

Abstract
Alterations in neurotrophic signaling pathways may contribute to the changes in the mesolimbic dopamine system induced by chronic morphine exposure. In a rat model of morphine dependence, we previously identified increased levels of phospholipase C gamma-1 (PLCγ1) immunoreactivity specifically within the ventral tegmental area (VTA) following chronic morphine treatment. Using an antibody specific for the tyrosine-phosphorylated, activated form of PLCγ1, we now show that chronic morphine also significantly upregulates PLCγ1 activity in the VTA, as well as in the nucleus accumbens and hippocampus, regions which are also implicated in the reinforcing properties of morphine. In contrast, no increase in PLCγ1 activity was found in the substantia nigra or dorsal striatum. HSV-mediated overexpression of PLCγ1 in PC12 cells induced ERK activation via a mechanism dependent, in part, on both MAP-ERK kinase (MEK) and protein kinase C. PLCγ1 overexpression in the VTA similarly induced ERK activation in the VTA in vivo. As chronic morphine treatment has been shown to increase ERK activity within the VTA, the current results suggest that increased PLCγ1 activity may be an upstream mediator of this effect.
Keywords: Opiate, Phospholipase Cγ, Neurotrophin, Tyrosine Phosphorylation, Ventral Tegmental Area, Herpes Simplex Virus
1. Introduction
Persistent changes in the structure and function of the mesolimbic dopamine system play an important role in mediating the addictive properties of opiates and other drugs of abuse (Nestler and Aghajanian, 1997; Koob et al., 1998; Pierce and Kumaresan, 2006). This system is composed of dopaminergic neurons arising in the midbrain ventral tegmental area (VTA) and the projections of these neurons to several regions of the limbic forebrain. Given their importance to many forms of neural plasticity, neurotrophic factors (NTFs) and their signaling pathways are likely to be involved in these long-lasting changes (Nestler et al., 1996; Nawa et al., 1997; Bolaños and Nestler, 2004; Janak et al., 2006). Many of the morphological and biochemical alterations induced in the VTA by chronic morphine resemble the expected consequences of trophic factor deprivation (Nestler, 1992), and direct infusion of NTFs into the VTA can oppose these drug-induced changes (Berhow et al., 1995; Messer et al., 2000). Exogenous administration of NTFs (or truncated receptors which act as NTF antagonists), or genetic knockout of NTFs, also alters behavioral sensitivity to the reinforcing and locomotor-sensitizing effects of drugs of abuse (Horger et al., 1999; Messer et al., 2000; Pierce and Bari, 2001; Hall et al., 2003; Lu et al., 2004).
This suggests that opiate and NTF actions have convergent effects on the phenotype and function of mesolimbic dopamine neurons. While convergent effects could occur at multiple levels, interactions at the level of intracellular second messengers are likely to be particularly important. Opiates bind and activate specific G protein-coupled receptors (GPCRs); dopamine, released from opiate-disinhibited VTA neurons, also binds GPCRs. While GPCRs have long been known to signal through adenylyl cyclase and phospholipase Cβ, among other second messenger pathways, more recent studies have shown that GPCRs can also activate the tyrosine kinase signaling cascades best known for their role in NTF action (Rajagopal et al., 2004, Luttrell, 2005). NTF-activated tyrosine kinases utilize a relatively small set of conserved signaling pathways to regulate neuronal survival, differentiation, and synaptic plasticity. The best characterized of these signaling pathways include: one that contains phospholipase Cγ (PLCγ), which subsequently regulates the phosphatidylinositol pathway leading to intracellular calcium release and protein kinase C (PKC) activation; another, termed the MAP-kinase pathway, which involves Ras, Raf, MEK (MAP-ERK kinase), and ERK (extracellular signal regulated kinase); and a third that leads, via IRS (insulin receptor substrate) adaptor proteins, to the activation of phosphatidylinositol-3-kinase (PI-3-K) and AKT (Russell, 1995; Segal, 2003; Huang and Reichardt, 2003; Thomas and Huganir, 2004).
Prior studies from our laboratory have demonstrated that chronic opiate administration differentially regulates the abundance and activity of NTF signaling proteins. Chronic morphine increases the activity of ERK specifically within the VTA, without any change in the abundance of ERK protein or the abundance of proteins known to activate ERK, including Ras, Raf, and MEK (Berhow et al., 1996; Wolf et al., 1999). Chronic morphine also downregulates IRS protein levels and the activity of downstream AKT signaling, and upregulates PLCγ1 protein levels within the VTA (Wolf et al., 1999; Russo et al., 2007).
PLCγ1 is part of the proximal signal transduction mechanism activated by multiple tyrosine kinase receptors, including those with potent trophic effects on midbrain dopaminergic neurons, such as BDNF, NT-4, and GDNF (Widmer et al., 1992; Zirrgiebel et al., 1995; Rhee, 2001, Lee et al., 2006). Like other PLC enzymes, PLCγ1 catalyzes the production of diacylglycerol (DAG) and inositol triphosphate (IP3), which are known to serve as potent second messengers in the regulation of cell function (Berridge, 1993). However, PLCγ1 is the only tyrosine kinase-regulated PLC expressed widely in brain, and as such probably serves a substantially different functional role than other PLCs. Its ability to mediate NTF-induced intracellular calcium elevations suggests that regulation of PLCγ1 levels could have important effects on NTF-mediated neuronal plasticity. While the embryonic lethality of the PLCγ1 knockout mouse indicates a unique and essential role in development, it unfortunately also prevents this model from providing any insight into the role of PLCγ1 signaling in the adult CNS (Ji et al., 1997). Viral vectors provide another approach to studying the role of PLCγ1 in the brain: viral-mediated overexpression of PLCγ1 in the rat VTA has been shown to potently regulate the rewarding and locomotor-activating effects of morphine (Bolaños et al., 2003, 2005). However, the ability to interpret these findings is limited, since it remains unclear whether the morphine-induced increase in PLCγ1 protein levels is associated with a net increase in PLCγ1 signaling.
The current studies address the functional impact of morphine-induced upregulation of PLCγ1 protein levels. The effect of chronic morphine treatment on PLCγ1 activity was examined using an antibody specific for the tyrosine-phosphorylated, activated form of PLCγ1. In order to explore the relationship of increased PLCγ1 levels to other aspects of NTF signaling implicated in addictive processes, the effects of PLCγ1 overexpression on ERK activity were studied in PC12 cells and in vivo, using a herpes simplex virus vector encoding wild-type PLCγ1 (HSV-PLCγ1).
2. Results
2.1. Upregulation of PLCγ1 tyrosine phosphorylation by chronic morphine
We generated a polyclonal anti-peptide antibody specific for PLCγ1 phosphorylated on Tyr783, as described in Experimental Procedures. To demonstrate the expected utility of this antibody, primary striatal cultures were treated with BDNF (50 ng/ml) and PC12 cells were treated with NGF (50 ng/ml); both neurotrophic factors (NTFs) are known to activate PLCγ1 in these cell types via Trk family tyrosine kinase receptors. Western blotting of lysates from striatal cultures or PC12 cells demonstrated dramatic NTF-induced upregulation of a 145 kD band representing tyrosine-phosphorylated PLCγ1 (data from striatal cultures shown in Fig. 1A,B). As a control, the phospho-specific antibody was pre-incubated with the phopho-peptide used to generate it; this pre-incubation blocked the detection of the BDNF-induced band by western blotting (Fig. 1A). To confirm the identity of this band, PC12 cell lysates were immunoprecipitated with a highly specific commercial monoclonal PLCγ1 antibody, and these immunoprecipitates were western-blotted with the phospho-specific antibody, revealing a band at the same position as the one upregulated by NTF treatment (Fig. 1C). Phospho-PLCγ1 western blots were reprobed with a commercial antibody against total PLCγ1, which demonstrated that there was no difference in total PLCγ1 levels between control and NTF-treated cell lysates (Fig. 1B).
Fig. 1.
