

Abstract
OpcA from Neisseria meningitidis, the causative agent of meningococcal meningitis and septicemia, is an integral outer membrane protein that facilitates meningococcal adhesion through binding the proteoglycan receptors of susceptible cells. Two structures of OpcA have been determined by x-ray diffraction to 2 Å resolution, revealing dramatically different conformations in the extracellular loops—the protein domain implicated in proteoglycan binding. In the first structure, a positively charged crevice formed by loops 1 and 2 was identified as the site for binding proteoglycans, whereas in the second structure the crevice was not evident as loops 1 and 2 adopted different conformations. To reconcile these results, molecular-dynamics simulations were carried out on both structures embedded in a solvated lipid bilayer membrane. Free of crystal contacts and crystallization agents, the loops were observed to undergo large structural transformations, suggesting that the conformation of the loops in either x-ray structure is affected by crystallization. Subsequent simulations of both structures in their crystal lattices confirmed this conclusion. Based on our molecular-dynamics trajectories, we propose a model for OpcA that combines stable structural features of the available x-ray structures. In this model, all five extracellular loops of OpcA have stable secondary structures. The loops form a funnel that leads to the base of the β-barrel and that includes Tyr-169 on its exposed surface, which has been implicated in proteoglycan binding.
INTRODUCTION
It is hard to conceive of modern cell biology in the absence of x-ray crystallography. The advent of this method brought about atomic-detail structures of biomolecules, allowing the mechanisms of cellular function to be understood at the molecular level. To obtain such structures, many copies of biomolecules are forced to assemble into crystals that produce a diffraction pattern when exposed to x-ray radiation (1). Most biomolecules, however, do not naturally assemble into crystals under physiological conditions. Hence, there is always a possibility that the conformation of a biomolecule resolved by the x-ray diffraction method will differ from that associated with a living cell. Molecular dynamics (MD) (2,3) is a computational method that can animate atomic-scale models of biomolecules (4–10), and thereby adapt the structures resolved in a protein crystal to physiological conditions. In this study, we deploy MD to refine the structure of a membrane protein OpcA, for which two recent x-ray structures revealed dramatically different conformations of its functionally important domain.
OpcA (formerly called Opc) is an integral membrane protein found in the outer membrane of Neisseria meningitidis (11), the causative agent of meningococcal meningitis and septicemia, that facilitates adhesion and subsequent internalization of unencapsulated meningococci by the host cells (12). Cell-surface proteoglycans, such as heparin and heparan sulfate, have been identified as prime receptors of OpcA in epithelial cells (13). OpcA can also bind to the serum glycoprotein vitronectin, leading to meningococcal invasion in endothelial cells (14). The physiological role of OpcA in N. gonorrhoeae, the causative agent of gonorrhea, is not clear (15). OpcA of N. meningitidis is thought to be a functional homolog of the unrelated (by sequence) opacity-associated (Opa) proteins that mediate tight interaction of Neisseria pathogens with human cells and are responsible for an opaque phenotype associated with agar-grown colonies (16).
As with most outer membrane proteins (OMP) (17–19), OpcA has a β-barrel architecture (20). The β-barrel of OpcA has 10 strands with five loops protruding into the extracellular space. The extracellular loops of the protein are thought to harbor a binding site for sialic acid-terminated proteoglycan receptors of the host cells (21), although the recognition mechanism and the specific location of the binding site are unknown (22). The specificity of the proteoglycan binding by OpcA was recently examined using a fluorescence-based binding assay, which demonstrated that sialic acid binding reduced the intrinsic fluorescence of resonance energy transfer from tyrosine to tryptophan residues located at the top of the β-barrel, close to the external loop regions (22). Hence, the conformation of the OpcA loops is thought to be critical to the molecular mechanism of proteoglycan recognition and binding.
Two atomic-resolution structures of OpcA are available currently. The first structure was solved by the x-ray diffraction method using crystals formed by the surfactant-solubilized protein (21,23). In this structure, the extracellular loops 1 and 2 form a crevice, which was claimed to bind the mono- or di-saccharide moieties of proteoglycan receptors (21). However, the second x-ray structure of OpcA (24), obtained using crystals grown by the cubic mesophase technique (25,26), revealed an alternative arrangement of loops 1 and 2, in which the putative binding site was no longer evident. The former and the latter structures are referenced hereafter as the in surfo and in meso structures, respectively.
Both in surfo and in meso protein crystals have layered packing of the protein. In each layer, the OpcA molecules are held together mainly by the contacts between their β-barrels. The contacts between the protein layers are mediated mostly by the extracellular loops, labeled L1–L5. Fig. 1 illustrates the protein contacts in both structures. In the in surfo structure (Fig. 1 a), zinc ions, essential to protein crystallization (23), mediate crystal contacts between the loops. In the in meso structure, which was obtained in the absence of zinc ions, L2 protrudes to the base of the β-barrel of the adjacent molecule (Fig. 1 b). Hence, it is conceivable that in both structures the conformation of the loops is strongly affected by the crystal contacts and that neither structure represents the conformation of OpcA in vivo.
FIGURE 1.

Crystal contacts of the OpcA loops in the in surfo (a) and in the in meso (b) structures. Different copies of the protein are shown in different colors. Individual loops are labeled. Zinc ions in the in surfo model are shown as spheres. The β-barrels of the protein copies shown in orange are aligned.
Below, we deploy the MD method to reconcile the in surfo and in meso structures of OpcA and suggest, to the extent possible, a model for the OpcA conformation in vivo. Each x-ray structure is embedded in a solvated lipid bilayer membrane and equilibrated for ∼20 ns, while the conformation of the loops is closely monitored. For comparison, both OpcA structures are equilibrated in their respective crystal lattices. Comparing the structural dynamics of the four systems reveals the structural elements affected by the crystal contacts and allows a consensus model, incorporating stable elements of both structures, to be suggested. Further, the structural dynamics of the model is investigated by MD, which confirms that the consensus model is stable in a lipid bilayer. The model is characterized by computing the distribution of the electrostatic potential and the osmotic permeability to water. A funnel formed by the loops in the consensus structure is proposed to lead to the site of proteoglycan recognition and binding.
