CM, Nathan J, Kreitman M. Functional evolution of a cis-regulatory module. PLoS Biol. 2005;3:e93. doi: 10.1371/journal.pbio.0030093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Laskowski RA, Thornton JM. NUCPLOT: a program to generate schematic diagrams of protein-nucleic acid interactions. Nucleic Acids Res. 1997;25:4940–4945. doi: 10.1093/nar/25.24.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamane Y, Heylbroeck C, Genin P, Algarte M, Servant MJ, LePage C, DeLuca C, Kwon H, Lin R, Hiscott J. Interferon regulatory factors: the next generation. Gene. 1999;237:1–14. doi: 10.1016/s0378-1119(99)00262-0. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, Wathelet MG. Structure and function of the interferon-β enhanceosome. Cold Spring Harb Symp Quant Biol. 1998;63:609–620. doi: 10.1101/sqb.1998.63.609. [DOI] [PubMed] [Google Scholar]
- Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merika M, Thanos D. Enhanceosomes. Curr Opin Genet Dev. 2001;11:205–208. doi: 10.1016/s0959-437x(00)00180-5. [DOI] [PubMed] [Google Scholar]
- Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol Cell. 1998;1:277–287. doi: 10.1016/s1097-2765(00)80028-3. [DOI] [PubMed] [Google Scholar]
- Morin P, Braganca J, Bandu MT, Lin R, Hiscott J, Doly J, Civas A. Preferential binding sites for interferon regulatory factors 3 and 7 involved in interferon-A gene transcription. J Mol Biol. 2002;316:1009–1022. doi: 10.1006/jmbi.2001.5401. [DOI] [PubMed] [Google Scholar]
- Muller CW, Rey FA, Sodeoka M, Verdine GL, Harrison SC. Structure of the NF-kappa B p50 homodimer bound to DNA. Nature. 1995;373:311–317. doi: 10.1038/373311a0. [DOI] [PubMed] [Google Scholar]
- Munshi N, Agalioti T, Lomvardas S, Merika M, Chen G, Thanos D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science. 2001;293:1133–1136. doi: 10.1126/science.293.5532.1133. [DOI] [PubMed] [Google Scholar]
- Munshi N, Yie Y, Merika M, Senger K, Lomvardas S, Agalioti T, Thanos D. The IFN-β enhancer: a paradigm for understanding activation and repression of inducible gene expression. Cold Spring Harb Symp Quant Biol. 1999;64:149–159. doi: 10.1101/sqb.1999.64.149. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Panne D, Maniatis T, Harrison SC. Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-beta enhancer. EMBO J. 2004;23:4384–4393. doi: 10.1038/sj.emboj.7600453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M. A Genetic Switch. 3. New York: Cold Spring Harbor Laboratory Press; 2004. [Google Scholar]
- Qin BY, Liu C, Srinath H, Lam SS, Correia JJ, Derynck R, Lin K. Crystal structure of IRF-3 in complex with CBP. Structure. 2005;13:1269–1277. doi: 10.1016/j.str.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Small S, Kraut R, Hoey T, Warrior R, Levine M. Transcriptional regulation of a pair-rule stripe in Drosophila. Genes Dev. 1991;5:827–839. doi: 10.1101/gad.5.5.827. [DOI] [PubMed] [Google Scholar]
- Struhl K. Gene regulation. A paradigm for precision. Science. 2001;293:1054–1055. doi: 10.1126/science.1064050. [DOI] [PubMed] [Google Scholar]
- Thanos D, Du W, Maniatis T. The high mobility group protein HMG I(Y) is an essential structural component of a virus-inducible enhancer complex. Cold Spring Harb Symp Quant Biol. 1993;58:73–81. doi: 10.1101/sqb.1993.058.01.011. [DOI] [PubMed] [Google Scholar]
- Thanos D, Maniatis T. The high mobility group protein HMG I(Y) is required for NF-kappa B- dependent virus induction of the human IFN-beta gene. Cell. 1992;71:777–789. doi: 10.1016/0092-8674(92)90554-p. [DOI] [PubMed] [Google Scholar]
- Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–1100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- Thomas JO. HMG1 and 2: architectural DNA-binding proteins. Biochem Soc Trans. 2001;29:395–401. doi: 10.1042/bst0290395. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. Journal of Applied Crystallography. 1997;30:1022–1025. [Google Scholar]
- Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell. 1998;1:507–518. doi: 10.1016/s1097-2765(00)80051-9. [DOI] [PubMed] [Google Scholar]
- Yie J, Senger K, Thanos D. Mechanism by which the IFN-beta enhanceosome activates transcription. Proc Natl Acad Sci U S A. 1999;96:13108–13113. doi: 10.1073/pnas.96.23.13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Structure of the IRF-3:DNA complex. (A) Ribbons representation of the complex containing four copies of IRF-3 bound to a sequence-optimized enhancer. (B) Sequence of the wild-type and the optimized enhancer. The positions of the four Positive Regulatory Domains (PRDI-IV) are shown below the enhancer sequence. The two sites containing a nucleotide change A89G and the deletion G75 are indicated by arrows. (C) Alignment of the four IRF binding sites of the wild-type and mutant sequence.
Figure S2: Electrophoretic mobility shift assays.