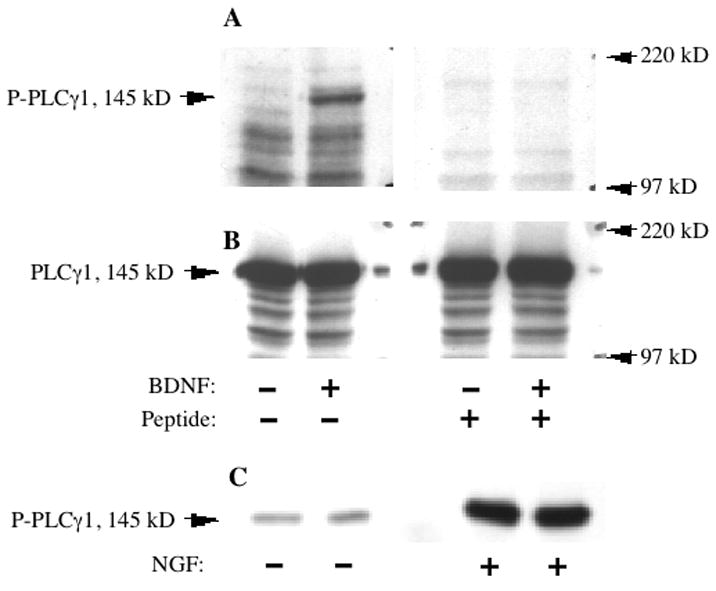
Specific detection of neurotrophin-activated PLCγ1 by a phospho-PLCγ1 antibody. A: Western blots of cell lysate from primary striatal neurons, treated with or without 50 ng/mL BDNF for 10 min. Both blots were probed with the phospho-PLCγ1 antibody (P-PLCγ1), but the antibody used on the right-hand blot was pre-incubated with the antigenic phospho-peptide used to generate it. This pretreatment blocked the detection of BDNF induced PLCγ1 phosphorylation observed in the blot on the left. B: The same membrane as in A, reprobed with an antibody against total PLCγ1, demonstrating the abundant and equal presence of total PLCγ1 in each lane. C: Duplicate lanes of lysate from PC12 cells treated with or without 50 ng/mL NGF for 10 min, and then immunoprecipated with a highly specific mixed monoclonal PLCγ1 antibody. Immunoprecipitated PLCγ1 is detected by the phospho-PLCγ1 antibody.
Having confirmed its function in western blotting experiments, this phospho-PLCγ1 antibody was next used to examine the regulation of PLCγ1 tyrosine phosphorylation by chronic morphine treatment (Fig. 2). Analysis of phospho-PLCγ1 levels in brain regions from control and morphine-treated rats revealed a significant morphine-induced increase in phospho-PLCγ1 within the VTA (+55 ± 13%, P= 0.01, t=3.23, df=10); the SN, in contrast, revealed no significant change in phospho-PLCγ1 levels (−27±9%, P=0.11, t=1.77, df=10). The hippocampus demonstrated the most dramatic increase in levels of phospho-PLCγ1 (+144±5%, P< 0.001, t=4.69, df=10); there was also a significant increase in the NAc (+86±25%, P=0.04, t=2.37, df=10), without any statistically significant change in the striatum (+37±15%, P=0.11, t=1.76, df=10). Concomitant treatment with naltrexone, an opioid receptor antagonist, eliminated the morphine-induced upregulation in phospho-PLCγ1 levels in these regions, including the VTA (naltrexone+morphine compared to controls: VTA −2±1%, P=0.51, t=0.68, df=10; other regions all <5% change, P>.1) indicating a specific opioid receptor-mediated effect. Total PLCγ1 protein levels were not significantly regulated by morphine in any of the brain regions examined in this experiment, including the VTA (+8%, P=0.08, t=1.95, df=10; all other regions <5%, P>.1). The large increase in PLCγ1 phosphorylation therefore cannot be explained by increased total PLCγ1 levels, but instead reflects a true increase in the degree of phosphorylation, which serves as a marker for enzyme activity.
Fig. 2.
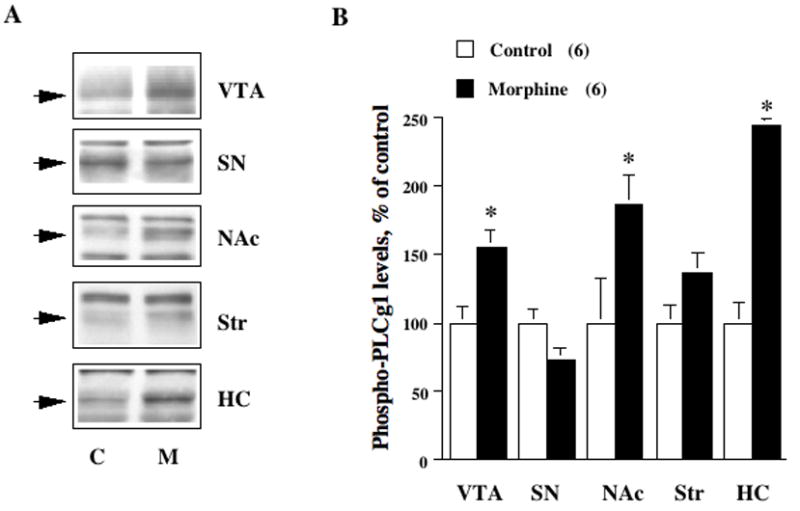
Chronic morphine upregulates PLCγ1 in several limbic brain regions involved in opiate reward. A: Tissue samples from five brain regions in control (C) or morphine-treated (M) rats (n=6) were immunoblotted using an antibody that specifically recognizes the tyrosine phosphorylated, activated form of PLCγ1 (P-PLCγ1). Shown here are pairs (C and M) of representative bands from each region; these bands, at an apparent molecular weight of 145 kD, correspond to phospho-PLCγ1 (indicated by arrows). Note non-specific bands are unregulated. B: Graphical representation of the mean change in phospho-PLCγ1 levels (±SEM) in each brain region, compared to the control average in that region, considered as 100%. There is a statistically significant (* denotes P<0.05) upregulation of phospho-PLCγ1 in the VTA, NAc, and hippocampus (HC), but not SN or dorsal striatum (Str).
2.2. Generation of HSV vectors expressing wild-type PLCγ1
To examine causal relationships between increased PLCγ1 activity and other biochemical consequences of chronic morphine treatment, we used HSV vectors expressing wild-type PLCγ1 or control proteins such as LacZ or GFP (Bolaños et al., 2003, 2005). To validate the HSV-PLCγ1 vector, PC12 cells were infected with the vector for 4 hr, and cell lysates from infected and uninfected cells were analyzed by western blot. Probing with an antibody specific for PLCγ1 revealed the expected 145 kD PLCγ1 band in uninfected PC12 cells; this band was several-fold stronger in HSV-PLCγ1 infected cells, but not in cells infected with a control virus (Fig. 3B). Immunohistochemical analysis revealed viral overexpression of PLCγ1 in the cytoplasm but not the nucleus (data not shown), consistent with the expression pattern of endogenous PLCγ1 (Diakonova et al., 1997).
Fig. 3.
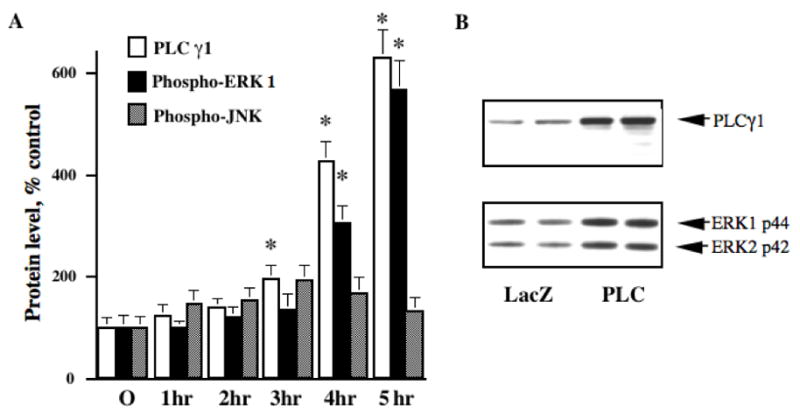
HSV-mediated overexpression of PLCγ1 in PC12 cells induces ERK activation. A: Graphical representation of the timecourse of HSV-mediated PLCγ1 overexpression and ERK activation in PC12 cells, as detected by immunoblotting of infected PC12 cell lysates with antibodies against PLCγ1 and the phosphorylated, activated form of ERK. The timecourse of ERK activation increase over five hours of infection, paralleling the timecourse of PLCγ1 overexpression; in contrast, levels of activated JNK, an ERK homolog associated with cellular stress and apoptosis, increased early and then decreased, never increasing to a statistically significant degree. Values are mean ±SEM, relative to average level at time 0 considered as 100%; * indicates P<0.05, n=4. B: Representative lanes (in duplicate) from western blots of PC12 cell lysates infected for 4 hr with either HSV-PLCγ1 (PLC) or a control virus (LacZ); the top blot was probed with PLCγ1 antibody, detecting the expected 145 kD band; the bottom blot was probed with an antibody specific for the phosphorylated, activated form of ERK, and detects both ERK1 and ERK2 at the expected apparent molecular weights. Both ERK isoforms are activated by PLCγ1 overexpression; phospho-ERK1 levels were used for quantitation in PC12 experiments.