METHODS
Microscopic models of OpcA in a lipid bilayer membrane
We built three models of OpcA in its native environment, a lipid bilayer membrane. For the first model, the atomic coordinates of the protein were taken from the in meso x-ray structure (Protein DataBank access code No. 2J9S). The second model was built using the atomic coordinates of the in surfo structure (Protein DataBank access code No. 1K24). The residues absent in the in meso structure were modeled using the corresponding fragments of the in surfo structure. The first four residues missing in both structures were not modeled. The third model was built using the fragments of the equilibrated in surfo and in meso structures. This model is described in detail under Results. The atomic coordinates of the third model are provided in the Supplementary Material.
In addition to water resolved in the x-ray structures, ∼50 water molecules were placed into the internal cavities of the protein using the Dowser program (27). After that, a 3 Å layer of water was created around the protein using the Solvate program (28). The protein was merged with a patch of a preequilibrated and solvated POPC lipid bilayer comprising 154 lipid molecules. The protein was oriented in the membrane with its β-barrel forming a 6° angle with the z axis, normal to the lipid bilayer plane. The location of the protein in the membrane and its orientation were chosen according to the Orientations of Proteins in Membranes database (29). All lipid molecules that overlapped with the protein were removed, along with all water molecules around the protein that overlapped with the lipid bilayer. Neutral protonation states were assigned to all histidine residues, unless specified otherwise.
The protein-lipid complex was solvated in a rectangular volume of preequilibrated TIP3P (30) water molecules. Corresponding to a solution concentration of 0.12 M Na+ and Cl− ions were added at random positions. The final systems measured ∼76 × 76 × 104 Å3, included over 58,000 atoms, and had a zero total charge. One of the final systems is shown in Fig. 2 a.
FIGURE 2.

Microscopic models of OpcA. (a) The in meso model of OpcA in a lipid bilayer membrane. Parts of the OpcA protein resolved in the in meso structure are shown in orange; the missing residues that were rebuilt using the in surfo structure (1K24) are shown in blue. The water inside the transmembrane part of the protein is shown as van der Waals spheres; bulk water is partially transparent. The POPC molecules making up the bilayer are shown as brown lines. Sodium and chloride ions are shown as yellow and cyan spheres, respectively. (b) The in meso model of OpcA in a crystal lattice. The unit cell is shown as a rectangular box. There are four copies of the OpcA molecule in a unit cell. Individual proteins are colored (orange, red, blue, and green). Octane, water, and sulfate ions resolved in the x-ray structure are shown as van der Waals spheres. Bulk water, filling the remaining volume of the unit cell, is not shown.
To remove possible steric clashes that might have been introduced during the assembly process, each system underwent 3000 steps of minimization using a conjugate-gradients method. After that, the systems were equilibrated at 310 K in the NpT ensemble. In the case of the in surfo model, the protein backbone was restrained during the first 0.5 ns of the equilibration. To ensure that the modeled parts of the in meso structure were properly aligned with the rest of the protein, these systems were simulated for the first 0.5 ns having all but the modeled part of the protein backbone restrained. The rest of the simulations were carried out in the NpT ensemble without deploying any restraints.
Microscopic models of OpcA in a crystal lattice
To construct a microscopic model of the in meso structure in a crystal lattice, four copies of OpcA were produced using the crystallographic symmetry transformations of the P212121 space group. The resulting unit cell, shown in Fig. 2 b, measured 37.9 × 42.5 × 150.4 Å3. In addition to 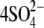 ions, 4 octanes, and 572 water molecules resolved in the x-ray structure, 44 Cl− ions were added to ensure the model's electric neutrality. Bulk water was added to fill the volume of the unit cell, increasing the number of atoms in the system to ∼50,000. The resulting structure was minimized for 3000 steps, followed by equilibration in the NVT ensemble at 295 K—the temperature at which the protein crystals were obtained.
ions, 4 octanes, and 572 water molecules resolved in the x-ray structure, 44 Cl− ions were added to ensure the model's electric neutrality. Bulk water was added to fill the volume of the unit cell, increasing the number of atoms in the system to ∼50,000. The resulting structure was minimized for 3000 steps, followed by equilibration in the NVT ensemble at 295 K—the temperature at which the protein crystals were obtained.
The preparations and simulations of the in surfo structure in a crystal lattice were done following the same procedures as in the case of the in meso model. Four copies of OpcA were produced using the symmetries of the P21212 space group. The resulting unit cell measured 96.9 × 46.3 × 74.0 Å3. Zinc ions, resolved in the in surfo crystal structure, were preserved. The protonation states of the histidines adjacent to the ions were adjusted to minimize local electrostatic energy between the histidines and the ions. Seventy-six chlorine ions were added to reduce the system's total charge to zero.
MD methods
All MD simulations were performed using the program NAMD (31), the CHARMM27 force field (32), periodic boundary conditions, particle-mesh Ewald full electrostatics, and multiple time-stepping (33). The particle-mesh Ewald electrostatics was computed over a 64 × 64 × 84 grid. The temperature was kept constant by applying Langevin forces (34) to all nonhydrogen atoms; the Langevin damping constant was set to 1 ps−1. The integration time-step chosen was 1 fs. The equilibration in the NpT ensemble was performed using Nosé-Hoover Langevin piston pressure control (35) at 1 bar. The van der Waals energies were calculated using a smooth (10–12 Å) cutoff. Restraints were imposed through harmonic forces using the spring constant of 1 kcal/(Å2 × mol).
RESULTS
Conformational dynamics of the in surfo structure
To facilitate the crystallization of OpcA by the in surfo method, zinc ions were introduced into the protein solution (23). As a side effect of this procedure, three zinc ions were embedded in the resulting x-ray structure: two in the protein loops, and one in the β-barrel (Fig. 3 a). The crevice between L1 and L2 in this structure (Fig. 3 b) was proposed to harbor a binding site for proteoglycans (21). To investigate the role of zinc ions and crystal contacts in stabilizing this particular conformation of OpcA, we carried out MD simulations of the in surfo structure in a lipid bilayer membrane and in a protein crystal with and without the zinc ions.
FIGURE 3.

Transformation of the in surfo structure of OpcA simulated in a lipid bilayer membrane. (a and b) The conformation of OpcA in the in surfo x-ray structure. L1 and L2 create a crevice that was proposed to harbor a binding site for proteoglycans (21). Zinc ions (orange), resolved in the x-ray structure, are shown as van der Waals spheres. (c and d) The conformation of the in surfo model after 16-ns equilibration in a lipid bilayer carried out in the absence of the zinc ions. In this simulation, the crevice between L1 and L2 disappeared after ∼8 ns. The protein structure is shown in cartoon (a and c) and molecular surface (b and d) representations. The loops are shown in different colors.