(A) Comparison of NFκB and a IRF-3 fusion protein (IRF-3)2binding to the 57mer DNA duplex used for crystallization. The DNA contains the mutant interferon-β enhancer sequence as shown in Figure S1. In lanes 1–4, 10 μM NFκB heterodimer was preincubated with 20 μM substrate DNA for 10 minutes before addition of increasing concentrations of (IRF-3)2. In lanes 5–8, (IRF-3)2 was mixed directly with 20 μM substrate DNA. (IRF-3)2 concentrations were 6 μM (lanes 1, 5), 12 μM (lanes 2, 6), 24 μM (lanes 3, 7), 48 μM (lane 4, 8). Note the relative homogeneous species (lane 4) containing NFκB and two (IRF-3)2 molecules. (B) NFκB binding in the presence of either the IRF-3 fusion protein (IRF-3)2 (lanes 1–4) or the IRF-7/IRF-3 fusion protein (IRF-7-3)2 (lanes 5–8) to the 35mer DNA duplex used for crystallization (Figure 2). In lanes 1–4, 15 μM (IRF-7-3)2; and in lanes 5–8, 15 μM (IRF-3)2 was preincubated with 20 μM substrate DNA for 10 minutes before addition of increasing concentrations of NFκB 4.8 μM (lanes 1, 5), 9.6 μM (lanes 2, 6), 20 μM (lanes 3, 7), 39 μM (lanes 4, 8). Note the absence of cooperative binding of NFκB with either (IRF-3)2 or (IRF-7-3)2. (C) Electrophoretic mobility shift assay of the five part fusion protein NFκB:IRF-7:IRF-3:IRF-7:IRF-3 binding to a 60mer DNA duplex. The following amounts of fusion protein 1.25 μM (lane 1), 2.5 μM (lane 2), 5 μM (lanes 3), 10 μM (lane 4) were incubated for 10 minutes with 10 μM substrate DNA and then deposited on the gel.
Figure S3: Sequence alignment of the DNA-binding domains of human IRF proteins. Helices are marked with boxes; β-sheets, with arrows and loops with lines above the sequences. Residues that are conserved in most IRF proteins are shown in red; chemically conserved residues, in blue. Residues involved in recognition of the core AANNGAAA sequence of the binding site are marked with an asterisk (*) above the sequence. Residues involved in additional direct DNA contacts in IRF-3 and IRF-7 are marked with #. Arg7, involved in contacts between IRF-3A and IRF-3C, is indicated by +.
Figure S4: Interface between loop L1 of p50 and loop L2 of IRF-7D. For comparison of IRF loop conformations, IRF-3 (green) has been superposed on IRF-7D (yellow). The two domains superpose with a root mean square deviation (rmsd) of 0.98Å. For comparison of p50 loop comparisons, the dimerization domains (RHR-C domains) of published NFκB:DNA complexes can be superposed on the RHR-C domains in our structure. These superpositions reveal rigid body displacements of the p50 and RelA RHR-N domains with respect to the RHR-C dimer module, similar to shifts described previously (Berkowitz et al., 2002; Escalante et al., 2002b). Thus, the RHR-C domains of NFκB bound to the PRDII site (1LE5) superpose on those in our structure with a rmsd of 0.65Å, and in this superposition, the RHR-N domains of p50 in the two complexes are displaced from each other by ~5.6Å (shown in red), and those of RelA, by ~6.5Å. For comparison, the RHR-C domains of NFκB bound to the Ig-κB site (1VKX) superpose on those in our structure with a rmsd of 0.81Å, and in this superposition, the RHR-N domains of p50 in the two complexes are displaced from each other by ~4.2Å, and those of RelA, by ~1.0Å. Similarly, the RHR-C domains of NFκB bound to the HIV-κB site (1LEI) superpose on those in our structure with a rmsd of 0.65Å, and in this superposition, the RHR-N domains of p50 are displaced from each other by ~2.8Å, and those of RelA, by ~5.0Å. In short, complexes crystallized on identical DNA binding sites (the present structure and 1LE5) exhibit conformational differences that are as large as those crystallized on different DNA sites (the present structure and 1VKX or 1LEI). The superposed p50 subunit of the p50:RelA:PRDII complex (1LE5) is shown in red, and IRF-3, in green. Loop L2 of IRF-7D avoids overlap with loop L1 of p50, which has a different conformation than it does in the 1LE5 structure, shifted by about 5.6Å towards IRF-7D, as indicated by the arrows. The absence of a predicted clash between loop L2 of the superposed IRF-3 and L1 of p50 is due to a difference between the DNA conformations in the structure of NFκB bound to PRDII in isolation and in that of the enhanceosome complex.
Figure S5: DNA conformation in the enhanceosome: Plots of the variations in widths of the major and minor grooves in the fully assembled enhanceosome.
Figure S6: Sequence alignment of the INF-β enhancer from different species. Conserved residues are shown in red. The core binding sites for each protein are boxed.
Figure 7S: Superposition of the NFκB:IRF-7:IRF-3:PRDI-II structure on that of ATF-2:c-Jun:2(IRF-3):PRDIV-III (1T2K). The two structures contain overlapping DNA nucleotides, one set of which was removed in the final assembly shown in Figure 4 and in the upper part of this figure. The relevant region of overlap between the two structures is boxed. In the lower part of this figure, nucleotides and phosphate backbone from the NFκB:IRF-7:IRF-3:DNA structure are shown in grey. Nucleotides from the ATF-2:c-Jun:2(IRF-3):DNA structure are shown in brown, and the phosphate backbone, in blue. There is excellent superposition of the overlapping nucleotides. The final enhanceosome model shown in Figure 4 contains the IRF-7D chain from the NFκB:IRF-7:IRF-3:DNA structure superposed onto (and substituted for) the IRF-3B chain of the ATF-2:c-Jun:2(IRF-3):DNA structure. These two chains superpose with a rmsd of 0.98 Å. IRF-3B is shown in green; the superposed IRF-7 is shown in yellow (IRF-7B).