2.3. Effects of PLCγ1 overexpression on ERK activity in PC12 cells
Infection of PC12 cells with HSV-PLCγ1 led to an almost three-fold increase in PLC activity associated with PLCγ1 immunoprecipitates, as compared to immunoprecipitates from PC12 cells infected with a control virus (control: 231±47 counts per minute [cpm], PLCγ1: 585±113 cpm; n=4, P=0.03, t=2.86, df=6). HSV-PLCγ1 infected cells also demonstrated increased levels of tyrosine phosphorylated PLCγ1, as detected by western blotting using the phospho-PLCγ1 antibody described in Experimental Procedures (data not shown). Together, these results demonstrate that the HSV-PLCγ1 vector induces an increase in levels of tyrosine phosphorylated, catalytically active PLCγ1.
Infection of PC12 cells with HSV-PLCγ1 led to a significant increase in phospho-ERK levels, as compared to levels in cells infected with either HSV-GFP or HSV-LacZ control viruses. This PLCγ1-induced activation of ERK increased with longer infection times, in parallel with the increasing levels of PLCγ1 itself (Fig. 3A). In contrast, levels of phospho-JNK (an ERK homolog whose activation is associated with cellular stress and apoptosis but not NTF signaling) were not significantly increased at any timepoint; nor did the shape of the phospho-JNK timecourse parallel that of PLCγ1 and phospho-ERK. Neither HSV-PLC nor the control virus produced any change in total ERK levels within this timeframe, and total ERK levels after 5 hours of infection did not differ between PLC and control virus conditions (difference +2.06 ± 1.3%, P = .28, t=2.23, df=10 for total ERK1).
2.4. Pharmacological regulation of PLCγ1-induced ERK activation in PC12 cells
In an attempt to identify the signaling pathways mediating the PLCγ1-induced ERK activity, PC12 cells were infected with HSV-PLCγ1 and treated with a variety of pharmacological inhibitors specific for different signaling pathways (Fig. 4). The immediate upstream activator of ERK is MEK; the MEK inhibitor PD98509 dramatically reduced the levels of phospho-ERK in cells infected with HSV-PLCγ1 (−74±7%, n=6, P<0.001, t=4.82, df=10).
Fig. 4.

Graphical summary of the effects of various pharmacological agents on ERK activation induced by HSV-mediated PLCγ1 overexpression in PC12 cells. ERK activation was measured by immunoblot analysis using an antibody specific for the phosphorylated, activated form of ERK (P-ERK), and drug-induced changes in P-ERK levels are shown here as a percentage of control cells; both control and drug-treated cells were infected with HSV-PLCγ1. Viral and drug treatments were started simultaneously, lasting 4 hr. Agents included: MEK inhibitor PD98509 (50 μM, n=6), PKC inhibitor calphostin (1 μM, n=4, black bar; 100 nM, n=4, grey bar), PKC downregulator PMA (24 hr treatment, 1 μM, n=6), CaM-kinase inhibitor KN93 (1 μM, n=8), PKA inhibitor H89 (5 μM, n=8), intracellular calcium releaser thapsigargin (1 μM, n=8), and intracellular calcium chelator BAPTA-AM (50 μM, n=8). Inhibition of MEK or PKC significantly decreased ERK activation by HSV-PLCγ1, while inhibitors of CaM-kinase or PKA had no significant effect. The calcium chelator BAPTA-AM significantly increased P-ERK levels; the mechanism of this unexpected effect is unknown. * indicates P<0.05.
PLCγ1, like other PLC enzymes, is known to activate PKC via increases in both DAG and intracellular calcium. PKC, in turn, is capable in some systems of producing ERK activation. Overnight treatment of cultured cells with phorbol esters downregulates PKC enzymes and inhibits PKC-dependent signaling. HSV-PLCγ1 induced phospho-ERK activity was significantly reduced (−25±5%, n=6, P=0.002, t=4.21, df=10) in phorbol ester (PMA)-treated cells compared to untreated cells. Calphostin (1 μM), a highly specific inhibitor of PKC, likewise inhibited the ability of HSV-PLCγ1 to induce ERK activation (−81±3%, n=4, P=0.003, t=4.82, df=6), but did not have a statistically significant effect at a lower dose of 100 nM (−30±6%, n=4, p=0.14, t=1.71, df=6). Together, these data suggest that PKC activity plays a significant, but partial role in the induction of ERK activity by HSV-PLCγ1.
In contrast to the effect of PKC inhibition, treatment with KN93, an inhibitor of CaM-kinases (which could also potentially be activated by PLC-induced calcium release), had no effect on phospho-ERK induction by HSV-PLCγ1 (−2±6%, n=8). PKA inhibition by H89 also had no significant effect of PLCγ1-induced ERK activity (+19±14%, n=8, P=0.32, t=1.04, df=14).
PLCγ1, like other PLC enzymes, leads to intracellular calcium release via the production of IP3; increased levels of intracellular calcium can lead to ERK activation in primary neurons and PC12 cells (Rosen et al., 1994; Xia et al., 1996). Thapsigargin treatment depletes intracellular calcium stores, and thereby inhibits further release of intracellular calcium. However, thapsigargin did not affect the induction of phospho-ERK by HSV-PLCγ1 (−2±15%, n=8). Furthermore, treatment with BAPTA, an intracellular calcium chelator, increased the degree of ERK activity in PLCγ1-infected cells (+58±15%, n=8, P=0.005, t=3.37, df=14). BAPTA is frequently used to block calcium-induced ERK activation; however, BAPTA has also been reported to increase ERK activity in some cell types (Maloney et al., 1999), and different doses of BAPTA can produce opposite effects on PC12 cells (Kozak and Yavin, 1992). Together, our observations suggest that the PLCγ1-induced activation of ERK, at least in PC12 cells, is not dependent upon the ability of PLCγ1 to raise intracellular calcium levels. Rather, our data suggest that PLCγ1-mediated production of diacylglycerol (DAG) may be more significant, possibly activating calcium-independent PKC isoforms.
2.5. Effects of PLCγ1 overexpression on ERK activity in rat VTA in vivo
Finally, we determined whether PLCγ1 overexpression could similarly regulate ERK activity in the VTA in vivo. HSV-PLCγ1 or HSV-GFP were injected into the rat VTA and 3 days later, when transgene is maximal, VTA tissue was analyzed for phospho- and total ERK by western blotting. As shown in Fig. 5, PLCγ1 overexpression (+43±4%, n=5, P< 0.001, t=5.13, df=8) significantly increased levels of phospho-ERK in the VTA (pERK1 +53±16%, P=0.046, t=2.36, df=8; pERK2 +49±12%, P=0.004, t=4.01, df=8), without affecting total levels of the enzyme (ERK1 −7±5%, P=0.29, t=1.14, df=8; ERK2 −2±2%, P=0.47, t=0.77, df=8).
Fig. 5.
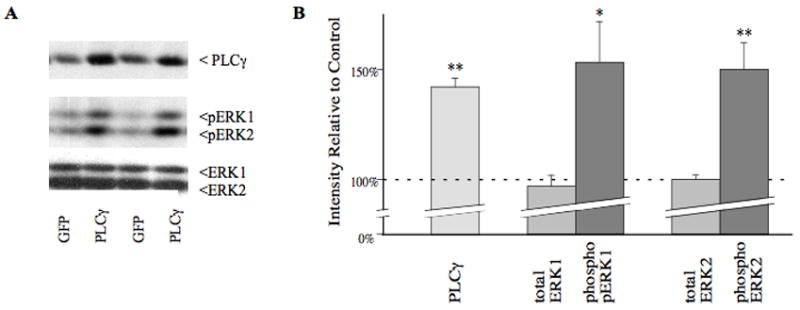
HSV-mediated overexpression of PLCγ1 in the rat VTA in vivo induces ERK activation. A: Representative lanes (in duplicate) from western blots of rat VTA, 3 days after midline injection of 2 μL of either HSV-PLCγ1 (PLC) or a control virus expressing green fluorescent protein (GFP); the top blot was probed with PLCγ1 antibody, detecting the expected 145 kD band; the middle blot is probed with an antibody specific for the phosphorylated, activated form of ERK, and detects both ERK1 and ERK2 at the expected apparent molecular weights; and the bottom blot is probed with an antibody to ERK protein independent of phosphorylation. B: Graphical representation of the mean change in PLCγ1, phosphoERK, and total ERK levels (±SEM) following HSV-PLCγ1 injection in the VTA, compared to the control average in that region, considered as 100% (n=5 per group). There is a statistically significant (* denotes P< 0.05, ** denotes P< 0.005) upregulation of PLCγ1 as expected, and significant increases in activated forms of ERK1 and ERK2, without any change in total ERK levels.