Fig. 4 a illustrates the root mean-square deviation (RMSD) (36) of the OpcA backbone from the x-ray coordinates in a 16-ns simulation carried out in a lipid bilayer having no zinc ions embedded in the protein. The RMSD of the protein backbone saturates at ∼2.5 Å after 8 ns. The RMSD of the β-barrel part saturates at ∼1.2 Å, indicating that the conformation of the β-barrel is stable. The RMSD of the loops is much higher (>4 Å), suggesting instability of the loops' structure.
FIGURE 4.

RMSD of the in surfo structure simulated in a lipid bilayer (gray) and in a protein crystal (black). (a) Time-dependence of the RMSD of the protein backbone from the x-ray coordinates of the β-barrel, extracellular loops, and the entire protein. (b) Per-residue RMSD of the protein backbone from the x-ray coordinates, averaged over the last nanosecond of each simulation. The extracellular loops and periplasmic turns, respectively, are labeled L1–L5 and T1–T4. The shaded region indicates residues missing in the x-ray structure.
The conformation of the in surfo structure at the end of the 16-ns equilibration is shown in Fig. 3, c and d. L2–L4, which were connected by a zinc ion in the x-ray structure, moved apart. L2 moved from a position on top of the center of the barrel toward L1, while L1 moved toward the barrel's center. Meanwhile, residues Glu-67 in L2 and Lys-27 in L1 formed a salt-bridge that brought L1 and L2 even closer. Hence, by the end of the simulation, the crevice formed by L1 and L2 in the x-ray structure disappeared (Fig. 3 d). As L4 was moving away from L2 and L5, the α-helical part of L4 elongated by two residues; the L4 helix tilted away from the barrel (Fig. 3 c). In this simulation, L3 and L5 maintained their secondary structures, although the location and the tilt of L5 changed considerably. Our observations of L2 mobility qualitatively agree with the results of a recent computational study carried out using a united-atom force field (37). Although in that study the OpcA protein was embedded in a different type of lipid bilayer membrane (DMPC), the results demonstrated that, in the absence of zinc, L2 drifts away from the barrel center, forming a pathway for sialic acid among L2–L5. The same study, however, reported unfolding of a short helix in L4, which was not observed in our simulations.
The transformation of the OpcA structure is dissected in the per-residue RMSD plot (Fig. 4 b), which was computed against the x-ray coordinates for the last nanosecond of the equilibration. The per-residue RMSD is high for both extracellular loops (L1–L5) and periplasmic turns (T1–T4), indicating that these parts of the protein are mobile. On average, the extracellular loops of OpcA are more flexible than the periplasmic turns because the latter are shorter. The RMSD values for the shortest loop (L3) are as small as those for the turns. For other loops, high RMSD values (>5 Å) are consistent with observed changes of the loops' conformation.
For comparison, we carried out MD simulations of the in surfo structure in a crystal lattice, preserving zinc ions at their crystallographic positions. As shown in Fig. 4 a, the RMSD (averaged over four copies) of the protein backbone from the x-ray coordinates saturates quickly after ∼6 ns. The RMSD of the protein backbone is smaller in a crystal lattice than in a lipid bilayer membrane (Fig. 4 a). The RMSD of the barrel reaches approximately the same value as in a lipid bilayer. However, the averaged RMSD (Fig. 4 a) of the loops is ∼1.3 Å smaller in the crystal environment, indicating that the loops are less mobile. The RMSD of the averaged (over the four copies of the protein in the unit cell) structure attains similar values. Such RMSD plots are available in the Supplementary Material.
Individual mobility of the residues in the crystal lattice is characterized by the per-residue RMSD plot (Fig. 4 b). For the majority of the loop residues, the RMSD values are smaller in the crystal lattice simulation, particularly for residues in contact with the zinc ions, which are identified along the x axis in Fig. 4 b.
So far, we have demonstrated that removing zinc ions and placing the in surfo structure in a lipid bilayer destabilizes the conformation of the OpcA loops. The observed instability of loop conformation can be explained by the electrostatic unbalance introduced upon the removal of the zinc ions. Fig. 5 illustrates the zinc binding sites in the loops of the in surfo structure. The first binding site (Fig. 5 a) is formed by residues Asp-69 (L2), His-128 (L3), and Thr-176 (L4) from the same OpcA copy and Glu-223 (L5) from an adjacent copy. In a lipid bilayer, removing this zinc ion leads to a sudden increase in the local electrostatic energy because of the high density of negative charge. The residues coordinating this zinc binding site repel each other. Consequently, L2 and L4 move in opposite directions, as shown in Fig. 3 c. When the in surfo structure was simulated having His-128 in a fully protonated state (total charge +1e), L2 and L4 did not separate within 10 ns. In the second binding site (Fig. 5 b), a zinc ion connects Glu-28 and His-30 of two adjacent copies, stabilizing the conformation of L1 resolved in the protein crystal. Consequently, when simulated in a lipid bilayer and without zinc, L1 can move toward the barrel.
FIGURE 5.

Stabilization of the crystal contacts by zinc ions in the in surfo structure of OpcA. (a) Zinc binding site between L2, L3, and L4 of the same protein and L5 of the adjacent copy. In each copy, L2–L5 are shown in cyan, orange, green, and blue, respectively. The rest of the protein is shown in yellow. (b) Zinc binding site between L1s of neighboring copies of OpcA. The conformation of L1 in the in meso structure (white) is shown for comparison. Snapshots of the OpcA structure were obtained from a 14-ns equilibration performed in a crystal lattice. The residues coordinating the zinc ions are shown in atomic bond representation and colored according to the atom names. Zinc ions are shown as van der Waals spheres.
We have also simulated the in surfo model in a crystal lattice without including the zinc ions. L2 was observed to move in the same direction as in the simulation carried out in a lipid bilayer. Interestingly, without zinc ions, the crystal contacts between L1 of the adjacent protein copies were stabilized by two salt-bridges formed by Glu-28 from one copy and Lys-29 from the other. In this simulation, we used a different protonation state of His-128 than in the simulations that included zinc ions. The following two cases were considered: a proton was located at the nitrogen atom nearest to the position of the zinc ion in the x-ray structure, and His-128 was fully protonated (total charge +1e). Such protonations of His-128 reduced the electrostatic repulsion between L3 and the other two loops, L2 and L4. Nevertheless, the conformation of the residues near the (empty) binding site was unstable and the loops moved apart, which is consistent with the fact that the in surfo structure could not be crystallized in the absence of zinc ions.