3. Discussion
A major finding of the current study is that chronic morphine treatment significantly upregulates PLCγ1 tyrosine phosphorylation in the VTA, NAc, and hippocampus. In contrast, no significant changes were observed in the SN or dorsal striatum. This regional specificity appears to correlate with the reinforcing properties of morphine, which is self-administered into the VTA, NAc, and hippocampus, but not the SN or dorsal striatum (McBride et al., 1999).
There have been few studies examining the regulation of PLCγ1 phosphorylation or activity within the brain. Most studies of PLC activity in brain fail to distinguish between different PLC isoforms and, in those that do, most have not directly demonstrated changes in PLCγ1 activity or phosphorylation (Katsura et al., 1994; Fukuda et al., 1994; Buckley and Caldwell, 2004; Dwivedi et al., 2005). Lee et al. (1993) did identify increased PLCγ1 tyrosine phosphorylation within the hippocampus following electroconvulsive seizure. Another exception, and the only other study we are ware of that demonstrates in vivo regulation of brain PLCγ1 by opioid administration, reported increased phospho-PLCγ1 immunoreactivity within the mouse periaqueductal gray region following acute morphine treatment (Narita et al., 2003). Consistent with our findings, that study also noted an increased phospho-PLCγ1 immunoreactivity within the nucleus accumbens; however, results in VTA and hippocampus were not reported, and the effects of chronic treatment were not evaluated. The current study is thus among the first to demonstrate regulation of PLCγ1 activity within the brain, and the first to demonstrate its regulation by chronic opiate treatment within a set of brain regions linked to addiction.
The mechanism underlying increased PLCγ1 tyrosine phosphorylation is unknown. It could reflect increased levels of total PLCγ1, which were described in a previous publication (Wolf et al., 1999). However, in the present study we observed only a small, statistically insignificant increase in PLCγ1 that cannot explain the much larger increase observed in phospho-PLCγ1. The reason for the less robust effect on total PLCγ1 in the present study is not known. The current findings therefore imply increased activation of an upstream tyrosine kinase, or decreased action of a tyrosine phosphatase, as also important for induction of PLCγ1 activity. Non-receptor tyrosine kinases such as Src and Pyk2 can be activated by GPCRs (Luttrell, 2005), and both Src and focal adhesion kinase, a Pyk2 homolog, have been reported to promote PLCγ1 activity, (Zhang et al., 1999; Tao et al., 2005). Upregulation of neutrophic factors or their receptors would be expected to increase PLCγ1 activity. However, while regulation of endogenous NTFs has been implicated in the VTA following chronic stimulant administration (Horger et al, 1999; Messer et al., 2000; Freeman and Pierce, 2002; Mueller et al., 2006; Pu et al., 2006), and in the locus coeruleus following chronic morphine treatment (Numan et al., 1998; Akbarian et al., 2002), it has not been identified to date in the VTA using chronic morphine paradigms.
Previous studies have shown that local increases in total levels of PLCγ1 in the VTA, achieved via HSV gene transfer as in this study, potently regulate the rewarding and locomotor-activating effects of morphine, with reduced morphine responses seen with PLGγ1 overexpression in caudal VTA where dopaminergic neurons predominate (Bolaños et al., 2003, 2005). Results of the present study, therefore, suggest that induction of PLCγ1 activity in the VTA by chronic morphine represents a mechanism of drug tolerance. Further studies are needed to explore the functional significance of the observed chronic morphine-induced upregulation of PLCγ1 activity in the NAc and hippocampus.
Long-term potentiation (LTP), a form of synaptic plasticity important in learning and memory, has been best characterized in the hippocampus, but also occurs in the VTA and NAc, where it may contribute to addictive processes (Kombien and Malenka, 1994; Bonci and Malenka, 1999). In the hippocampus, neurotrophic factors and tyrosine kinases are essential for normal LTP (Patterson et al., 1996; Kojima et al., 1997, Brahmam and Messaoudi, 2005), and recent studies have demonstrated an important role for PLCγ1 in mediating LTP and long-term memory formation (Minichiello et al., 2002; Blum and Dash, 2004; Gartner et al., 2006). Opiates can enhance LTP in the hippocampus by a disinhibition of GABAergic interneurons similar to that induced by morphine in the VTA (Svoboda and Lupica, 1998; Jin and Chavkin, 1999; Harrison et al., 2002). Thus, the observed increase in PLCγ1 signaling in the VTA, NAc, and hippocampus may contribute to morphine-induced synaptic plasticity in those regions.
Elevation of intracellular calcium levels (via IP3) and activation of PKC enzymes (via DAG and Ca2+) are the best characterized consequences of PLCγ1 activity. Calcium signaling plays a critical role in LTP and other forms of neuronal plasticity (Ghosh and Greenberg, 1995), as does PKC signaling (Soderling and Derkach, 2000, Boehm et al., 2006). Furthermore, both Ca2+ and PKC can regulate opiate responses, at the cellular and behavioral levels. Elevation of intracellular Ca2+ levels via glutamate receptors in the VTA has been implicated in sensitization to the rewarding and locomotor activating effects of morphine (Carlezon et al., 1997). PKC has been shown to desensitize opioid receptor signaling and contribute to the development of opiate tolerance (Chen and Yu, 1994; Narita et al., 1995; Fundytus and Coderre, 1996). Mice lacking PLCβ3 exhibit enhanced sensitivity to morphine analgesia, apparently due to reduced negative feedback from a PLCβ3/PKC pathway (Xie et al., 1999). However, intraventricular antisense inhibition of PLCγ1 reduced morphine analgesia in mice (Narita et al., 2003), indicating that downstream signaling pathways of different PLC isoforms may mediate distinct opiate effects.
In addition to the well-known role of PLCγ1 in activating calcium and PKC signaling pathways, the current study demonstrates that in PC12 cells and in the VTA in vivo, PLCγ1 activation is sufficient to cause activation of ERK. The PLCγ1-induced ERK activation in PC12 cells was dependent, in part, on the activity of MEK and PKC, and was affected in a complex way by inhibitors of Ca2+ signaling. It is well-known that both elevated intracellular calcium (Xia et al., 1996) and PKC activation (Kolch et al., 1993) can lead to increased ERK activity, and multiple studies have identified a role for PLCγ1 in activating ERK in non-neuronal cell lines (Bassa et al., 1999; Zhang et al., 2000; Mariappan et al., 2005). However, ERK activation has not generally been considered a downstream effector of PLCγ1 in neurons. Consistent with our report, another study in PC12 cells demonstrated a causal link between PLCγ1 levels and ERK activation in PC12 cells, but did not explore the intermediate signaling mechanisms (Choi et al., 2004). In cultured cortical neurons, Matsumoto et al. (2006) reported that BDNF activated ERK via PLCγ1, although the pharmacological PLC inhibitor (U73122) used in that study is not specific to PLCγ1. To our knowledge, the current study is the first to directly demonstrate the ability of PLCγ1 to activate ERK in brain in vivo. Interestingly, Buckley and Caldwell (2004) demonstrated a physical association of PLCγ1 with ERK2 induced by fear conditioning in vivo in the hippocampus, and also showed PLCγ1 is phosphorylated in vitro by ERK2, suggesting the potential for PLCγ1-ERK feedback loops.
Chronic morphine treatment has been shown to increase ERK activity in the VTA (Berhow et al., 1996), in the absence of any identified upregulation of NTFs, NTF receptors, or elements of the upstream Ras-Raf-MEK pathway. The results presented here demonstrate that PLCγ1 activity is also increased in the VTA by chronic morphine, and provide in vitro and in vivo evidence that increased PLCγ1 function can activate ERK. The morphine-induced increase in ERK activity in the VTA may therefore be, at least in part, a downstream consequence of the upregulation in PLCγ1. In turn, ERK has been shown to be an important regulator of synaptic plasticity and gene expression in several brain regions in morphine and other addiction models (e.g., Sgambato et al., 1998; Impey et al., 1999; Jordan et al., 2001; Eitan et al., 2003; Schulz et al., 2004; Valjent et al., 2005). Thus, by regulating Ca2+, PKC, and ERK signaling pathways, increased PLCγ1 activity could play a significant role in the mesolimbic neural plasticity underlying opiate addiction. PLCγ1 and ERK pathways have also been implicated in regulation of mood and anxiety (Lovlie et al., 2001; Bolaños et al., 2003; Duman et al., 2007; Nestler and Carlezon, 2006), suggesting potential signficance of these results for other psychiatric disorders as well.