The in surfo structure was also simulated in a lipid bilayer having all three zinc ions embedded in the structure (24). In that case, Asp-69 in L2 was strongly attracted to a nearby zinc ion, while L1, bound to another zinc ion, moved further away from the barrel. The latter can be explained by the fact that the surface of the crevice formed by L1 and L2 is positively charged in the vicinity of residues Lys-60, Lys-61, and Lys-80, which belong to L2. Hence, having a zinc ion bound to L1, the electrostatic force between L1 and L2 is repulsive. Therefore, in the absence of the crystal contact stabilizing L1, the crevice becomes unstable and widens.
In the last system, the protonation state of (neutral) His-128 was found to be important for the stability of the zinc binding site formed by L2–L4 (Fig. 3 a). Placing a proton on the nitrogen nearest to the zinc ions (CHARMM topology entry HSE) was observed to disrupt the zinc binding pocket. However, when the proton was placed on the nitrogen furthest from the zinc ion (CHARMM topology entry HSD), the structure of this zinc binding site was stable within the simulation timescale (5 ns). Changing the protonation state of His-128 did not affect widening of the crevice between L1 and L2.
These MD simulations indicate that the conformations of L1 and L2, and, to a smaller extent, of L4 and L5 in the in surfo structure are affected by the crystal contacts and by the zinc ions. The crevice between L1 and L2 is not stable when the crystal contacts are removed. Neither the presence of zinc in a lipid bilayer membrane simulation, nor the crystal contacts in the absence of zinc in the crystal lattice simulation, could stabilize the structure resolved by the x-ray diffraction. As zinc ions are not required for physiological function of OpcA, significance of the conformation of the loops resolved in the in surfo structure is not clear.
Conformational dynamics of the in meso structure
The in meso x-ray structure was determined from membrane protein crystals that had a record high fraction (57%) of the unit cell volume occupied by the protein (24). Hence, it is conceivable that the in meso conformation of OpcA is also influenced by its crystal environment. For example, the tip of L2 is stretched away from the body of the protein by almost one diameter of the β-barrel (Fig. 1 b). To identify the parts of the structure affected by protein contacts, we performed a 22-ns MD simulation of the protein in a lipid bilayer membrane, and an 8-ns MD simulation in a protein crystal.
Fig. 6 a shows the average RMSD of the protein backbone from the x-ray coordinates for the simulation carried out in a lipid bilayer. After 5 ns, the RMSD saturates at ∼4 Å. As this value is much larger than the length of the atomic bonds in the protein backbone, the plot suggests large transformations of the OpcA structure. The parts of the structure that underwent structural transformations can be discerned from the per-residue RMSD plot (Fig. 6 c). The RMSD of residues in L2 and L5 are considerably larger than those in the other loops. Thus, for example, the RMSD of the L2 tip exceeds 15 Å. To check whether the structure had reached a new metastable state, the averaged RMSD of the protein backbone was computed against the state obtained 8 ns after the beginning of the simulation. As shown in Fig. 6 a, the RMSD value saturates at ∼2 Å for the rest of the simulation. The per-residue RMSDs relative to the 8-ns state are also much smaller (Fig. 6 c). These plots indicate that the in meso structure, when placed in a lipid bilayer environment, evolved into a new, relatively stable conformation.
FIGURE 6.

RMSD of the in meso structure simulated in a lipid bilayer membrane and in a crystal lattice. (a) RMSD of the protein backbone in a lipid bilayer from the x-ray coordinates (solid), and from the state obtained after the first 8 ns of the equilibration (shaded). (b) RMSD of the protein backbone in a crystal lattice from the x-ray coordinates during an 8-ns equilibration. The RMSD values shown were averaged over the four copies of the protein (see also Fig. 2 b). (c) Per-residue RMSD of the protein backbone in a lipid bilayer from the x-ray coordinates (dashed line), and from the state obtained after the first 8 ns of the equilibration (shaded). Per-residue RMSD of the protein backbone in a crystal lattice from the x-ray coordinates (solid). All per-residue RMSD plots were obtained by averaging over the last nanosecond of the MD trajectory. The extracellular loops are labeled L1–L5 whereas the inner turns are labeled T1–T4. The shaded regions indicate residues missing in the in meso x-ray structure.
The details of the structural transformation are shown in Fig. 7. Starting from the x-ray structure (Fig. 7, a and b), L2 was observed to move toward the center of the β-barrel, while L5 moved closer to L2. The upper cord of L2 (Lys-75 to Lys-80) rotated by ∼45° toward the extracellular side (Fig. 7, a and c), while the lower cord (Leu-66 to Thr-68) rotated just a little (<15°). After the motion of the cords, the helix in L2 (Asp-69 to Gly-74) rotated by ∼90°. When viewed from the extracellular side, the helix also rotated by 45° counterclockwise about the barrel axis, along with the upper cord (Fig. 7, b and d). In the final state, the loops are much closer to each other than in the in meso x-ray structure. We also carried out a short (5 ns) simulation of this model without rebuilding any missing residues and observed similar loop motion.
FIGURE 7.

Transformation of the in meso structure of OpcA simulated in a lipid bilayer membrane. (a and b) Side and top views of the protein at the beginning of the MD simulation. (c and d) Side and top views of the protein after a 22-ns equilibration. (a and b) Direction of the structural rearrangement is shown schematically by the curved arrows. In this simulation, the protein reached a stable conformation after 8 ns. For the remaining 14 ns, the protein conformation did not change significantly (see also Fig. 6).
Compared to the in surfo model, the RMSD values of L1 in the in meso model are considerably smaller, because the unstructured part of this loop is much shorter. The RMSD of L4 and L5 are greater in the in meso structure, likely because several residues of these loops were not resolved in the in meso structure, and were rebuilt using the in surfo fragments (see Fig. 2 a). Although L3 was also rebuilt, its residues attain smaller RMSD values in the in meso structure. In contrast, the β-barrel residues in the in meso structure have RMSD values as small as 1 Å, which indicates that the conformation of the β-barrel is, most likely, very similar to that in a lipid bilayer membrane.