4. Experimental procedures
4.1. Morphine treatments
For all animal experiments, procedures were in accordance with guidelines of the Yale Animal Care and Use Committee, and used methods designed to minimize any pain or discomfort. Rats used in this study were outbred male Sprague-Dawley animals weighing 150–170 g when obtained from the supplier (Charles River Laboratory, Wilmington, MA). Morphine was administered chronically to rats with initial weights of 170–195 g. The opiate treatment paradigm used followed protocols previously described by Rasmussen and colleagues (1990). Rats (n=6) were lightly anesthetized with halothane and implanted with a single morphine pellet (75 mg of morphine base; National Institute on Drug Abuse) subcutaneously (s.c.), daily for 5 days. Control rats (n=6) received sham surgery, identical to the experimental group except no morphine pellets were implanted. This morphine treatment paradigm has been shown to produce profound states of tolerance and dependence and to result in characteristic biochemical adaptations within the VTA and nucleus accumbens (NAc) (Rasmussen et al., 1990; Beitner-Johnson et al., 1992). On day 6, the rats were killed by decapitation and the brain regions of interest collected by rapid dissection in ice-cold artificial CSF and frozen on dry ice. The VTA and substantia nigra (SN) were dissected from 1 mm thick coronal brain slices using a 15 gauge syringe needle; the NAc and dorsal striatum were dissected with a 12 gauge needle; and the hippocampus was obtained by gross dissection. To test the specificity of the opiate response, in a third group of rats (n=6) the opiate antagonist naltrexone was given daily, concurrently with the morphine treatment: 50 mg/kg in an emulsion (light mineral oil/mannide oleate/saline 1:1:1) was given s.c., and 50 mg/kg in saline solution was given intraperitoneally (i.p.), as previously described (Beitner-Johnson et al., 1993). Rats in the non-naltrexone groups (control, morphine) received emulsion vehicle s.c. and saline i.p. No naltrexone-only group was included.
4.2. Western blotting
Frozen brain samples were solubilized by probe sonication in a buffer containing 1% SDS, 10 mM Tris (pH 7.4), 150 mM NaCl, 5 mM each of EDTA and EGTA, 10 μg/ml each of the protease inhibitors aprotinin and leupeptin, and 1 μg/ml pepstatin A, and phosphatase inhibitors sodium orthovanadate (1 mM) and sodium fluoride (10 mM). Protein concentration was determined in triplicate by the Lowry method (Lowry et al., 1951).
Cultured cells were harvested by scraping into a lysis buffer identical to the one above except containing 1% Triton X-100 instead of 1% SDS; the lysate was rocked for 30′ and spun at 14,000 r.p.m. in a microfuge for 10′ at 4°C, and the supernatant removed into a new tube and stored briefly at −20°C, while the pellet was discarded.
Solubilized brain or cell samples were boiled for 5 min in sample dye containing SDS and the reducing agent β-mercaptoethanol, subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to nitrocellulose membranes for immunoblotting. Membranes were blocked in 5% milk for 1 hr, incubated overnight in primary antibody at 4°C, re-blocked in 2% milk for 1 hr, incubated with appropriate horseradish peroxidase (HRP)-linked secondary antibody (in 2% milk) for 30 min, and then washed three times for 15 min each in buffer. For phospho-antibodies, all steps were performed in phosphate-free buffer (10 mM Tris-buffered saline, with 150 mM NaCl and 0.05% w/v Tween 20); for other antibodies, 10 mM phosphate buffered saline (PBS, with 138 mM NaCl, 2.7 mM KCl, and 0.05% Tween 20) was used. Immunolabeled bands were then visualized with enhanced chemiluminescence (ECL, Amersham, Piscataway, NJ) and Kodak XAR film (Rochester, NY). Luminescence of specific bands was quantified on a Macintosh-based image analysis system with NIH Image software. The data, in arbitrary densitometry units, from control and experimental groups were compared using an unpaired two-tailed Student’s t-test.
The amount of protein loaded for each protein was determined by pilot experiments to lie within the linear range of detection. 5 μg/lane was loaded for PLCγ1, and ERK1 and ERK2. 50–75 μg/lane was used for blots probed with phospho-PLCγ1, phospho-ERK, and phospho-JNK. These aliquots were obtained from the same sonicated samples and run on identical gels and immunoblotted at the same time under identical conditions.
For western blots assessing phospho-ERK regulation, all phospho-ERK blots were reprobed with an antibody to total ERK to confirm that changes in phospho-ERK were not due to changes in total ERK or to variations in the amount of total protein added; total ERK levels were in fact roughly equal in all samples. While the lower half (30–90 kD) of each membrane was probed for phospho-ERK, the upper half (90–200 kD) was probed for PLCγ1 to confirm the expected levels of viral-mediated PLCγ1 expression.
The following antibodies were used for western blotting studies: ERK1, ERK2, phospho-JNK (Santa Cruz Biotechnology, Santa Cruz, CA); PLCγ1 (Upstate Biotechnology, Inc., Lake Placid, NY); GFP (Clontech, Palo Alto, CA); phospho-ERK (1:10,000, Promega, Madison, WI); and phospho-PLCγ 1 (1:2000) was generated for these studies as described below. Peroxidase-linked goat anti-rabbit and rabbit anti-mouse antibodies, used at a 1:2000 dilution, were obtained from Vector Labs (Burlingame, CA). Unless otherwise noted, all primary antibodies were used at 1:1000 dilution in Tris-buffered saline solution containing 3% bovine serum albumin (BSA).
4.3. Creation of a phospho-PLCγ1 polyclonal antibody
To create an anti-peptide antibody specific for the activated form of PLCγ1, a 15 amino acid sequence EGRNPGF(pY)VEANPMP was selected, surrounding tyrosine 783 (shown in bold). This choice was based upon published work demonstrated a strong correlation between phosphorylation at this site and increases in PLCγ1 catalytic activity (Nishibe et al., 1990; Kim et al., 1991), as well as sequence homology comparisons suggesting antibodies to this peptide would not cross-react strongly with PLCγ2, other PLC isoforms, or any other proteins in the public database. The phospho-peptide was coupled to the carrier protein bovine serum albumin (BSA); the BSA-peptide conjugate was then used to immunize rabbits according to a standard immunization protocol. Immune sera from both rabbits were affinity-purified and tested by ELISA assay for their ability to discriminate the phosphorylated peptide from an unphosphorylated version; the sera were also tested by western blot for the ability to detect PLCγ1 in neurotrophin-treated PC12 cells. Affinity-purified antibody from one of the rabbits proved more sensitive in detecting PLCγ1 by western blot, and this preparation was used for further experiments, as described in the Results section. Of the steps described above, peptide selection and western blot testing of the sera and affinity-purified antibodies were performed in our laboratory, while the rest were carried out commercially (Quality Controlled Biochemicals, Hopkinton, MA).
4.4. Immunoprecipitations
For immunoprecipitation studies of PLCγ1 in PC12 cells, equal numbers of PC12 cells were plated in 35 mm culture dishes, grown until 75–85% confluent, and then treated with NGF or HSV-PLCγ1 as described below. Cells were then lysed in 400 μL 1% Triton buffer and 200 μL of cleared supernatant was used for immunoprecipitating with 10 μg/mL of a highly specific mixed-monoclonal PLCγ1 antibody (Upstate Biotechnology, Inc., Lake Placid, NY). After rocking for 1 hr at 4°C, the immunoprecipitates were collected on protein G-agarose beads. The beads were washed 5 times with 1 ml each of the Triton buffer. The immune complexes were then either used for PLC activity assays (described below) or solubilized in 1x sample buffer and subjected to western blotting with the PLCγ1 antibody, as described above.
4.5. Generation and in vivo injection of the HSV-PLCγ1 viral vector
Rat brain PLCγ1 cDNA (generous gift of S. G. Rhee, NIH, Bethesda, MD) was inserted into the HSV-PrPUC amplicon and packaged into highly purified and concentrated (titer 1×108/ml in PBS with 10% sucrose) HSV-PLCγ1 viral preparations as described (Neve et al., 1997), in R. Neve’s laboratory (Harvard, Belmont, MA). Control vectors (HSV-LacZ, HSV-GFP) were also generated using identical procedures.