The results of the simulation carried out in a protein crystal are summarized in Fig. 6, b and c. After ∼4 ns, the RMSD from the x-ray coordinates of the loops, the β-barrel, and the entire protein reach a constant value (Fig. 6 b). All RMSD values shown were averaged over the four copies of the protein (see Fig. 2 b). The RMSD values of the β-barrel residues are ∼0.9 Å, on average. The RMSD values of the loops are approximately twice as large, but still are much smaller than the values obtained from the simulation in a lipid bilayer membrane (Fig. 6 b). The RMSD of the entire protein backbone saturates at ∼1.3 Å, which is also much smaller than in a lipid bilayer membrane (Fig. 6 a).
Another outer membrane protein, OmpA, was simulated in both the DMPC lipid bilayer (38) and the crystal environment (39) using a united-atom force field. In that study, the RMSD of the barrel was also observed to be much smaller than that of the loops. It seems that the rigid barrel and flexible loops are common features of OMPs. When simulated in the crystal lattice, the average RMSD of the OmpA α-carbons reached ∼3.5 Å. Our calculations suggest that the in meso structure of OpcA is much less mobile in the crystal environment than OmpA, most likely because of the dense packing of OpcA in the in meso protein crystal (17).
In Fig. 6 c, per-residue RMSDs are plotted for the simulations carried out in a lipid bilayer membrane and in a protein crystal. These RMSD values were obtained by averaging over the last nanoseconds of the respective MD trajectories against the in meso x-ray structure. Due to the crystal contacts, per-residue RMSD values of the in meso model are considerably smaller in a protein crystal than in a lipid bilayer membrane, particularly for the extracellular loops and the periplasmic turns. Residues missing in the original structure, but that were rebuilt using the in surfo fragments, show large RMSD values in both simulations. L2 and L5 were not observed to move considerably in the crystal environment, as they did when the crystal contacts were removed. The per-residue RMSD values of these loops (Fig. 6 c) are consistent with this observation.
These simulations demonstrate that, although the conformation of L2 in the in meso structure is strongly affected by crystal contacts, when the crystal contacts are removed, L2 adopts a new, stable conformation; the α-helix in L2 does not unfold. The conformation of L1 does not appear to be affected by the crystal contacts in the in meso structure.
The x-ray structures converge in a lipid bilayer membrane
Given enough time, one can expect that the two x-ray structures of OpcA simulated in a lipid bilayer might converge to the same conformation. At present, the range of MD is limited to tens of nanoseconds, which usually is not sufficiently long to observe spontaneous folding of a protein, or large conformational transitions, but is long enough to observe instability of the secondary structure. In our case, however, spontaneous transformations in the OpcA structures were dramatic enough to demonstrate the approach to convergence of the two simulations.
Fig. 8 shows the conformations of the in meso and in surfo structures before (Fig. 8 a; x-ray structures) and after (Fig. 8 b) equilibration in a lipid bilayer. The two structures were aligned using the coordinates of their β-barrels. Before the equilibration, L2 is located on top of the β-barrel in the in surfo structure, while, in the in meso structure, the loop is stretched away from the barrel. After several nanoseconds of MD simulation, L2 in the in surfo structure drifts toward the edge of the barrel, while L2 in the in meso structure moves toward the barrel. At some point, the conformation of L2 from the two different structures overlaps, as shown in Fig. 8 b.
FIGURE 8.

Convergence of the in meso (blue) and in surfo (yellow) structures of OpcA simulated in a lipid bilayer membrane. (a) The original x-ray structures of both models. (b) The structures of both models obtained after equilibration in a lipid bilayer. (c) RMSD of the protein backbone between any two frames of the in surfo and in meso trajectories.
Fig. 8 c shows the RMSD of the protein backbone between every two frames of the in meso and in surfo trajectories. The initial RMSD of the two x-ray structures is ∼6 Å (at the origin of the plot). The RMSD decreases with time for each trajectory. The minimum RMSD value between the trajectories is ∼4 Å.
Despite the convergence, the conformation of L1 relative to L2 in both structures is still quite different. In the in meso model, L1 and L2 form a β-sheet; L1 is short and ordered. L1 in the in surfo crystal is less ordered and forms a crystal contact with another L1 of an adjacent protein, as shown in Fig. 5 b. During equilibration in a lipid bilayer, L1 of the in surfo structure moves toward L2, which is consistent with the conformation of L1 in the in meso model. However, for L1 and L2 to form a β-sheet in the in surfo structure, the side chains of Glu-28 and His-30 in L1 should flip by 180° to face the interior of the β-barrel (Fig. 5 b). Within the timescale of MD simulation, such motion was not observed. As the conformation of Glu-28 and His-30 in the in surfo model is strongly affected by the crystal environment (Fig. 5 b), we conclude that the ordered β-sheet structure of L1 in the in meso model, stabilized by hydrogen bonds with the neighboring strands of the β-barrel, is likely to be close to the structure of L1 in a lipid bilayer.
L3–L5 in both simulations have similar conformations and secondary structures, partly because the residues missing in the in meso structure were modeled according to the in surfo structure. The conformation of L3 is the same in both models. The short helix in L4 was not stable in the in meso structure, as that loop was built using fragments of the in surfo structures. For the same reason, L5 in the equilibrated in meso structure has a shorter β-sheet than in the in surfo structure.
Most probable conformation of OpcA in a lipid bilayer
Our MD simulations of the in surfo and in meso x-ray structures have revealed the influence of the crystal contacts and the crystallization agents on the resolved conformations of the protein. Although the global conformations of the loops were observed to converge to a common structure in the simulations carried out in a lipid bilayer (Fig. 8), the local conformation of L1, L2, L4, and L5 in the in surfo and in meso structures remained different. Using the MD trajectories of both structures in a lipid bilayer, we attempted to reconstruct the conformation of OpcA in a lipid bilayer membrane. The resulting consensus structure is shown in Fig. 9. The atomic coordinates of the consensus structure are provided in the Supplementary Material.
FIGURE 9.

The consensus model of OpcA derived from MD simulations of the in meso and in surfo structures. The upper portion of the protein that projects above the membrane is shown in cartoon representation (orange). Tyr-169 and the lipid bilayer are shown as atomic bonds and as van der Waals spheres, respectively, colored according to the atom names.
Like other OMPs (9,20), OpcA has a rigid β-barrel. When simulated in a lipid bilayer membrane, the RMSD of the β-barrel residues from the crystal structure is only ∼1 Å, which is in accordance with the small B-factor values measured experimentally (21,24). The resolved β-barrel parts of the in meso and in surfo structures have very similar conformations. Due to our choice of the L1 structure (see below), the β-barrel part of the consensus structure was modeled according to the in meso structure. As the dynamics of the periplasmic turns are very similar in both equilibrated crystal structures, the structure of the turns was taken from the in meso model as well.