Intra-VTA injections of HSV-PLCγ1 or HSV-GFP (as a control) were carried out according to published procedures (Bolaños et al., 2003). Injection coordinates were −5.3 anteroposterior, midline, 7.5 mm below dura (Paxinos and Watson, 1997). HSV vectors cause maximal transgene expression 3–4 days after viral injection and are not associated with toxicity greater than that seen with vehicle injections alone (see Carlezon et al., 1997; Bolaños et al., 2003). VTA dissection and immunoblotting analysis were performed as described above.
4.6. Cell culture, viral and pharmacological treatments
PC12 cells were used to examine the consequences of PLCγ1 overexpression on neurotrophic signaling pathways in vitro. Cells were grown at 5% CO2, in RPMI media with 10% fetal calf serum and 1% Pen/Strep. For viral and pharmacological treatments, equal numbers of PC12 cells were plated into 6-well or 24-well dishes, and grown until 75–85% confluent.
4 μL of HSV-PLCγ1 or one of the control viruses (HSV-LacZ or HSV-GFP), infecting roughly 80–90% of the roughly 500,000 cells in each well of the 24 well plate. 4 hr infection times were used; robust overexpression of PLCγ1 was observed by this point, and cells still retained normal morphology (after 6 hrs, at the viral concentrations used, an increasing percentage of cells began to show morphological abnormalities). Pharmacological agents (except PMA) were added to the cells at the time of initial viral infection, so that the length of treatment was also 4 hr. Pharmacological agents used included: MEK inhibitor PD98509 (50 μM), PKC downregulator PMA (overnight treatment, 1 μM), PKC inhibitor calphostin (1 μM), CaM-kinase inhibitor KN93 (1μM), protein kinase A (PKA) inhibitor H89 (5 μM), intracellular calcium releaser thapsigargin (1μM), and intracellular calcium chelator BAPTA-AM (50 μM). All agents were from Calbiochem (San Diego, CA), except for PD98509 (Alexis Corp, San Diego, CA) and BAPTA-AM (Molecular Probes, Eugene, Oregon).
Acute neurotrophin treatments were for 10 min, using 50 ng/mL BDNF or NGF (both kindly provided by R. Lindsay, Regeneron Pharmaceuticals, Tarrytown, NY). NGF was used to activate Trk signaling in PC12 cells, while BDNF was used to activate Trk signaling in primary cultures of embryonic striatal neurons.
Primary cultures of striatal neurons were prepared from embyronic day 18 Sprague-Dawley rats. Dissected striatal tissue was suspended in Neurobasal medium (GibcoBRL) supplemented with 20 mL/L B27 (Gibco BRL), and dissociated with a fire-narrowed Pasteur pipette. Cells were plated in six-well plates at a density of 2×106 cells/well. Treatment with BDNF was performed on in vitro day 4 or 5.
4.7. Phospholipase C activity assay
To assess the catalytic activity of virally-expressed PLCγ1, a standard in vitro PLC activity assay (Wahl et al., 1992) was performed on PLCγ1 purified by immunoprecipitation with a highly specific PLCγ1 antibody from lysates of cells infected with either HSV- PLCγ1 or a control virus (HSV-GFP), as described above. Immunoprecipitates from HSV-PLCγ1 infected cells were resuspended in PLC activity assay buffer containing 200 μM PIP2 (Boehringer-Mannheim), 20 nM tritiated PIP2 (NEN-Dupont), 0.15% n-octyl-B-glucopyranoside, 0.05% Triton X-100, 0.8 mM EGTA, 0.8 mM CaCl2, 35 mM NaH2P04, 70 mM KCl, in a total volume of 50 μL. The reaction was incubated at 35°C for 15 minutes, and stopped by addition of 10% trichloracetic acid (TCA). The samples were centrifuged and supernatants added to scintillation fluid for quantitation of tritiated phospho-inositols, the expected products of PLC catalytic activity. As negative controls, the assay was also performed on a mock-immunoprecipitate done without any PLCγ1 antibody, as well as on a sample of lysis buffer containing no cellular material.
Acknowledgments
This work was supported by grants from the National Institute on Drug Abuse, National Institute of Mental Health, and by the Abraham Ribicoff Research Facilities of the State of Connecticut Department of Mental Health and Addiction Services.
Abbreviations used
- BDNF
brain-derived neurotrophic factor
- DAG
diacylglycerol
- ERK
extracellular signal regulated kinase
- GDNF
glial cell line-derived neurotrophic factor
- GFP
green fluorescent protein
- HSV
herpes simplex virus
- IP3
inositol triphosphate
- IRS
insulin receptor substrate
- MEK
map-erk kinase
- NAc
nucleus accumbens
- NGF
nerve growth factor
- NT-4
neurotrophin-4
- NTF
neurotrophic factor
- PI
phosphatidyl inositol
- PI-3-K
phosphatidyl inositol-3-kinase
- PKC
protein kinase C
- PLC
phospholipase C
- SDS
sodium dodecyl sulfate
- SN
substantia nigra
- VTA
ventral tegmental area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akbarian S, Rios M, Liu RJ, Gold SJ, Fong HF, Zeiler S, Coppola V, Tessarollo L, Jones KR, Nestler EJ, Aghajanian GK, Jaenisch R. Brain-derived neurotrophic factor is essential for opiate-induced plasticity of noradrenergic neurons. J Neurosci. 2002;22:4153–4162. doi: 10.1523/JNEUROSCI.22-10-04153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassa BV, Roh DD, Vaziri ND, Kirschenbaum MA, Kamanna VS. Lysophosphatidylcholine activates mesangial cell PKC and MAP kinase by PLC gamma-1 and tyrosine kinase-Ras pathways. Am J Physiol. 1999;277:F328–F337. doi: 10.1152/ajprenal.1999.277.3.F328. [DOI] [PubMed] [Google Scholar]
- Beitner-Johnson D, Guitart X, Nestler EJ. Neurofilament proteins and the mesolimbic dopamine system: common regulation by chronic morphine and chronic cocaine in the rat ventral tegmental area. J Neurosci. 1992;12:2165–2176. doi: 10.1523/JNEUROSCI.12-06-02165.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitner-Johnson D, Guitart X, Nestler EJ. Glial fibrillary acidic protein and the mesolimbic dopamine system: regulation by chronic morphine and Lewis-Fischer strain differences in the rat ventral tegmental area. J Neurochem. 1993;61:1766–1773. doi: 10.1111/j.1471-4159.1993.tb09814.x. [DOI] [PubMed] [Google Scholar]
- Berhow MT, Russell DS, Terwilliger RZ, Beitner-Johnson D, Self DW, Lindsay RM, Nestler EJ. Influence of neurotrophic factors on morphine- and cocaine- induced biochemical changes in the mesolimbic dopamine system. Neuroscience. 1995;68:969–979. doi: 10.1016/0306-4522(95)00207-y. [DOI] [PubMed] [Google Scholar]
- Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (Extracellular Signal Regulated Kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M. Inositol trisphophate and calcium signaling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Blum S, Dash PK. A cell-permeable phospholipase Cgamma1-binding peptide transduces neurons and impairs long-term spatial memory. Learn Mem. 11:239–243. doi: 10.1101/lm.74104. [DOI] [PubMed] [Google Scholar]
- Boehm J, Kang MG, Johnson RC, Esteban J, Huganir RL, Malinow R. Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron. 2006;51:213–225. doi: 10.1016/j.neuron.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Bolaños CA, Perrotti LI, Edwards S, Eisch AJ, Barrot M, Olson VG, Russell DS, Neve RL, Nestler EJ. Viral-mediated expression of phospholipase Cγ in distinct regions of the ventral tegmental area differentially modulates mood-related behaviors. J Neurosci. 2003;23:7569–7576. doi: 10.1523/JNEUROSCI.23-20-07569.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolaños CA, Nestler EJ. Neurotrophic mechanisms in drug addiction. J Neuromol Med. 2004;5:69–83. doi: 10.1385/NMM:5:1:069. [DOI] [PubMed] [Google Scholar]
- Bolaños CA, Neve RL, Nestler EJ. Phospholipase Cγ in distinct regions of the ventral tegmental area differentially regulates morphine-induced locomotor activity. Synapse. 2005;56:166–169. doi: 10.1002/syn.20136. [DOI] [PubMed] [Google Scholar]
- Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Buckley CT, Caldwell KK. Fear conditioning is associated with altered integration of PLC and ERK signaling in the hippocampus. Pharmacol Biochem Behav. 2004;79:633–640. doi: 10.1016/j.pbb.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Boundy V, Haile C, Lane S, Kalb RG, Neve R, Nestler EJ. Sensitization to morphine induced by viral-mediated gene transfer. Science. 1997;277:812–814. doi: 10.1126/science.277.5327.812. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yu L. Differential regulation by cAMP-dependent protein kinase and protein kinase C of the mu opioid receptor coupling to a G protein-activated K+ channel. J Biol Chem. 1994;269:7839–7842. [PubMed] [Google Scholar]
- Choi JH, Park JB, Bae SS, Yun S, Kim HS, Hong WP, Kim IS, Kim JH, Han MY, Ryu SH, Patterson RL, Snyder SH, Suh PG. Phospholipase C-gamma1 is a guanine nucleotide exchange factor for dynamin-1 and enhances dynamin-1-dependent epidermal growth factor receptor endocytosis. J Cell Sci. 2004;117:3785–3795. doi: 10.1242/jcs.01220. [DOI] [PubMed] [Google Scholar]
- Diakonova M, Chilov D, Arnaoutov A, Alexeyev V, Nikolsky N, Medvedeva N. Intracellular distribution of phospholipase C gamma1 in cell lines with different levels of transformation. Eur J Cell Biol. 1997;73:360–367. [PubMed] [Google Scholar]
- Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS. A role for MAP Kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry. 2007;61:661–670. doi: 10.1016/j.biopsych.2006.05.047. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Mondal AC, Rizavi HS, Shukla PK, Pandey GN. Single and repeated stress-induced modulation of phospholipase C catalytic activity and expression: role in LH behavior. Neuropsychopharmacology. 2005;30:473–483. doi: 10.1038/sj.npp.1300605. [DOI] [PubMed] [Google Scholar]
- Eitan S, Bryant CD, Saliminejad N, Yang YC, Vojdani E, Keith D, Jr, Polakiewicz R, Evans CJ. Brain region-specific mechanisms for acute morphine-induced mitogen-activated protein kinase modulation and distinct patterns of activation during analgesic tolerance and locomotor sensitization. J Neurosci. 2003;23:8360–8369. doi: 10.1523/JNEUROSCI.23-23-08360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman AY, Pierce RC. Neutralization of neutrophin-3 in the ventral tegmental area or nucleus accumbens differentially modulates cocaine-induced behavioral plasticity in rats. Synapse. 2002;46:57–65. doi: 10.1002/syn.10123. [DOI] [PubMed] [Google Scholar]
- Fukuda H, Nishida A, Saito H, Shimizu M, Yamawaki S. Imipramine stimulates phospholipase C activity in rat brain. Neurochem Int. 1994;25:567–571. doi: 10.1016/0197-0186(94)90155-4. [DOI] [PubMed] [Google Scholar]
- Fundytus ME, Coderre TJ. Chronic inhibition of intracellular Ca2+ release or protein kinase C activation significantly reduces the development of morphine dependence. Eur J Pharmacol. 1996;300:173–181. doi: 10.1016/0014-2999(95)00871-3. [DOI] [PubMed] [Google Scholar]
- Gartner A, Polnau DG, Staiger V, Sciarretta C, Minichiello L, Thoenen H, Bonhoeffer T, Korte M. Hippocampal long-term potentiation is supported by presynaptic and postsynaptic tyrosine receptor kinase B-mediated phospholipase Cgamma signaling. J Neurosci. 2006;26:3496–3504. doi: 10.1523/JNEUROSCI.3792-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- Hall FS, Drgonova J, Goeb MK, Uhl GR. Reduced behavioral effects of cocaine in heterozygous brain-derived neurotrophic factor (BDNF) knockout mice. Neuropsychopharmacology. 2003;28:1485–1490. doi: 10.1038/sj.npp.1300192. [DOI] [PubMed] [Google Scholar]
- Harrison JM, Allen RG, Pellegrino MJ, Williams JT, Manzoni OJ. Chronic morphine treatment alters endogenous opioid control of hippocampal mossy fiber synaptic transmission. J Neurophysiol. 2002;87:2464–2470. doi: 10.1152/jn.2002.87.5.2464. [DOI] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR. Enhancement of locomotor activity and conditioned reward to cocaine by Brain-Derived Neurotrophic Factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- Janak PH, Wolf FW, Heberlein U, Pandey SC, Logrip ML, Ron D. BIG news in alcohol addiction: new findings on growth factor pathways BDNF, insulin, and GDNF. Alcohol Clin Exp Res. 2006;30:214–221. doi: 10.1111/j.1530-0277.2006.00026.x. [DOI] [PubMed] [Google Scholar]
- Ji Q, Winnier G, Niswender K, Horstman D, Wisdom R, Magnuson M, Carpenter G. Essential role of the tyrosine kinase substrate phospholipase C-gamma1 in mammalian growth and development. Proc Natl Acad Sci USA. 1997;94:2999–3003. doi: 10.1073/pnas.94.7.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Chavkin C. Mu opioids enhance mossy fiber synaptic transmission indirectly by reducing GABAB receptor activation. Brain Res. 1999;821:286–293. doi: 10.1016/s0006-8993(99)01089-6. [DOI] [PubMed] [Google Scholar]
- Jordan BA, Trapaidze N, Gomes I, Nivarthi R, Devi LA. Oligomerization of opioid receptors with beta 2-adrenergic receptors: a role in trafficking and mitogen-activated protein kinase activation. Proc Natl Acad Sci USA. 2001;98:343–348. doi: 10.1073/pnas.011384898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura M, Ohkuma S, Chen D, Kuriyama K. Ethanol-induced alteration in activities of cerebral phosphatidlyinositol-4,5-bisphosphate-specific and cytosolic phospholipase C in the brain: analysis using NG 108-15 cells and brains from ethanol inhaled mice. Neurochem Int. 1994;24:541–547. doi: 10.1016/0197-0186(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Kim H, Kim J, Zilberstein A, Margolis B, Kim J, Schlessinger J, Rhee SG. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-gamma1 phosphorylation on tyrosine residues 783 and 1254. Cell. 1991;65:435–441. doi: 10.1016/0092-8674(91)90461-7. [DOI] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, Kandel ER. Rescuing impairment of long-term potentiation in Fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci USA. 1997;94:4761–4765. doi: 10.1073/pnas.94.9.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolch W, Heldecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marmé D, Rapp UF. Protein kinase C alpha activates Raf-1 by direct phosphorylation. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- Kombien SB, Malenka RC. Simultaneous LTP of non-NMDA- and LTD of NMDA-receptor-mediated responses in the nucleus accumbens. Nature. 1994;368:242–246. doi: 10.1038/368242a0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- Kozak A, Yavin E. Isolation and characterization by cell density adjustment of a PC12 pheochromocytoma variant with altered Ca2+ homeostasis. J Mol Neurosci. 1992;3:203–212. doi: 10.1007/BF03380140. [DOI] [PubMed] [Google Scholar]
- Lee RH, Wong WL, Chan CH, Chan SY. Differential effects of glial cell line-derived neurotrophic factor and neurturin in RET/GFRalpha1-expressing cells. J Neurosci Res. 2006;83:80–90. doi: 10.1002/jnr.20701. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ryu S, Suh P, Park J, Ahn Y, Kim Y. Tyrosine phosphorylation of PLC-gamma1 induced by electroconvulsive shock in the rat hippocampus. Biochem Biophys Res Comm. 1993;194:665–669. doi: 10.1006/bbrc.1993.1873. [DOI] [PubMed] [Google Scholar]
- Lovlie R, Berle JO, Stordal E, Steen VM. The phospholipase C-gamma1 gene (PLCG1) and lithium-responsive bipolar disorder: re-examination of an intronic dinucleotide repeat polymorphism. Psychiatr Genet. 2001;11:41–43. doi: 10.1097/00041444-200103000-00008. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM. Composition and function of G protein-coupled receptor signalsomes controlling mitogen-activated protein kinase activity. J Mol Neurosci. 2005;26:253–264. doi: 10.1385/JMN:26:2-3:253. [DOI] [PubMed] [Google Scholar]
- Maloney JA, Tsygankova OM, Yang L, Li Q, Szot A, Baysal K, Williamson JR. Activation of ERK by Ca2+ store depletion in rat liver epithelial cells. Am J Physiol. 1999;276:C221–C230. doi: 10.1152/ajpcell.1999.276.1.C221. [DOI] [PubMed] [Google Scholar]