L1 in the in surfo model extends away from the barrel and forms a crystal contact with the L1 of an adjacent protein via a zinc ion (Fig. 5 b). Our simulations of the in surfo model, limited by the timescale of several tens of nanoseconds, did not reveal a better conformation. However, L1 in the in meso structure has an ordered β-sheet structure that is stable in a lipid bilayer. Hence, for the consensus model, the structure of L1 was taken from the equilibrated in meso structure.
The conformation of L2 in both x-ray structures is affected by the crystal environment. During the equilibration in a lipid bilayer, L2 of both structures were observed to drift toward the edge of the β-barrel (Fig. 8 b). The α-helix formed by Asp-69 to Gly-74 in the in meso structure, is also present but is of lesser extent (Asp-69 to Lys-72) in the in surfo structure. This helix was stable during the 22-ns equilibration of the in meso structure, but not during the 16-ns equilibration of the in surfo structure, most likely because, in the latter, the stabilizing zinc ion was removed. During the equilibration of the in meso structure, the helix in L2 was stabilized, in addition to hydrogen bonds within the helix, by two salt-bridges (Glu-70–Lys-75 and Glu-67–Lys-72) formed by the charged residues of the helix (Glu-70 and Lys-72) and the charged residues of the upper (Lys-75) and lower (Glu-67) cords. An additional 15-ns simulation of this helix alone in a water box confirmed the stability of the helix. Therefore, for the consensus model, the conformation of L2 was taken from the equilibrated in meso structure.
Because not all residues of L3–L5 were resolved in the in meso structure, in the consensus model, these loops were built according to the equilibrated in surfo structure (Fig. 3 c). Consequently, in our model, L3 is a short cord, L4 has both α-helical (from Leu-172 to Leu-178) and β-sheet (from Asp-181 to Lys-185) fragments, and L5 has a β-sheet (from Ser-221 to Ile-237) connected to the β-barrel by two short cords. Note that the structures of L3–L5 in the consensus model are different from those of the in meso model; in the latter, residues missing from L3–L5 were rebuilt using the fragment of the in surfo x-ray structure, but in the consensus model, entire loops were built using the fragments of the equilibrated in surfo structure.
Characterization of the consensus model
To test the structural stability of the new model, it was embedded in a lipid bilayer membrane and equilibrated for ∼20 ns following the same protocols as in the case of the in surfo and in meso structures. A movie illustrating the simulated trajectory is available in the Supplementary Material. During this simulation, the location of L1–L4 relative to the β-barrel did not change, while L5 was observed to move sporadically between the edge and the center of the β-barrel. This motion is illustrated in Fig. 10. When L5 is located near the barrel's edge (snapshots at 7 and 16 ns), the loops form a funnel wide enough for sialic acid and other saccharides to enter. When L5 is located above the barrel's center (snapshot at 12 ns), L3 bends toward L5, and the passage is closed.
FIGURE 10.

Snapshots of the consensus model of OpcA simulated in a lipid bilayer membrane and viewed from the extracellular space. The starting conformation of this simulation is shown in Fig. 9. The funnel formed by the loops can be either open (7 and 16 ns) or closed (12 ns), mainly depending on the conformation of L5. The protein structure is shown in cartoon (top) and molecular surface (bottom) representations.
The secondary structure of OpcA during a 9-ns fragment of the MD trajectory is shown in Fig. 11 a. This fragment covers the time interval from 7 to 16 ns shown in Fig. 10, in which the sporadic back-and-forth motion of L5 was observed. In the rest of the trajectory, the conformation of L5 remains similar to the initial one (Fig. 9). The secondary structure analysis program STRIDE (40) identified T1–T4 and the short loops L1 and L3 as turns. The secondary structure plot demonstrates that the α-helices in L2 and L4 are stable, as indeed are the β-sheets in L4 and L5.
FIGURE 11.

Secondary structure and RMSF of the consensus model of OpcA simulated in a lipid bilayer. (a) The secondary structure of OpcA during the 9-ns segment of the MD trajectory. The plot covers the time interval from 7 to 16 ns referred to in Fig. 10. The y axis of the plot indicates the OpcA residue by number, while the x axis shows the simulation time. The following secondary structure elements are identified by color: β-sheet (yellow), cord (white), turn (cyan), and α-helix (pink). The secondary structure analysis was performed using STRIDE (40). (b) RMSF of the protein α-carbon atoms during the ∼9-ns of the simulation. Loops are labeled L1–L5.
Although the loops in the consensus model maintain stable secondary structures, they are still more mobile than the β-barrel itself. The root mean-square fluctuation (RMSF) (36) of the α-carbon atoms of the protein computed for the same 9-ns segment of the simulation trajectory are shown in Fig. 11 b. The RMSF plot peaks at L5, which is consistent with the motion depicted in Fig. 10. In the starting conformation, L5 of the consensus model is weakly connected to the other loops, while the same conformation of L5 in the crystal is stabilized by crystal contacts with neighboring copies of OpcA (Fig. 1). Therefore, despite the stable secondary structure, L5 is still very flexible and can move back and forth between the edge and the center of the β-barrel. The simulated B-factor computed from the RMSF values are compared with the crystallographic B-factors in the Supplementary Material.
If the OpcA adhesin binds negatively charged moieties of proteoglycans, as previously suggested (21,24), the distribution of the electrostatic potential, especially in the loop region of OpcA, must have important implications for the binding mechanism. By analyzing the MD trajectory resulting from the simulation of the consensus model in a lipid bilayer membrane, we determined the average distribution of the electrostatic potential for the loop part of the protein in the open conformation. These calculations were performed using a 7-ns fragment of the trajectory in which the open conformation was stable. The resulting electrostatic potential is displayed in Fig. 12. The loops in the open conformation form a funnel. The electrostatic potential at the surface of the funnel is mostly positively biased. The surface of the funnel includes Tyr-169 that was implicated in sialic acid-binding changes in the fluorescence of the protein (Fig. 12 (22,24)). The entrance to the funnel is decorated with positively charged residues Lys-77 and Lys-80 (L2), Lys-127 (L3), and Lys-229 (L5). The only negatively charged residue at the entrance of the funnel is Asp-232 (L5). Hence, it is conceivable that this particular distribution of the electrostatic potential could facilitate the entrance of the negatively charged moieties into the funnel and onto the binding site.
FIGURE 12.

The distribution of the electrostatic potential in the loop region of the consensus model of OpcA. An open conformation of OpcA is shown in cartoon (left) and molecular surface (right) representations, colored according to the values of the electrostatic potential. The distribution of the electrostatic potential was obtained by averaging instantaneous snapshots of the electrostatic potential over a 7-ns MD trajectory. Tyr-169 is shown in the atomic bond representation. The view is from the extracellular medium.
To characterize the permeability of OpcA to small solutes, the osmotic permeability to water was computed for all three models simulated in a lipid bilayer membrane. The method used to compute the osmotic permeability is described elsewhere (41); the details of the calculations are presented in the Supplementary Material. The osmotic permeability to water of OpcA was determined to be 1.4 × 10−14 cm3/s, which is two orders-of-magnitude less than that of the α-hemolysin channel (42), and five times less than that of aquaporin (43). The permeability to water varied very little with different loop conformations in all trajectories, as the barrier to water permeation is located inside the β-barrel (see Supplementary Material for more details).
CONCLUSION
MD simulations of OpcA were carried out in a lipid bilayer membrane and in a crystal lattice to reconcile the two x-ray structures obtained using different protein crystallization methods (21,24). The simulations revealed that the x-ray structures of OpcA incorporate conformations in the extracellular loop region that arise due to crystal contacts (both structures) and the protein crystallization agents (zinc ions, in surfo structure only). When placed in a lipid bilayer membrane, both structures were observed to evolve toward a common conformation, although the loops did not converge on the exact same structure likely due to the timescale limitation of the MD method. The simulations clearly demonstrated that, in the in meso structure, L2, which starts out, swung out and away from the protein drifts toward the center of the β-barrel when the crystal contacts are removed (Fig. 7). In the case of the in surfo structure, removing the crystal contacts leads to closure of the crevice between L1 and L2 (Fig. 3), which calls the proposed molecular mechanism of proteoglycan binding (21) into question.
The structural transformations observed in our MD simulations were used to construct a consensus model of OpcA (Fig. 9) that, we believe, better represents the conformation of OpcA in vivo than either x-ray structure. Having the advantage of comparing structural dynamics of the two x-ray models in a lipid bilayer membrane and in a crystal lattice, we could choose for the new model structural parts least affected by the crystal contacts. Subsequent MD simulations proved that the model proposed is stable in a lipid bilayer membrane. The conformation of the loops in the new model reveals a pathway toward a binding site, presumably located at the confluence of the loops and the barrel (22,24). Thus, the loops form a funnel wide enough for sialic acid and other saccharides to access the base of the loops and to pass Tyr-169, whose fluorescence is affected by sialic acid binding (22).
Our results do not rule out the possibility that further rearrangements of the protein structure can occur upon changing the external conditions, or binding a polysaccharide ligand. Thus, as the motion of the OpcA loops is largely driven by the electrostatic interactions of its charged residues, it is possible that the loops will adopt a different conformation at a higher salt concentration, which was also suggested in Bond et al. (37). Another factor that was not considered in our study is the presence of lipopolysaccharides in the outer leaflet of the bacterial membrane, which could affect the conformational dynamics of the protein. Our simulations of OpcA in the crystal lattices did not include the crystallographically observed detergent/salt mixture, which could be important to accurately reproduce structural dynamics of a protein crystal having low protein density (39). Nevertheless, this work clearly demonstrates that MD, although limited by the nanosecond timescale, can be successfully deployed to adopt x-ray structures of membrane proteins affected by the “tyranny of the lattice” to physiological conditions.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This work is supported by grants from the National Institutes of Health (grant No. PHS 5 P41 RR05969) and the startup funds provided by the Department of Physics at the University of Illinois at Urbana-Champaign. Authors gladly acknowledge supercomputer time provided by the Pittsburgh Supercomputer Center and the National Center for Supercomputing Applications via Large Resources Allocation Committee grant No. MCA05S028. Grant support for M.C. was provided by Science Foundation Ireland (No. 02-IN1-B266), the National Institute of Health (Nos. GM61070 and GM75915), and the National Science Foundation (No. IIS-0308078).
Editor: Peter Tieleman.
References
- 1.Drenth, J. 2006. Principles of Protein X-Ray Crystallography, 3rd Ed. Springer Verlag, Berlin.
- 2.Allen, M. P., and D. J. Tildesley. 1987. Computer Simulation of Liquids. Oxford University Press, New York.
- 3.Adcock, S. A., and J. A. McCammon. 2006. Molecular dynamics: survey of models for simulating the activity of proteins. Chem. Rev. 106:1589–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gumbart, J., Y. Wang, A. Aksimentiev, E. Tajkhorshid, and K. Schulten. 2005. Molecular dynamics simulations of proteins in lipid bilayers. Curr. Opin. Struct. Biol. 15:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarek, M. 2005. Membrane electroporation: a molecular dynamics simulation. Biophys. J. 88:4045–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernèche, S., and B. Roux. 2001. Energetics of ion conduction through the K+ channel. Nature. 414:73–77. [DOI] [PubMed] [Google Scholar]
- 7.Anishkin, A., and S. Sukharev. 2004. Water dynamics and dewetting transitions in the small mechanosensitive channel MscS. Biophys. J. 86:2883–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garofoli, S., and P. Jordan. 2003. Modeling permeation energetics in the KcsA potassium channel. Biophys. J. 84:2814–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bond, P. J., and M. S. P. Sansom. 2004. Simulation approach to bacterial outer membrane proteins. Mol. Microbiol. 21:151–161. [DOI] [PubMed] [Google Scholar]