- Mariappan MM, Senthil D, Natarajan KS, Choudhury GG, Kasinath BS. Phospholipase Cgamma-Erk axis in vascular endothelial growth factor-induced eukaryotic initiation factor 4E phosphorylation and protein synthesis in renal epithelial cells. J Biol Chem. 2005;280:28402–28411. doi: 10.1074/jbc.M504861200. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Numakawa T, Yokomaku D, Adachi N, Yamagishi S, Numakawa Y, Kunugi H, Taguchi T. Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: different dependency on signaling pathways and neuronal activity. Mol Cell Neurosci. 2006;31:70–84. doi: 10.1016/j.mcn.2005.09.002. [DOI] [PubMed] [Google Scholar]
- McBride WJ, Murphy JM, Ikemoto S. Localization of brain reinforcement mechanisms: intracranial self-administration and intracranical place-conditioning studies. Behavioral Brain Res. 1999;101:129–152. doi: 10.1016/s0166-4328(99)00022-4. [DOI] [PubMed] [Google Scholar]
- Messer CJ, Eisch A, Carlezon WA, Whisler K, Shen L, Wolf DH, Westphal H, Collins F, Russell DS, Nestler EJ. Role for GDNF in biochemical and behavioral adaptations to drugs of abuse. Neuron. 2000;26:247–257. doi: 10.1016/s0896-6273(00)81154-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Mueller D, Chapman CA, Stewart J. Amphetamine induces dendritic growth in ventral tegmental area dopaminergic neurons in vivo via basic fibroblast growth factor. Neuroscience. 2006;137:727–735. doi: 10.1016/j.neuroscience.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Narita M, Narita M, Mizoguchi H, Tseng LF. Inhibition of protein kinase C, but not of protein kinase A, blocks the development of acute antinociceptive tolerance to an intrathecally administered mu-opioid receptor agonist in the mouse. Eur J Pharmacol. 1995;280:R1–R3. doi: 10.1016/0014-2999(95)00322-c. [DOI] [PubMed] [Google Scholar]
- Narita M, Ohnishi O, Narita M, Aoki T, Suzuki M, Yajima Y, Funahashi H, Shioda S, Suzuki T. Direct evidence for the activation of phospholipase C gamma 1 by in vivo treatment with morphine in the mouse periaqueductal gray matter. Brain Res. 2003;970:140–148. doi: 10.1016/s0006-8993(03)02301-1. [DOI] [PubMed] [Google Scholar]
- Nawa H, Saito M, Nagano T. Neurotrophic factors in brain synaptic plasticity. Crit Rev Neurobiol. 1997;11:91–100. doi: 10.1615/critrevneurobiol.v11.i1.50. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular mechanisms of drug addiction. J Neurosci. 1992;12:2439–2450. doi: 10.1523/JNEUROSCI.12-07-02439.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Berhow MT, Brodkin ES. Molecular mechanisms of drug addiction: adaptations in signal transduction pathways. Mol Psych. 1996;1:190–199. [PubMed] [Google Scholar]
- Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Neve RL, Howe JR, Hong S, Kalb RG. Introduction of the glutamate receptor subunit 1 into motor neurons in vitro and in vivo using a recombinant herpes simplex virus. Neuroscience. 1997;79:435–447. doi: 10.1016/s0306-4522(96)00645-8. [DOI] [PubMed] [Google Scholar]
- Nishibe S, Wahl M, Hernandez-Sotomayor S, Tonks N, Rhee SG, Carpenter G. Increase of the catalytic activity of phospholipase C-gamma1 by tyrosine phosphorylation. Science. 1990;250:1253–1256. doi: 10.1126/science.1700866. [DOI] [PubMed] [Google Scholar]
- Numan S, Lane-Ladd SB, Zhang L, Lundgren KH, Russell DS, Seroogy KB, Nestler EJ. Differential regulation of neurotrophin and trk receptor mRNAs in catecholaminergic nuclei during chronic opiate treatment and withdrawal. J Neurosci. 1998;18:10700–10708. doi: 10.1523/JNEUROSCI.18-24-10700.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 3. Academic Press; San Diego: 1997. [Google Scholar]
- Pierce RC, Bari AA. The role of neurotrophic factors in psychostimulant-induced behavioral and neuronal plasticity. Rev Neurosci. 2001;12:95–110. doi: 10.1515/revneuro.2001.12.2.95. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Kumaresan V. The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev. 2006;30:215–238. doi: 10.1016/j.neubiorev.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Rajagopal R, Chen ZY, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci. 2004;24:6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen K, Beitner-Johnson D, Krystal JH, Aghajanian GK, Nestler EJ. Opiate withdrawal and the rat locus coeruleus: behavioral, electrophysiological, and biochemical correlates. J Neurosci. 1990;10:2308–2317. doi: 10.1523/JNEUROSCI.10-07-02308.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- Russell DS. Neurotrophins: mechanisms of action. The Neuroscientist. 1995;1:3–6. [Google Scholar]
- Russo SJ, Bolaños CA, Theobald DE, DeCarolis NA, Renthal WR, Kumar A, Winstanley CA, Renthal NE, Wiley MD, Self DW, Russell DS, Neve RL, Eisch AJ, Nestler EJ. The IRS2-Akt pathway in midbrain dopaminergic neurons regulates behavioral and cellular responses to opiates. Nature Neurosci. 2007;10:93–99. doi: 10.1038/nn1812. [DOI] [PubMed] [Google Scholar]
- Schulz R, Eisinger DA, Wehmeyer A. Opioid control of MAP kinase cascade. Eur J Pharmacol. 2004;500:487–497. doi: 10.1016/j.ejphar.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Segal RA. Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci. 2003;26:299–330. doi: 10.1146/annurev.neuro.26.041002.131421. [DOI] [PubMed] [Google Scholar]
- Sgambato V, Pages C, Rogard M, Besson M, Caboche J. Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. 1998;18:8814–8825. doi: 10.1523/JNEUROSCI.18-21-08814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- Svoboda KR, Lupica CR. Opioid inhibition of hippocampal interneurons via modulation of potassium and hyperpolarization-activated cation (Ih) currents. J Neurosci. 1998;18:7084–7098. doi: 10.1523/JNEUROSCI.18-18-07084.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Q, Spring SC, Terman BI. Comparison of the signaling mechanisms by which VEGF, H2O2, and phosphatase inhibitors activate endothelial cell ERK1/2 MAP-kinase. Microvasc Res. 2005;69:36–44. doi: 10.1016/j.mvr.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nature Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Herve D, Girault JA. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MI, Jones GA, Nishibe S, Rhee SG, Carpenter G. Growth factor stimulation of phospholipase C-gamma1 activity. J Biol Chem. 1992;267:10447–10456. [PubMed] [Google Scholar]
- Widmer H, Knusel B, Hefti F. Stimulation of phosphatidylinositol hydrolysis by brain-derived neurotrophic factor and neurotrophin-3 in rat cerebral cortical neurons developing in culture. J Neurochem. 1992;59:2113–2124. doi: 10.1111/j.1471-4159.1992.tb10102.x. [DOI] [PubMed] [Google Scholar]
- Wolf DH, Numan S, Nestler EJ, Russell DS. Regulation of phospholipase Cgamma in the mesolimbic dopamine system by chronic morphine administration. J Neurochem. 1999;73:1520–1528. doi: 10.1046/j.1471-4159.1999.0731520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Samorski GM, McLaughlin JP, Romoser VA, Smrcka A, Hinkle PM, Bidlack JM, Gross RA, Jiang H, Wu D. Genetic alteration of phospholipase beta3 expression modulates behavioral and cellular responses to mu opioids. Proc Natl Acad Sci USA. 1999;96:10385–10390. doi: 10.1073/pnas.96.18.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Trible RP, Zhu M, Liu SK, McGlade CJ, Samelson LE. Association of Grb2, Gads, and phospholipase C-gamma 1 with phosphorylated LA tyrosine residues. Effect of LAT tyrosine mutations on T cell angigen receptor-mediated signaling. J Biol Chem. 2000;275:23355–23361. doi: 10.1074/jbc.M000404200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chattopadhyay A, Ji Q, Owen JD, Ruest PJ, Carpenter G, Hanks SK. Focal adhesion kinase promotes phospholipase C-gamma1 activity. Proc Natl Acad Sci USA. 1999;96:9021–9026. doi: 10.1073/pnas.96.16.9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirrgiebel U, Ohga Y, Carter B, Berninger B, Inagaki N, Thoenen H, Lindholm D. Characterization of TrkB receptor-mediated signaling pathways in rat cerebellar granule neurons: involvement of protein kinase C in neuronal survival. J Neurochem. 1995;65:2241–2250. doi: 10.1046/j.1471-4159.1995.65052241.x. [DOI] [PubMed] [Google Scholar]