- 10.Tieleman, D. P. 2006. Computer simulations of transport through membranes: passive diffusion, pores, channels and transporters. Clin. Exp. Pharmacol. Physiol. 33:893–903. [DOI] [PubMed] [Google Scholar]
- 11.Zhu, P., M. J. Klutch, J. P. Derrick, S. M. Prince, R. S. W. Tsang, and C. Tsai. 2003. Identification of OpcA gene in Neisseria polysaccharea, interspecies diversity of Opc protein family. Gene. 307:31–40. [DOI] [PubMed] [Google Scholar]
- 12.Nassif, X., C. Pujol, P. Morand, and E. Eugene. 1999. Interactions of pathogenic Neisseria with host cells. Is it possible to assemble the puzzle? Mol. Microbiol. 32:1124–1132. [DOI] [PubMed] [Google Scholar]
- 13.de Vries, F. P., R. Cole, J. Dankert, M. Frosch, and J. P. M. van Putten. 1998. Neisseria meningitidis producing the Opc adhesin binds epithelial cell proteoglycan receptors. Mol. Microbiol. 27:1203–1212. [DOI] [PubMed] [Google Scholar]
- 14.Virji, M., K. Makepeace, and E. R. Moxon. 1994. Distinct mechanisms of interactions of Opc-expressing meningococci at apical and basolateral surfaces of human endothelial cells; the role of integrins in apical interactions. Mol. Microbiol. 14:173–184. [DOI] [PubMed] [Google Scholar]
- 15.Zhu, P., G. Morelli, and M. Achtman. 1999. The OpcA and ΨOpcB regions in Neisseria: genes, pseudogenes, deletions, insertion elements and DNA islands. Mol. Microbiol. 33:635–650. [DOI] [PubMed] [Google Scholar]
- 16.Hauck, C. R., and T. M. Meyer. 2003. “Small” talk: Opa proteins as mediators of Neisseria-host-cell communication. Curr. Opin. Microbiol. 6:43–49. [DOI] [PubMed] [Google Scholar]
- 17.Cherezov, V., E. Yamashita, W. Liu, M. Zhalnina, W. A. Cramer, and M. Caffrey. 2006. In meso structure of the cobalamin transporter, BtuB, at 1.95 Å resolution. J. Mol. Biol. 364:716–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pautsch, A., and G. E. Schulz. 2000. High-resolution structure of the OmpA membrane domain. J. Mol. Biol. 298:273–282. [DOI] [PubMed] [Google Scholar]
- 19.Ferguson, A. D., W. Welte, E. Hofmann, B. Lindner, O. Holst, J. W. Coulton, and K. Diederichs. 2000. A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure. 8:585–592. [DOI] [PubMed] [Google Scholar]
- 20.Koebnik, R., K. Locher, and P. V. Gelder. 2000. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol. Microbiol. 37:239–253. [DOI] [PubMed] [Google Scholar]
- 21.Prince, S. M., M. Achtman, and J. P. Derrick. 2002. Crystal structure of the OpcA integral membrane adhesin from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA. 99:3417–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore, J., S. E. S. Bailey, Z. Benmechernene, C. Tzitzilonis, N. J. E. Griffiths, M. Virji, and J. P. Derrick. 2005. Recognition of saccharides by the OpcA, OpaD, and OpaB outer membrane proteins from Neisseria meningitidis. J. Biol. Chem. 280:31489–31497. [DOI] [PubMed] [Google Scholar]
- 23.Prince, S. M., C. Feron, D. Janssens, Y. Lobet, M. Achtman, B. Kusecek, P. A. Bullough, and J. P. Derrick. 2001. Expression, refolding and crystallization of the OpcA invasin from Neisseria meningitidis. Acta Crystallogr. 57:1164–1166. [DOI] [PubMed] [Google Scholar]
- 24.Cherezov, V., W. Liu, J. Derrick, B. Luan, A. Aksimentiev, V. Katritch, and M. Caffrey. In meso crystal structure and docking simulations suggest an alternative proteoglycan binding site in the OpcA outer membrane adhesin. Proteins: Struct., Funct., Bioinf. In press. [DOI] [PubMed]
- 25.Landau, E. M., and J. P. Rosenbusch. 1996. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA. 93:14532–14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caffrey, M. 2003. Membrane protein crystallization. J. Struct. Biol. 142:108–132. [DOI] [PubMed] [Google Scholar]
- 27.Zhang, L., and J. Hermans. 1996. Hydrophilicity of cavities in proteins. Proteins Struct. Funct. Gen. 24:433–438. [DOI] [PubMed] [Google Scholar]
- 28.Grubmüller, H., B. Heymann, and P. Tavan. 1996. Ligand binding and molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- 29.Lomize, M. A., A. L. Lomize, and H. I. M. I. D. Pogozheva. 2006. OPM: orientations of proteins in membranes database. Bioinformatics. 22:623–625. [DOI] [PubMed] [Google Scholar]
- 30.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 31.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacKerell, A. D., Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 33.Batcho, P. F., D. A. Case, and T. Schlick. 2001. Optimized particle-mesh Ewald/multiple-time step integration for molecular dynamics simulations. J. Chem. Phys. 115:4003–4018. [Google Scholar]
- 34.Brünger, A. T. 1992. X-PLOR, Version 3.1: A System for X-Ray Crystallography and NMR. The Howard Hughes Medical Institute and Department of Molecular Biophysics and Biochemistry, Yale University.
- 35.Martyna, G. J., D. J. Tobias, and M. L. Klein. 1994. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101:4177–4189. [Google Scholar]
- 36.Faraldo-Gómez, J. D., and B. Roux. 2004. Electrostatics of ion stabilization in a ClC chloride channel homologue from Escherichia coli. J. Mol. Biol. 339:981–1000. [DOI] [PubMed] [Google Scholar]
- 37.Bond, P. J., J. P. Derrick, and M. S. P. Sansom. 2007. Membrane simulations of OpcA: gating in the loops? Biophys. J. 92:L23–L25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bond, P. J., and M. S. P. Sansom. 2003. Membrane protein dynamics versus environment: simulations of OmpA in a micelle and in a bilayer. J. Mol. Biol. 329:1035–1053. [DOI] [PubMed] [Google Scholar]
- 39.Bond, P. J., J. D. Faraldo-Gómez, S. S. Deol, and M. S. P. Sansom. 2006. Membrane protein dynamics and detergent interactions within a crystal: a simulation study of OmpA. Proc. Natl. Acad. Sci. USA. 103:9518–9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frishman, D., and P. Argos. 1995. Knowledge-based secondary structure assignment. Proteins Struct. Funct. Gen. 23:566–579. [DOI] [PubMed] [Google Scholar]
- 41.Zhu, F., E. Tajkhorshid, and K. Schulten. 2004. Theory and simulation of water permeation in aquaporin-1. Biophys. J. 86:50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aksimentiev, A., and K. Schulten. 2005. Imaging α-hemolysin with molecular dynamics: ionic conductance, osmotic permeability and the electrostatic potential map. Biophys. J. 88:3745–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu, F., E. Tajkhorshid, and K. Schulten. 2002. Pressure-induced water transport in membrane channels studied by molecular dynamics. Biophys. J. 83:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.